Retinoblastoma (RB) is an intraocular malignancy
occurring in young children. For the vast majority of cases, RB
develops as a result of biallelic inactivation of the RB
transcriptional corepressor 1 (RB1) gene in the developing
retina, followed by genetic and epigenetic alterations during tumor
progression (1–3). Since RB1 mutations are
required for RB initiation and are also frequently found in other
human cancers, especially during cancer progression, extensive
research efforts have been made to elucidate the functions of RB
protein (pRB) in tumor suppression for the past decades. This has
unraveled the multifaceted roles of pRB in a wide variety of
cellular events, ranging from canonical cell cycle regulation at
local gene promoters to organization of higher-order chromatin
structures and chromosomes (4–7).
Furthermore, a deeper understanding of pRB functions in the context
of cancers has enabled envisioning of novel therapeutic strategies
for cancers with RB1 loss, which frequently develop therapy
resistance by varied mechanisms (8,9).
Genomic instability is a characteristic of most
human malignancies and the impact of genome maintenance mechanisms
on neoplastic transformation and subsequent development of cancer
has been well established (10).
Notably, most of the chromatin-associated functions exerted by pRB
at diverse genomic locations bear important relevance to the
maintenance of genomic stability (5,11).
Functional inactivation or gene disruption experimental models have
revealed at least three major mechanisms by which pRB participates
in genome maintenance: First, pRB is recruited to DNA double-strand
breaks (DSBs) and directly promotes DNA repair (12,13).
A study reported that pRB interacts with repair factors X-ray
repair cross complementing (XRCC)5 and XRCC6 at DSBs to facilitate
canonical nonhomologous end-joining (C-NHEJ) repair, while another
study demonstrated that pRB recruits BRM/SWI2-related gene 1 (BRG1)
chromatin remodeler to alter the chromatin structure and stimulate
DNA end resection for homologous recombination (HR) repair
(12,13). Notably, E2 factor (E2F)
transcription factor 1 (E2F1) has been demonstrated to be required
for the recruitment of pRB and BRG1 to DNA breaks for HR repair,
which suggests a transcription-independent function of E2F1 in DNA
repair (13,14). Second, pRB ensures the fidelity of
DNA replication and chromosome segregation (15–20).
Inactivation of RB family proteins by human papilloma virus
oncoprotein E7 causes the stalling of replication forks prevalently
at repetitive regions of the genome and results in DSBs (15). Furthermore, pRB has been reported
to be essential for recruitment of condensin II and cohesin, which
are involved in structural maintenance of chromatin during DNA
replication and mitosis, and pRB loss-driven defects in the
recruitment of such factors results in aberrant replication
followed by chromosome segregation errors, which can directly
contribute to aneuploidy and facilitate tumor development (16–20).
Third, pRB serves critical roles in silencing repetitive sequences
across the genome and maintaining heterochromatin by recruiting
repressive histone modifiers such as enhancer of zeste 2 polycomb
repressive complex 2 subunit, suppressor of variegation 4–20 and
histone deacetylase (HDAC) complexes for stable maintenance of the
genomic regions via deposition of distinct histone modification
marks (21–24). This illustrates that the roles of
pRB in genome maintenance are further extended to the maintenance
of epigenetic stability in genomes. As aforementioned for HR
repair, some of these genome maintenance functions exerted by pRB
have been demonstrated to be dependent on the presence of E2F1 at
the sites in a sequence-independent manner (19,21).
Thus, these findings further support the notion that E2F1 may
actively participate in genome-wide functions of pRB in chromatin
regulation, independently of its canonical roles in cell cycle
control and transcription.
As pRB protects against genomic instability, and
functional pRB is deficient following initiation of RB due to
biallelic inactivation of RB1, the present review begins
with an overview of RB genomic analysis results to understand the
relationship between the intrinsic RB1 loss and the status
of genome stability observed in primary RB tumors. Subsequently,
previous findings on genome maintenance mechanisms in RB are
presented, revealing the possibility of novel therapeutic
opportunities. Finally, advantages and challenges for exploiting
the newly identified therapeutic vulnerabilities in RB are
discussed.
In contrast to the widespread roles of pRB in genome
maintenance as demonstrated by RB1 gene mutation and pRB
depletion in the aforementioned experimental models, whole-genome
sequencing (WGS) of human RB tumors has revealed that RB genomes
are relatively stable compared with those of other cancer types
(25). Although only four RB
specimens were used for the WGS analysis, the study also
demonstrated that, despite multiple passaging over a prolonged
time, orthotopic xenografts of the same human RB displayed only a
modest increase in passenger mutations without gross defects in
chromosome stability, suggesting that human RB genomes are
maintained stably in vivo and massive genome instability may
not be a strong driver for RB progression. Subsequent genomic
analyses in larger RB cohorts have employed WGS, exome sequencing
and targeted next-generation sequencing (NGS) (26–31).
These studies consistently identified driver mutations in MYCN
proto-oncogene, bHLH transcription factor (MYCN) and BCL6
corepressor (BCOR), and verified recurrent copy number
alterations on chromosomes 1q, 2p, 6p and 16q that had also been
identified by previous cytogenetic analyses (3,32).
Notably, two molecular subtypes of human RB tumors identified by a
recent multi-omics approach were also associated with these genomic
characteristics, represented by subtype 1 harboring few genetic
alterations other than RB1 mutations and subtype 2
presenting MYCN amplification or recurrent 1q gain and/or
16q loss (33). In addition to
these known genomic changes, the targeted NGS approach on
cancer-related gene panels led to the identification of several
genetic alterations beyond RB1 inactivation. Although these
additional gene mutations were found to occur at low frequency
except for BCOR (14–23%), the presence of the non-RB1
alterations was demonstrated to be associated with aggressive
histopathologic features and poor prognosis when combined with the
corresponding clinicopathological data (29,30).
Given the limited cohort size in most studies, further
investigations are required to verify the clinical significance of
the non-RB1 mutations as biomarkers for prognosis. Of
particular relevance, if the RB subtypes identified by the recent
multi-omics approach and their close association with the known
genomic attributes can be validated in larger cohorts, this may
impact the therapeutic decision-making process through
subtype-based patient stratification, since subtype 2 tumors have
been demonstrated to possess stemness features and a higher
predilection for metastasis (33).
Since targeted NGS studies interrogate specific
genes and select genomic regions, assessment of overall genome
stability in the RB specimens may be limited (29,30).
A recent WGS study on 21 RB samples has revealed that the overall
mutation burden is consistently low in these tumors, as evidenced
by the average count of 275 substitutions at a frequency of 0.085
per Mb, 70 small insertions/deletions with a frequency of 0.021 per
Mb and 17 structural rearrangements at a frequency of 0.005 per Mb
(27). This low somatic mutational
burden in RB was also demonstrated by exome sequencing of 71 RB
samples, suggesting that RB is among the least mutated cancer types
(28). Notably, this genomic
feature appears to be common in a number of pediatric neoplasms as
a pan-cancer genomic analysis of 24 types of childhood cancer has
demonstrated that overall somatic mutation frequencies of pediatric
cancers are markedly lower than those of adult cancers (34). The low mutational burden in
pediatric malignancies may be related to the age at diagnosis or
tumor resection as somatic mutations tend to accumulate with age by
DNA replication errors and environmental factors throughout life
(35). Indeed, albeit being low in
terms of overall mutation frequency, the total number of
substitution mutations in RB exhibited a positive association with
the age of enucleation, indicative of the absence of specific
mutational mechanisms other than cell division-related mutational
processes (27). In agreement with
this interpretation, another recent study reported that variability
in RB genomic alterations is associated with patient age at
diagnosis but not with the possession of germline RB1
mutations (36). Not only for
single nucleotide variants but also for somatic copy number
alterations (SCNAs), RB genomes have been revealed to harbor
relatively few SCNAs compared with other cancer types (28). Considering that most genomic
analyses have been carried out with advanced tumors from enucleated
eyes, the observation of low mutation burden and fewer SCNAs in
these specimens suggests that RB genomes are relatively stable and
there may be genome maintenance mechanisms operating in RB to
counteract or compensate for the risk of RB1
deficiency-driven genomic instability.
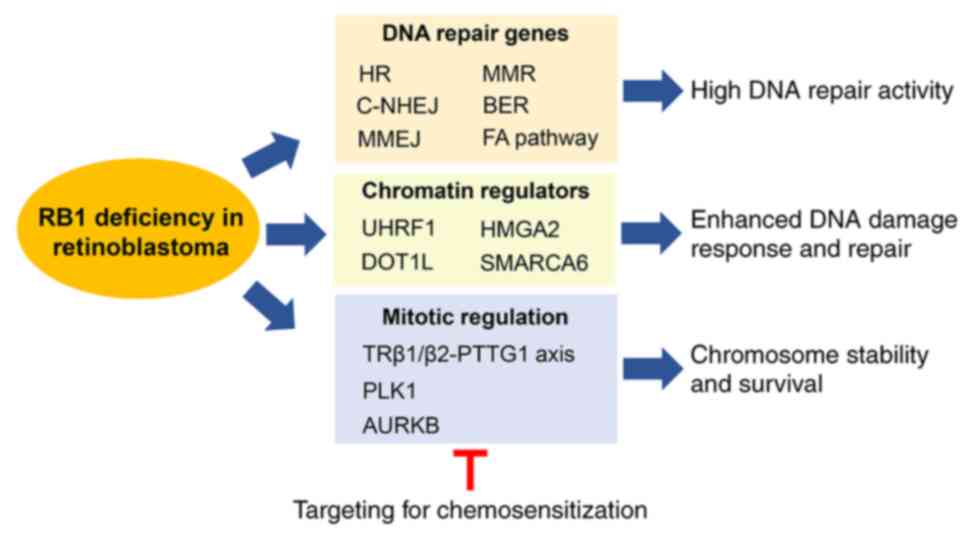
The next section presents an overview of mechanisms
and factors contributing to genome maintenance in RB, which include
the gene signature associated with the DNA repair/DNA damage
pathway in primary RB tumors, emerging roles of chromatin
regulators in DNA damage response/repair, and protein factors which
have been proposed to be important for maintaining chromosome
stability and promoting survival in RB.
The DNA damage response serves a pivotal role in
ensuring genome integrity throughout the cell cycle by sensing DNA
damages, activating cell cycle checkpoints, and engaging multiple
DNA repair pathways or apoptosis (55). Defects in DNA damage response and
repair could be detrimental to cancer cells, particularly in cancer
cells with a functional p53 signaling pathway. As shown in Table I, RB tumors exhibit high expression
levels of genes implicated in DNA damage response and diverse DNA
repair pathways, which may facilitate the repair of DNA lesions and
subsequent cell cycle progression. Efficient DNA damage detection
and repair also requires close cooperation with
chromatin-associating proteins to alter the local chromatin
environment near the DNA lesions and promote recruitment of DNA
repair factors (56). Notably, RB
tumors harbor a number of aberrantly expressed chromatin
regulators, which are not expressed in the normal retina, and some
of these chromatin regulators are direct E2F1 targets (57).
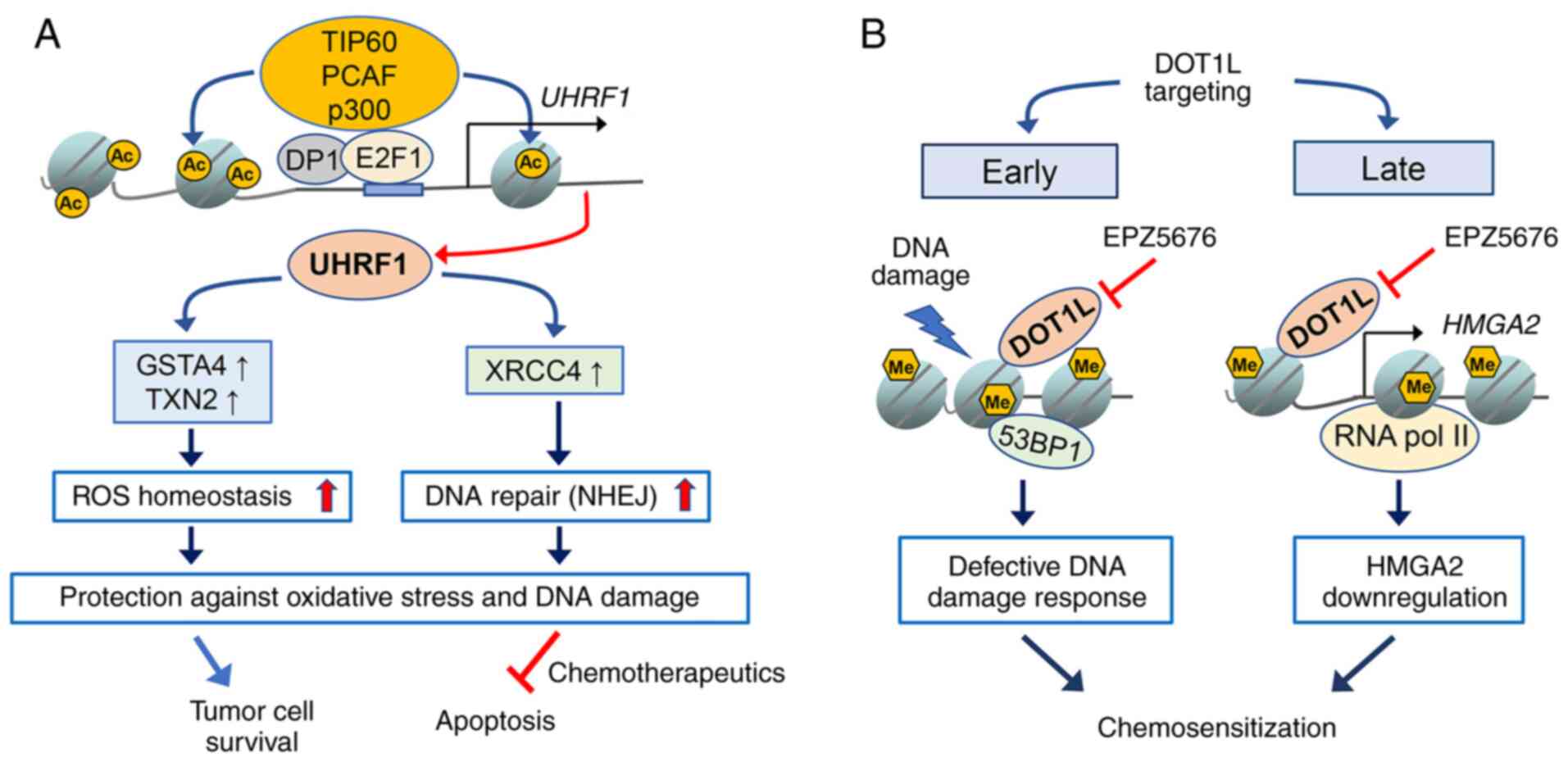
Ubiquitin-like with PHD and RING finger domains 1
(UHRF1) is an epigenetic regulator that is frequently upregulated
in cancer and promotes tumor development by altering gene
expression through changes in DNA methylation and histone
modifications via recruitment of various chromatin modifiers
(58,59). Furthermore, several studies have
reported that UHRF1 is implicated in diverse aspects of DNA damage
response and repair by sensing DNA damages such as interstrand
crosslinks, interacting with relevant repair factors and regulating
the cell cycle-dependent choice of DSB repair pathways (60–63).
All of these findings may mechanistically explain the observation
that Uhrf1-null embryonic stem (ES) cells are more sensitive
to genotoxic insults induced by irradiation and DNA-damaging agents
than Uhrf1+/+ and Uhrf1+/− ES
cells (64). Our previous study
demonstrated that UHRF1 knockdown sensitizes RB cells to
chemotherapeutic drugs by impeding DNA repair via downregulation of
XRCC4 involved in C-NHEJ repair and consequential impairment of DNA
ligase IV loading onto damaged chromatin (65). In addition, another recent study
revealed that UHRF1 depletion in RB cells increases the sensitivity
to HDAC inhibitors by enhancing oxidative stress-mediated apoptosis
via downregulation of the redox-responsive genes encoding
glutathione S-transferase α4 and thioredoxin 2 (66). In agreement with the results in a
cell study, UHRF1 depletion in RB cells increased the therapeutic
efficacy of the HDAC inhibitor MS-275 in murine orthotopic
xenografts (66). The detailed
underlying mechanisms of how UHRF1 modulates these distinct sets of
effector genes in DNA repair and redox homeostasis remain to be
elucidated; however, both studies point to the role of UHRF1 in
genome maintenance in RB cells by functionally linking NHEJ to
redox homeostasis since oxidative stress even at low levels has
been demonstrated to induce DSBs and NHEJ repair-deficient cells
are hypersensitive to oxidative stress (67,68).
Therefore, the findings support the hypothesis that enhanced DNA
repair capacity and ROS homeostasis driven by UHRF1 may protect RB
cells against endogenous DNA damage or chemotherapeutics-induced
cell death (Fig. 1A). Furthermore,
in contrast to normal retina lacking UHRF1 expression, its
constitutive expression in RB as a direct E2F1 target gene makes
UHRF1 an attractive therapeutic target for RB treatment (57).
Similar to the case of UHRF1, disruptor of telomeric
silencing 1-like (DOT1L) is highly and exclusively expressed in RB
although it is thus far unknown whether DOT1L is also an E2F1
target (69). DOT1L is the only
known histone methyltransferase catalyzing H3K79 methylation, which
is considered mostly as an activating mark for gene transcription
(70,71). Notably, DOT1L and H3K79 methylation
have been demonstrated to be indispensable for ionizing
radiation-induced tumor protein p53 binding protein 1 foci
formation during G1/G2 phase, and
pharmacological inhibition of DOT1L in combination with
DNA-damaging agents further decreased the proliferation of
colorectal cancer cells and mixed-lineage leukemia (MLL)-rearranged
leukemia cells (72–74). Consistent with the findings in
other cancer cells, DOT1L targeting by EPZ5676 (pinometostat)
sensitized RB cells to chemotherapeutic drugs by impairing the DNA
damage response and thereby enhancing apoptosis, while it was
largely inefficacious as a single-agent therapy in both RB cells
and an orthotopic xenograft model (69). In addition to verifying the role of
DOT1L in DNA damage response and chemosensitization in RB cells,
the study also revealed that high mobility group AT-hook 2 (HMGA2)
is a novel DOT1L target gene and its expression is epigenetically
upregulated by DOT1L. Notably, HMGA2 has been reported to promote
RB cell proliferation and participate in the regulation of DNA
damage response in cancer cells (75–78).
HMGA2 depletion reduces checkpoint kinase 1 phosphorylation during
the etoposide-induced DNA damage response and potentiates the drug
sensitivity in RB cells (69). The
aforementioned study suggested that DOT1L targeting has a dual role
in chemosensitization of RB cells by immediately hindering the
early DNA damage response mediated by DOT1L itself upon genotoxic
insults, and also by downregulating HMGA2 expression as a late
effect of DOT1L inhibition (Fig.
1B).
In addition to the aforementioned epigenetic
regulators, several chromatin remodelers may also exhibit
chemosensitization properties in RB cells upon their co-inhibition
in combination with conventional genotoxic drugs. Chromatin
remodelers, including BRG1, helicase, lymphoid specific (HELLS) and
SWI/SNF-related, matrix-associated actin-dependent regulator of
chromatin, subfamily a, containing DEAD/H box 1, are known to be
recruited to DSBs and facilitate HR repair (13,79,80).
In particular, HELLS (also known as SMARCA6) is an E2F1 target gene
and has been demonstrated to be crucial for RB tumor initiation and
progression in genetically engineered mouse models (81). Given the importance of HELLS for RB
development and high dependence of RB cells on HR repair for their
survival, it would be of great interest to investigate the
functions of HELLS in the context of the DNA damage response and
repair in RB cells as well as its chemosensitization properties in
preclinical animal models.
In addition to alterations at the level of DNA bases
and small stretches of DNA, aneuploidy generated by gains and
losses of whole chromosomes is a major indicator of genomic
instability, which is a common feature of a number of cancer cells
such as ovarian, breast and prostate cancer, and is often related
to RB1 loss (82). However,
RB tumors appear to maintain overall chromosome stability with a
few recurrent chromosome arm-level alterations but limited
whole-chromosome aneuploidy (25).
This raises the question of how RB cells can achieve chromosomal
stability despite the RB1 loss from the initiation of
tumors. One study attempted to investigate this question by
examining genes expressed prominently in cones, under the
hypothesis that RB cells may use the intrinsic molecular network of
the cell-of-origin to restrain RB1 deficiency-associated
chromosome instability for their survival and proliferation
(83). The authors revealed that
thyroid hormone receptor β1 and 2 (TRβ1 and TRβ2), which are highly
expressed in both cones and RB cells, inhibit the expression of
PTTG1 regulator of sister chromatid separation, securin (PTTG1).
Since PTTG1 prevents separase from promoting sister chromatid
separation (84), PTTG1
accumulation in RB cells upon TRβ1 and TRβ2 knockdown led to an
increase in polyploidy, demonstrating the role of the TRβ1/β2-PTTG1
signaling pathway in maintaining chromosome stability in RB cells
(83). Although the aforementioned
study reported that both TRβ1 and TRβ2 knockdown resulted in E2F1
accumulation in RB cells and E2F1 depletion led to a decrease in
PTTG1 expression, it remains unclear whether E2F1 acts on the same
pathway mediated by TRβ1 and TRβ2. Furthermore, PTTG1 has
been found to be one of mitotic genes that are highly expressed in
primary RB tumors compared with normal retinal tissues (43,44),
which requires a comparative analysis of PTTG1 expression in
purified retinal cone cells relative to whole retinal tissues and
RB tumors in order to firmly establish the role of TRβ1 and TRβ2 in
suppression of polyploidy by PTTG1 downregulation.
Defects in mitotic checkpoint signaling are one of
the prime causal factors for chromosome missegregation and
consequential generation of aneuploidy (85). Notably, inhibition of mitotic
kinases, such as aurora kinase A and B (AURKA and AURKB), has been
found to be synthetic lethal with RB1 deficiency in cancer
cells in pharmacological or CRISPR/CRISPR associated protein 9
(Cas9)-based screens (86,87). Since AURKA and AURKB contribute to
correct mitotic spindle assembly and chromosome segregation
(88), these kinases serve
critical roles in ensuring mitotic fidelity and their inhibition is
lethal for RB1-deficient cancer cells, which upregulate a
number of mitotic genes as a result of E2F deregulation (50,51,82).
As is the case with other RB1-deficient cancer cells,
primary RB tumors display high expression levels of several mitotic
genes, including AURKB, polo-like kinase 1 (PLK1),
mitotic arrest deficient 2 like 1 and BUB1 mitotic checkpoint
serine/threonine kinase (43,44,89,90).
Two recent studies have demonstrated that pharmacological
inhibition of AURKB and PLK1 in RB cells resulted in cell cycle
arrest and increased apoptosis, whereas the effects of the
inhibitors on a nontumoral retinal pigment epithelial cell line
(ARPE-19) were negligible under identical conditions, which was
indicative of a higher sensitivity of RB cells to these inhibitors
(89,91). Although both studies have not
examined whether inhibition of these upregulated mitotic kinases
causes chromosomal aberrations that may eventually lead to cell
death, the results support the possibility that cancer cells with
hyperactive mitotic checkpoint signaling due to RB1 loss
might depend on AURKB and PLK1 for efficient mitotic exit and
survival, establishing a synthetic lethal relationship with
RB1 deficiency upon their inhibition. Notably, PLK1
targeting by ON 01910.Na (rigosertib) has been found to be
efficacious for local therapy in orthotopic xenografts of RB
(91). Since PLK1 is known to have
other genome maintenance functions beyond mitosis, in particular
during DNA replication and the DNA damage response (92), further studies are required to
achieve an improved mechanistic understanding of PLK1 targeting in
RB.
In RB, targeted therapies are currently lacking as a
standard treatment option in clinics. For past decades, research
efforts have been directed toward the identification of potential
driver genes or pathways that promote RB development and can also
be targeted therapeutically (93).
This has led to numerous discoveries in RB cells and the proposal
of potential therapeutic targets involved in diverse cellular
processes (93); however, at
present, none of the proposed targets has advanced into clinical
trials and some of these targets, including microRNAs, are not
amenable to specific targeting by small-molecule inhibitors
(94). Although conventional
genotoxic drugs, which are widely used for chemotherapy in RB, are
efficacious in saving eyes and lives upon early diagnosis and
timely treatment, high doses of such non-specific genotoxic drugs
would be detrimental to young children and may result in multiple
adverse effects during treatment or later in their life, as
exemplified by ocular toxicities such as maculopathy and uveal
effusion, ocular motility restriction due to fibrosis of orbital
tissues, and rare incidence of secondary leukemia associated with
cumulative doses and high-intensity treatment schedules (95–97).
In this regard, strategies to selectively sensitize RB tumors to
conventional chemotherapeutics may serve as a practical and viable
approach to achieve the same therapeutic outcome with lower doses
of the drugs, while minimizing any undesired toxicity in normal
cells. An approach that can be taken for such endeavors would be to
exploit the known genome maintenance mechanisms in RB and leverage
them to sensitize RB cells to chemotherapy in a selective manner.
As aforementioned, the identification of BRCA1 and
RAD51 as the most critical genes for RB cell survival from a
recent functional RNAi screen in RB1null
and RB1wt;
MYCNamp orthotopic xenografts (46) provides strong evidence that genome
maintenance mechanisms serve a pivotal role in RB cell survival,
and these attributes can be exploited therapeutically to develop
more effective chemosensitization strategies. Furthermore, the
tumor-promoting functions of the identified genes were associated
with DNA repair but not with other known functions, such as
centrosome duplication and heterochromatin integrity, and RAD51
targeting by a small-molecule inhibitor engaged the classical
p53-mediated apoptotic pathway and synergized with topoisomerase
inhibitors, which suggests that targeting of these factors may not
involve other unknown cellular pathways, which can potentially
complicate the assessment of therapeutic effects (46). Notably, the DNA-repair hub has been
found to be overlapping for survival of both RB1-inactivated
tumors and MYCN-amplified tumors harboring intact RB1
gene (46), which implies a
wide-range applicability of the chemosensitization strategies
toward different RB subtypes. In line with this notion, poly
(ADP-ribose) polymerase (PARP) inhibition was revealed to be
efficacious for proliferation inhibition of RB1-mutated
osteosarcoma cells, and the hypersensitivity to PARP inhibitors was
associated with rapid activation of DNA replication checkpoint
signaling, while no apparent defects in HR repair were observed in
the RB1-mutated cells (98). These findings collectively suggest
that the therapeutic vulnerabilities identified to be associated
with RB1 loss hinge on DNA damage response and repair,
albeit with variability in the detailed mechanisms of action.
When common genome maintenance mechanisms, such as
DNA repair pathways, are directly targeted for therapies, a key
consideration for effective therapy is how to minimize their
non-selective toxicity to normal cells, while eliciting a favorable
response to therapy. A series of dosing and drug combination
schemes has to be tested to achieve optimal therapeutic regimens.
Alternatively, co-targeting of molecules involved in DNA damage
response and repair, which are expressed exclusively in RB tumors,
may enable more effective therapy by selectively sensitizing RB
cells to chemotherapy. As aforementioned, chromatin regulators,
including UHRF1, DOT1L and HMGA2, are exclusively expressed in RB
without any detectable expression in normal retina, and their
targeting by gene knockdown or pharmacological inhibition
sensitizes RB cells to chemotherapeutics by employing diverse
mechanisms involved in DNA damage response and repair (65,66,69).
Currently, only DOT1L has several types of small-molecule
inhibitors available for clinical trials; however, their poor
pharmacokinetic properties limit the therapeutic efficacy and
necessitate combinations with other drugs (99–101). Since pinometostat, a DOT1L
inhibitor, has been reported to be generally safe in patients with
MLL-rearranged leukemia even after prolonged continuous intravenous
infusion (101), local
combination therapies with a DOT1L inhibitor by intra-arterial or
intravitreal chemotherapy for patients with RB may reduce the
effective dose of standard chemotherapeutic drugs, thereby
preventing systemic toxicity and mitigating any adverse effects in
the eyes (102–104). Given the proven effectiveness of
local therapies for the management of RB as both primary care and
secondary treatment (102–104),
this approach for DOT1L inhibitors appears to hold great promise as
a novel therapy.
Development of small-molecule inhibitors for other
chromatin regulators may also benefit a wide range of patients with
cancer as these epigenetic regulators are upregulated in a number
of cancer types of different cellular origins and their genome
maintenance functions are conserved across various cancer cells
including breast, lung, and colorectal cancer cells (57,105,106). In particular, UHRF1 is a known
E2F1 target (107), which allows
its constitutive expression in RB1-deficient tumors,
including RB, and thereby obviates the patient selection process
for UHRF1 targeting. UHRF1 has also been identified as one of the
top 21 synthetic lethal genes in a recent CRISPR/Cas9 screen in
RB1−/− small cell lung cancer cells (87). Therefore, development of UHRF1
inhibitors may impact the cancer therapy beyond RB if specificity
and toxicity profiles of the inhibitors are in acceptable ranges
for clinical trials. Since most epigenetic regulators are
considered to be druggable (108), a complete understanding of their
functions and comprehensive validation of clinical relevance would
be a prerequisite to prioritize the targets that are amenable to
selective inhibition by small-molecule inhibitors.
Another potentially important group of therapeutic
targets in RB is mitotic kinases, some of which have a synthetic
lethal relationship with RB1 deficiency in other cancer
cells (86,87). Although rigosertib, a PLK1
inhibitor, does not target RB cells selectively and is also known
to have a short half-life and rapid clearance, it has shown
remarkably less eye toxicity upon local therapy in orthotopic
xenografts than melphalan, which is most commonly used for
intravitreal therapy in clinics (91,103,109). Therefore, despite the mixed
results in clinical trials with advanced solid tumors (110–112), comprehensive preclinical studies
with rigosertib as a single agent or in combination with other
drugs by local administration may result in promising outcomes,
which could encourage clinical trials for RB.
Given the well-known functions of pRB in the
maintenance of genome stability, lack of functional pRB by
biallelic inactivation of the RB1 gene in the vast majority
of human RB cases suggests that human RB genomes would display a
high degree of genomic instability. However, several whole-genome
analyses (25–27) have revealed that RB genomes are
relatively stable, characterized by low mutation burden and certain
recurrent chromosomal alterations associated with somatic copy
number changes. These findings have brought a novel perspective on
the genome maintenance mechanisms in RB that may operate actively
to attenuate the RB1 deficiency-associated risk of genomic
instability and thereby avoid any catastrophic genomic defects that
would jeopardize survival and growth of RB. As summarized in
Fig. 2, RB tumors possess multiple
mechanisms to invigorate their DNA damage response and repair
processes in response to genotoxic insults (including the examples
shown in Fig. 1) (42–45,65,66,69),
and to prevent chromosome instability, such as aneuploidy (83). Although these genome maintenance
mechanisms might have been evolved and adapted to promote RB cell
survival and proliferation, the dependency of RB cells on these
mechanisms may expose their unique vulnerability to chemotherapy,
particularly when the genome maintenance mechanisms are tumor
cell-specific. In order to achieve tumor cell-specific
chemosensitization by selective targeting of the genome maintenance
machineries, a thorough understanding of their functions and
comprehensive evaluation of therapeutic efficacy in preclinical
models, as well as development of an efficient methodology for
monitoring toxicity in normal tissues, are required. This
combination-based therapeutic approach exploiting genome
maintenance mechanisms in RB as susceptibility factors may improve
the efficacy of current chemotherapy, and may at least partially
compensate for the lack of targeted therapies in RB by enabling
more efficient control of treatment-related toxicity.
As massive induction of DNA damages and genomic
instability to a lethal level is the basis for chemotherapy. Loss
of pRB in cancer cells has been associated with inherent
sensitivity to DNA-damaging agents due to the roles of pRB in
promoting DNA repair and genome stability (8). While RB1-deficient cancer
types respond to genotoxic drug-based therapies, findings in RB
(25,27) suggest that the sensitivity is not
strictly based on the compromised genome stability driven by pRB
loss that would affect the sensitivity threshold to genotoxic
drugs. The difference observed in RB may be related to the timing
and prevalence of RB1 loss. RB is initiated by biallelic
inactivation of the RB1 gene, and thus, the frequency of
RB1 mutations is exceptionally high, whereas the majority of
human cancer types acquire RB1 mutations during cancer
progression and the mutation frequency is relatively low (4). In the case of RB with a functional
p53 signaling pathway (38),
heightened genome maintenance mechanisms may be indispensable for
tumor initiation and progression by preventing the occurrence of
any lethal genomic defects, while tolerable genomic alterations may
still be allowed to occur for an improved chance of survival and
outgrowth. This suggests that RB1 deficiency in cancer does
not necessarily indicate a high degree of genome instability in the
cancer, and the context of RB1 loss during the course of
tumor development and the presence of other gene mutations should
be considered to better understand the etiology of the disease. The
information obtained for RB may provide novel insights into the
understanding of the biology for other cancer types with early pRB
loss and may guide toward improved therapeutic strategies for such
cancer types.
Not applicable.
Funding: No funding was received.
Not applicable.
CL and JKK conceived the idea for this review,
performed literature search, and contributed to writing and editing
of the manuscript. Data authentication is not applicable. Both
authors have read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Dimaras H, Corson TW, Cobrinik D, White A,
Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F
and Gallie BL: Retinoblastoma. Nat Rev Dis Primers. 1:150212015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Benavente CA and Dyer MA: Genetics and
epigenetics of human retinoblastoma. Annu Rev Pathol. 10:547–562.
2015. View Article : Google Scholar
|
|
3
|
Theriault BL, Dimaras H, Gallie BL and
Corson TW: The genomic landscape of retinoblastoma: A review. Clin
Exp Ophthalmol. 42:33–52. 2014. View Article : Google Scholar
|
|
4
|
Burkhart DL and Sage J: Cellular
mechanisms of tumour suppression by the retinoblastoma gene. Nat
Rev Cancer. 8:671–682. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dick FA, Goodrich DW, Sage J and Dyson NJ:
Non-canonical functions of the RB protein in cancer. Nat Rev
Cancer. 18:442–451. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Talluri S and Dick FA: Regulation of
transcription and chromatin structure by pRB: Here, there and
everywhere. Cell Cycle. 11:3189–3198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Uchida C: Roles of pRB in the regulation
of nucleosome and chromatin structures. Biomed Res Int.
2016:59597212016. View Article : Google Scholar
|
|
8
|
Knudsen ES, Pruitt SC, Hershberger PA,
Witkiewicz AK and Goodrich DW: Cell cycle and beyond: Exploiting
New RB1 controlled mechanisms for cancer therapy. Trends Cancer.
5:308–324. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Linn P, Kohno S, Sheng J, Kulathunga N, Yu
H, Zhang Z, Voon D, Watanabe Y and Takahashi C: Targeting RB1 loss
in cancers. Cancers (Basel). 13:37372021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar
|
|
11
|
Velez-Cruz R and Johnson DG: The
Retinoblastoma (RB) tumor suppressor: Pushing back against genome
instability on multiple fronts. Int J Mol Sci. 18:17762017.
View Article : Google Scholar
|
|
12
|
Cook R, Zoumpoulidou G, Luczynski MT,
Rieger S, Moquet J, Spanswick VJ, Hartley JA, Rothkamm K, Huang PH
and Mittnacht S: Direct involvement of retinoblastoma family
proteins in DNA repair by non-homologous end-joining. Cell Rep.
10:2006–2018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Velez-Cruz R, Manickavinayaham S, Biswas
AK, Clary RW, Premkumar T, Cole F and Johnson DG: RB localizes to
DNA double-strand breaks and promotes DNA end resection and
homologous recombination through the recruitment of BRG1. Genes
Dev. 30:2500–2512. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Manickavinayaham S, Velez-Cruz R, Biswas
AK, Chen J, Guo R and Johnson DG: The E2F1 transcription factor and
RB tumor suppressor moonlight as DNA repair factors. Cell Cycle.
19:2260–2269. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bester AC, Roniger M, Oren YS, Im MM,
Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS and Kerem B:
Nucleotide deficiency promotes genomic instability in early stages
of cancer development. Cell. 145:435–446. 2011. View Article : Google Scholar
|
|
16
|
Longworth MS, Herr A, Ji JY and Dyson NJ:
RBF1 promotes chromatin condensation through a conserved
interaction with the Condensin II protein dCAP-D3. Genes Dev.
22:1011–1024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Manning AL, Longworth MS and Dyson NJ:
Loss of pRB causes centromere dysfunction and chromosomal
instability. Genes Dev. 24:1364–1376. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Manning AL, Yazinski SA, Nicolay B, Bryll
A, Zou L and Dyson NJ: Suppression of genome instability in
pRB-deficient cells by enhancement of chromosome cohesion. Mol
Cell. 53:993–1004. 2014. View Article : Google Scholar
|
|
19
|
Coschi CH, Ishak CA, Gallo D, Marshall A,
Talluri S, Wang J, Cecchini MJ, Martens AL, Percy V, Welch I, et
al: Haploinsufficiency of an RB-E2F1-Condensin II complex leads to
aberrant replication and aneuploidy. Cancer Discov. 4:840–853.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coschi CH, Martens AL, Ritchie K, Francis
SM, Chakrabarti S, Berube NG and Dick FA: Mitotic chromosome
condensation mediated by the retinoblastoma protein is
tumor-suppressive. Genes Dev. 24:1351–1363. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ishak CA, Marshall AE, Passos DT, White
CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch
ID, et al: An RB-EZH2 complex mediates silencing of repetitive DNA
Sequences. Mol Cell. 64:1074–1087. 2016. View Article : Google Scholar
|
|
22
|
Gonzalo S, Garcia-Cao M, Fraga MF, Schotta
G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T
and Blasco MA: Role of the RB1 family in stabilizing histone
methylation at constitutive heterochromatin. Nat Cell Biol.
7:420–428. 2005. View Article : Google Scholar
|
|
23
|
Isaac CE, Francis SM, Martens AL, Julian
LM, Seifried LA, Erdmann N, Binne UK, Harrington L, Sicinski P,
Berube NG, et al: The retinoblastoma protein regulates pericentric
heterochromatin. Mol Cell Biol. 26:3659–3671. 2006. View Article : Google Scholar
|
|
24
|
Montoya-Durango DE, Ramos KA, Bojang P,
Ruiz L, Ramos IN and Ramos KS: LINE-1 silencing by retinoblastoma
proteins is effected through the nucleosomal and remodeling
deacetylase multiprotein complex. BMC Cancer. 16:382016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang J, Benavente CA, McEvoy J,
Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J,
et al: A novel retinoblastoma therapy from genomic and epigenetic
analyses. Nature. 481:329–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McEvoy J, Nagahawatte P, Finkelstein D,
Richards-Yutz J, Valentine M, Ma J, Mullighan C, Song G, Chen X,
Wilson M, et al: RB1 gene inactivation by chromothripsis in human
retinoblastoma. Oncotarget. 5:438–450. 2014. View Article : Google Scholar
|
|
27
|
Davies HR, Broad KD, Onadim Z, Price EA,
Zou X, Sheriff I, Karaa EK, Scheimberg I, Reddy MA, Sagoo MS, et
al: Whole-Genome sequencing of retinoblastoma reveals the diversity
of rearrangements disrupting RB1 and uncovers a treatment-related
mutational signature. Cancers (Basel). 13:7542021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kooi IE, Mol BM, Massink MP, Ameziane N,
Meijers-Heijboer H, Dommering CJ, van Mil SE, de Vries Y, van der
Hout AH, Kaspers GJ, et al: Somatic genomic alterations in
retinoblastoma beyond RB1 are rare and limited to copy number
changes. Sci Rep. 6:252642016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Afshar AR, Pekmezci M, Bloomer MM, Cadenas
NJ, Stevers M, Banerjee A, Roy R, Olshen AB, Van Ziffle J, Onodera
C, et al: Next-Generation sequencing of retinoblastoma identifies
pathogenic alterations beyond RB1 inactivation that correlate with
aggressive histopathologic features. Ophthalmology. 127:804–813.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Francis JH, Richards AL, Mandelker DL,
Berger MF, Walsh MF, Dunkel IJ, Donoghue MTA and Abramson DH:
Molecular changes in retinoblastoma beyond RB1: Findings from
next-generation sequencing. Cancers (Basel). 13:1492021. View Article : Google Scholar
|
|
31
|
Mendonca V, Evangelista AC, P Matta B, M
Moreira MÂ, Faria P, Lucena E and Seuanez HN: Molecular alterations
in retinoblastoma beyond RB1. Exp Eye Res. 211:1087532021.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Corson TW and Gallie BL: One hit, two
hits, three hits, more? Genomic changes in the development of
retinoblastoma. Genes Chromosomes Cancer. 46:617–634. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu J, Ottaviani D, Sefta M, Desbrousses
C, Chapeaublanc E, Aschero R, Sirab N, Lubieniecki F, Lamas G,
Tonon L, et al: A high-risk retinoblastoma subtype with stemness
features, dedifferentiated cone states and neuronal/ganglion cell
gene expression. Nat Commun. 12:55782021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grobner SN, Worst BC, Weischenfeldt J,
Buchhalter I, Kleinheinz K, Rudneva VA, Johann PD, Balasubramanian
GP, Segura-Wang M, Brabetz S, et al: The landscape of genomic
alterations across childhood cancers. Nature. 555:321–327. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alexandrov LB, Jones PH, Wedge DC, Sale
JE, Campbell PJ, Nik-Zainal S and Stratton MR: Clock-like
mutational processes in human somatic cells. Nat Genet.
47:1402–1407. 2015. View Article : Google Scholar
|
|
36
|
Polski A, Xu L, Prabakar RK, Gai X, Kim
JW, Shah R, Jubran R, Kuhn P, Cobrinik D, Hicks J and Berry JL:
Variability in retinoblastoma genome stability is driven by age and
not heritability. Genes Chromosomes Cancer. 59:584–590. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kato MV, Shimizu T, Ishizaki K, Kaneko A,
Yandell DW, Toguchida J and Sasaki MS: Loss of heterozygosity on
chromosome 17 and mutation of the p53 gene in retinoblastoma.
Cancer Lett. 106:75–82. 1996. View Article : Google Scholar
|
|
38
|
Kondo Y, Kondo S, Liu J, Haqqi T, Barnett
GH and Barna BP: Involvement of p53 and WAF1/CIP1 in
gamma-irradiation-induced apoptosis of retinoblastoma cells. Exp
Cell Res. 236:51–56. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu XL, Fang Y, Lee TC, Forrest D,
Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH and
Cobrinik D: Retinoblastoma has properties of a cone precursor tumor
and depends upon cone-specific MDM2 signaling. Cell. 137:1018–1031.
2009. View Article : Google Scholar
|
|
40
|
Xu XL, Singh HP, Wang L, Qi DL, Poulos BK,
Abramson DH, Jhanwar SC and Cobrinik D: Rb suppresses human
cone-precursor-derived retinoblastoma tumours. Nature. 514:385–388.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Laurie NA, Donovan SL, Shih CS, Zhang J,
Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, et al:
Inactivation of the p53 pathway in retinoblastoma. Nature.
444:61–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chakraborty S, Khare S, Dorairaj SK,
Prabhakaran VC, Prakash DR and Kumar A: Identification of genes
associated with tumorigenesis of retinoblastoma by microarray
analysis. Genomics. 90:344–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ganguly A and Shields CL: Differential
gene expression profile of retinoblastoma compared to normal
retina. Mol Vis. 16:1292–1303. 2010.PubMed/NCBI
|
|
44
|
Kapatai G, Brundler MA, Jenkinson H,
Kearns P, Parulekar M, Peet AC and McConville CM: Gene expression
profiling identifies different sub-types of retinoblastoma. Br J
Cancer. 109:512–525. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rajasekaran S, Nagarajha Selvan LD, Dotts
K, Kumar R, Rishi P, Khetan V, Bisht M, Sivaraman K, Krishnakumar
S, Sahoo D, et al: Non-coding and coding transcriptional profiles
are significantly altered in pediatric retinoblastoma tumors. Front
Oncol. 9:2212019. View Article : Google Scholar
|
|
46
|
Aubry A, Pearson JD, Huang K, Livne-Bar I,
Ahmad M, Jagadeesan M, Khetan V, Ketela T, Brown KR, Yu T, et al:
Functional genomics identifies new synergistic therapies for
retinoblastoma. Oncogene. 39:5338–5357. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Markey MP, Bergseid J, Bosco EE, Stengel
K, Xu H, Mayhew CN, Schwemberger SJ, Braden WA, Jiang Y, Babcock
GF, et al: Loss of the retinoblastoma tumor suppressor:
Differential action on transcriptional programs related to cell
cycle control and immune function. Oncogene. 26:6307–6318. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mayhew CN, Carter SL, Fox SR, Sexton CR,
Reed CA, Srinivasan SV, Liu X, Wikenheiser-Brokamp K, Boivin GP,
Lee JS, et al: RB loss abrogates cell cycle control and genome
integrity to promote liver tumorigenesis. Gastroenterology.
133:976–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Black EP, Huang E, Dressman H, Rempel R,
Laakso N, Asa SL, Ishida S, West M and Nevins JR: Distinct gene
expression phenotypes of cells lacking Rb and Rb family members.
Cancer Res. 63:3716–3723. 2003.PubMed/NCBI
|
|
50
|
Ren B, Cam H, Takahashi Y, Volkert T,
Terragni J, Young RA and Dynlacht BD: E2F integrates cell cycle
progression with DNA repair, replication, and G(2)/M checkpoints.
Genes Dev. 16:245–256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bracken AP, Ciro M, Cocito A and Helin K:
E2F target genes: Unraveling the biology. Trends Biochem Sci.
29:409–417. 2004. View Article : Google Scholar
|
|
52
|
Mun JY, Baek SW, Park WY, Kim WT, Kim SK,
Roh YG, Jeong MS, Yang GE, Lee JH, Chung JW, et al: E2F1 promotes
progression of bladder cancer by modulating RAD54L involved in
homologous recombination repair. Int J Mol Sci. 21:90252020.
View Article : Google Scholar
|
|
53
|
Sakthivel KM and Hariharan S: Regulatory
players of DNA damage repair mechanisms: Role in cancer
chemoresistance. Biomed Pharmacother. 93:1238–1245. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hosoya N and Miyagawa K: Targeting DNA
damage response in cancer therapy. Cancer Sci. 105:370–388. 2014.
View Article : Google Scholar
|
|
55
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar
|
|
56
|
Jeggo PA and Downs JA: Roles of chromatin
remodellers in DNA double strand break repair. Exp Cell Res.
329:69–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee C and Kim JK: Chromatin regulators in
retinoblastoma: Biological roles and therapeutic applications. J
Cell Physiol. 236:2318–2332. 2021. View Article : Google Scholar
|
|
58
|
Bronner C, Krifa M and Mousli M:
Increasing role of UHRF1 in the reading and inheritance of the
epigenetic code as well as in tumorogenesis. Biochem Pharmacol.
86:1643–1649. 2013. View Article : Google Scholar
|
|
59
|
Alhosin M, Omran Z, Zamzami MA, Al-Malki
AL, Choudhry H, Mousli M and Bronner C: Signalling pathways in
UHRF1-dependent regulation of tumor suppressor genes in cancer. J
Exp Clin Cancer Res. 35:1742016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tian Y, Paramasivam M, Ghosal G, Chen D,
Shen X, Huang Y, Akhter S, Legerski R, Chen J, Seidman MM, et al:
UHRF1 contributes to DNA damage repair as a lesion recognition
factor and nuclease scaffold. Cell Rep. 10:1957–1966. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liang CC, Zhan B, Yoshikawa Y, Haas W,
Gygi SP and Cohn MA: UHRF1 is a sensor for DNA interstrand
crosslinks and recruits FANCD2 to initiate the Fanconi anemia
pathway. Cell Rep. 10:1947–1956. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang H, Liu H, Chen Y, Yang X, Wang P,
Liu T, Deng M, Qin B, Correia C, Lee S, et al: A cell
cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand
break repair pathway choice. Nat Commun. 7:102012016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mancini M, Magnani E, Macchi F and
Bonapace IM: The multi-functionality of UHRF1: Epigenome
maintenance and preservation of genome integrity. Nucleic Acids
Res. 49:6053–6068. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Muto M, Kanari Y, Kubo E, Takabe T,
Kurihara T, Fujimori A and Tatsumi K: Targeted disruption of Np95
gene renders murine embryonic stem cells hypersensitive to DNA
damaging agents and DNA replication blocks. J Biol Chem.
277:34549–34555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
He H, Lee C and Kim JK: UHRF1 depletion
sensitizes retinoblastoma cells to chemotherapeutic drugs via
downregulation of XRCC4. Cell Death Dis. 9:1642018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kim JK, Kan G, Mao Y, Wu Z, Tan X, He H
and Lee C: UHRF1 downmodulation enhances antitumor effects of
histone deacetylase inhibitors in retinoblastoma by augmenting
oxidative stress-mediated apoptosis. Mol Oncol. 14:329–346. 2020.
View Article : Google Scholar
|
|
67
|
Sharma V, Collins LB, Chen TH, Herr N,
Takeda S, Sun W, Swenberg JA and Nakamura J: Oxidative stress at
low levels can induce clustered DNA lesions leading to NHEJ
mediated mutations. Oncotarget. 7:25377–25390. 2016. View Article : Google Scholar
|
|
68
|
Karanjawala ZE, Murphy N, Hinton DR, Hsieh
CL and Lieber MR: Oxygen metabolism causes chromosome breaks and is
associated with the neuronal apoptosis observed in DNA
double-strand break repair mutants. Curr Biol. 12:397–402. 2002.
View Article : Google Scholar
|
|
69
|
Mao Y, Sun Y, Wu Z, Zheng J, Zhang J, Zeng
J, Lee C and Kim JK: Targeting of histone methyltransferase DOT1L
plays a dual role in chemosensitization of retinoblastoma cells and
enhances the efficacy of chemotherapy. Cell Death Dis. 12:11412021.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Feng Q, Wang H, Ng HH, Erdjument-Bromage
H, Tempst P, Struhl K and Zhang Y: Methylation of H3-lysine 79 is
mediated by a new family of HMTases without a SET domain. Curr
Biol. 12:1052–1058. 2002. View Article : Google Scholar
|
|
71
|
Wood K, Tellier M and Murphy S: DOT1L and
H3K79 methylation in transcription and genomic stability.
Biomolecules. 8:112018. View Article : Google Scholar
|
|
72
|
Wakeman TP, Wang Q, Feng J and Wang XF:
Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA
damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J.
31:2169–2181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kari V, Raul SK, Henck JM, Kitz J, Kramer
F, Kosinsky RL, Ubelmesser N, Mansour WY, Eggert J, Spitzner M, et
al: The histone methyltransferase DOT1L is required for proper DNA
damage response, DNA repair, and modulates chemotherapy
responsiveness. Clin Epigenetics. 11:42019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu W, Deng L, Song Y and Redell M: DOT1L
inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS One.
9:e982702014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chau KY, Manfioletti G, Cheung-Chau KW,
Fusco A, Dhomen N, Sowden JC, Sasabe T, Mukai S and Ono SJ:
Derepression of HMGA2 gene expression in retinoblastoma is
associated with cell proliferation. Mol Med. 9:154–165. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Palmieri D, Valentino T, D'Angelo D, De
Martino I, Postiglione I, Pacelli R, Croce CM, Fedele M and Fusco
A: HMGA proteins promote ATM expression and enhance cancer cell
resistance to genotoxic agents. Oncogene. 30:3024–3035. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Natarajan S, Hombach-Klonisch S, Droge P
and Klonisch T: HMGA2 inhibits apoptosis through interaction with
ATR-CHK1 signaling complex in human cancer cells. Neoplasia.
15:263–280. 2013. View Article : Google Scholar
|
|
78
|
Nalini V, Deepa PR, Raguraman R, Khetan V,
Reddy MA and Krishnakumar S: Targeting HMGA2 in Retinoblastoma
Cells in vitro using the aptamer strategy. Ocul Oncol Pathol.
2:262–269. 2016. View Article : Google Scholar
|
|
79
|
Kollarovic G, Topping CE, Shaw EP and
Chambers AL: The human HELLS chromatin remodelling protein promotes
end resection to facilitate homologous recombination and
contributes to DSB repair within heterochromatin. Nucleic Acids
Res. 48:1872–1885. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Costelloe T, Louge R, Tomimatsu N,
Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry
A, Burma S, et al: The yeast Fun30 and human SMARCAD1 chromatin
remodellers promote DNA end resection. Nature. 489:581–584. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zocchi L, Mehta A, Wu SC, Wu J, Gu Y, Wang
J, Suh S, Spitale RC and Benavente CA: Chromatin remodeling protein
HELLS is critical for retinoblastoma tumor initiation and
progression. Oncogenesis. 9:252020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Manning AL and Dyson NJ: RB: Mitotic
implications of a tumour suppressor. Nat Rev Cancer. 12:220–226.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pappas L, Xu XL, Abramson DH and Jhanwar
SC: Genomic instability and proliferation/survival pathways in
RB1-deficient malignancies. Adv Biol Regul. 64:20–32. 2017.
View Article : Google Scholar
|
|
84
|
Salehi F, Kovacs K, Scheithauer BW, Lloyd
RV and Cusimano M: Pituitary tumor-transforming gene in endocrine
and other neoplasms: A review and update. Endocr Relat Cancer.
15:721–743. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Holland AJ and Cleveland DW: Boveri
revisited: Chromosomal instability, aneuploidy and tumorigenesis.
Nat Rev Mol Cell Biol. 10:478–487. 2009. View Article : Google Scholar
|
|
86
|
Gong X, Du J, Parsons SH, Merzoug FF,
Webster Y, Iversen PW, Chio LC, Van Horn RD, Lin X, Blosser W, et
al: Aurora A kinase inhibition is synthetic lethal with loss of the
RB1 tumor suppressor gene. Cancer Discov. 9:248–263. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Oser MG, Fonseca R, Chakraborty AA, Brough
R, Spektor A, Jennings RB, Flaifel A, Novak JS, Gulati A, Buss E,
et al: Cells lacking the RB1 tumor suppressor gene are
hyperdependent on Aurora B kinase for survival. Cancer Discov.
9:230–247. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Willems E, Dedobbeleer M, Digregorio M,
Lombard A, Lumapat PN and Rogister B: The functional diversity of
Aurora kinases: A comprehensive review. Cell Div. 13:72018.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Borah NA, Sradhanjali S, Barik MR, Jha A,
Tripathy D, Kaliki S, Rath S, Raghav SK, Patnaik S, Mittal R and
Reddy MM: Aurora Kinase B expression, its regulation and
therapeutic targeting in human retinoblastoma. Invest Ophthalmol
Vis Sci. 62:162021. View Article : Google Scholar
|
|
90
|
Singh L, Pushker N, Sen S, Singh MK,
Chauhan FA and Kashyap S: Prognostic significance of polo-like
kinases in retinoblastoma: Correlation with patient outcome,
clinical and histopathological parameters. Clin Exp Ophthalmol.
43:550–557. 2015. View Article : Google Scholar
|
|
91
|
Ma H, Nie C, Chen Y, Li J, Xie Y, Tang Z,
Gao Y, Ai S, Mao Y, Sun Q and Lu R: Therapeutic Targeting PLK1 by
ON-01910.Na is effective in local treatment of retinoblastoma.
Oncol Res. 28:745–761. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Takaki T, Trenz K, Costanzo V and
Petronczki M: Polo-like kinase 1 reaches beyond
mitosis-cytokinesis, DNA damage response, and development. Curr
Opin Cell Biol. 20:650–660. 2008. View Article : Google Scholar
|
|
93
|
Sun J, Xi HY, Shao Q and Liu QH:
Biomarkers in retinoblastoma. Int J Ophthalmol. 13:325–341. 2020.
View Article : Google Scholar
|
|
94
|
Chai P, Jia R, Li Y, Zhou C, Gu X, Yang L,
Shi H, Tian H, Lin H, Yu J, et al: Regulation of epigenetic
homeostasis in uveal melanoma and retinoblastoma. Prog Retin Eye
Res. Dec 1–2021.(Epub ahead of print). View Article : Google Scholar
|
|
95
|
Chan HS, Gallie BL, Munier FL and Beck
Popovic M: Chemotherapy for retinoblastoma. Ophthalmol Clin North
Am. 1855–63. (viii)2005. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gombos DS, Hungerford J, Abramson DH,
Kingston J, Chantada G, Dunkel IJ, Antoneli CB, Greenwald M, Haik
BG, Leal CA, et al: Secondary acute myelogenous leukemia in
patients with retinoblastoma: Is chemotherapy a factor?
Ophthalmology. 114:1378–1383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mulvihill A, Budning A, Jay V, Vandenhoven
C, Heon E, Gallie BL and Chan HS: Ocular motility changes after
subtenon carboplatin chemotherapy for retinoblastoma. Arch
Ophthalmol. 121:1120–1124. 2003. View Article : Google Scholar
|
|
98
|
Zoumpoulidou G, Alvarez-Mendoza C, Mancusi
C, Ahmed RM, Denman M, Steele CD, Tarabichi M, Roy E, Davies LR,
Manji J, et al: Therapeutic vulnerability to PARP1,2 inhibition in
RB1-mutant osteosarcoma. Nat Commun. 12:70642021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Basavapathruni A, Jin L, Daigle SR, Majer
CR, Therkelsen CA, Wigle TJ, Kuntz KW, Chesworth R, Pollock RM,
Scott MP, et al: Conformational adaptation drives potent, selective
and durable inhibition of the human protein methyltransferase
DOT1L. Chem Biol Drug Des. 80:971–980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Waters NJ: Preclinical Pharmacokinetics
and pharmacodynamics of pinometostat (EPZ-5676), a First-in-Class,
small Molecule S-Adenosyl methionine competitive inhibitor of
DOT1L. Eur J Drug Metab Pharmacokinet. 42:891–901. 2017. View Article : Google Scholar
|
|
101
|
Stein EM, Garcia-Manero G, Rizzieri DA,
Tibes R, Berdeja JG, Savona MR, Jongen-Lavrenic M, Altman JK,
Thomson B, Blakemore SJ, et al: The DOT1L inhibitor pinometostat
reduces H3K79 methylation and has modest clinical activity in adult
acute leukemia. Blood. 131:2661–2669. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Shields CL, Manjandavida FP, Lally SE,
Pieretti G, Arepalli SA, Caywood EH, Jabbour P and Shields JA:
Intra-arterial chemotherapy for retinoblastoma in 70 eyes: Outcomes
based on the international classification of retinoblastoma.
Ophthalmology. 121:1453–1460. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Munier FL, Gaillard MC, Balmer A, Soliman
S, Podilsky G, Moulin AP and Beck-Popovic M: Intravitreal
chemotherapy for vitreous disease in retinoblastoma revisited: From
prohibition to conditional indications. Br J Ophthalmol.
96:1078–1083. 2012. View Article : Google Scholar
|
|
104
|
Ghassemi F, Shields CL, Ghadimi H,
Khodabandeh A and Roohipoor R: Combined intravitreal melphalan and
topotecan for refractory or recurrent vitreous seeding from
retinoblastoma. JAMA Ophthalmol. 132:936–941. 2014. View Article : Google Scholar
|
|
105
|
Lee JE and Kim MY: Cancer epigenetics:
Past, present and future. Semin Cancer Biol. Mar 31–2021.(Epub
ahead of print). View Article : Google Scholar
|
|
106
|
He L and Lomberk G: Collateral Victim or
Rescue Worker?-The role of histone methyltransferases in DNA damage
repair and their targeting for therapeutic opportunities in cancer.
Front Cell Dev Biol. 9:7351072021. View Article : Google Scholar
|
|
107
|
Unoki M, Nishidate T and Nakamura Y:
ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG
through its SRA domain. Oncogene. 23:7601–7610. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ganesan A, Arimondo PB, Rots MG, Jeronimo
C and Berdasco M: The timeline of epigenetic drug discovery: From
reality to dreams. Clin Epigenetics. 11:1742019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chun AW, Cosenza SC, Taft DR and Maniar M:
Preclinical pharmacokinetics and in vitro activity of ON 01910.Na,
a novel anti-cancer agent. Cancer Chemother Pharmacol. 65:177–186.
2009. View Article : Google Scholar
|
|
110
|
Ohnuma T, Lehrer D, Ren C, Cho SY, Maniar
M, Silverman L, Sung M, Gretz HF III, Benisovich V, Navada S, et
al: Phase 1 study of intravenous rigosertib (ON 01910.Na), a novel
benzyl styryl sulfone structure producing G2/M arrest and
apoptosis, in adult patients with advanced cancer. Am J Cancer Res.
3:323–338. 2013.PubMed/NCBI
|
|
111
|
O'Neil BH, Scott AJ, Ma WW, Cohen SJ,
Aisner DL, Menter AR, Tejani MA, Cho JK, Granfortuna J, Coveler L,
et al: A phase II/III randomized study to compare the efficacy and
safety of rigosertib plus gemcitabine versus gemcitabine alone in
patients with previously untreated metastatic pancreatic cancer.
Ann Oncol. 26:1923–1929. 2015. View Article : Google Scholar
|
|
112
|
Bowles DW, Diamond JR, Lam ET, Weekes CD,
Astling DP, Anderson RT, Leong S, Gore L, Varella-Garcia M, Vogler
BW, et al: Phase I study of oral rigosertib (ON 01910.Na), a dual
inhibitor of the PI3K and Plk1 pathways, in adult patients with
advanced solid malignancies. Clin Cancer Res. 20:1656–1665. 2014.
View Article : Google Scholar : PubMed/NCBI
|