Introduction
Bone and soft tissue sarcomas (STSs) are malignant
mesenchymal neoplasms. They represent 1% of all malignant diseases
(1). The majority of sarcomas
arise from soft tissue (75%), followed by gastrointestinal stromal
tumors (GISTs; ~15%) and bone sarcomas (10%) (2). STSs are classified into 120
histological groups. Moreover, a recent World Health Organization
classification described an even greater number of molecular
subsets, comprising of a heterogeneous group of tumors with rare
and ultrarare subcategories of STSs (3). The most common histological types are
liposarcoma and leiomyosarcoma, with an incidence of less than 1
case in 100,000 individuals/year, which highlights the rarity of
sarcomas (4).
Bone sarcomas have distinct patterns of incidence,
with no more than 0.3 cases in 100,000 individuals/year for each
bone sarcoma subtype (5).
Osteosarcoma (OST) and Ewing sarcoma (ES) are most common in young
individuals, with a high incidence rate in individuals <20 years
old, whereas chondrosarcoma (CS) is more common in older adults
(6). Extremities and trunk areas
are the most common locations for the majority of bone sarcomas and
approximately half of STSs. However, both bone and STSs can also
develop in the head and neck, retroperitoneum, gastrointestinal
tract and genitourinary tract (7,8).
A multidisciplinary approach is strongly recommended
for sarcoma treatment due to the rarity and heterogeneity of bone
sarcomas and STSs. Complete surgical resection remains the
treatment of choice in regimens with curative intent. Cytotoxic
chemotherapy is the backbone of the systemic treatment approach,
but the discovery of various molecular signaling pathways
implicated in sarcomagenesis has paved the way for targeted
therapeutics (7–10). The targeting of tyrosine kinases is
currently studied in clinical trials but this approach has also
been used in clinical practice.
Tyrosine kinases are important molecules that
cross-talk and regulate the activity of several intracellular
signaling pathways. They are divided into two subtypes: i) receptor
tyrosine kinases (RTKs); and ii) non-RTKs (NRTKs). RTKs are
transmembrane glycoproteins that regulate proliferation, survival,
migration, apoptosis and cell adhesion upon ligand binding. NRTKs
act downstream of several signaling molecules and are located
either in the cytoplasm or the nucleus. Tyrosine kinase receptors
that have been considered as potential therapeutic targets in
sarcomas include VEGFR, platelet-derived growth factor receptor
(PDGFR), insulin-like growth factor receptor, cellular (c)-receptor
tyrosine kinase KIT (KIT), fibroblast growth factor receptor
(FGFR), mesenchymal epithelial transition (MET) and AXL receptor
tyrosine kinase (AXL). Tyrosine kinase inhibitors (TKIs) are small
molecules that inhibit these receptor tyrosine kinases (11,12).
The aim of the present systematic review was to
discuss the role of TKIs in the treatment of patients with locally
advanced, unresectable, or metastatic STSs and bone sarcomas.
Methods
This present review was conducted and reported in
accordance with the Preferred Reporting Items for Systematic
Reviews and Meta-Analyses (PRISMA) statement and the common
practices in the field. Eligible articles were identified by
performing a search of the Medical Literature Analysis and
Retrieval System Online bibliographical database for the period
between the 1 January 2000 and 30 August 2021. The search strategy
included the following keywords: STS (neoplasm or cancer or
sarcoma), bone sarcoma (neoplasm or cancer or sarcoma) and TKI
(chemotherapy or systemic therapy or management). Only articles in
English were included in the present review. Reviews, expert
opinions and prospective and retrospective studies were also
included, whereas case reports were excluded from the analysis.
Moreover, manuscripts that did not state the name of the authors
were excluded. Additional articles were identified from the
reference lists of the retrieved articles.
Results of the literature meta-analysis
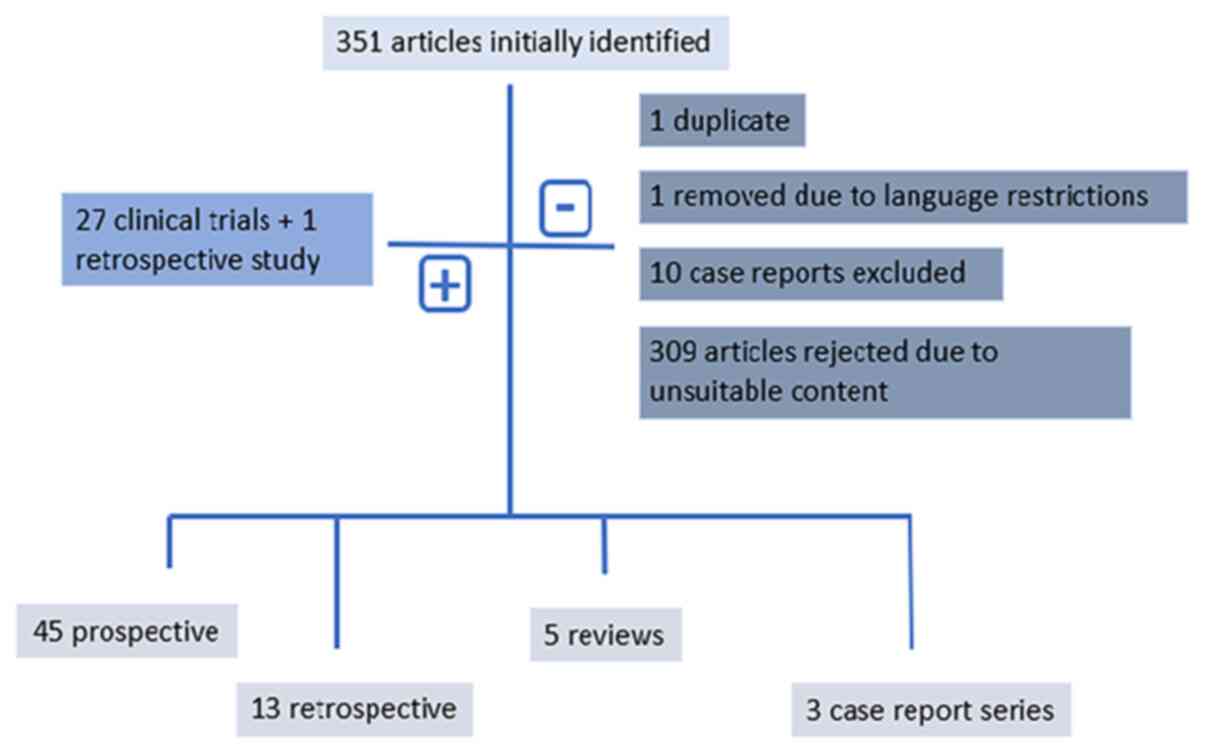
The initial literature search resulted in the
identification of 351 articles, from which one duplicate was
removed. Moreover, one article was excluded due to language
restrictions and 10 case reports were excluded; however, three case
report series were included. Furthermore, 309 additional articles,
which focused on sarcoma diagnosis, prognosis and biology, and
articles presenting surgical approaches and primary or adjuvant
sarcoma treatment or preclinical assays, or those referring to
GISTs, were considered to be outside the scope of the present
review and were therefore also excluded. Overall, 28 studies (27
prospective clinical trials-two phase I, one phase Ib/II and 24
phase II studies, of which five were randomized trials and one was
a retrospective study) were recovered from the reference lists of
rejected articles. Overall, 58 studies were considered eligible for
inclusion in the final analysis. The research strategy is presented
in Fig. 1.
The results identified 45 prospective clinical
trials published between 2000 and 2021, among which six were phase
I trials, two were phase Ib/II trials, 36 were phase II trials and
one was a phase III trial. All studies were single arm, with the
exception of seven previous, randomized, placebo-controlled trials
(13–20). Moreover, 13 retrospective studies
were analyzed (21–33). Furthermore, five reviews, one
systematic literature review (34), one expert review (35) and three reviews of clinical trials
(36–38) were identified.
Pazopanib
Pazopanib is a small molecule TKI. It primarily
targets VEGFR-1/2/3, PDGFR-α/β and KIT (13).
The use of pazopanib in STSs was approved based on
the results of the double-blinded, placebo-controlled, randomized,
pazopanib explored in STS (PALETTE) phase III trial. This clinical
trial demonstrated a significant improvement in the
progression-free survival (PFS) of patients with non-adipocytic STS
who were treated with pazopanib. The median PFS was 4.6 months for
patients treated with pazopanib compared with 1.6 months for
patients treated with the placebo (P<0.0001). However, no
significant difference was observed in overall survival (OS; 12.5
vs. 10.7 months for the pazopanib and placebo arms, respectively)
(14). Patients with liposarcoma
were excluded from the PALETTE study based on the previous results
of the European Organization for Research and Treatment of Cancer
(EORTC) 62043 phase II study, in which patients in the liposarcoma
cohort demonstrated a discouraging progression-free point at 12
weeks (39). However, preclinical
investigations and prospective clinical trials focusing on
dedifferentiated liposarcoma have reported that pazopanib has
potential antitumor activity (40). In a phase II study of pazopanib for
adipocytic sarcomas, a 68.3% PFS rate (PFSR) at 12 weeks and a
median PFS of 4.4 months were demonstrated; well-differentiated
liposarcomas were excluded from this study (41).
In a retrospective analysis based on two EORTC
clinical trials, pazopanib was studied in uterine and nonuterine
sarcomas. Pazopanib activity in patients with uterine sarcomas was
similar to that in patients with nonuterine STSs. The PFS was 3.0
months (95% CI, 2.5-4.7 months) in uterine vs. 4.5 months (95% CI,
3.7-5.1 months) in nonuterine STS. Furthermore, the median OS was
17.5 months in the uterine STS population (95% CI, 11.1-19.6
months) vs. 11.1 months (95% CI, 10.2-12.0 months) (P=0.352) in the
nonuterine STS population (21).
The activity of pazopanib in angiosarcoma (AS) is
also comparable to its reported activity in other STS subtypes, as
noted in a retrospective analysis (22). The median PFS and median OS were 3
(95% CI, 2.1-4.4 months) and 9.9 months (95% CI, 6.5-11.3 months),
respectively, in AS. In this previous study, the activity of
pazopanib was similar in cutaneous/non-cutaneous and
radiation/non-radiation-associated AS. Furthermore, another study
demonstrated that pazopanib exhibited promising activity in
epithelioid hemangioendothelioma (EH) and intimal sarcoma (IS), two
rare sarcomas with limited treatment options. The response rates
were 8/40 (20%), 2/10 (20%) and 2/2 (100%) patients in the AS, EH
and IS subtype groups, respectively.
The SPIRE study examined pazopanib for compassionate
use in heavily pretreated patients with advanced STS. The treatment
duration was influenced by histological subtype, with certain
patients exhibiting long responses. Long responses were reported
for perivascular epithelioid cell tumors (8.2 months), aggressive
fibromatosis (AF; 8.0 months), alveolar soft part sarcoma (ASPS;
7.1 months), desmoplastic small round cell tumors (5.7 months) and
synovial sarcoma (5.1 months). Histological subtypes with the
highest percentage of clinical benefit [complete response (CR) +
partial response (PR) + stable disease (SD)] at any time, included
nonuterine leiomyosarcoma (45%; 18/40 patients), uterine
leiomyosarcoma (43%; 17/40 patients), synovial sarcoma (54%; 13/24
patients), undifferentiated sarcoma (42%; 8/19 patients), AS (38%;
6/16 patients) and solitary fibrous tumors (54%; 7/13 patients)
(23).
Pazopanib also benefits patients with surgically
unresectable or metastatic CS. The disease control rate (DCR) was
achieved in 43% of patients following 16 weeks of pazopanib
treatment (95% CI, 28-58%) and the median OS was 17.6 months (95%
CI, 11.3-35.0 months). Even though only 43% of patients reached the
DCR, the results of this previous study are clinically meaningful
as they offer the option of pazopanib in a traditionally
chemotherapy-resistant disease (36,42).
Several case reports have also demonstrated PRs in
patients with ES, with acquired resistance being developed with
prolonged use (37). A single
reference center case series report demonstrated that 2/4 patients
with unresectable or metastatic chordoma, who were treated with
pazopanib, derived clinical benefits and SD was achieved for 14 and
15 months (43).
The Pediatric Preclinical Testing Program evaluation
of pazopanib revealed a statistically prolonged event-free survival
(EFS) in ES xenografts but no objective responses were exhibited
(44). Currently, in advanced OST,
only case reports have been published with pazopanib (45).
Coadministration of TKIs with other targeted
therapeutics and cytotoxic agents has also been investigated in
retrospective studies. The safety and efficacy of pazopanib has
been analyzed in combination with vorinostat, everolimus, lapatinib
or trastuzumab. and MEK inhibitors in patients with advanced
sarcoma. Pazopanib administration in combination with other agents
is safe; however, pazopanib combinations do not reverse resistance.
In a previous case series report, the inhibition of VEGFR with
everolimus initially resulted in SD for the majority of patients
with advanced sarcomas. The addition of everolimus resulted in a
clinical benefit at 4 months for 3/6 patients (24). Moreover, a study on the addition of
sirolimus in patients with metastatic high-grade STS, who
progressed after previous clinical benefit on pazopanib, suggested
that the combination of sirolimus and pazopanib may serve as a
potential treatment to reverse resistance and extend the
chemotherapy-free window (25).
However, the small sample size of these aforementioned studies is a
limitation that influences the statistical significance of these
observations. Therefore, prospective studies are needed support the
conclusions of these aforementioned studies. Furthermore, the
combination of pazopanib with chemotherapy is associated with
greater toxicity (26). Table I summarizes pazopanib clinical
trials.
 | Table I.Pazopanib. |
Table I.
Pazopanib.
| Study type | Phase | Patient number | Subtype | PFS (months) | OS (months) | Outcomes | (Refs.) |
|---|
| Randomized | III | 372 | Non-adipocytic
STS | 4.6 | 12.5 | Approved as 2nd
line treatment | (14) |
| Prospective | II | 41 | Liposarcoma | 4.4 | 12.6 | Promising
activity | (41) |
| Retrospective |
| 44 | Uterine
sarcomas | 3 | 17.5 | Promising
activity | (21) |
| Retrospective |
| 42 | Vascular
sarcomas | 3 | 9.9 | Promising
activity | (22) |
| Retrospective |
| 211 | Advanced STS | 3 | 11.1 | Activity in
compassionate use setting | (23) |
| Prospective | II | 47 | Chondrosarcoma | 7.9 | 17.6 | Negative | (42) |
| Retrospective |
| 9 | Advanced
sarcoma | 3.1 | - | Disease stability
with the combo of pazopanib + everolimus | (24) |
| Retrospective |
| 8 | Advanced STS | 5.5 | - | Promising
activity | (25) |
| Retrospective |
| 44 | Advanced
sarcoma | 2.4 | 9 | Negative | (26) |
Apatinib
Apatinib is a small-molecule receptor TKI with
potential antiangiogenic and antineoplastic activities. It
selectively targets VEGFR2 and therefore inhibits endothelial cell
migration and proliferation and further influences tumor
microvascular density (46).
In a single-arm, phase II trial, apatinib exhibited
encouraging objective efficacy and manageable toxicity in patients
with metastatic STS who were previously unable to receive
chemotherapy. At 12 weeks, the PFSR, objective response rate (ORR)
and DCR were 70, 26.32 and 86.84%, respectively. The median PFS was
7.87 months and the median OS was 17.55 months. Apatinib was
reported to be well tolerated, with the main adverse effects
including, hypertension, palmar plantar erythrodysesthesia,
anorexia, proteinuria, pain, fatigue and diarrhea, which were
adequately controlled following symptomatic treatment or dose
reduction to 375 mg and subsequently 250 mg (47). In an observational study, patients
with leiomyosarcoma treated with apatinib did not show significant
survival benefits compared with patients with other histological
subtypes. The median PFS was 3.79 months (95% CI, 0.96-7.24 months)
in patients with leiomyosarcoma vs. 4.35 months (95% CI, 2.22-5.58
months) in patients with other histological subtypes (P=0.3170).
The median OS was 8.17 months (95% CI, 1.56 months-not estimated)
months in patients with leiomyosarcoma vs. 11.22 months (95% CI,
6.64-18.72 months) in patients with other histological subtypes
(P=0.9219) (27).
The results from a prospective phase II trial
demonstrated that the administration of apatinib in patients with
unresectable, locally advanced or metastatic OST, which was
progressing upon prior treatment with chemotherapy agents, resulted
in tumor shrinkage of at least 30% in 16/37 patients (43.24%).
However, this result only had a short response duration, as the
median duration response was 5.07 months (95% CI, 2.70-6.53 months)
(48). In a recently published
retrospective study, a regime of a combination of apatinib with
ifosfamide and etoposide exhibited clinically meaningful antitumor
activity in patients with recurrent or refractory OST, as the 4-
and 6-month EFS rates were 90.9 (95% CI, 74.4-97.0%) and 78.5% (95%
CI, 60.0-89.1%), respectively. However, the adverse effects of the
combination were severe and the majority of grade 3 and 4
toxicities included myelosuppression, bronchial infection,
pneumothorax, anorexia and posterior leukoencephalopathy syndrome,
which resulted in dose reductions (28).
In an off-label study of apatinib in patients with
advanced, previously treated bone and STS, toxicity was more severe
than results reported in clinical trials. The ORR (CR + PR) was
40.9% (9/22 patients) for OST, 70% (7/10 patients) for ES, 100%
(3/3 patients) for CS and 71.4% (15/21 patients) for STS. Moreover,
the median duration of response was 8.8 months (95% CI, 4.3-11.5
months) for malignant peripheral nerve sheath tumors and 5.6 months
(95% CI, 1.3-9.8 months) for undifferentiated pleomorphic sarcoma
(UPS) (29).
In preclinical studies, apatinib reduces programmed
death-ligand 1 (PD-L1) expression in OST cells, which suggested
that it may act as an immunotherapy modulator in patients with
sarcoma (49,50). An open-label, phase II trial
studying the combination of apatinib with camrelizumab (an
anti-programmed cell death 1 antibody) revealed prolongation of PFS
compared with apatinib only in treating advanced OST (51). However, this previous clinical
trial did not reach the prespecified target of a 6-month PFS of
60%. This study also suggested that patients with high PD-L1
expression levels and pulmonary metastases exhibited a longer PFS
than those with bone lesions (P=0.017). Table II summarizes the clinical trials
that have studied apatinib.
| Table II.Apatinib. |
Table II.
Apatinib.
| Study type | Phase | Patient number | Subtype | PFS (months) | OS months) | Outcomes | (Refs.) |
|---|
| Prospective | II | 42 | Advanced STS | 7.87 | 17.55 | Promising
activity | (47) |
| Retrospective | II | 31 | Advanced STS | 4.25 | 9.43 | Promising
activity | (27) |
| Prospective | II | 37 | Advanced OS | 4.5 | 9.87 | Moderate
activity | (48) |
| Retrospective |
| 79 | Advanced OS | 12.6 | 19.8 | High toxicity | (28) |
| Prospective | II | 43 | Advanced OS | 6.2 | - | Negative | (51) |
Sunitinib
Sunitinib malate is a multitargeted TKI with
activity against VEGFR1/2/3, PDGFR-α/β, KIT, Fms-like tyrosine
kinase 3 (FLT3), RET and colony stimulating factor 1 (CSF1)
(52). Sunitinib also has both
antiangiogenic and antitumor activities.
George et al (53) reported a multicenter, single-arm,
phase II study of sunitinib in metastatic or locally advanced
non-GIST STS, in which 53 patients were enrolled and 48 of them
were eligible for response assessment. An imaging assessment
demonstrated a median PFS of 1.8 months, with 11/48 patients (22%)
exhibiting SD at 12 weeks and seven patients (14%) maintaining SD
after 24 weeks of treatment (53).
In another phase II study, 48 patients with documented unresectable
or metastatic STS (liposarcoma, leiomyosarcoma and UPS), in which
other therapeutic approaches had failed, were treated with
sunitinib malate. The median PFS and OS for liposarcoma,
leiomyosarcoma and malignant fibrous histiocytoma were 3.9 and
18.6, 4.2 and 10.1 and 2.5 and 13.6 months, respectively. The
safety profile did not reveal any new toxicities, with the most
common adverse effects being fatigue/asthenia and other
gastrointestinal complaints at grade 1 or 2 (54).
A further small, nonrandomized, open-label,
prospective, phase II trial of sunitinib was performed by Jo et
al (55), in which 19 patients
with advanced AF, which was not amenable to surgery, were treated
with 37.5 mg sunitinib once daily. Following treatment, five
patients (26.3%) achieved a PR and eight (42.1%) presented with SD.
With a median follow-up time of 20.3 months (range, 1.8-50.7
months), the 2-year rates for PFS and OS were 74.7 and 94.4%,
respectively. However, 3/12 patients in this trial with mesenteric
AF experienced serious adverse effects, including mesenteric mass
bleeding (n=1), bowel perforation (n=1) and bowel fistula (n=1),
which were likely to be related to tumor necrosis. Therefore,
sunitinib may be useful for the management of non-mesenteric AF
(55). Sunitinib has also been
tested in patients with metastatic ASPS in a small case series. In
nine patients with progressive/advanced ASPS, treated with
sunitinib, five patients (55%) had a PR, based on the Response
Evaluation Criteria in Solid Tumors (RECIST), and an additional
three patients (33%) exhibited SD (56). When sunitinib was given to 31
patients with progressive advanced solitary fibrous tumors, of
which 25 patients were pretreated with conventional
chemotherapeutic regimens, disease control was achieved in 18/31
patients (58%) with a median PFS of 6 months (30). In a retrospective case series of 10
patients with extra-skeletal myxoid CS treated with sunitinib, 6/10
patients (60%) had a PR, which was determined using RECIST, two
patients presented with SD (20%) and two patients exhibited disease
progression (20%) (31). The
single-arm, nonrandomized design of these studies limited any
definitive conclusions regarding the efficacy of sunitinib in STS.
However, the activity of sunitinib in specific subtypes is very
promising despite the often-indolent nature of these tumors, such
as AF.
Sunitinib has previously been co-administered with
nivolumab in patients with refractory, advanced sarcomas. The
ImmunoSarc trial evaluated the efficacy of sunitinib in combination
with nivolumab as assessed by PFSR at 6 months. The 6-month PFSR
was 48% according to the central assessment, whereas the median PFS
was 5.6 months (3–8.1 months). One CR was registered in a patient
with AS and a PR was described in patients with ASPS, AS, synovial
sarcoma and extra-skeletal myxoid CS (57). The bone sarcoma group demonstrated
similar results, with a 6-month PFSR of 32% and a modified (m)PFS
of 3.7 months (95% CI, 3.4–4.0 months). Moreover, the median OS was
14.2 months (95% CI, 7.1–21.3 months), inducing durable disease
control in 55% of patients and a PR in one patient with OST
(58). Table III summarizes the trials that
have studied sunitinib.
| Table III.Sunitinib. |
Table III.
Sunitinib.
| Study type | Phase | Patient number | Subtype | PFS (months) | OS | Outcomes | (Refs.) |
|---|
| Prospective | II | 53 | Advanced STS | 1.8 | - | Activity in DC
(SD) | (53) |
| Prospective | II | 48 | Liposarcoma, | 3.9 (LS) | 18.6 (LS) | Moderate
activity | (54) |
|
|
|
|
leiomyosarcoma, | 4.4 (LMS) | 10.1 (LMS) |
|
|
|
|
|
| malignant
fibrous | 2.5 (MFH) | 13.6 (MFH) |
|
|
|
|
|
| histiocytoma |
|
|
|
|
| Prospective | II | 19 | Advanced aggressive
fibromatosis | 74.7% (median
follow-up time of 20.3 months) | 94.4% (median
follow-up time of 20.3 months) | Promising
activity | (55) |
| Retrospective |
| 35 | Advanced solitary
fibrous tumor | 6 | 16 | Promising activity
with a long-lasting response | (30) |
| Retrospective |
| 10 | Extraskeletal
myxoid chondrosarcoma | Not reached | - | Statistically
non-significant | (31) |
| Prospective | Ib/II | 68 | Advanced STS | 5.6 | 24 | Promising
activity | (57) |
| Prospective | II | 40 | Advanced OS | 3.7 | 14.2 | Promising
activity | (58) |
Regorafenib
Regorafenib is an oral multikinase inhibitor that
targets VEGFR1/2/3, FGFR1, PDGFR-α/β, CSF1 receptor and c-KIT
(59).
A randomized placebo-controlled phase II trial,
regorafenib in metastatic STS (REGOSARC), was performed for
non-GIST STSs in which the response and survival benefit of
regorafenib were evaluated in four cohorts (leiomyosarcoma,
synovial sarcoma, liposarcoma and other histologies). Three
cohorts, excluding the patients with liposarcoma, exhibited PFS
prolongation compared with the placebo arm. PFS was 1.1 months with
regorafenib (95% CI, 0.9–2.3 months) vs. 1.7 months (0.9–1.8) with
the placebo [hazard ratio (HR), 0.89; 95% CI, 0.48-1.64 months]
(P=0.70) in the liposarcoma cohort. Furthermore, in the
leiomyosarcoma cohort PFS was 3.7 months (95% CI, 2.5-5.0 months)
with regorafenib vs. 1.8 months (95% CI, 1.0-2.8 months) with the
placebo (HR, 0.46; 95% CI, 0.46-0.80 months) (P=0.0045). In the
synovial sarcoma cohort, PFS was 5.6 months (95% CI, 1.4-11.6
months) with regorafenib vs. 1.0 months (95% CI, 0.8-1.4 months)
with the placebo (HR, 0.10; 95% CI, 0.03-0.35 months)
(P<0.0001). Finally, in the other histologies sarcoma cohort,
PFS was reported to be 2.9 months (95% CI, 1.0-7.8) with
regorafenib vs. 1.0 months (95% CI, 0.9-1.9) with the placebo (HR,
0.46; 95% CI, 0.25-0.81 months) (P=0.0061) (15). The REGOSARC trial also demonstrated
the benefits of quality-adjusted survival (60). The survival benefits of regorafenib
in the REGOSARC trial were similar to those of pazopanib. The
median PFS of the regorafenib-treated patients with non-liposarcoma
was limited to 4 months and the OS was limited to 13.4 months.
Moreover, regorafenib treatment for liposarcoma, similar to
pazopanib, failed to result in PFS prolongation (38). The most common clinically
significant grade 3 or higher adverse events exhibited included
arterial hypertension (19%), hand and foot skin reactions (15%) and
asthenia (13%).
The results of a prospective, open-label,
single-arm, nonrandomized phase II trial verified that regorafenib
is active in patients with non-adipocytic pretreated advanced STS
(leiomyosarcoma, synovial sarcoma and vascular sarcoma), as 13/21
(62%) patients were progression-free at 8 weeks (61). Therefore, regorafenib proved to
have a clinically meaningful antitumor effect in non-adipocytic
STSs by improving PFS.
The randomized, double-blind, placebo-controlled
regorafenib in patients with metastatic bone sarcomas (REGOBONE)
and SARC024 clinical trials, in relapsed progressive metastatic
OST, demonstrated the benefit of regorafenib in patients with bone
sarcoma. In the REGOBONE trial, 38 patients with advanced bone
sarcoma were randomized (2:1) to receive either regorafenib or the
placebo. This study demonstrated that 17/26 patients (65%;
one-sided 95% CI, 47%) in the regorafenib group were nonprogressive
at 8 weeks compared with no patients in the placebo group,
resulting in a median PFS of 4.1 months (95% CI, 8.0-27.3 months)
vs. 1.0 month (95% CI, 3.0-5.7 months), respectively (16). Similarly, in the North American
trial, among the 22 patients treated with regorafenib, a median PFS
of 3.6 months was achieved vs. 1.7 months in the placebo group (HR,
0.42; 95% CI, 0.21-0.85 months; P=0.017). However, regarding OS,
there was no statistically significant difference (11.1 vs. 13.4
months for regorafenib and placebo, respectively; P=0.62) (17).
SARC024 also assessed the efficacy of regorafenib in
30 patients with advanced ES, in which it was noted that the median
PFS was 3.6 months (95% CI, 2.8-3.8 months) and the median duration
of response was 5.5 months (95% CI, 2.9-8.0 months). This study met
its primary endpoint and the toxicity of the drug was similar to
that seen previously; no grade 4 adverse effects were noted
(18,37). In the same study, the OST cohort
included 42 patients and regorafenib resulted in a mPFS of 3.6 (95%
CI, 2-7.6 months) vs. 1.7 months (95% CI, 1.2-1.8 months) for the
placebo. There was no benefit to OS. Table IV summarizes the trials that have
studied regorafenib.
| Table IV.Regorafenib. |
Table IV.
Regorafenib.
| Study type | Phase | Patient number | Subtype | PFS (months) | OS (months) | Outcomes | (Refs.) |
|---|
| Randomizee | II | 182 | Liposarcoma,
leiomyosarcoma, synovial sarcoma, other histologies | 1.1 (LS) 3.7 (LMS)
5.6 (SVS) 2.9 (other) | - | Promising activity
except from LS | (15) |
| Prospective | II | 21 | Advanced STS | 3.8 | 14.8 | Clinical
activity | (61) |
| Randomized | II | 38 | Advanced OS | 4.1 | - | Clinical
activity | (16) |
| Randomized | II | 42 | Advanced OS | 3.6 | - | Clinical
activity | (17) |
| Randomized | II | 30 | Ewing sarcoma | 3.6 | - | Clinical
activity | (18) |
Sorafenib
Sorafenib targets Raf, VEGFR2/3 and PDGFR-β, and
therefore inhibits tumor cell proliferation and angiogenesis
(37).
In a prospective multicenter open-label
nonrandomized phase II trial, 101 patients with advanced STS,
pretreated with anthracycline-based chemotherapy, received
sorafenib (400 mg) twice daily for 28 days. Even though the primary
endpoint of the PFSR at 6 months was not reached by the entire
population, patients with leiomyosarcoma achieved a 6-month PFSR of
38.4%, which confirmed the activity of sorafenib in this subset of
patients (62). In the French
Sarcoma Group study, which assessed the response to sorafenib in 41
patients with advanced AS, the primary end point was PFS at 9
months, which was assessed by RECIST. There were no responses
reported in the chemotherapy naïve group, but there was a 40% tumor
control rate and a 23% response rate in pretreated patients
(63). Similar results were seen
in another phase II trial of 51 patients with advanced STS, in
which five patients with vascular sarcoma (63%), eight patients
with leiomyosarcoma (42%) and two patients with dedifferentiated
liposarcoma (20%) had SD, resulting in a median PFS of 5 months for
patients with vascular sarcoma compared with 2-3 months for the
patients with liposarcoma and leiomyosarcoma (64).
Regarding bone sarcomas, sorafenib has also
demonstrated activity in patients with OST. The Italian Sarcoma
Group designed a single-arm phase II study of sorafenib as a single
treatment agent in patients with relapsed and unresectable OST. PFS
at four months was 46%, whereas OS was 7 months. An objective
response was seen in 14% of patients and 29% of patients had SD
(65). Another previous
nonrandomized phase II trial used a combination of sorafenib and
everolimus in 38 patients with high-grade, nonresectable OST. This
combination demonstrated the greatest antitumor activity as a
second- and third-line treatment for OST, as 17/38 patients (45%;
95% CI, 28-61) were progression free at 6 months. However, the
trial did not reach the prespecified threshold of activity (6-month
PFS ≥50%) with a 6-month PFS of 45% (66).
Cabozantinib
Cabozantinib inhibits the activity of multiple
tyrosine kinases that are expressed in STSs, such as MET, VEGFR,
AXL and TYRO3 protein tyrosine kinase (67).
In a phase II study, 20 heavily-treated patients
with relapsed uterine leiomyosarcoma received a combination of
temozolomide and bevacizumab without (n=9) or with cabozantinib
(n=6). Cabozantinib, in combination with temozolomide and
bevacizumab, increased the clinical benefit rate (CBR) from 67 to
100%, without providing any additional benefits to the ORR (33% for
both cohorts) (68).
In the phase II cabozantinib in patients with
advanced ES or OS clinical trial, heavily pretreated patients with
OST (n=45) or ES (n=45) were enrolled and patients were treated
with cabozantinib (60 mg) once daily in adults or 40
mg/m2 once daily in children (<16 years). In patients
with OST and ES, the median PFS was 6.7 (95% CI, 5.4-7.9 months)
and 4.4 months (95% CI, 3.7-5.6 months), respectively, and the
median OS was 10.6 (95% CI, 7.4-12.5 months) and 10.2 months (95%
CI, 8.5-18.5 months), respectively, which demonstrated promising
activity for cabozantinib. However, 61/90 (68%) patients presented
with at least one serious adverse event. No deaths due to
drug-related toxic effects were reported (69).
Cediranib
Cediranib is a receptor TKI that targets VEGFR1/2/3,
KIT and PDGFRs (70).
A phase II study, including GISTs and sarcomas,
reported the activity of cediranib in metastatic ASPS. Of the six
patients with ASPS, two achieved PR and four exhibited SD (71). Further investigation in a
double-blind, placebo-controlled, randomized, phase II trial
confirmed cediranib's activity in ASPS (19). A total of 48 patients with advanced
ASPS were recruited and randomly assigned to cediranib treatment
(n=32) or placebo (n=16) groups. The primary endpoint was the
percentage change in the sum of the longest diameters of target
marker lesions between the baseline and week 24, or progression if
this was sooner. The median PFS was 10.1 months (95% CI, 5.3-19.0
months) with cediranib and 4.9 months (95% CI, 1.9-20.0 months)
with the placebo.
A pediatric phase I study of cediranib for children
and adolescents with refractory solid tumors defined the maximum
tolerated monotherapy dose as 12 mg/m2/dose administered
orally, once daily, continuously. Objective responses were observed
in pediatric patients with ES, synovial sarcoma and OST, which
resulted in a reduction in primary tumor size and pulmonary
metastatic lesions (72). As a
result, a phase II study of cediranib in children with metastatic
ASPS was developed. This study did not reach the primary endpoint
and cediranib as a single agent was found to be inactive in ASPS in
the pediatric cohort compared with the adult PR rate of 35%.
Therefore, the role of cediranib in the treatment of children and
adolescents with ASPS remains unclear (73).
Crizotinib
Crizotinib is a small molecule that targets MET,
anaplastic lymphoma kinase (ALK) and ROS proto-oncogene 1 receptor
tyrosine kinase.
The EORTC initiated a multinational, multi-tumor,
prospective phase II clinical trial, cross-tumoral phase II with
crizotinib (‘CREATE’), and evaluated the efficacy and safety of
crizotinib in patients with advanced tumors characterized by
abnormal MET and/or ALK expression. In this study 26/43 enrolled
patients with clear-cell sarcoma were treated with crizotinib. The
study design focused on MET+ disease with documented rearrangement
of the EWS RNA binding protein 1 gene confirmed via fluorescence
in situ hybridization. The primary end point of the trial,
the ORR, was not met, as only one objective PR was observed in 26
MET+ patients (ORR, 3.8%; 95% CI, 0.1-19.6%). However, disease
control was achieved in 18/26 MET+ patients (DCR, 69.2%; 95% CI,
48.2-85.7%) and the median PFS was 131 days (95% CI, 49-235 days).
This trial demonstrated that crizotinib could potentially provide
clinical benefits to patients with locally advanced or metastatic
MET+ clear-cell sarcoma (74). The
‘CREATE’ trial also demonstrated that crizotinib is an active
compound for patients with advanced or metastatic ASPS with central
determination of rearrangement of transcription factor binding to
IGHM enhancer 3. The DCR determined in this histotype-specific
trial was 90% (36/40 MET+ patients; 95% CI, 76.3-97.2%) and the
PFSR at 1 year was 37.5% (95% CI, 22.9-52.1%) (75). Crizotinib as a single agent in
patients with advanced metastatic alveolar rhabdomyosarcomas was
well tolerated but lacked clinically meaningful activity, with a
median PFS of 1.3 months (95% CI, 0.5-1.5 months) and a median OS
of 5.6 months (95% CI, 0.7-7.0 months) (76). Crizotinib could be considered as
the standard of care for patients with locally advanced or
metastatic ALK-positive inflammatory myofibroblastic tumors, as
6/12 ALK+ patients (50%; 95% CI, 21.1-78.9) and 1/7 ALK-negative
patients (14%; 95% CI, 0.0-57.9) achieved an objective response
according to RECIST 1.1 in the ‘CREATE’ trial (77).
Axitinib
Axitinib is a selective small molecule inhibitor of
VEGFR1/2/3 that binds to the inactive conformation of the catalytic
domain of VEGF RTKs.
A phase I trial of young patients with refractory
solid neoplasms demonstrated the safety and tolerability of
axitinib. One patient with ASPS had a confirmed PR that lasted for
6 months and both patients with OST and ½ patients with ES were
progression free at 6 months of axitinib treatment (78).
Moreover, the combination of axitinib plus
pembrolizumab was used in 33 patients with advanced sarcomas,
including ASPS, in a single-arm, phase II trial. This treatment
combination was reported to result in 3-month PFS rates of 65.6%
(95% CI, 46.6-79.3%) for all evaluable patients, especially
leiomyosarcoma and UPS, and 72.7% (95% CI, 37.1-90.3%) for patients
with ASPS, with a manageable toxicity profile (79).
Anlotinib
Anlotinib inhibits angiogenesis signaling via
selectively targeting VEGFR1/2/3 and FGFR1/2/3/4. Furthermore, it
targets and decreases the activity of PDGFR-α/β, c-KIT and RET and
therefore significantly inhibits tumor cell proliferation in
preclinical studies (80).
In a phase I, open-label study, at a dose of 12 mg
once daily with a 2-1 treatment schedule, anlotinib displayed
significant antitumor effects in patients with advanced refractory
solid tumors, including STS (80).
A phase II clinical trial demonstrated that 166 patients with
advanced STS had a 12-week PFSR of 68.42% and an ORR of 12.65%
following treatment with anlotinib. PFS and OS were 5.63 and 12.33
months, respectively (81). A
placebo-controlled trial of 233 patients with recurrent advanced
STS presented with a PFS of 6.27 months vs. 1.47 months
(P<0.0001) for anlotinib compared with the placebo (82). A retrospective single-center
analysis studying the combination of chemotherapy with anlotinib
and anlotinib maintenance in patients with metastatic STS, reported
that the treatment had good efficacy and a favorable survival
benefit. The PFS at 3 and 6 months was 81 and 69%, respectively
(32).
Imatinib
Imatinib is known to inhibit c-KIT, the breakpoint
cluster region-ABL fusion protein and PDGFRs (83).
In a phase II multicenter trial, the efficacy of
imatinib was tested in patients with different subtypes of advanced
sarcoma. However, although CBRs, defined as SD + PR + CR, were
exhibited in the subgroups of patients with liposarcomas
(CBR=24.1%) and leiomyosarcomas (CBR=21.4%), imatinib was not
considered an active agent in patients with advanced STS (84).
In a collaborative Italian-Swiss, prospective, phase
II clinical trial, 56 patients with advanced chordoma expressing
PDGFR-β and/or platelet-derived growth factor subunit B (PDGFB)
were treated with imatinib. Confirmed SD (RECIST PR + SD) was
observed in 72% of patients, with a median PFS of 9 months and a
64% CBR, defined as RECIST CR + PR + SD, which was greater than
that exhibited at 6 months (85).
Trials with imatinib have demonstrated promising results,
especially in PDGFB+ or PDGFR-β+ chordomas, whereby SD has been
exhibited in previously progressive advanced chordomas in up to 70%
of cases. However, no benefit in survival rates was noted (33).
Imatinib as a single agent has also been tested in
young patients with relapsed or refractory ES and OST but it did
not confer any objective benefit (86).
Erlotinib
Erlotinib is a highly potent inhibitor of the EGFR
tyrosine kinase with significant but lesser inhibitory activity
against Erb-B2 receptor tyrosine kinase 2.
Erlotinib and temozolomide form another tolerable
double regimen that has been tested in patients <22 years old
with osteogenic sarcoma, rhabdomyosarcoma and STS. However, it was
not demonstrated to be effective against recurrent OST and STS
(87).
Gefitinib
Gefitinib is an EGFR TKI. The results of a phase II
study in which gefitinib was used as a monotherapy for patients
with advanced HER1-expressing synovial sarcoma, as
doxorubicin-containing regimens were ineffective, did not
demonstrate sufficient activity in this tumor subtype. These
results suggested that HE-1 was not a critical protein in tumor
progression in this disease (88).
A phase I trial that combined gefitinib and
irinotecan treatment reported no benefit for patients with OST or
ES, despite exhibiting activity in other tumor types (89).
Nintedanib
Nintedanib is an oral TKI that targets PDGFR-α/β,
FGFR1/2/3, VEGFR1/2/3 and FLT3. A prospective, multicentric,
randomized, open-label phase II trial assessed the efficacy and
safety of nintedanib compared with the intravenous cytotoxic
compound ifosfamide. Patients had advanced, inoperable and/or
metastatic STS following the failure of systemic
non-oxazaphosphorine-based first-line chemotherapy. However, the
trial was stopped early as nintedanib did not prove to be
beneficial as a second-line therapy and resulted in a mPFS of 2.5
months compared with 4.4 months for ifosfamide (20).
Lenvatinib
Lenvatinib is a synthetic, orally available
inhibitor of VEGFR2 with antineoplastic activity. A single-arm
phase Ib/II study reported on the combination of lenvatinib and
eribulin in advanced adipocytic sarcoma and leiomyosarcoma. This
previous study was designed to investigate the safety and efficacy
of this treatment in 20 patients with inoperable or metastatic
liposarcoma and leiomyosarcoma. The ORR, determined using RECIST
1.1, was 27% (5/18 patients; 95% CI, 10-53%) and the median PFS and
6-month PFSR were 56 weeks (95% CI, 25 weeks-not reached) and 72%,
respectively. Even though no benefit was observed in OS, the
combination of lenvatinib and eribulin exhibited promising efficacy
in advanced leiomyosarcomas and liposarcomas (90).
Discussion
In the present study, a systematic literature review
of the available clinical evidence regarding the role of TKIs in
the treatment of patients with locally advanced, unresectable or
metastatic STS and bone sarcoma, was performed. For this purpose,
available scientific reports that were published between 2000 and
2021 were explored. The results identified 45 prospective clinical
trials, 13 retrospective studies and five reviews of clinical
studies that fit into the aforementioned criteria and the results
of these were therefore discussed in the present review.
Advanced, unresectable and/or metastatic sarcomas,
both soft tissue and bone, have a relatively poor outcome, with
very few systemic therapies demonstrating clinical benefits in
terms of PFS and OS prolongation. Current practice for advanced,
unresectable, or metastatic STSs includes anthracycline-based
chemotherapy regimens as first-line treatment, which present with a
median PFS of 4.5 months and a median OS of 12-18 months. It has
previously been reported that a combination of doxorubicin and
ifosfamide improves the mPFS (7.4 vs. 4.6 months) and ORR (26 vs.
14%) compared with doxorubicin alone. However, toxicity is
increased with the combination without being associated with an OS
benefit compared with the doxorubicin alone. (91) TKIs are included in the algorithm
for the treatment of STSs and bone sarcomas, as demonstrated in the
National Comprehensive Cancer Network and European Society for
Medical Oncology guidelines (7–10).
However, the fact that first-line therapy is still based on
anthracyclines, which have become the gold standard over the last
35 years, highlights the necessity for the development of new
treatment options for this heterogeneous group of tumors.
The results of the PALETTE study were the
foundations for the approval of pazopanib for STSs, which was the
first molecular targeted therapy for STS (14). After the introduction of pazopanib,
numerous clinical trials have been performed for other
antiangiogenic TKIs (34,35). The majority of TKIs that display
clinical activity against sarcomas offer a 6-month PFSR of ~40-50%,
which is close to a recent strict limit of 42% that is recommended
from the EORTC meta-analysis for leiomyosarcomas (92). These results provide an important
rational to further investigate TKIs in the treatment of sarcomas
as a second-line treatment and beyond. Furthermore, the combination
of TKIs with immune checkpoint inhibitors has demonstrated
promising results in pretreated sarcomas both in STSs and bone
tumors (57,58,79).
Moreover, an understanding of the molecular mechanisms underlying
mesenchymal tumor responses to immunotherapy could potentially
support the potential activity of immunotherapy agents in
combination with TKIs. However, a 6-month PFSR of >70%, at least
for leiomyosarcomas, is very high for the implementation of TKIs in
a first-line setting.
Despite improvements in treatment approaches,
unresectable and/or metastatic bone and STSs remain a therapeutic
challenge, as the median OS of patients with sarcomas is <2
years. Several preclinical studies and investigations of sarcoma
genomics and mutations of signaling pathways have indicated
potential therapeutic targets. The molecular biology of both STSs
and bone sarcomas demonstrates the importance of several signaling
pathways for the oncogenesis of these tumors (35). It can therefore be hypothesized
that the addition of TKIs that interfere with these cellular
signaling pathways may have an important clinical impact. The
results of numerous phase II trials support this hypothesis.
However, it should be noted that the results of trials testing
TKIs, either in monotherapy or in combination with other
treatments, have not exhibited unequivocal superiority compared
with other treatments. Moreover, the rarity and heterogeneity of
sarcomas highlights the relatively low number of randomized studies
and phase III trials with TKIs. Therefore trials of this design
should be performed in the future. The active trials using
combinations of TKIs are summarized in Table V.
| Table V.Active trials. |
Table V.
Active trials.
| Trial | Study type | Phase | Interventions | Primary outcome
measures |
|---|
| NCT03798106 | A study of
pazopanib and durvalumab for metastatic soft tissue sarcoma | II | Durvalumab +
Pazopanib | PFR at 12
weeks |
| NCT04741438 | Efficacy of the
combination of nivolumab and ipilimumab as a treatment in patients
with sarcoma of rare subtype (RAR-Immune) | III,
Randomized | Nivolumab +
Ipilimumab OR Pazopanib | PFS up to 36
months |
| NCT04351308 | Comparison of MAPI
+ camrelizumab vs. API + apatinib vs. MAPI in patients with a poor
response to preoperative chemotherapy for newly diagnosed
high-grade osteosarcoma (MAPAC) | II, Randomized | MAPI chemotherapy
OR Apatinib Mesylate OR Camrelizumab | EFSR |
| NCT03475953 | A phase I/II study
of regorafenib plus avelumab in solid tumors (REGOMUNE) | I/II | Regorafenib +
Avelumab | Objective
response |
| NCT04803877 | SARC038: Phase 2
study of regorafenib and nivolumab in osteosarcoma | II | Regorafenib +
Nivolumab | PFSR at 16
weeks |
Furthermore, the design of these trials should not
only compare the study drug with the placebo but should also
compare the study drug with at least one active agent already used
in sarcoma treatment. Due to the heterogeneity of sarcomas, these
agents can be chosen according to the sarcoma histotype, which
would provide more representative data regarding the activity of
the tested TKI.
It is important to highlight that TKIs may cause
numerous adverse effects. TKI toxicities may systematically affect
the gastrointestinal tract, cutaneous system, cardiovascular system
and may result in biochemical abnormalities or general symptoms,
such as fatigue. When TKIs are given in combination with other
agents, the toxicity profile seems to be unchanged; however,
clinicians need to specify the causality of each adverse event. The
wide use of TKIs in the treatment of numerous types of tumor has
ensured that medical oncologists have the necessary experience to
cope effectively with the toxicity profile of these drugs, which
therefore contributes to good tolerability in patients.
The present review demonstrated that TKIs are an
important therapeutic option in the treatment of both STSs and bone
sarcomas, especially in second-line settings. Furthermore, the
results of recent trials have highlighted the potential beneficial
activity of combinations of TKIs with immunotherapy in patients
with sarcoma. The molecular and genetic background of sarcomas
further supports the implementation of TKIs in their treatment. New
clinical trials with histotype specificity, even with a lower
number of patients, are needed for this heterogeneous and lethal
group of tumors.
Conclusion
In conclusion, STSs and bone sarcomas are
significant neoplasms; however, their rarity and heterogeneity have
contributed to a relatively low number of studies. Molecular
targeted therapy development has brought a new era of drug
treatments for bone and STSs. Future studies will shed light on the
underlying pathophysiology of sarcoma, providing patients and
physicians with more treatment options.
Acknowledgements
Not applicable.
Funding
This study was funded by HESMO (Hellenic Society of Medical
Oncology; grant no. 8036/25-09-2020).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AK, LEG were the writers of the article. AK and LEG
were the two investigators, who performed the literature search and
data extraction from all studies examined. Data acquisition was
performed by AK, IK, MP, IFN, MA, GK, APa and IC. PE, IK and AK
contributed to conception and design of the study. Manuscript
editing and the revision were performed by PE and APs. All authors
have read and approved the final manuscript. Data sharing is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Burningham Z, Hashibe M, Spector L and
Schiffman JD: The epidemiology of sarcoma. Clin Sarcoma Res.
2:142012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stiller CA, Trama A, Serraino D, Rossi S,
Navarro C, Chirlaque MD and Casali PG; RARECARE Working Group, :
Descriptive epidemiology of sarcomas in Europe: Report from the
RARECARE project. Eur J Cancer. 49:684–695. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
World Health Organization (WHO), . Soft
Tissue and Bone Tumours. WHO Classification of Tumours. 5th
edition. Vol 3. IARC; Lyon: 2020, https://publications.iarc.fr/588
|
|
4
|
Gatta G, Capocaccia R, Botta L, Mallone S,
De Angelis R, Ardanaz E, Comber H, Dimitrova N, Leinonen MK,
Siesling S, et al: Burden and centralized treatment in Europe of
rare tumors: Results of RARECAREnet-a population-based study.
Lancet Oncol. 18:1022–1039. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valery PC, Laversanne M and Bray F: Bone
cancer incidence by morphological subtype: A global assessment.
Cancer Causes Control. 26:1127–1139. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
National Comprehensive Cancer Network
(NCCN), . Bone Cancer (ver. 2.2021). http://www.nccn.org/professionals/physician_gls/pdf/bone.pdfJuly
26–2021
|
|
8
|
National Comprehensive Cancer Network
(NCCN), . Soft Tissue Sarcoma (ver. 2.2021). https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdfJuly
26–2021
|
|
9
|
Gronchi A, Miah AB, Dei Tos AP, Abecassis
N, Bajpai J, Bauer S, Biagini R, Bielack S, Blay JY, Bolle S, et al
ESMO Guidelines Committee EURACAN and GENTURIS, : Soft tissue and
visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol.
32:1348–1365. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Casali PG, Bielack S, Abecassis N, Aro HT,
Bauer S, Biagini R, Bonvalot S, Boukovinas I, Bovee JVMG, Brennan
B, et al ESMO Guidelines Committee, PaedCan and ERN EURACAN, : Bone
sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for
diagnosis, treatment and follow-up. Oncol. 29 (Suppl 4):iv79–iv95.
2018.PubMed/NCBI
|
|
11
|
Rettew AN, Getty PJ and Greenfield EM:
Receptor tyrosine kinases in osteosarcoma: Not just the usual
suspects. Adv Exp Med Biol. 804:47–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu P, Nielsen TE and Clausen MH:
FDA-approved small-molecule kinase inhibitors. Trends Pharmacol
Sci. 36:422–439. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hamberg P, Verweij J and Sleijfer S:
(Pre)clinical pharmacology and activity of pazopanib, a novel
multikinase angiogenesis inhibitor. Oncologist. 15:539–547. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Van der Graaf WTA, Blay JY, Chawla SP, Kim
DW, Bui-Nguyen B, Casali PG, Schöffski P, Aglietta M, Staddon AP,
Beppu Y, et al: Pazopanib formetastatic soft-tissue sarcoma
(PALETTE): A randomized, double-blind, placebo-controlled phase 3
trial. Lancet. 379:1879–1886. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mir O, Brodowicz T, Italiano A, Wallet J,
Blay JY, Bertucci F, Chevreau C, Piperno-Neumann S, Bompas E, Salas
S, et al: Safety and efficacy of regorafenib in patients with
advanced soft tissue sarcoma (REGOSARC): A randomized,
double-blind, placebo-controlled, phase 2 trial. Lancet Oncol.
17:1732–1742. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duffaud F, Mir O, Boudou-Rouquette P,
Piperno-Neumann S, Penel N, Bompas E, Delcambre C, Kalbacher E,
Italiano A, Collard O, et al: Efficacy and safety of regorafenib in
adult patients with metastatic osteosarcoma: A noncomparative,
randomized, double-blind, placebo-controlled, phase 2 study. Lancet
Oncol. 20:120–133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davis LE, Bolejack V, Ryan CW, Ganjoo KN,
Loggers ET, Chawla S, Agulnik M, Livingston MB, Reed D, Keedy V, et
al: Randomized double-blind phase II study of regorafenib in
patients with metastatic osteosarcoma. J Clin Oncol. 37:1424–1431.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Attia S, Bolejack V, Ganjoo KN, George S,
Agulnik M, Rushing DA, Loggers ET, Livingston MB, Wright JA, Chawla
SP, et al: A phase 2 trial of regorafenib (REGO) in patients with
advanced Ewing sarcoma and related tumors of soft tissue and bone:
SARC0024 trial results. J Clin Oncol. 35 (Suppl 15):S110052017.
View Article : Google Scholar
|
|
19
|
Judson I, Morden JP, Kilburn L, Leahy M,
Benson C, Bhadri V, Campbell-Hewson Q, Cubedo R, Dangoor A, Fox L,
et al: Cediranib in patients with alveolar softpart sarcoma
(CASPS): A double-blind, placebo-controlled, randomized, phase 2
trial. Lancet Oncol. 20:1023–1034. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schöffski P, Toulmonde M, Estival A,
Marquina G, Dudzisz-Śledź M, Brahmi M, Steeghs N, Karavasilis V, de
Haan J, Wozniak A, et al: Randomized phase 2 study comparing the
efficacy and safety of the oral tyrosine kinase inhibitor
nintedanib with single agent ifosfamide in patients with advanced,
inoperable, metastatic soft tissue sarcoma after failure of
first-line chemotherapy: EORTC-1506-STBSG ‘ANITA’. Eur J Cancer.
152:26–40. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Benson C, Ray-Coquard I, Sleijfer S,
Litière S, Blay JY, Le Cesne A, Papai Z, Judson I, Schöffski P,
Chawla S, et al: Outcome of uterine sarcoma patients treated with
pazopanib: A retrospective analysis based on two European
organisation for research and treatment of cancer (EORTC) soft
tissue and bone sarcoma group (STBSG) clinical trials 62043 and
62072. Gynecol Oncol. 142:89–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kollár A, Jones RL, Stacchiotti S,
Gelderblom H, Guida M, Grignani G, Steeghs N, Safwat A, Katz D,
Duffaud F, et al: Pazopanib in advanced vascular sarcomas: An EORTC
soft tissue and bone sarcoma group (STBSG) retrospective analysis.
Acta Oncol. 56:88–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gelderblom H, Judson IR, Benson C,
Merimsky O, Grignani G, Katz D, Freivogel KW, Stein D, Jobanputra
M, Mungul A, et al: Treatment patterns and clinical outcomes with
pazopanib in patients with advanced soft tissue sarcomas in a
compassionate use setting: Results of the SPIRE study. Acta Oncol.
56:1769–1775. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
ElNaggar AC, Hays JL and Chen JL: Addition
of everolimus post VEGFR inhibition treatment failure in advanced
sarcoma patients who previously benefited from VEGFR inhibition: A
case series. PLoS One. 11:e01569852016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Katz D, Azraq Y, Eleyan F, Gill S, Peretz
T and Merimsky O: Pazolimus: Pazopanib plus sirolimus following
progression on pazopanib, a retrospective case series analysis. BMC
Cancer. 16:6162016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dembla V, Groisberg R, Hess K, Fu S,
Wheler J, Hong DS, Janku F, Zinner R, Piha-Paul SA, Ravi V, et al:
Outcomes of patients with sarcoma enrolled in clinical trials of
pazopanib combined with histone deacetylase, mTOR, Her2, or MEK
inhibitors. Sci Rep. 7:159632017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu B, Li J, Xie Q, Diao L, Gai L and Yang
W: Efficacy and safety of apatinib monotherapy in advanced bone and
soft tissue sarcoma: An observational study. Cancer Biol Ther.
19:198–204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie L, Xu J, Sun X, Li X, Liu K, Liang X,
Zhou Z, Zhuang H, Sun K, Wu Y, et al: Apatinib plus ifosfamide and
etoposide for relapsed or refractory osteosarcoma: A retrospective
study in two centers. Oncol Lett. 22:5522021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie L, Guo W, Wang Y, Yan T, Ji T and Xu
J: Apatinib for advanced sarcoma: Results from multiple
institutions' off-label use in China. BMC Cancer. 18:3962018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stacchiotti S, Negri T, Libertini M,
Palassini E, Marrari A, De Troia B, Gronchi A, Dei Tos AP, Morosi
C, Messina A, et al: Sunitinib malate in solitary fibrous tumor
(SFT). Ann Oncol. 23:3171–3179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stacchiotti S, Pantaleo MA, Astolfi A,
Dagrada GP, Negri T, Dei Tos AP, Indio V, Morosi C, Gronchi A,
Colombo C, et al: Activity of sunitinib in extraskeletal myxoid
chondrosarcoma. Eur J Cancer. 50:1657–1664. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang HY, Chu JF, Zhang P, Wang JQ, Yan Z,
Yao SN, Yao ZH and Liu YY: Safety and efficacy of chemotherapy
combined with anlotinib plus anlotinib maintenance in Chinese
patients with advanced/metastatic soft tissue sarcoma. Onco Targets
Ther. 13:1561–1568. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hindi N, Casali PG, Morosi C, Messina A,
Palassini E, Pilotti S, Tamborini E, Radaelli S, Gronchi A and
Stacchiotti S: Imatinib in advanced chordoma: A retrospective case
series analysis. Eur J Cancer. 51:2609–2614. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smolle MA, Szkandera J, Andreou D,
Palmerini E, Bergovec M and Leithner A: Treatment options in
unresectable soft tissue and bone sarcoma of the extremities and
pelvis-a systematic literature review. EFORT Open Rev. 5:799–814.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Constantinidou A, Pollack S, Loggers E,
Rodler E and Jones RL: The evolution of systemic therapy in
sarcoma. Expert Rev Anticancer Ther. 13:211–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Polychronidou G, Karavasilis V, Pollack
SM, Huang PH, Lee A and Jones RL: Novel therapeutic approaches in
chondrosarcoma. Future Oncol. 13:637–648. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bailey K, Cost C, Davis I, Glade-Bender J,
Grohar P, Houghton P, Isakoff M, Stewart E, Laack N, Yustein J, et
al: Emerging novel agents for patients with advanced Ewing sarcoma:
A report from the Children's Oncology Group (COG) New Agents for
Ewing Sarcoma Task Force. F1000Res. 8:F1000Faculty Rev. –493. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakano K and Takahash S: current molecular
targeted therapies for bone and soft tissue sarcomas. Int J Mol
Sci. 19:7392018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sleijfer S, Ray-Coquard I, Papai Z, Le
Cesne A, Scurr M, Schöffski P, Collin F, Pandite L, Marreaud S, De
Brauwer A, et al: Pazopanib, a multikinase angiogenesis inhibitor,
in patients with relapsed or refractory advanced soft tissue
sarcoma: A phase II study from the European organization for
research and treatment of cancer-soft tissue and bone sarcoma group
(EORTC Study 62043). J Clin Oncol. 27:3126–3132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li H, Wozniak A, Sciot R, Cornillie J,
Wellens J, Van Looy T, Vanleeuw U, Stas M, Hompes D, Debiec-Rychter
M and Schöffski P: Pazopanib, a receptor tyrosine kinase inhibitor,
suppresses tumor growth through angiogenesis in dedifferentiated
liposarcoma xenograft models. Transl Oncol. 7:665–671. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Samuel BL, Chawla SP, Somaiah N, Staddon
AP, Skubitz KM, Milhem MM, Kaiser PE, Portnoy DC, Priebat DA,
Walker MS and Stepanski EJ: Results of a prospective phase 2 study
of pazopanib in patients with advanced intermediate-grade or
high-grade liposarcoma. Cancer. 123:4640–4647. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chow W, Frankel P, Ruel C, Araujo DM,
Milhem M, Okuno S, Hartner L, Undevia S and Staddon A: Results of a
prospective phase 2 study of pazopanib in patients with surgically
unresectable or metastatic chondrosarcoma. Cancer. 126:105–111.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lipplaa A, Dijkstra S and Gelderblom H:
Efficacy of pazopanib and sunitinib in advanced axial chordoma: A
single reference centre case series. Clin Sarcoma Res. 6:192016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Keir ST, Morton CL, Wu J, Kurmasheva RT,
Houghton PJ and Smith MA: Initial testing of the multitargeted
kinase inhibitor pazopanib by the pediatric preclinical testing
program. Pediatr Blood Cancer. 59:586–588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Umeda K, Kato I, Saida S, Okamoto T and
Adachi S: Pazopanib for second recurrence of osteosarcoma in
pediatric patients. Pediatr Int. 59:937–938. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu X, Xu J, Li F, Liao Z, Ren Z, Zhu L,
Shi Y, Zhao G, Bai X, Zhao J, et al: Efficacy and safety of the
VEGFR2 inhibitor Apatinib for metastatic soft tissue sarcoma:
Chinese cohort data from NCT03121846. Biomed Pharmacother.
122:1095872020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xie L, Xu J, Sun X, Tang X, Yan T, Yang R
and Guo W: Apatinib for advanced osteosarcoma after failure of
standard multimodal therapy: An open label phase II clinical trial.
Oncologist. 24:e542–e550. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zheng B, Ren T, Huang Y and Guo W:
Apatinib inhibits migration and invasion as well as PD-L1
expression in osteosarcoma by targeting STAT3. Biochem Biophys Res
Commun. 495:1695–1701. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li F, Liao Z, Zhang C, Zhao J, Xing R,
Teng S, Zhang J, Yang Y and Yang J: Apatinib as targeted therapy
for sarcoma. Oncotarget. 9:24548–24560. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xie L, Xu J, Sun X, Guo W, Gu J, Liu K,
Zheng B, Ren T, Huang Y, Tang X, et al: Apatinib plus camrelizumab
(anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO)
progressing after chemotherapy: A single-arm, open-label, phase 2
trial. J Immunother Cancer. 8:e0007982020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chow LQ and Eckhardt SG: Sunitinib: From
rational design to clinical efficacy. J Clin Oncol. 25:884–896.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
George S, Merriam P, Maki RG, Van den
Abbeele AD, Yap JT, Akhurst T, Harmon DC, Bhuchar G, O'Mara MM,
D'Adamo DR, et al: Multicenter phase II trial of sunitinib in the
treatment of nongastrointestinal stromal tumor sarcomas. J Clin
Oncol. 27:3154–3160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mahmood ST, Agresta S, Vigil CE, Zhao X,
Han G, D'Amato G, Calitri CE, Dean M, Garrett C, Schell MJ, et al:
Phase II study of sunitinib malate, a multitargeted tyrosine kinase
inhibitor in patients with relapsed or refractory soft tissue
sarcomas. Focus on three prevalent histologies: Leiomyosarcoma,
liposarcoma and malignant fibrous histiocytoma. Int J Cancer.
129:1963–1969. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jo JC, Hong YS, Kim KP, Lee JL, Lee J,
Park YS, Kim SY, Ryu JS, Lee JS and Kim TW: A prospective
multicenter phase II study of sunitinib in patients with advanced
aggressive fibromatosis. Invest New Drugs. 32:369–376. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jagodzińska-Mucha P, Świtaj T, Kozak K,
Koseła-Paterczyk H, Klimczak A, Ługowska I, Rogala P, Wągrodzki M,
Falkowski S and Rutkowski P: Long-term results of therapy with
sunitinib in metastatic alveolar soft part sarcoma. Tumori.
103:231–235. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Martin-Broto J, Hindi N, Grignani G,
Martinez-Trufero J, Redondo A, Valverde C, Stacchiotti S,
Lopez-Pousa A, D'Ambrosio L, Gutierrez A, et al: Nivolumab and
sunitinib combination in advanced soft tissue sarcomas: A
multicenter, single-arm, phase Ib/II trial. J Immunother Cancer.
8:e0015612020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Palmerini E, Lopez-Pousa A, Grignani G,
Redondo A, Hindi N, Stacchiotti S, Sebio A, Lopez-Martin JA,
Morales CMV, Martinez-Trufero J, et al: IMMUNOSARC: A collaborative
Spanish (GEIS) and Italian (ISG) sarcoma groups phase I/II trial of
sunitinib and nivolumab in advanced soft tissue and bone sarcoma:
Results from the phase II part, bone sarcoma cohort. J Clin Oncol.
38 (Suppl 15):Se115222020. View Article : Google Scholar
|
|
59
|
Mross K, Frost A, Steinbild S, Hedbom S,
Büchert M, Fasol U, Unger C, Krätzschmar J, Heinig R, Boix O and
Christensen O: A phase I dose-escalation study of regorafenib (BAY
73-4506), an inhibitor of oncogenic, angiogenic, and stromal
kinases, in patients with advanced solid tumors. Clin Cancer Res.
18:2658–2667. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Berry V, Basson L, Bogart E, Mir O, Blay
JY, Italiano A, Bertucci F, Chevreau C, Clisant-Delaine S,
Liegl-Antzager B, et al: REGOSARC: Regorafenib versus placebo in
doxorubicin-refractory soft-tissue sarcoma-A Quality-adjusted time
without symptoms of progression or toxicity analysis. Cancer.
123:2294–2302. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Marrari A, Bertuzzi A, Bozzarelli S,
Gennaro N, Giordano L, Quagliuolo V, De Sanctis R, Sala S,
Balzarini L and Santoro A: Activity of regorafenib in advanced
pretreated soft tissue sarcoma: Results of a single-center phase II
study. Medicine (Baltimore). 99:e207192020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Santoro A, Comandone A, Basso U, Soto
Parra H, De Sanctis R, Stroppa E, Marcon I, Giordano L, Lutman FR,
Boglione A and Bertuzzi A: Phase II prospective study with
sorafenib in advanced soft tissue sarcomas after
anthracycline-based therapy. Ann Oncol. 24:1093–1098. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Valentin T, Fournier C, Penel N, Bompas E,
Chaigneau L, Isambert N and Chevreau C: Sorafenib in patients with
progressive malignant solitary fibrous tumors: A subgroup analysis
from a phase II study of the French Sarcoma Group (GSF/GETO).
Invest New Drugs. 31:1626–1627. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
von Mehren M, Rankin C, Goldblum JR,
Demetri GD, Bramwell V, Ryan CW and Borden E: Phase 2 Southwest
Oncology Group-directed intergroup trial (S0505) of sorafenib in
advanced soft tissue sarcomas. Cancer. 118:770–776. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Grignani G, Palmerini E, Dileo P, Asaftei
SD, D'Ambrosio L, Pignochino Y, Mercuri M, Picci P, Fagioli F,
Casali PG, et al: A phase II trial of sorafenib in relapsed and
unresectable high-grade osteosarcoma after failure of standard
multimodal therapy: An Italian Sarcoma Group study. Ann Oncol.
23:508–516. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Grignani G, Palmerini E, Ferraresi V,
D'Ambrosio L, Bertulli R, Asaftei SD, Tamburini A, Pignochino Y,
Sangiolo D, Marchesi E, et al: Sorafenib and everolimus for
patients with unresectable high-grade osteosarcoma progressing
after standard treatment: A nonrandomized phase 2 clinical trial.
Lancet Oncol. 16:98–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schöffski P, Blay JY and Ray-Coquard I:
Cabozantinib as an emerging treatment for sarcoma. Curr Opin Oncol.
32:321–331. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ikeda S, Kudoh K, Sasaki N, Takano M, Goto
T, Sakamoto RK, Kita T, Susumu N, Aoki D, Kouta H and Kikuchi Y:
Synergistic effects of cabozantinib to temozolomide and bevacizumab
in patients with heavily pretreated relapsed uterine
leiomyosarcoma. J Clin Oncol. 33 (15_Suppl):S55902015. View Article : Google Scholar
|
|
69
|
Italiano A, Mir O, Mathoulin-Pelissier S,
Penel N, Piperno-Neumann S, Bompas E, Chevreau C, Duffaud F,
Entz-Werlé N, Saada E, et al: Cabozantinib in patients with
advanced Ewing sarcoma or osteosarcoma (CABONE): A multicenter,
single-arm, phase 2 trial. Lancet Oncol. 21:446–455. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Morabito A, De Maio E, Di Maio M, Normanno
N and Perrone F: Tyrosine kinase inhibitors of vascular endothelial
growth factor receptors in clinical trials: Current status and
future directions. Oncologist. 11:753–764. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Brahmi M, Vanacker H and Dufresne A: Novel
therapeutic options for alveolar soft part sarcoma: Antiangiogenic
therapy, immunotherapy and beyond. Curr Opin Oncol. 32:295–300.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Fox E, Aplenc R, Bagatell R, Chuk MK,
Dombi E, Goodspeed W, Goodwin A, Kromplewski M, Jayaprakash N,
Marotti M, et al: A phase 1 trial and pharmacokinetic study of
cediranib, an orally bioavailable pan-vascular endothelial growth
factor receptor inhibitor, in children and adolescents with
refractory solid tumors. J Clin Oncol. 28:5174–5181. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cohen JW, Widemann BC, Derdak J, Dombi E,
Goodwin A, Dompierre J, Onukwubiri U, Steinberg SM, O'Sullivan
Coyne G, Kummar S, et al: Cediranib phase-II study in children with
metastatic alveolar soft-part sarcoma (ASPS). Pediatr Blood Cancer.
66:e279872019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Schöffski P, Wozniak A, Stacchiotti S,
Rutkowski P, Blay JY, Lindner LH, Strauss SJ, Anthoney A, Duffaud
F, Richter S, et al: Activity and safety of crizotinib in patients
with advanced clear-cell sarcoma with MET alterations: European
organization for research and treatment of cancer phase II trial
90101 ‘CREATE’. Ann Oncol. 28:3000–3008. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Schöffski P, Wozniak A, Kasper B, Aamdal
S, Leahy MG, Rutkowski P, Bauer S, Gelderblom H, Italiano A,
Lindner LH, et al: Activity and safety of crizotinib in patients
with alveolar soft part sarcoma with rearrangement of TFE3:
European organization for research and treatment of cancer (EORTC)
phase II trial 90101 ‘CREATE’. Ann Oncol. 29:758–765. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Schöffski P, Wozniak A, Leahy MG, Aamdal
S, Rutkowski P, Bauer S, Richter S, Grünwald V, Debiec-Rychter M,
Sciot R, et al: The tyrosine kinase inhibitor crizotinib does not
have clinically meaningful activity in heavily pre-treated patients
with advanced alveolar rhabdomyosarcoma with FOXO rearrangement:
European Organisation for Research and Treatment of Cancer phase 2
trial 90101 ‘CREATE’. Eur J Cancer. 94:156–167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Schöffski P, Sufliarsky J, Gelderblom H,
Blay JY, Strauss SJ, Stacchiotti S, Rutkowski P, Lindner LH, Leahy
MG, Italiano A, et al: Crizotinib in patients with advanced,
inoperable inflammatory myofibroblastic tumours with and without
anaplastic lymphoma kinase gene alterations (European Organisation
for Research and Treatment of Cancer 90101 CREATE): A multicentre,
single-drug, prospective, non-randomised phase 2 trial. Lancet
Respir Med. 6:431–441. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Geller JI, Fox E, Turpin BK, Goldstein SL,
Liu X, Minard CG, Kudgus RA, Reid JM, Berg SL and Weigel BJ: A
study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in
children and adolescents with recurrent or refractory solid tumors:
A children's oncology group phase 1 and pilot consortium trial
(ADVL1315). Cancer. 124:4548–4555. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wilky BA, Trucco MM, Subhawong TK, Florou
V, Park W, Kwon D, Wieder ED, Kolonias D, Rosenberg AE, Kerr DA, et
al: Axitinib plus pembrolizumab in patients with advanced sarcomas
including alveolar soft-part sarcoma: A single-center, single-arm,
phase 2 trial. Lancet Oncol. 20:837–848. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sun Y, Niu W, Du F, Du C, Li S, Wang J, Li
L, Wang F, Hao Y, Li C and Chi Y: Safety, Pharmacokinetics, and
antitumor properties of anlotinib, an oral multitarget tyrosine
kinase inhibitor, in patients with advanced refractory solid
tumors. J Hematol Oncol. 9:1052016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chi Y, Fang Z, Hong X, Yao Y, Sun P, Wang
G, Du F, Sun Y, Wu Q, Qu G, et al: Safety and efficacy of
anlotinib, a multikinase angiogenesis inhibitor, in patients with
refractory metastatic soft-tissue sarcoma. Clin Cancer Res.
24:5233–5238. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Tang L, Wang Y, Zhang J, Yu W, Huang Y and
Yao Y: Efficacy and safety of anlotinib in advanced soft tissue
sarcoma: Results from one of multi-centers in a phase IIB trial
(ALTER0203). J Clin Oncol. 37 (Suppl 15):e225182019. View Article : Google Scholar
|
|
83
|
Pindolia VK and Zarowitz BJ: Imatinib
mesylate, the first molecularly targeted gene suppressor.
Pharmacotherapy. 22:1249–1265. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chugh R, Wathen JK, Maki RG, Benjamin RS,
Patel SR, Meyers PA, Priebat DA, Reinke DK, Thomas DG, Keohan ML,
et al: Phase II multicenter trial of imatinib in 10 histologic
subtypes of sarcoma using a bayesian hierarchical statistical
model. J Clin Oncol. 27:3148–3153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Stacchiotti S, Longhi A, Ferraresi V,
Grignani G, Comandone A, Stupp R, Bertuzzi A, Tamborini E, Pilotti
S, Messina A, et al: Phase II study of imatinib in advanced
chordoma. J Clin Oncol. 30:914–920. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Bond M, Bernstein ML, Pappo A, Schultz KR,
Krailo M, Blaney SM and Adamson PC: A phase II study of imatinib
mesylate in children with refractory or relapsed solid tumors: A
Children's oncology group study. Pediatr Blood Cancer. 50:254–258.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jakacki RI, Hamilton M, Gilbertson RJ,
Blaney SM, Tersak J, Krailo MD, Ingle AM, Voss SD, Dancey JE and
Adamson PC: Pediatric phase I and pharmacokinetic study of
erlotinib followed by the combination of erlotinib and
temozolomide: A Children's oncology group phase I consortium study.
J Clin Oncol. 26:4921–4927. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ray-Coquard I, Le Cesne A, Whelan JS,
Schoffski P, Bui BN, Verweij J, Marreaud S, van Glabbeke M,
Hogendoorn P and Blay JY: A phase II study of gefitinib for
patients with advanced HER-1 expressing synovial sarcoma refractory
to doxorubicin-containing regimens. Oncologist. 13:467–473. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Brennan RC, Furman W, Mao S, Wu J, Turner
DC, Stewart CF, Santana V and McGregor LM: Phase I dose escalation
and pharmacokinetic study of oral gefitinib and irinotecan in
children with refractory solid tumors. Cancer Chemother. Pharmacol.
74:1191–1198. 2014.PubMed/NCBI
|
|
90
|
Chen TWC, Yu CW, Hong RL, Yen CC, Guo JC,
Chen SC, Lee JC, Chen ML, Chang HF, Hsu MC and Kung TF: An Ib/II
study of the combination of lenvatinib (L) and eribulin (E) in
advanced liposarcoma (LPS) and leiomyosarcoma (LMS) (LEADER). J
Clin Oncol. 38 (15 _Suppl):S115072020. View Article : Google Scholar
|
|
91
|
Judson I, Verweij J, Gelderblom H,
Hartmann JT, Schöffski P, Blay JY, Kerst JM, Sufliarsky J, Whelan
J, Hohenberger P, et al: Doxorubicin alone versus intensified
doxorubicin plus ifosfamide for first-line treatment of advanced or
metastatic soft-tissue sarcoma: A randomized controlled phase 3
trial. Lancet Oncol. 15:415–423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kantidakis G, Litière S, Neven A, Vinches
M, Judson I, Schöffski P, Wardelmann E, Stacchiotti S, D'Ambrosio
L, Marréaud S, et al: Efficacy thresholds for clinical trials with
advanced or metastatic leiomyosarcoma patients: A European
organization for research and treatment of cancer soft tissue and
bone sarcoma group meta-analysis based on a literature review for
soft-tissue sarcomas. Eur J Cancer. 154:253–268. 2021. View Article : Google Scholar : PubMed/NCBI
|