Introduction
Liver cancer is one of the most common malignant
tumors of the digestive system and exhibits characteristics of
high-grade malignancy, rapid progression, high recurrence rates and
a high probability of metastasis (1,2).
According to statistics, liver cancer will become a global public
health challenge for >1 million patients in 2025 (3). It is generally acknowledged that the
Warburg effect (4), and metabolic
disturbances of glutamine (5) and
fatty acids (6), are able to
accelerate the progression of cancer and the reprogramming of cell
metabolism leads to uncontrollable proliferation activity. In
primary liver cancer, metabolic diseases, such as obesity (7), non-alcoholic fatty liver disease
(8) and type 2 diabetes mellitus
(9) have been listed as high-risk
factors. Of note, whether in the initiation or promotion stages of
hepatocarcinogenesis, the stability of the metabolic environment
determines the final outcome.
As a member of the apolipoprotein family,
apolipoprotein M (ApoM) participates in the synthesis of
high-density lipoprotein (HDL) and the reverse transport of
cholesterol (10). In view of the
contribution of ApoM in protecting against insulin resistance
(11), exhibiting
antiatherosclerotic functions (12) and reducing liver lipid accumulation
(13), it is considered to be an
important factor in regulating glucose and lipid metabolism. In
recent years, accumulating evidence has indicated that ApoM is
associated with the occurrence and development of primary liver
cancer. For instance, the mRNA and protein levels of ApoM in liver
cancer tissues are significantly reduced compared with those in
neighboring tissues (14);
hsa-microRNA (miR)-573, as a potential target of ApoM, is able to
reduce ApoM expression levels with an accompanying reduction in
hepatoma cell apoptosis (15). In
addition, ApoM functions as a tumor suppressor to inhibit the
growth and metastasis of SMMC7721 cells via vitamin D receptor
signaling (16). In summary, ApoM
exhibits a positive effect in suppressing tumor progression. Of
note, results of a previous study by our group demonstrated that
ApoM-knockout mice formed tumors faster under the induction of
N-nitrosodiethylamine, which may indicate that ApoM also has an
important inhibitory effect in the process of liver cancer
(17).
Based on the above viewpoints, ApoM is an
apolipoprotein that inhibits the occurrence and development of
liver cancer, and it is closely related to the body's glucose and
lipid metabolism. At present, the metabolic level and tumor
metabolic microenvironment are still the focus of cancer related
research. But whether ApoM affects the development of liver cancer
through glycolipid metabolism remains unclear. Of note, the results
of a previous study by our group demonstrated that deficiency of
the ApoM gene causes damage to autophagy activity in the liver and
eventually leads to lipid accumulation (18). Furthermore, the expression levels
of sterol regulatory element-binding protein 1 (SREBF1; also known
as SREBP1) are markedly upregulated in this process (18). ApoM has been proven to regulate
various biological processes, including lipid biosynthesis
(19), insulin resistance
(20) and tumor growth (21). Considering that glycolysis is a
typical metabolic pathway in cancer cells, the present study aimed
to investigate whether ApoM regulates glycolysis through the SREBF1
pathway, thus affecting the progression of liver cancer. The
present study aimed to further elucidate the potential association
between ApoM, glycolysis and primary liver cancer and explored the
potential mechanism by which ApoM inhibits the occurrence and
development of liver cancer.
Materials and methods
Cell culture
Huh-7 cells (BeNa Culture Collection) and Mhcc97h
cells (Guangzhou Saiku) were cultured in high-sugar DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(Shanghai ExCell Biology, Inc.) and 1% penicillin/streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) and were incubated at 37°C
in an atmosphere with 5% CO2. As for the reason for
choosing Huh7 and Mhcc97h cells, it was observed that SREBF1 was
highly expressed in Huh7 and Mhcc97h cells (22). Small interfering (si)RNA (Guangzhou
RiboBio Co., Ltd.) was transfected into cells using
Lipofectamine® 3000 reagent (cat. no. L3000150; Thermo
Fisher Scientific, Inc.). The sequences were as follows:
si-negative control (NC), 5′-GGCTCTAGAAAAGCCTATGC-3′ (this control
was non-targeting), si-SREBF1, 5′-CGGAGAAGCTGCCTATCAA-3′ and
si-ApoM, 5′-GAGCACAGATCTCAGAACT-3′.
Dual-luciferase activity
detection
The Ensembl database (http://asia.ensembl.org/index.html) was used to query
the promoter sequence of ATP-dependent 6-phosphofructokinase, liver
type (PFKL). The JASPAR 2022 database (https://jaspar.genereg.net/) found that SREBF1 may be
a transcription factor of PFKL and has a binding site. The highest
scoring sequences were selected an constructed synthetically into
pgl3-basic (Nanjing Qingke Co., Ltd.). The pgl3-basic and
pEnCMV-SREBF1 (human)-HA plasmids (Nanjing Qingke Co., Ltd.) were
then co-transfected into 293T cells. Dual-Luciferase Reporter Gene
Assay kit (cat. no. E1910; Glomax; Promega Corporation) was used to
detect both the firefly and Renilla luciferase gene
activity.
EDU staining assay
Each group of cells (si-NC, si-SREBF1, pcDNA3.1-NC,
pcDNA3.1-SREBF1, si-ApoM−/−+si-SREBF1 and
si-SREBF1+pcDNA3.1-PFKL) was stained according to the instructions
of the EDU Cell Proliferation kit (cat. no. C10310-1; Guangzhou
RiboBio Co., Ltd.). Images were obtained using an inverted
fluorescence microscope (Olympus Corporation). ImageJ software
V1.8.0.112 [National Institutes of Health (NIH)] was used for data
analysis.
Western blot analysis
RIPA lysis buffer (cat. no. BL651A; Biosharp Life
Sciences) and PMSF (cat. no. BL507A; Biosharp Life Sciences) were
used for tissue and cell protein extraction, and the protein
concentration was measured using a NanoDrop® 2000
mini-spectrophotometer (Thermo Fisher Scientific, Inc.). Proteins
(60 µg) were separated using SDS-PAGE (10 or 12% gel), transferred
to a PVDF membrane (MilliporeSigma) and subsequently blocked with
blocking solution (cat. no. P0023B; Beyotime Institute of
Biotechnology) for 10 min at room temperature. Subsequently,
samples were incubated with the appropriate primary antibody
solution overnight at 4°C. The next day, the PVDF membranes were
incubated in secondary antibody solution for 2 h at room
temperature (both 1:3,000 dilution; cat. nos. BL001A and BL003A;
Biosharp Life Sciences). ECL chemiluminescent fluid (cat. no.
BL520A; Biosharp Life Sciences) and an imaging system
(ShanghaiTanon-5200 Co., Ltd.) were used for exposure. The
following antibodies were used: Anti-ApoM (cat. no. A5336; 1:1,000
dilution; ABclonal Biotech Co., Ltd.), anti-PFKL (cat. no. A7708;
1:1,000 dilution; ABclonal Biotech Co., Ltd.) and anti-SREBP1 (cat.
no. ab138663; 1:1,000 dilution; Abcam) antibody were used to react
with human hepatoma cells, while anti-SREBP1 (cat. no. ab28481;
1:1,000 dilution; Abcam) antibody was used to react with mouse
tissue proteins and β-actin (A1978; 1:5,000 dilution;
MilliporeSigma). ImageJ software V1.8.0.112 (NIH) was used for
statistical analysis.
Transwell migration and invasion
assays
Following cell transfection with si-NC, si-SREBF1,
pcDNA3.1-NC, pcDNA3.1-SREBF1, si-ApoM−/−+si-SREBF1 or
si-SREBF1+ pcDNA3.1-PFKL plasmids for 48 h at 37°C, the cell
concentration was adjusted to 1×105 cells/ml using
serum-free cell culture medium and 200 µl cell suspension was added
to the upper chamber for the migration assay (cat. no. 3422;
Corning, Inc; PC membrane, 6.5 mm; pore size, 8.0 µm). A total of
600 µl cell culture medium with 20% serum was added to the lower
chamber and plates were cultured for 48 h. Cells that passed
through the membrane were stained with 4% paraformaldehyde (Phygene
Brotechnology Co., Ltd.) and 0.1% crystal violet solution (cat. no.
C0121; Beyotime Institute of Biotechnology.) for 10 min at room
temperature. Finally, use a cotton swab to wipe the cells that have
not crossed the membrane. Migrated cells were counted under a
microscope (Olympus Corporation). For the invasion assay, Dilute
Matrigel® to 200 µg/ml with PBS, and 50 µl
Matrigel® was added to the upper chamber prior to
incubation at 37°C for 2 h. The remaining steps were followed in an
identical manner to those of the migration assay above. ImageJ
software V1.8.0.112 (National Institutes of Health) was used for
data analysis.
Wound-healing assay
Following cell transfection with si-NC, si-SREBF1,
pcDNA3.1-NC, pcDNA3.1-SREBF1, si-ApoM−/−+ si-SREBF1 or
si-SREBF1+pcDNA3.1-PFKL plasmids for 48 h, a 200-µl pipette tip was
used to make a linear scratch on the cell monolayers. Cells were
subsequently washed three times with PBS and cultured in DMEM.
After 48 h of incubation, the width of the gap refilled by the
cells was measured and recorded, and the wound-healing rate was
calculated. ImageJ software V1.8.0.112 (NIH) was used for data
analysis.
Animals
The present study was approved by the Experimental
Animal Welfare and Ethics Committee of Wannan Medical College
(approval no. LLSC-2020-001). According to literature reports,
there are more new cases of liver cancer in Chinese males than in
females (23), and they are more
likely to get liver cancer. Therefore, male mice were selected to
establish the liver cancer induction model. C57BL/6J mice were
purchased from the Shanghai Model Organisms Center. The
experimental mice were reared in a specific pathogen-free area at
the Experimental Animal Center of Wannan Medical College and kept
at a constant temperature of 22–24°C, humidity of 38% and a 12-h
light/dark cycle. The animal cage and drinking water bottle were
subjected to high-temperature and high-pressure sterilization
(121°C, 30 min). Mice had free access to food and water and were
regularly observed and cared for every day. Furthermore, 40
8-week-old male WT mice (body weight, 20–23 g; Qinglongshan Animal
Farm) were randomly divided into two groups by intraperitoneal
injection of N-nitrosodiethylamine (35 mg/kg, Shanghai McLean Co.,
Ltd.) solution and an equal volume of normal saline once a week
until liver tumors developed. The same applies to the experimental
grouping of ApoM−/− mice. During this period, three mice
were randomly selected from each group every month. Mice were
anesthetized by intraperitoneal injection of 1% sodium
pentobarbital (50 mg/kg) and blood was collected from the medial
canthus. All animals were killed by cervical dislocation and a part
of the liver tissue was taken out and placed in 10% neutral
formalin solution (Jiangsu Siyan Biotechnology), and another part
was stored in a −80°C refrigerator. The experimental end-point was
the appearance of relevant indications of clinical symptoms (such
as the appearance of liver tumor). Selection of humane endpoints:
If the relevant indications of clinical symptoms in the experiment
had not yet appeared but the body weight of the mouse had decreased
by >20% and the mouse was hunched, trembled, distanced from the
group and unable to eat normally, the mouse was euthanized. Absence
of heartbeat and breathing, and the disappearance of reflexes were
used as the criteria for confirming death of the mice.
Detection of lactic acid (LA), ATP,
triglyceride (TG), total cholesterol (T-CHO), HDL cholesterol
(HDL-C) and low-density lipoprotein cholesterol (LDL-C) content in
tissues
The liver issues of WT healthy mice and WT
tumor-forming mice were obtained. The levels of LA, ATP, TG, T-CHO,
HDL-C and LDL-C were determined in tissues using kits purchased
from Beijing Solarbio Science & Technology Co., Ltd. (cat. no.
BC2235) and Nanjing Jiancheng Bioengineering Institute (cat. nos.
A095-1-1, A110-1-1, A111-1-1, A112-1-1 and A113-1-1), according to
the manufacturers' protocols.
H&E staining
Tissues were fixed in 10% formalin for 48 h at room
temperature, embedded in paraffin and cut into 4-µM sections.
Following deparaffinization and rehydration, the sections were
stained using H&E and the morphology was observed under a
microscope (Olympus Corporation).
Immunohistochemistry
The liver tissues of WT healthy mice and WT
tumor-forming mice were embedded in paraffin and the sections were
subsequently dissected into thin slices and deparaffinized in
xylene. Tissue slides were incubated with SREBF1 antibody (cat. no.
ab28481; 1:150 dilution; Abcam) overnight at 4°C. Following primary
incubation, the slides were incubated with the secondary antibody
(cat. no. GB23303; 1:200 dilution; Servicebio, Co., Ltd.) at 37°C
for 50 min. The slides were subsequently incubated with DAB (1:50
dilution; Servicebio, Co., Ltd.) to visualize the staining. Samples
were counterstained using hematoxylin solution (Servicebio, Co.,
Ltd.) for 90 sec and then differentiated using 1% hydrochloric acid
alcohol for several seconds at room temperature. The slides were
mounted using neutral gum (cat. no. G1403; Servicebio, Co., Ltd.)
prior to being placed under a microscope (Olympus Corporation) to
observe the expression of SREBF1 protein in each tissue.
Image-Pro-Plus 6.0 software (Media Cybernetics, Inc.) was used for
data analysis.
The Cancer Genome Atlas (TCGA) data
screening
R software (version 3.6.3) [DESeq2 (version 1.26.0)
25516281] was used to enter the Level 3 HTSeq-Counts RNASeqV2 data
in TCGA (https://portal.gdc.cancer.gov/). The hepatocellular
carcinoma project and the target molecule ApoM (ENSG00000204444)
were used for screening. The expression levels of SREBF1 and SREBF2
were calculated according to low and high expression levels of
ApoM.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 8.0 (GraphPad Software, Inc.) and SPSS 26.0 (IBM Corporation)
software. A total of three parallel experiments were set up in each
group and these were performed as three repeats. Values are
expressed as the mean ± standard deviation. The Kolmogorov-Smirnov
test was applied to determine compliance with a normal
distribution. Pairwise comparisons between groups in the presence
of multiple groups were performed using one-way ANOVA followed by
Tukey's post-hoc test. An unpaired Student's t-tests was used to
determine significant differences between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
N-nitrosodiethylamine induces high
expression of SREBF1 in liver cancer tissue derived from
tumor-forming mice
N-nitrosodiethylamine was used to induce liver
cancer in mice. Mice induced by N-nitrosodiethylamine developed up
to two tumors in the liver, but more frequently, one tumor was
formed. The tumor diameter in mice with liver tumors was 0.3-0.6 cm
(experimental period, 5–6 months) (Fig. 1A). In WT tumor-forming mice, it was
observed that the levels of LDL-C, T-CHO, TG and ATP increased,
while the levels of HDL-C decreased significantly (Fig. 1B-F), compared with the same
indicators in liver tissue of healthy WT mice. Western blot
analysis suggested that, compared with that in healthy WT mouse
liver tissue, the expression of SREBF1 in liver cancer tissue was
significantly increased and the expression of ApoM was
significantly decreased (Fig. 1J).
Furthermore, the results of the immunohistochemical analysis
further verified that WT tumor-forming mice exhibited increased
expression levels of SREBF1 compared with those of healthy WT mice
(Fig. 1H).
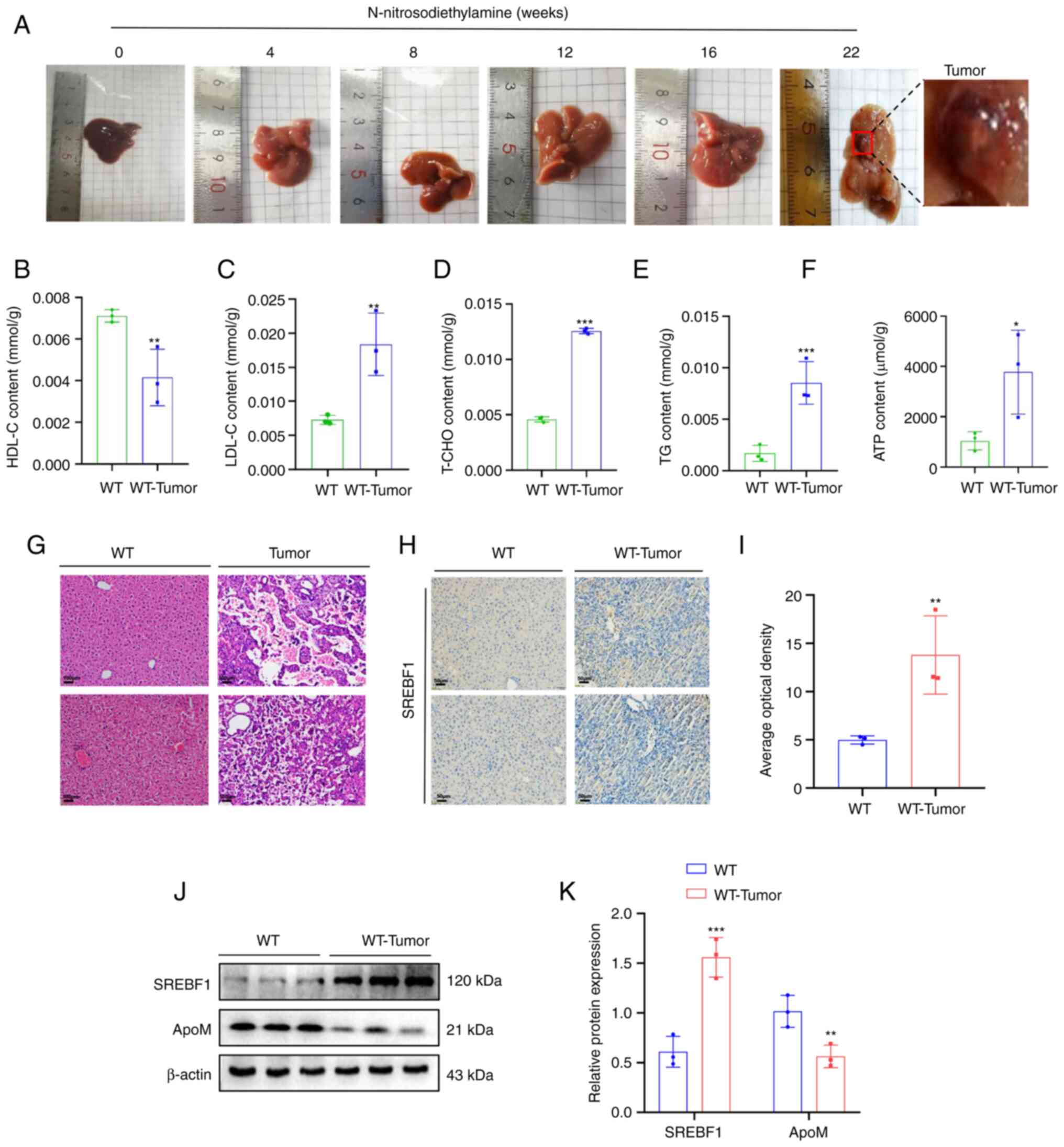 | Figure 1.High expression of SREBF1 in
N-nitrosodiethylamine-induced mouse hepatocellular carcinoma
tissues. (A) Images of livers from mouse models with
N-nitrosodiethylamine-induced liver cancer tumors. (B-F) A test kit
was utilized to detect the content of (B) HDL-C, (C) LDL-C, (D)
T-CHO, (E) TG and (F) ATP in liver cancer tissue of WT mice and
liver tissue from healthy mice. (G) H&E staining revealed the
morphology of liver tissue in mice prior to and after induction
with N-nitrosodiethylamine (scale bar, 100 µm). (H) SREBF1
expression levels were determined using immunohistochemistry (scale
bar, 50 µm). (J and K) Western blot analysis was performed to
evaluate the expression levels of SREBF1 and ApoM in liver cancer
tissue of WT mice and liver tissue from healthy mice. (J)
Representative western blots and (K) quantified results. Analysis
in each group was performed three times in parallel. *P<0.05,
**P<0.03, ***P<0.01 vs. WT. HDL-C, high-density lipoprotein
cholesterol; LDL-C, low-density lipoprotein cholesterol; T-CHO,
total cholesterol; TG, triglyceride; WT, wildtype; SREBF1, sterol
regulatory element-binding protein 1; ApoM, apolipoprotein M. |
ApoM knockout promotes SREBF1 to
regulate the expression of PFKL
Results of a previous study by our group
demonstrated that ApoM gene knockout in promoted the expression of
SREBF1 in the liver (18). In
addition, the levels of lactic acid in liver cancer tissue and
serum of mice with liver cancer in which the ApoM gene was
inhibited were significantly higher than those in WT mice with
liver cancer (Fig. S1). These
results suggested that the levels of glycolysis were increased. In
order to further explore whether ApoM affects the expression levels
of SREBF1 and PFKL in liver cancer cells, ApoM was silenced in
liver cancer cells and the expression levels of SREBF1 and PFKL
were markedly increased in Huh-7 and Mhcc97h cells (Fig. 2A-D). Following overexpression of
the ApoM gene, the expression levels of SREBF1 and PFKL were
decreased in Huh-7 and Mhcc97h cells (Fig. 2E-H). In addition, following SREBF1
gene knockout in liver cancer cells, the expression of PFKL in
Huh-7 cells and Mhcc97h cells was significantly decreased (Fig. 2I-L). Following overexpression of
the SREBF1 gene, the expression levels of PFKL were increased in
Huh-7 and Mhcc97h cells (Fig.
2M-P). Western blot analysis validated the PFKL overexpression
model in Huh-7 and Mhcc97h cells (Fig.
2Q-T). In order to explore whether the transcription factor
SREBF1 has the ability to regulate PFKL at the transcriptional
level, the JASPAR database was initially used to predict the
binding sites. Of note, the results of the luciferase-based gene
reporter assay demonstrated that SREBF1 enhanced PFKL promoter
activity (Fig. 2U and V).
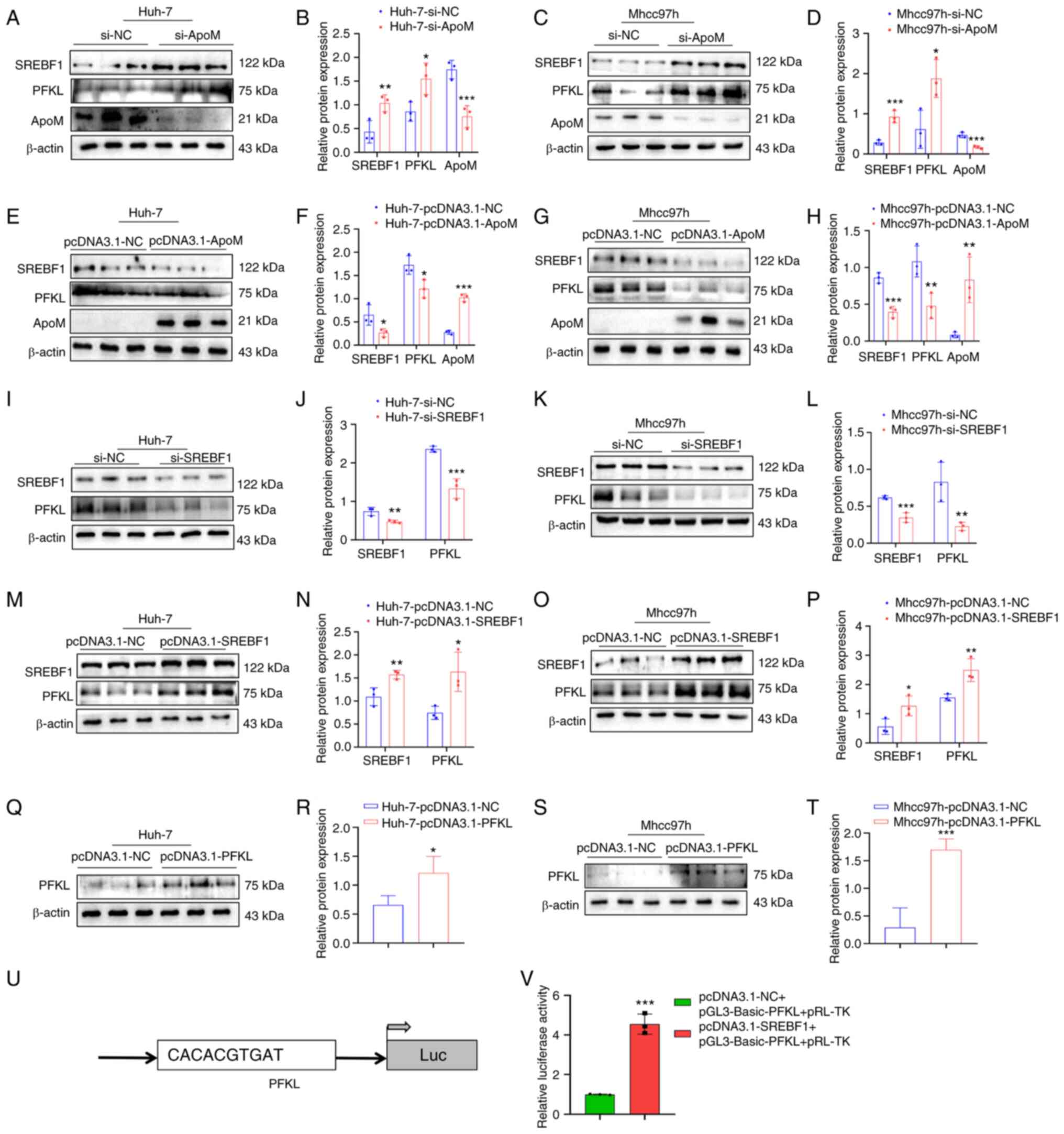 | Figure 2.SREBF1 is able to regulate the
expression of PFKL. (A-H) Detection of the expression levels of
SREBF1, PFKL and ApoM in Huh-7 and Mhcc97h cells using western blot
analysis after knockdown or overexpression of ApoM. (A)
Representative western blot image for Huh-7 cells with ApoM
knockdown and (B) quantified expression levels. (C) Representative
western blot image for Mhcc97h cells with ApoM knockdown and (D)
quantified expression levels. (E) Representative western blot image
for Huh-7 cells with ApoM overexpression and (F) quantified
expression levels. (G) Representative western blot image for
Mhcc97h cells with ApoM overexpression and (H) quantified
expression levels. (I-P) Detection of the expression levels of
SREBF1 and PFKL in Huh-7 and Mhcc97h cells using western blot
analysis after knockdown or overexpression of SREBF1. (I)
Representative western blot image for Huh-7 cells with SREBF1
knockdown and (J) quantified expression levels. (K) Representative
western blot image for Mhcc97h cells with SREBF1 knockdown and (L)
quantified expression levels. (M) Representative western blot image
for Huh-7 cells with SREBF1 overexpression and (N) quantified
expression levels. (O) Representative western blot image for
Mhcc97h cells with SREBF1 overexpression and (P) quantified
expression levels. (Q-T) Western blot analysis was used to validate
the PFKL overexpression model in Huh-7 cells and Mhcc97h cells. (Q)
Representative western blot image for Huh-7 cells with PFKL
overexpression and (R) quantified expression levels. (S)
Representative western blot image for Mhcc97h cells with PFKL
overexpression and (T) quantified expression levels. (U) Prediction
of the binding site of SREBF1 and PFKL. (V) Results of the
luciferase-based gene reporter assay used to detect the promoter
activity of PFKL through promoting SREBF1. Each group was set up
three times in parallel. *P<0.05, **P<0.03, ***P<0.01 vs.
NC group. ApoM, apolipoprotein M; SREBF1, sterol regulatory
element-binding protein 1; PFKL, ATP-dependent
6-phosphofructokinase, liver type; NC, negative control; si-, small
interfering RNA; Luc, luciferase. |
ApoM regulates PFKL through the
transcription factor SREBF1 to inhibit liver cancer cell
proliferation
Results of a previous study by our group
demonstrated that ApoM gene knockout significantly increased the
proliferation of liver cancer cells (17). These results demonstrated that ApoM
affects the expression of SREBF1 and thereby changes the promoter
activity of PFKL. Using EDU staining, the role of ApoM in the
proliferation of liver cancer cells through this pathway was
verified (Fig. 3). In Huh-7 cells,
the proliferation activity in the pcDNA3.1-SREBF1 group was
significantly increased compared with that in the pcDNA3.1-NC group
(Fig. 3A and C). Of note, the
levels of Mhcc97h cell proliferation were also increased (Fig. 3B and D). In the si-SREBF1 group,
the proliferation activity significantly decreased compared with
that in the si-NC group (Fig. 3A and
C). Consistent with these results, there was also a downward
trend in Mhcc97h cells (Fig. 3B and
D). The aforementioned results indicated that SREBF1
interference inhibited the proliferation activity of hepatoma
cells; on the contrary, SREBF1 overexpression promoted the
proliferation activity of liver cancer cells. However, there was no
significant difference between the proliferation activity in the
si-SREBF1+pcDNA3.1-PFKL group compared with that in the si-NC
group. Thus, it was hypothesized that the transcription factor
SREBF1 regulated PFKL to promote the proliferation of liver cancer
cells (Fig. 3A and C). These
results were consistent in both Mhcc97h and Huh-7 cells (Fig. 3B and D). In addition, there was no
significant difference between the proliferation activity of the
si-ApoM+si-SREBF1 group and the si-NC group (Fig. 3A and C). These results were also
consistent between both Mhcc97h and Huh-7 cells (Fig. 3B and D), suggesting that ApoM may
inhibit the proliferation of liver cancer cells through the
transcription factor SREBF1.
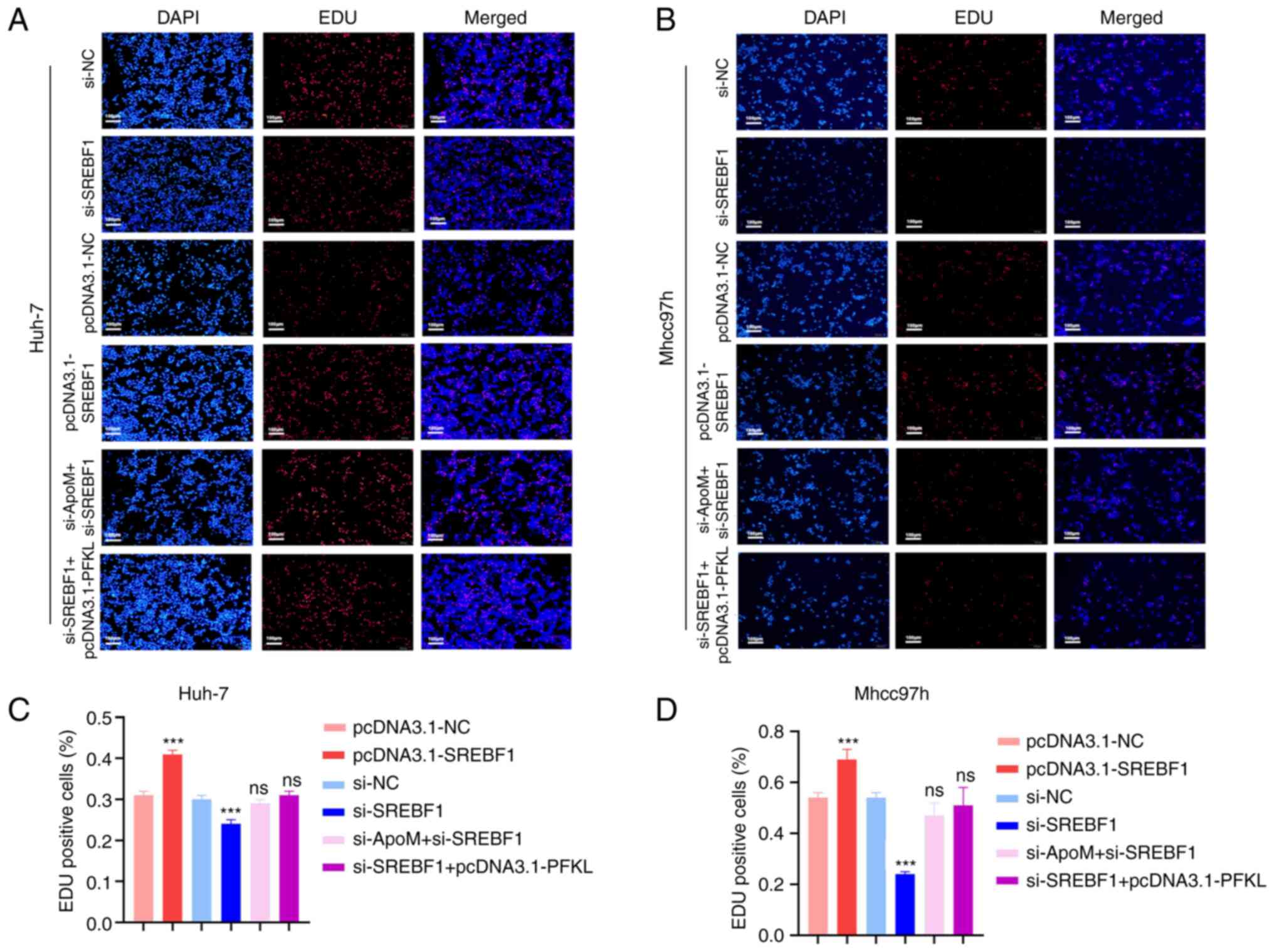 | Figure 3.Knockdown of ApoM promotes SREBF1 to
regulate PFKL to promote the proliferation of hepatoma cells. An
EDU staining assay was used to detect the proliferation activity of
(A) Huh-7 cells or (B) Mhcc97h cells in each group (pcDNA3.1-NC,
pcDNA3.1-SREBF1, si-NC, si-SREBF1, si-SREBF1+pcDNA3.1-PFKL and
si-ApoM+si-SREBF1; scale bar, 100 µm). Statistical analysis of the
results for (C) Huh-7 cells or (D) Mhcc97h cells using ImageJ. Each
group was set up three times in parallel. ns, no significance;
***P<0.01 vs. NC. NC, negative control; SREBF1, sterol
regulatory element-binding protein 1; siRNA, small interfering RNA;
PFKL, ATP-dependent 6-phosphofructokinase, liver type; ApoM,
apolipoprotein M. |
ApoM regulates PFKL through the
transcription factor SREBF1 to inhibit the migration and invasion
of liver cancer cells
Metastasis is one of the most important causes of
malignancy. Thus, Transwell assays were performed in the present
study to detect whether ApoM regulates PFKL through the
transcription factor SREBF1 and affects the migration and invasion
of liver cancer cells. First, the role of SREBF1 in the development
of liver cancer cells was identified (Fig. 4). The data suggested that
inhibiting the expression of SREBF1 gene in Huh-7 cells resulted in
a lower migration and invasion ability compared with the control
group (siSREBF1 vs. si-NC; Fig. 4A-D,
I and J), and the experimental results in Mhcc97h cells
exhibited the same trend (siSREBF1 vs. si-NC; Fig. 4E-H, K and L). On the contrary,
overexpression of SREBF1 significantly increased the migration and
invasion ability in the two groups of cells (pcDNA3.1-SREBF1 vs.
pcDNA3.1-NC; Fig. 4A-L).
Therefore, transcription factor SREBF1 has a positive role in the
development of hepatocellular carcinoma cells. A previous study by
our group proved that inhibition of ApoM expression increased the
expression of SREBF1 in the liver (18) and promoted the migration and
invasion of liver cancer cells (17). In the present study, in order to
verify whether the down-regulation of ApoM gene expression is able
to drive the progression of hepatocellular carcinoma mediated by
SREBF1, the expression of SREBF1 was inhibited by inhibiting ApoM
gene expression and it was observed whether this affected the
migration and invasion ability of liver cancer cells. Compared with
the control group, there was no significant change in the migration
or invasion ability of cells in both groups (si-ApoM + si-SREBF1
vs. si-NC; Fig. 4A-L). Therefore,
combined with the above conclusions, it was indicated that the
enhancement of the migration and invasion ability of liver cancer
cells caused by the downregulation of the ApoM gene was achieved by
promoting the expression of SREBF1. Finally, in order to explore
whether SREBF1 is a key factor for PFKL in promoting the
progression of liver cancer cells from another perspective, the
migration and invasion abilities of the si-SREBF1+ pcDNA3.1-PFKL
group and the siNC group were compared; the results indicated after
the SREBF1 gene was inhibited, even overexpression of the key
glycolysis enzyme PFKL did not enhance the ability of liver cancer
cells to migrate and invade (Fig.
4A-L). Taken together, these results suggest that ApoM
regulates PFKL by affecting the expression of SREBF1 and ultimately
mediate the progression of HCC cells.
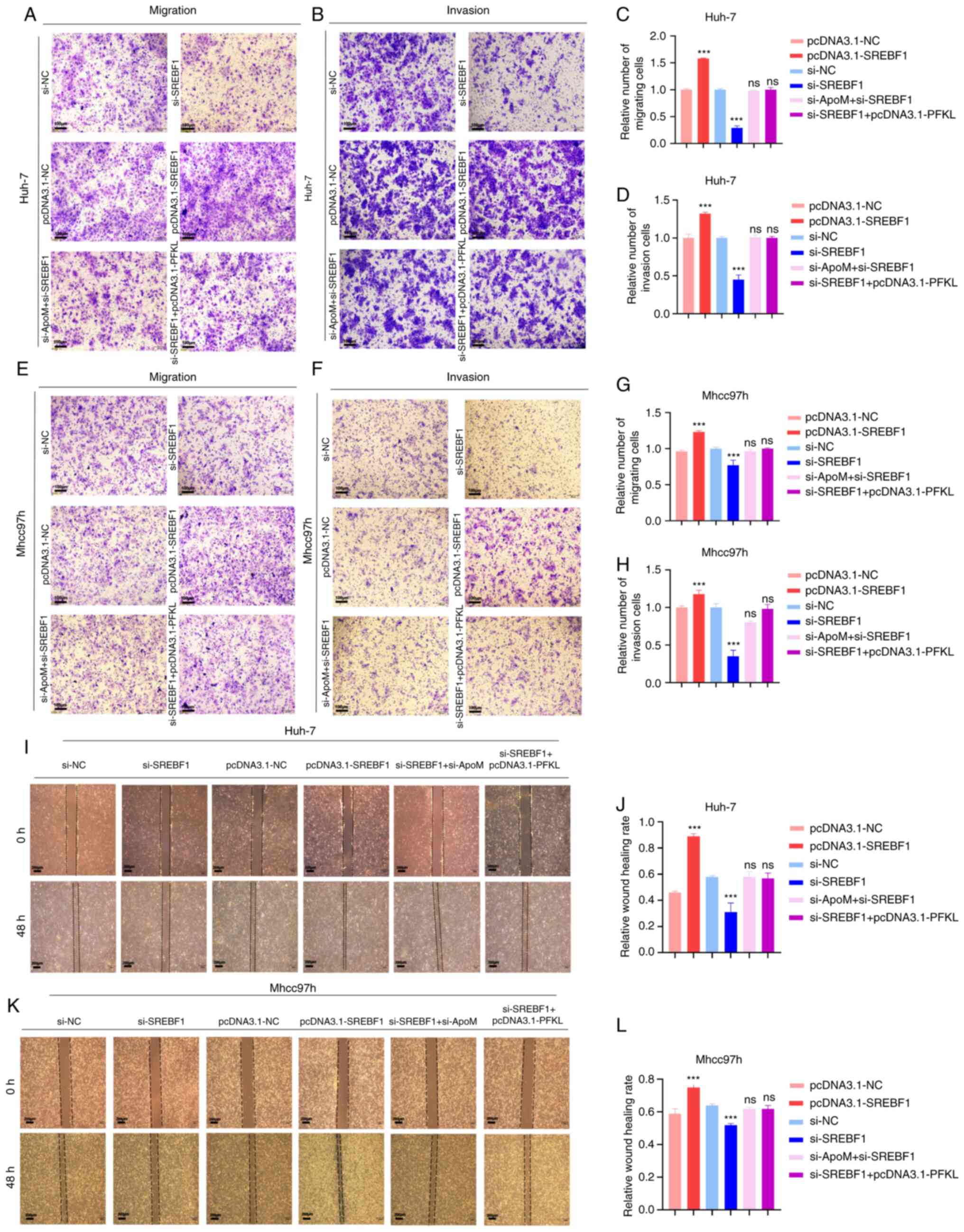 | Figure 4.Knockdown of ApoM promotes SREBF1 to
regulate PFKL to promote the migration and invasion of hepatoma
cells. Huh-7 cells or Mhcc97h cells were transfected with
pcDNA3.1-NC, pcDNA3.1-SREBF1, si-NC, si-SREBF1, si-SREBF1 +
pcDNA3.1-PFKL or si-ApoM + si-SREBF1. (A-H) Transwell assays.
Representative images of Huh-7 cells transgressed through the
membrane in the (A) migration and (B) invasion experiment and
quantified results for (C) migration and (D) invasion.
Representative images of Mhcc97h cells transgressed through the
membrane in the (E) migration and (F) invasion experiment and
quantified results for (G) migration and (H) invasion (scale bars,
100 µm). (I-L) Wound-healing assay. (I) Representative images of
migration of Huh-7 cells and (J) quantified results. (K)
Representative images of migration of Mhcc97h cells (scale bars,
200 µm) and (L) quantified results. Each group was set up three
times in parallel. ns, no significance; ***P<0.01 vs. NC. NC,
negative control; SREBF1, sterol regulatory element-binding protein
1; si-, small interfering RNA; PFKL, ATP-dependent
6-phosphofructokinase, liver type; ApoM, apolipoprotein M. |
Discussion
Results of previous studies have demonstrated that
ApoM is commonly associated with liver cancer. Previous reports
indicated differential ApoM mRNA levels and ApoM protein mass in
liver cancer tissue and adjacent tissues (14), and that loss of the ApoM gene
increased the proliferation activity (16) and decreased the level of apoptosis
(15). In summary, ApoM is
considered to be a potential protective factor that may inhibit the
occurrence and development of liver cancer.
Although ApoM has been confirmed to be involved in
glucose and lipid metabolism (11,24),
it has remained elusive whether ApoM affects liver cancer through
metabolic pathways. To abnormally proliferate, tumor cells exhibit
high levels of glycolysis, even in the presence of oxygen. This is
a phenomenon named the Warburg effect (25). Based on a previous study by our
group, the growth rate of tumors induced by diethylnitrosamine in
ApoM (−/-) mice was significantly higher than that in WT mice
(17). In a subsequent analysis,
the lactic acid levels in ApoM(−/-) mice were indicated to be
significantly higher than those in WT mice (Fig. S1). These data suggested that
downregulation of the ApoM gene promotes glycolysis in tumor
cells.
The glycolysis pathway contains three key
rate-limiting enzymes: Hexokinase, phosphofructokinase (PFK) and
pyruvate kinase. Results of previous studies have demonstrated that
PFK may act as the most important regulator in the glycolysis
pathway, including three PFK isoforms, platelet, liver and muscle
isoform (4,26). In the present study, inhibiting
ApoM expression upregulated the expression of PFKL in liver cancer
cells. By contrast, overexpression of ApoM in liver cancer cells
reduced PFKL expression. In accordance with the effects of ApoM on
lactate production in tumor cells, results of the present study
demonstrated that ApoM may attenuate the glycolytic pathway in
tumor cells by suppressing PFKL expression.
The present study also aimed to explore the
mechanism by which increased ApoM gene expression is negatively
correlated with PFKL expression. Results of a previous study
demonstrated that ApoM deficiency significantly suppressed the
autophagy function in the mouse liver and caused lipid
accumulation; furthermore, the expression levels of SREBF1 were
significantly increased (18). As
a transcription factor, SREBF1 has two different isoforms, SREBF1
and SREBF2 (also known as SREBP1 and SREBP2) (27). Using a bioinformatics analysis, the
present study demonstrated that the upregulation of SREBF1 gene
expression levels was more significant than that of SREBF2 in a
low-ApoM gene expression group, compared with that in an ApoM
high-expression group (Table SI).
Previous studies have further highlighted that SREBF1 regulates
lipid metabolism and it may provide energy for tumor cells through
the lipogenesis pathway (28). In
terms of glycolysis, it has been reported that SREBF1 and PKM2 are
closely associated with tumor growth; however, the association
between SREBF1 and PFKL has remained to be elucidated (29). To the best of our knowledge, the
present study was the first to confirm that ApoM inhibits the
expression of SREBF1 and PFKL. According to the results of the
dual-luciferase reporter gene assay, SREBF1 has the ability to bind
to PFKL and enhance its promoter activity. Furthermore, results of
the EDU cell proliferation and Transwell assays indicated that ApoM
may influence the growth and progression of liver cancer by
regulating PFKL through SREBF1.
In conclusion, the present study further elucidated
the potential mechanisms by which ApoM inhibits the development of
liver cancer by regulating glycolysis; however, further
investigations are required. Although metabolic enzymes are known
to regulate metabolic processes, their non-metabolic activities
require further investigation, which may include protein
interactions and the crosstalk between different compartments of
the signaling pathway. Similarly, this view is also particularly
important for SREBF1. The present study confirmed that SREBF1 as a
transcription factor may enhance the promoter activity of PFKL and
its expression level was affected by ApoM. With regard to the
question of how ApoM affects the expression of SREBF1, integration
of existing research results provides noteworthy and highly
relevant clues. First of all, SREBF1, as the central transcription
factor regulating lipid metabolism, mainly regulates the expression
of the factors required for fatty acid synthesis (30), which has recently been proved to be
the reason why PI3K-AKT-mTOR signaling pathway endows cancer cells
with the ability to resist iron death (31). Furthermore, ApoM is closely related
to the AKT-mTOR signaling pathway and is associated with
inflammation, apoptosis and insulin resistance (11,32,33).
Future work will aim to prove that ApoM affects SREBF1 through
certain pathways (at least the PI3K-AKT-mTOR signaling pathway). In
addition, with the application of gene chip and sequencing
technology, the downstream target genes of SREBF1 are gradually
being identified, whose target roles may be roughly divided into
direct regulation, indirect regulation and possible regulation.
Therefore, the advancement of research on ApoM-SREBF1 - downstream
effector will help to further expand and enrich the mechanism of
ApoM participating in the regulation of liver cancer
progression.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Natural Science
Research Project of Anhui Universities (grant no. KJ2020A0612) and
the Key Research and Development Program of Anhui, China (grant no.
1804h08020241).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and YB designed and supervised the study. WP
performed the data analysis, statistical analysis and completed the
manuscript writing. XZ, WZ and XL performed the experiments. XZ and
WP performed plausibility checks and confirmed the authenticity of
the raw data, which were further edited and later approved for
publication by all authors. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The use of the C57BL/6J mice required for the
experiment was approved by the Experimental Animal Welfare and
Ethics Committee of Wannan Medical College (Wuhu, China; approval
no. LLSC-2020-001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chao J, Zhao S and Sun H:
Dedifferentiation of hepatocellular carcinoma: molecular mechanisms
and therapeutic implications. Am J Transl Res. 12:2099–2109.
2020.PubMed/NCBI
|
|
2
|
Jiang H, Cao HJ, Ma N, Bao WD, Wang JJ,
Chen TW, Zhang EB, Yuan YM, Ni QZ, Zhang FK, et al: Chromatin
remodeling factor ARID2 suppresses hepatocellular carcinoma
metastasis via DNMT1-snail axis. Proc Natl Acad Sci USA.
117:4770–4780. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Rossi JZ and Finn
RS: Hepatocellular carcinoma. Nat Rev Dis Primers. 7:62021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li L, Li L, Li W, Chen T, Zou B, Zhao L,
Wang H, Wang X, Xu L, Liu X, et al: TAp73-induced
phosphofructokinase-1 transcription promotes the warburg effect and
enhances cell proliferation. Nat Commun. 9:46832018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hensley CT, Wasti AT and DeBerardinis RJ:
Glutamine and cancer: Cell biology, physiology, and clinical
opportunities. J Clin Invest. 123:3678–3684. 2013. View Article : Google Scholar
|
|
6
|
Carracedo A, Cantley LC and Pandolfi PP:
Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev
Cancer. 13:227–232. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li L and Wang H: Heterogeneity of liver
cancer and personalized therapy. Cancer Lett. 379:191–197. 2016.
View Article : Google Scholar
|
|
8
|
Center MM and Jemal A: International
trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers
Prev. 20:2362–2368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marengo A, Rosso C and Bugianesi E: Liver
Cancer: Connections with obesity, fatty liver, and cirrhosis. Annu
Rev Med. 67:103–117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu N and Dahlback B: A novel human
apolipoprotein (apoM). J Biol Chem. 274:31286–31290. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kurano M, Tsukamoto K, Shimizu T, Kassai
H, Nakao K, Aiba A, Hara M and Yatomi Y: Protection against insulin
resistance by apolipoprotein M/sphingosine-1-phosphate. Diabetes.
69:867–881. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kurano M and Yatomi Y: Sphingosine
1-phosphate and atherosclerosis. J Atheroscler Thromb. 25:16–26.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi Y, Lam SM, Liu H, Luo G, Zhang J, Yao
S, Li J, Zheng L, Xu N, Zhang X and Shui G: Comprehensive
lipidomics in apoM(−/-) mice reveals an overall state of metabolic
distress and attenuated hepatic lipid secretion into the
circulation. J Genet Genomics. 47:523–534. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang J, Wu C, Luo G, Zheng L, Chen L,
Zhang X and Xu N: Expression of apolipoprotein M in human
hepatocellular carcinoma tissues. Acta Histochem. 113:53–57. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu YW, Chen ZP, Hu XM, Zhao JY, Huang JL,
Ma X, Li SF, Qiu YR, Wu XJ, Sha YH, et al: The
miR-573/apoM/Bcl2A1-dependent signal transduction pathway is
essential for hepatocyte apoptosis and hepatocarcinogenesis.
Apoptosis. 20:1321–1337. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu M, Pan L, Sang C, Mu Q, Zheng L, Luo G
and Xu N: Apolipoprotein M could inhibit growth and metastasis of
SMMC7721 cells via vitamin D receptor signaling. Cancer Manag Res.
11:3691–3701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bai Y, Pei W, Zhang X, Zheng H, Hua C, Min
J, Hu L, Du S, Gong Z, Gao J and Zhang Y: ApoM is an important
potential protective factor in the pathogenesis of primary liver
cancer. J Cancer. 12:4661–4671. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Zhang P, Gao J and Huang Q:
Autophagy dysregulation caused by ApoM deficiency plays an
important role in liver lipid metabolic disorder. Biochem Biophys
Res Commun. 495:2643–2648. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shimano H and Sato R: SREBP-regulated
lipid metabolism: Convergent physiology-divergent pathophysiology.
Nat Rev Endocrinol. 13:710–730. 2017. View Article : Google Scholar
|
|
20
|
Tang JJ, Li JG, Qi W, Qiu WW, Li PS, Li BL
and Song BL: Inhibition of SREBP by a small molecule, betulin,
improves hyperlipidemia and insulin resistance and reduces
atherosclerotic plaques. Cell Metab. 13:44–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng C, Geng F, Cheng X and Guo D: Lipid
metabolism reprogramming and its potential targets in cancer.
Cancer Commun (Lond). 38:272018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin F, Feng F, Wang L, Wang X, Li Z and
Cao Y: SREBP-1 inhibitor betulin enhances the antitumor effect of
Sorafenib on hepatocellular carcinoma via restricting cellular
glycolytic activity. Cell Death Dis. 10:6722019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
An L, Zeng HM, Zheng RS, Zhang SW, Sun KX,
Zou XN, Chen R, Wang SM, Gu XY, Wei WW and He J: Liver cancer
epidemiology in China, 2015. Zhonghua Zhong Liu Za Zhi. 41:721–727.
2019.(In Chinese).
|
|
24
|
Borup A, Christensen PM, Nielsen LB and
Christoffersen C: Apolipoprotein M in lipid metabolism and
cardiometabolic diseases. Curr Opin Lipidol. 26:48–55. 2015.
View Article : Google Scholar
|
|
25
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schoneberg T, Kloos M, Brüser A,
Kirchberger J and Sträter N: Structure and allosteric regulation of
eukaryotic 6-phosphofructokinases. Biol Chem. 394:977–993. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dorotea D, Koya D and Ha H: Recent
insights Into SREBP as a direct mediator of kidney fibrosis via
lipid-independent pathways. Front Pharmacol. 11:2652020. View Article : Google Scholar
|
|
28
|
Xu C, Zhang L, Wang D, Jiang S, Cao D,
Zhao Z, Huang M and Jin J: Lipidomics reveals that sustained
SREBP-1-dependent lipogenesis is a key mediator of
gefitinib-acquired resistance in EGFR-mutant lung cancer. Cell
Death Discov. 7:3532021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tao T, Su Q, Xu S, Deng J, Zhou S, Zhuang
Y, Huang Y, He C, He S, Peng M, et al: Down-regulation of PKM2
decreases FASN expression in bladder cancer cells through
AKT/mTOR/SREBP-1c axis. J Cell Physiol. 234:3088–3104. 2019.
View Article : Google Scholar
|
|
30
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131. 2002.
View Article : Google Scholar
|
|
31
|
Yi J, Zhu J, Wu J, Thompson CB and Jiang
X: Oncogenic activation of PI3K-AKT-mTOR signaling suppresses
ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA.
117:31189–31197. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng Z, Zeng Y, Zhu X, Tan Y, Li Y, Li Q
and Yi G: ApoM-S1P modulates Ox-LDL-induced inflammation through
the PI3K/Akt signaling pathway in HUVECs. Inflammation. 42:606–617.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang L, Tang X and Li S: Propofol promotes
migration, alleviates inflammation, and apoptosis of
lipopolysaccharide-induced human pulmonary microvascular
endothelial cells by activating PI3K/AKT signaling pathway via
upregulating APOM expression. Drug Dev Res. 83:397–406. 2021.
View Article : Google Scholar : PubMed/NCBI
|














