Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a fatal
and hyperinflammatory disorder associated with excessive activation
of cytotoxic T cells, natural killer (NK) cells and macrophages.
Failure to control the immune response leads to marked elevation of
cytokines, inducing systemic inflammatory symptoms and signs
(1). While primary HLH is caused
by genetic defects in proteins affecting the release of cytolytic
granules, perforin1, unc-13 homolog D, syntaxin binding protein 2,
syntaxin 11, SH2 domain containing 1A, Rab27a or X-linked inhibitor
of apoptosis gene mutations are commonly detected among patients
with primary HLH (2). Secondary
HLH can occur due to environmental triggers, including severe
infectious and non-infectious factors (autoimmunity, malignancy,
drugs, pregnancy and others) (2,3).
Patients with HLH frequently present with inflammatory infiltration
manifestations characterized by fever, cytopenia, coagulopathy,
organomegaly (splenomegaly, hepatomegaly, lymphadenopathy), liver
dysfunction (elevated transaminases and bilirubin),
hemophagocytosis, hypertriglyceridemia, hyperferritinemia,
increased serum soluble interleukin-2 receptor/CD25 and diminished
NK cell function. The most widely accepted diagnostic criteria for
HLH are the HLH-2004 criteria (4),
which are based on the aforementioned clinical manifestations. As
the purpose of therapy is to limit the uncontrolled immune
response, the mainstay of therapy includes immunosuppressive and
myelosuppressive drugs, most frequently dexamethasone and
etoposide, according to two landmark studies performed in the
pediatric HLH population (4). It
is considered that timely recognition of the underlying disease and
accurate treatment to control the trigger are necessary to decrease
mortality rates (3).
Diffuse large B-cell lymphoma (DLBCL) accounts for
30–35% of newly diagnosed B-cell lymphomas (5). Although DLBCL is curable with
first-line treatment of rituximab plus cyclophosphamide,
doxorubicin, vincristine and prednisone (R-CHOP) in >60% of
patients, the remaining patients develop relapsed/refractory (RR)
disease. However, in transplant ineligible patients with RR,
management is almost palliative, and salvage regimens are limited
due to toxicities (6). Chimeric
antigen receptor T-cell therapy with anti-CD19 products has
obtained encouraging results, but notable toxicities and economic
challenges have restricted its usage. With the advent of
genome-wide molecular profiling, targeted strategies such as
ibrutinib, bortezomib (BTZ), and lenalidomide are promising, and
numerous trials exploring combinations of agents are underway
(7–10). Notably, a growing body of evidence
suggests that innovative approaches linking individual molecular
profiles to the efficacy of single agents or an ‘R-CHOP plus’
(R-CHOP with additional drugs) regimen underscore awareness about
how to select patients who will benefit from this type of extension
of the standard therapy (11).
As B-cell lymphoma with HLH is seldom reported, to
the best of our knowledge, this is the first report of a case of
HLH as a prelude to activated B-cell-like DLBCL (ABC DLBCL), which
was accompanied by plasmacytic differentiation and a MYD88 innate
immune signal transduction adaptor L265P (MYD88 L265P) mutation. It
was assumed that the mutation of MYD88 L265P and plasmacytic
differentiation resulted in activation of the endoplasmic reticulum
(ER) stress and NF-κB signaling pathways. Understanding the
pathogenesis enables researchers to develop novel intervention
strategies for patients with activated NF-κB and ER stress
signaling pathways, proposing a safe and feasible way to enhance
outcomes in this subset of patients.
Case report
Case
A 53-year-old male was admitted to Xiangya Hospital
(Central South University, Changsha, China) with a 1-month history
of hyperpyrexia in June 2018. Upon admission, the vital signs
included a temperature of 37.5°C, a heart rate of 96 beats/min, a
blood pressure of 110/50 mmHg, and a respiratory rate of 20
breaths/min. Physical examination revealed an anemic appearance but
no palpable lymphadenopathy. An initial workup showed acute
inflammatory deterioration [C reactive protein, 69.3 mg/l (normal,
0–8 mg/dl); procalcitonin, 0.88 ng/ml (normal, 0–0.1 ng/ml); anemia
[hemoglobin level, 5.4 g/dl (normal, 13–17.5 g/dl)],
thrombocytopenia [platelet level, 25×103/µl (normal,
100–300×103/µl)], hypertriglyceridemia [triglyceride
level, 3.47 mmol/l (normal, 0–1.7 mmol/l)] and increased ferritin
[>2,530 µg/l (normal, 16–400 µg/l)]. Moreover, the patient had
elevated immunoglobulin levels [IgM, 5,010 mg/l (normal, 400–2,800
mg/l)]. Simultaneously, both serum and urine electrophoresis
detected IgM κ chains. B-mode ultrasound imaging of the superficial
lymph nodes showed multiple lymph nodes on both sides of the
cervical region, axillary fossa and groin, but none were larger
than 13×4 mm. A computed tomography (CT) scan revealed multiple
mesentery lymph nodes and splenomegaly. Notably, bone marrow
aspiration showed hemophagocytosis (data not shown).
According to HLH-2004, a diagnosis of secondary HLH
could be made as the patient had 5 out of 8 parameters
(bicytopenia, splenomegaly, hypertriglyceridemia, hyperferritinemia
and hemophagocytosis) (4).
According to the HLH-1994 protocol, standard care for children with
HLH is a combination of etoposide and dexamethasone (12). The HLH-2004 chemotherapy protocol,
which was modified from HLH-1994, added cyclosporine A during the
initial therapy phase. Some previous studies have suggested that
adding cyclosporine offers no clinical advantage, and the present
patient started with the HLH-1994 protocol. After the first cycle
of therapy, the general condition of the patient improved
gradually, and no abnormal signs were observed during a physical
examination, except anemia. The values of the blood cells were
elevated markedly (hemoglobin level, 6.9 g/dl; platelet level,
10.2×104/µl). Therefore, the patient only underwent one
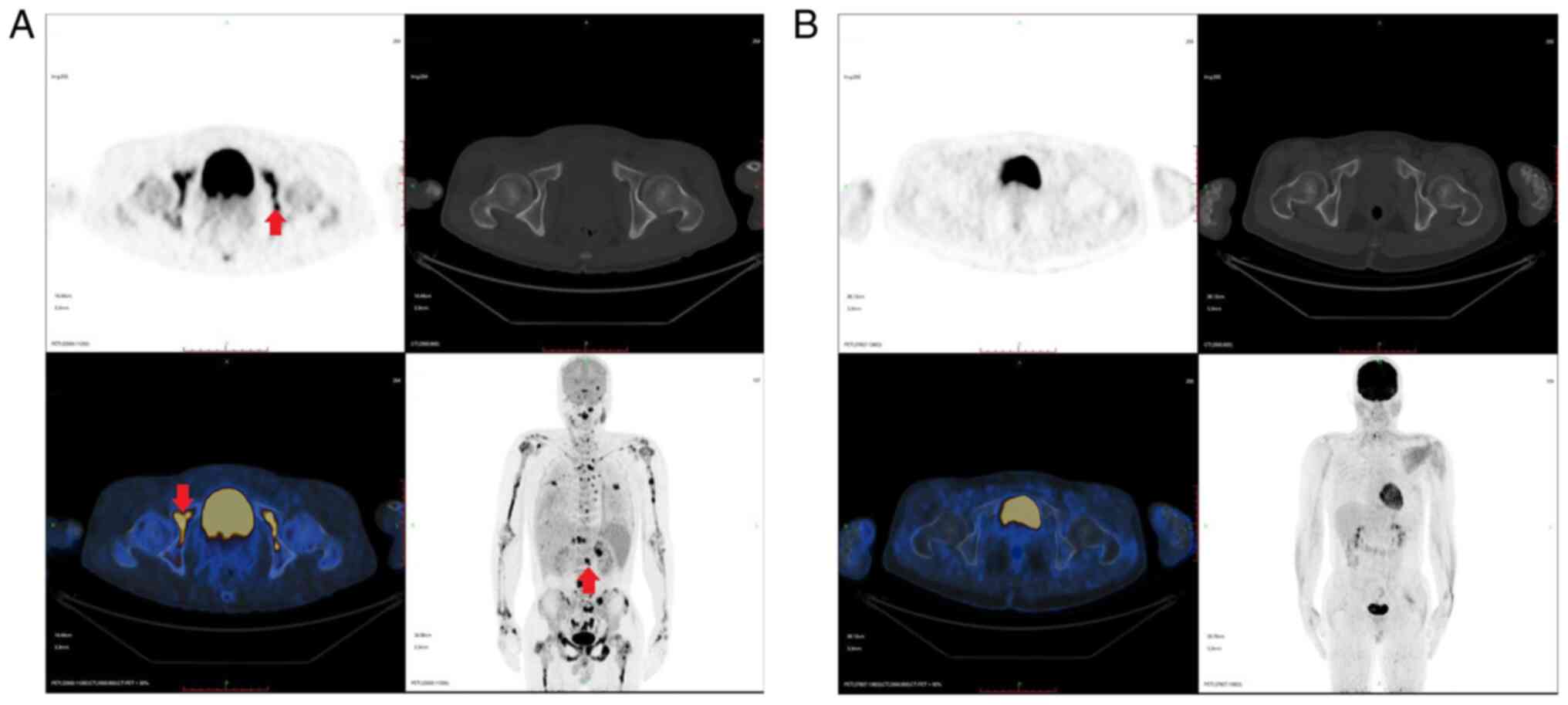
cycle of HLH-1994. Further investigations and positron emission
tomography (PET)/CT revealed multiple bone parenchyma, spleen and
both adrenal glands with increased glucose metabolism, which was
mostly due to lymphoma infiltration (Fig. 1A). According to the PET/CT results,
bilateral bone marrow biopsies were collected. While the
pathological results of the right posterior superior iliac spinal
biopsy revealed proliferative changes, the left side showed diffuse
large B-cell lymphoma that conformed to the ABC type described by
Hans et al (13) upon
immunohistochemical analysis, which showed the following results:
CD20+, paired box 5+, Bcl-6+,
multiple myeloma 1+, Bcl-2+ (50%),
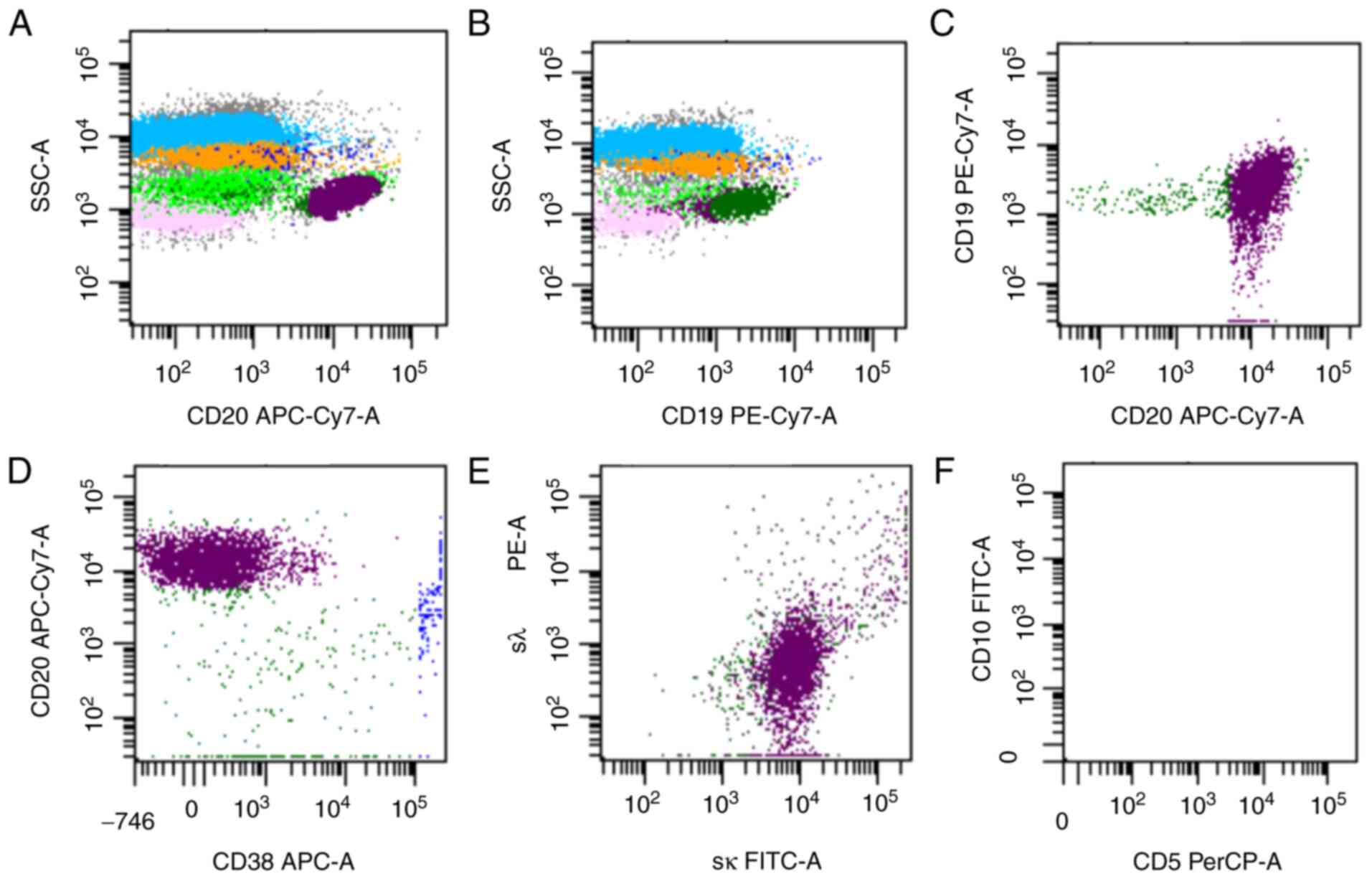
C-myc+ (10%) and Ki67+ (50%) (Fig. 2). Flow cytometry showed that there
were 3.1% mature clonal B cells among karyocytes, and these cells
were positive for CD45, CD19, CD20 and sκ, and negative for CD38,
CD10 and sλ (Fig. 3). The
chromosome karyotype analysis result was 46, XY. As IgM κ M
proteins were detected both in serum and urine electrophoresis,
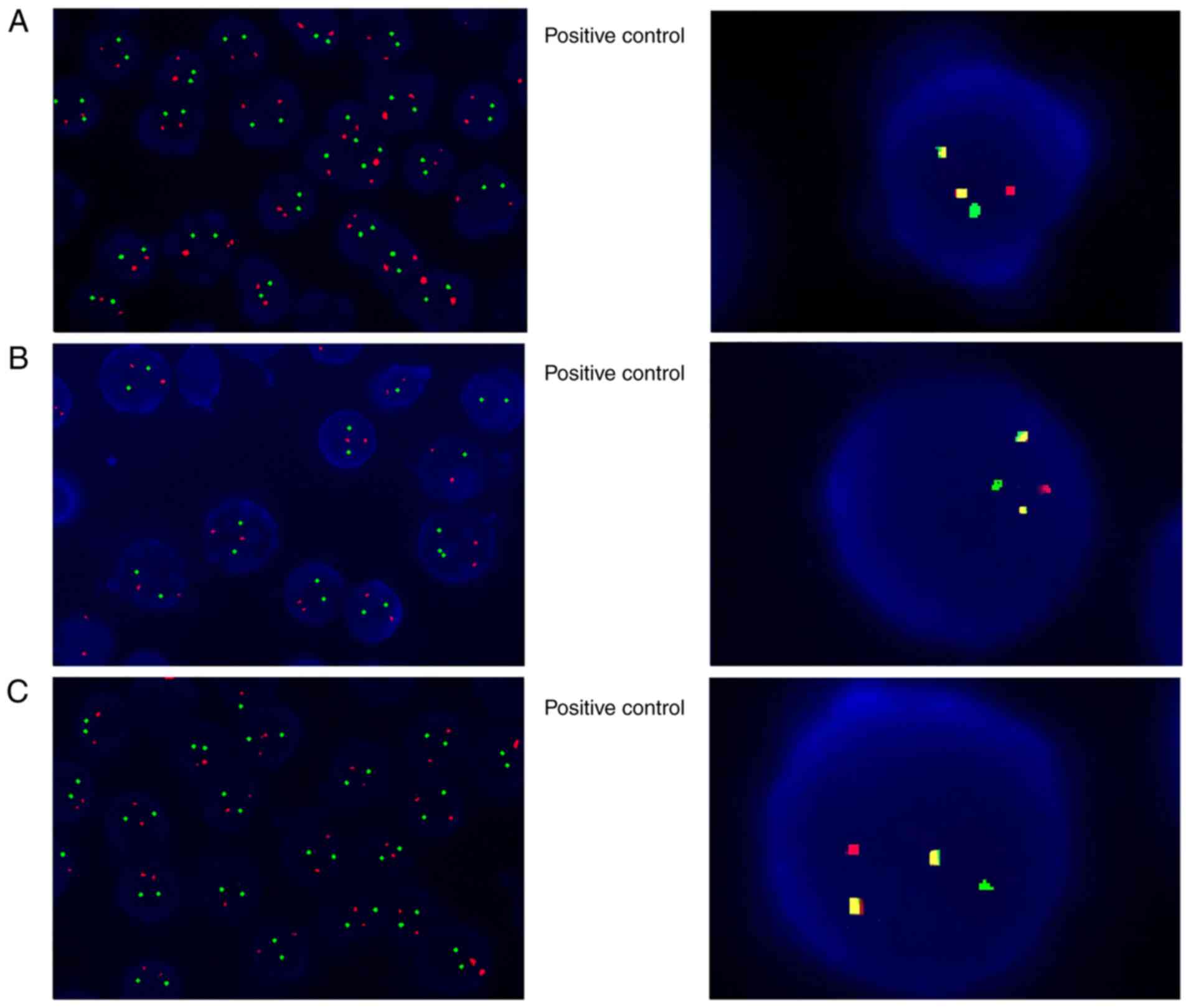
fluorescence in situ hybridization (FISH) analysis was
performed for multiple myeloma (MM) chromosomal translocations of
t(14;16)(q32;q23), t(4;14)(q16;q32) and t(11;14)(q13;q32), which
are likely to cause gene overexpression of MAF BZIP transcription
factor (MAF), FGFR3 and cyclin D1 (CCND1), respectively (14). All the aforementioned
translocations were negative (Fig.
4). Further FISH analysis results demonstrated negative results
for the 1q21/RB transcriptional corepressor 1 (RB1), D13S319/TP53,
immunoglobulin heavy locus (IGH), T-cell receptor α/δ (TCRAD) and
T-cell receptor β (TCRB) genes (Fig.
4). However, Ig VH, Ig DH, Ig K and Ig L gene rearrangements
were detected (data not shown). None of the explicit mutations
associated with hematological malignancies were revealed by
next-generation sequencing (NGS), but allele-specific PCR
discovered that the patient was positive for the MYD88 L265P gene
mutation. The inconformity of these two tests may be due to the
limitations of NGS, including analytic sensitivity of mutation
detection or areas of the genome that are difficult to sequence
(15). Consequently, the diagnosis
of the patient was DLBCL [ABC type, stage IVB; International
Prognostic Index (IPI) score, 4; CNS score, 5; plasmacytoid
differentiation; and MYD88 L265P mutation]. The patient therefore
did not undergo an HLH genetic study due to the increased
likelihood that HLH was secondary to DLBCL.
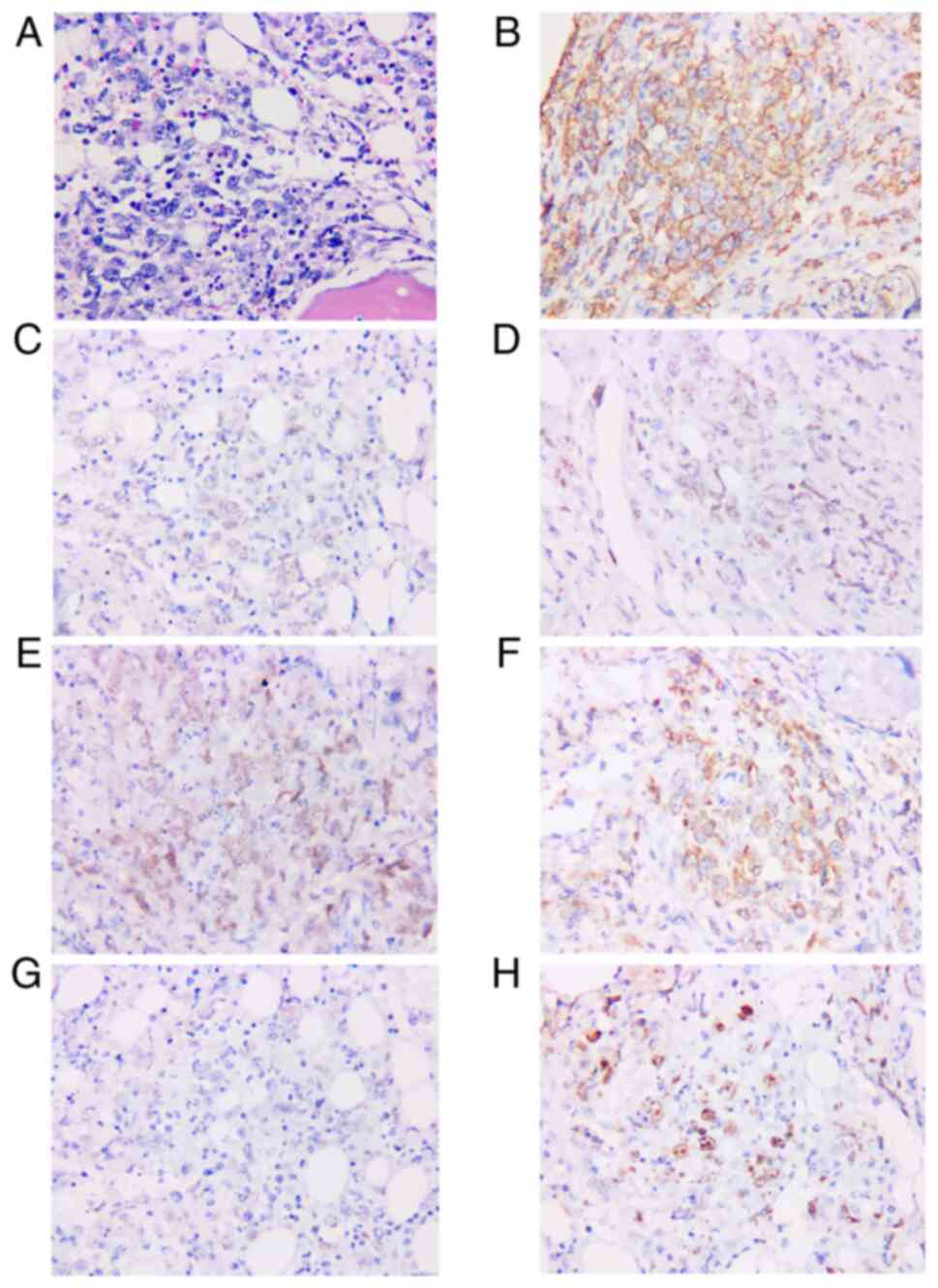 | Figure 2.Pathological findings of bone marrow
biopsy are diffuse large B-cell lymphoma. (A) Hematoxylin and eosin
staining patterns revealing diffuse, pleomorphic, medium to large
tumor cell infiltration (magnification, ×400). Immunohistochemical
staining patterns showing tumor cells positive for (B) CD20, (C)
paired box 5, (D) Bcl-6, (E) multiple myeloma ongogene 1, (F)
Bcl-2, (G) c-myc, and (H) Ki67 (magnification, ×400). |
Although R-CHOP is the frontline standard of care
for DLBCL, among patients with RR DLBCL, less favorable outcomes
have prompted efforts to improve first-line approaches. EPOCH-R, a
96-h continuous infusion regimen of R-CHOP that incorporates a
dynamic dose adjustment, has shown excellent efficacy in
Bcl-2-positive DLBCL (16). With
the consideration that the present study patient was Bcl-2-positive
with multiple organ infiltration, EPOCH-R was chosen for the
patient to prevent recurrence. As the lymphoma was likely to be
accompanied by plasmacytic differentiation, the patient received
intensive regimens of high-dose methotrexate (to prevent
intracranial involvement, as the abnormal adrenal gland was
presumed to be lymphoma infiltration) based on the combination
chemotherapy of rituximab and BTZ. The detailed timeline of
treatments is shown in Fig. 5.
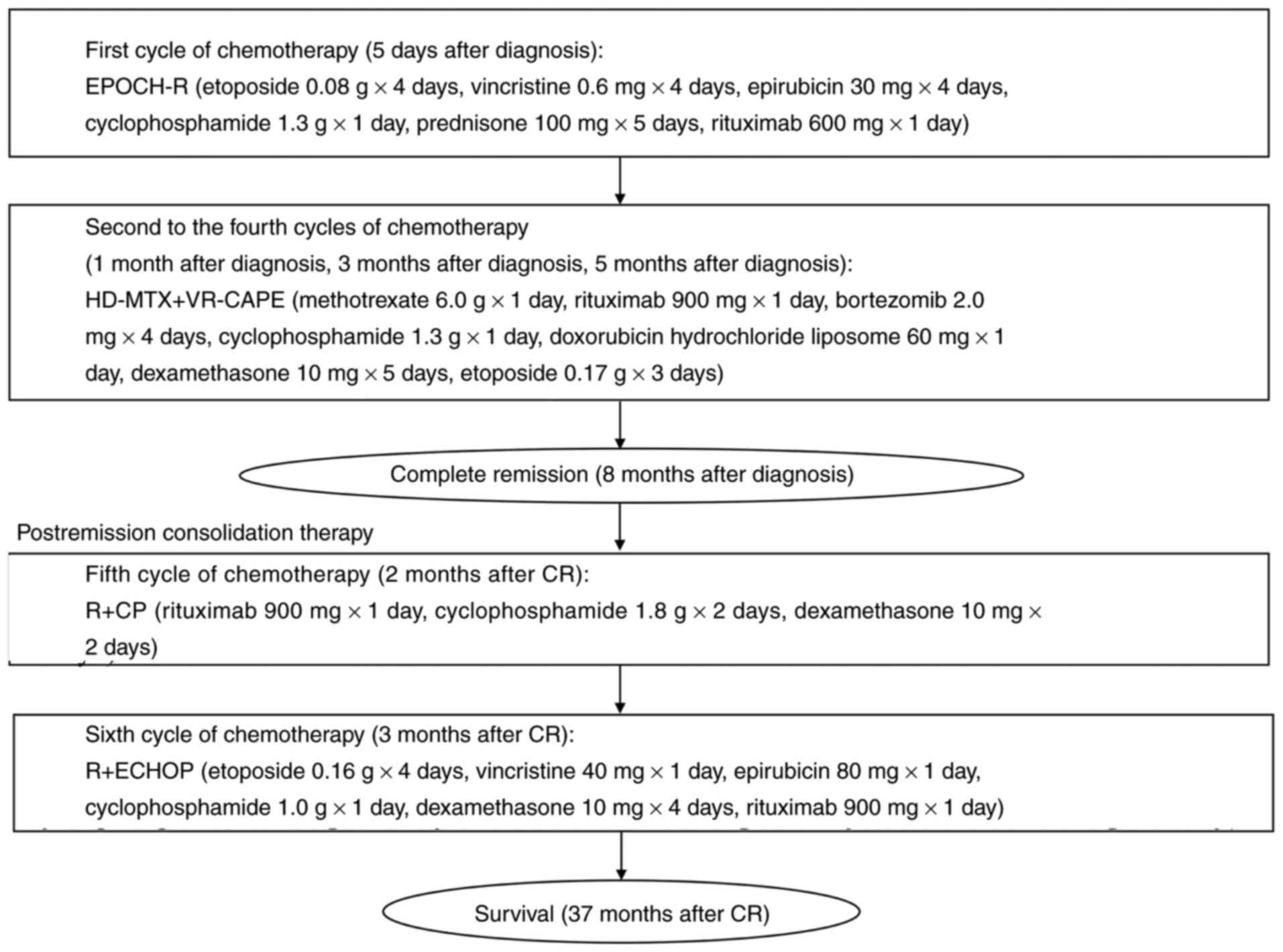 | Figure 5.Chemotherapy timeline. EPOCH-R,
etoposide, vincristine, epirubicin, cyclophosphamide, prednisone
and rituximab; HD-MTX + VR-CAPE, methotrexate, rituximab,
bortezomib, cyclophosphamide, doxorubicin hydrochloride liposome,
dexamethasone and etoposide; R+CP, rituximab, cyclophosphamide and
dexamethasone; R+ECHOP, etoposide, vincristine, epirubicin,
cyclophosphamide, dexamethasone and rituximab. |
Since the collection of stem cells from the bone
marrow failed, ibrutinib or lenalidomide administration was
recommended. The patient selected lenalidomide (25 mg daily) for
maintenance treatment. The last PET/CT performed in February 2019
showed that glucose metabolism, which was originally increased in
multiple bone parenchyma, the spleen and both adrenal glands, was
generally ameliorated, which indicated complete remission (CR) of
the lymphoma (Fig. 1B). The
patient is being followed up by the Department of Hematology in
Xiangya Hospital every 6 months, and currently remains free of
disease with quiescent HLH.
Imaging examinations
The plain and contrast-enhanced CT scans of the
chest, abdomen and pelvis were made with the Aquilion ONE
320-detector row CT scanner (Canon Medical Systems Corporation).
The CT parameters were as follows: 120 kV, automatic mAsec, 1.0-mm
thickness, 1.0-mm intervals and a matrix of 256×256. The PET/CT
scans were performed on a GE Discovery 690 PET/CT scanner (GE
Healthcare).
FISH analysis
Interphase FISH analysis was performed according to
the standards of the International System for Human Cytogenetic
Nomenclature 2016 (17). The
bacterial artificial chromosome (BAC) DNA clones were purchased
from Children's Hospital Oakland Research Institute and were
separated into individual colonies on a Luria Broth (LB) agar plate
(18). The colonies were
inoculated in 10 ml LB with appropriate antibiotics, incubated at
37°C and agitated at 250 rpm. The bacteria were collected by
centrifugation at 3,000 × g for 10 min at 4°C. Next, high-purity
BAC DNA was extracted using the NucleoBond PC20 kit (cat. no.
740519.20; Machery-Nagel). After digesting BAC plasmids with
restriction endonuclease BamHI (cat. no. 81295-09-2;
MilliporeSigma) at 37°C for >2 h, DNA bands were separated by
electrophoresis on a 1% agarose gel and using 1X Tris-acetate-EDTA
(TAE) buffer at 20–30 V to identify each clone by further FISH. The
BAC clones were stored in LB and glycerol at a ratio of 1:1 and
kept at −80°C. The BAC DNA was labeled by 50 µl reaction mixture
using a Nick Translation Reaction kit (cat. no. 07J00-001; Abbot
Pharmaceutical, Co., Ltd.), then incubated at 15°C for 1–2 h and at
70°C for 10 min. The Nick Translation products were separated with
1.5% agarose gel and 1X TAE buffer, at 70–100 V for ≥1 h. After
precipitation, DNA probes were collected at 17,000 × g in a
benchtop centrifuge at 4°C for 30 min, then washed and air-dried.
DNA probes were suspended in 100 µl in situ Hybridization
Kit BioAssay (cat. no. I7643-50H; United States Biological). After
denaturation at 75°C for 10 min, blocking of non-specific genomic
sequences was performed at 37°C for 30 min. Finally, the probes
were stored at −20°C. Bone marrow aspirate samples were mixed with
EDTA immediately once collected. RPMI-1640 medium + 5% fetal bovine
serum at a 1:1 or 1:5 ratio was added to bone marrow for removing
erythrocytes. Diluted cells were laid onto Histopaque-1077 medium
(cat. no. 397639117; MilliporeSigma). Next, cells were centrifuged
at 1,800-2,000 × g for 25 min at room temperature (RT). Leukocytes
were harvested, washed with RPMI-1640 medium and then counted. A
glass coverslip was used and washed with PBS once, and then 0.1 ml
of purified lymphocytes from the patient was added and centrifuged
for 5 min at 1,000 × g at RT. Plates were put on a clean bench for
30 min at RT. Slides were fixed at 37°C in a dry oven overnight and
stored at −20°C. For FISH, the chromosomal DNA was denatured on the
slides in 90% formamide/2X SSC solution at 37°C for 8 min. The
slides were dehydrated and then air-dried. The hybridization
mixture containing fluorochrome red and green probes was added to
the slides, covered with a cover slip and incubated in moist
plastic chambers at RT for 1 h. Slides were incubated in the
Hybrite thermal plate (MilliporeSigma) at 75°C for denaturation and
hybridization. Next, slides were washed, dried and then immersed in
1X PBS for 3 min at RT. The semidried slides were treated with 100
µl of 1:20 light chain (κ or λ) antibody staining dilute AMCA
goat-antibody (cat. no. L3065; Signalway Antibody LLC) and
incubated in humidity chamber at RT for 30 min. Slides were washed
with PBS, then immersed in 100 µl of FITC-conjugated goat
anti-rabbit antibody (cat. no. L30113; Signalway Antibody LLC)
(1:40 in dilution buffer) and incubated in the humidity chamber at
RT for 30 min. After the washing procedure, 10 µl antifade mounting
solution was added on each slide. Slides were covered with a
rectangle coverslip (20×60 cm) and stored at 4°C. The slides were
observed with a fluorescence microscope using a B2 or B-2A filter
cassette (Nikon Corporation). The FISH probes used in the present
study included IGH/MAF (14q32/16q23), IGH/FGFR3 (4q16/14q32),
IGH/CCND1 (11q13/14q32), glucagon-like peptide (GLP) 1q21/RB1
(1q21/13q14), GLP D13S319/TP53 (13q14.3/17p13.1), IGH (14q32),
TCRAD and TCRB probes. All of the FISH probes were purchased from
Wuhan Healthcare Biotechnology, Co., Ltd. A total of 200 cells were
evaluated under the fluorescence microscope. The cut-off points for
a positive test were 15% for probes of TCRAD or TCRB, and 8% for
other probes.
Flow cytometry
The flow cytometry analysis was performed in the
laboratory of the Department of Hematology, Xiangya Hospital, using
bone marrow aspirate specimens. A total of 3 ml
EDTA-anti-coagulated bone marrow samples were collected and kept on
ice (4°C). Samples (1.5 ml) were added into a conical centrifuge
tube with 10 ml erythrocyte-lysing solution (cat. no. 348202; BD
Biosciences). Next, the sample was incubated in the dark at 4°C for
5 min. After lysis procession, the cells were preserved using cell
preservation medium [composed of 500 ml RPMI1640, 25 ml 5% FBS
(cat. no. 164210-500; Procell Life Science & Technology Co.,
Ltd.) and 5 ml Penicillin-Streptomycin solution (cat. no. DXT-0503;
ScienCell Research Laboratories, Inc.)]. The cells were counted
after infiltration. The cell suspension concentration was adjusted
to 3.0×106/ml. A total of 100 µl cell suspension was
added to each tube, and then the fluorochrome-combined antibodies
were added to cell suspension for incubation at 4°C for 30 min.
During cell washing, the cells were centrifuged at 400 × g for 5
min at 4°C and resuspended in cold PBS three times. Finally, the
cells were preserved with incubation in the dark at 4°C to acquire
data as soon as possible. The fluorochrome-combined monoclonal
antibodies were as follows: Anti-CD7 (FITC-A), anti-CD56 (PE-A),
anti-CD8 (PE-Cy7-A), anti-CD3 (APC-A), anti-CD4 (APC-Cy7-A),
anti-CD45 (PerCP-A/APC-Cy7-A), anti-CD19 (PE-Cy7-A), anti-CD20
(APC-Cy7-A), anti-CD38 (APC-A), anti-CD34 (APC-A), anti-CD5
(PE-A/PerCP-A), anti-CD10 (FITC-A), sκ (FITC-A) and sλ (PE-A). All
of the fluorochromes and antibodies were acquired from
Becton-Dickinson and Company. Flow cytometry was performed by
FACSCanto II (BD Biosciences) and data were analyzed with FACSDiva
software (v7.0; BD Biosciences).
Biochemical examinations
The blood routine was analyzed by a Coulter LH 750
automatic blood cell analyzer. The biochemical tests including
hepatorenal function and ferritin analysis, were performed on
Beckman Coulter AU680 automatic biochemical analyzer (Beckman
Coulter, Inc.). All of the examinations were conducted and analyzed
by the Clinical Laboratory of Xiangya Hospital.
Immunohistochemistry (IHC) and
sequencing analysis
The bone marrow specimens collected from the patient
were fixed in 10% neutral formalin solution for further
pathological and immunohistochemistry (IHC) analysis, as well as
H&E staining, by Kindstar Global Company. The NGS of fusion
genes and gene mutations, FISH probes (bcl-2, bcl-6 and myc
rearrangements) and Ig gene rearrangements (PCR) were performed by
Kindstar Global Company. The MYD88 L265P mutation examination was
performed by Kindstar Global Company using allele-specific PCR.
Literature review
Search strategy
A comprehensive literature search was performed
using the databases of PubMed (https://pubmed.ncbi.nlm.nih.gov/), Ovid Medline
(https://www.wolterskluwer.com/en/solutions/ovid/ovid-medline-901),
Embase (https://www.elsevier.com/solutions/embase-biomedical-research),
Web of Science (http://webofscience.com) and Cochrane Library
(https://www.cochranelibrary.com/)
between January 2002 and January 2022 with the following terms:
‘Hemophagocytic syndrome(s)’ OR ‘hemophagocytic
lymphohistiocytosis’ OR ‘hematophagic histiocytosis’ OR ‘macrophage
activation syndrome(s)’ OR ‘cytokine release syndrome(s)’ OR ‘HLH’
AND ‘diffuse large B-cell lymphoma’ OR ‘non-Hodgkin lymphoma’ OR
‘DLBCL’.
Study identification
Studies were included if they met the following
criteria: i) Refer to secondary HLH in critically ill patients; ii)
concern patients with a diagnosis of histologically confirmed
DLBCL; iii) provide relevant information on clinical course,
treatments and outcomes; iv) are written in English; and v) contain
one or multiple clinical case reports or letters. Other recognized
LBCL variants, such as intravascular lymphoma,
T-cell/histiocyte-rich LBCL, primary diffuse LBCL of the central
nervous system (CNS) and Epstein-Barr virus-positive DLBCL were
excluded due to possible different pathophysiologies. Reviewers
resolved all discrepancies by discussion during the study
identification process.
Discussion
Among the malignancies associated with HLH, lymphoma
is the most common underlying condition. It was established that
patients with lymphoma who developed HLH exhibited immune
activation due to lymphoma cells or inhibitory immune function
failure from the disease or treatment-induced bone marrow
dysfunction (19). To the best of
our knowledge, most cases of lymphoma-associated hemophagocytic
syndrome (LAHS) are T-cell or NK-cell lymphomas, and HLH cases
secondary to B-cell lymphoma are rarely seen in Western countries
(19). Previous cohorts showed
that patients with B-cell LAHS were older, but they shared parallel
clinical features and laboratory data with patients with
T-cell/NK-cell LAHS (20,21). Patients with B-cell lymphoma had
longer survival times than those with T-cell lymphoma due to their
higher response rate to treatment and the use of rituximab
(22). However, HLH could be an
indicator of treatment resistance in patients with malignant
lymphoma, especially for patients with lymphoma originating from B
cells (1). The literature review
performed for the present study showed recently reported cases of
DLBCL with HLH, which were similar to the clinical manifestation of
the current patient, but none of them presented with plasmacytic
differentiation and MYD88 L265P mutation. Specifically, after
R-CHOP chemotherapy, few patients achieved remission (23,24),
and others achieved transient remission but relapsed soon after or
died immediately (25,26). In the summary of a clinical case
series, researchers suggested that after the combination of
HLH-directed therapy and malignancy-focused interventions,
treatment should be decided on a case-by-case basis (27). The detailed information of previous
cases is shown in Table I.
 | Table I.Clinical features of patients with
DLBCL and HLH. |
Table I.
Clinical features of patients with
DLBCL and HLH.
| First author,
year | Country | Age, years | Sex | Symptoms and
signs | Laboratory
examination | Subtype of
DLBCL | Clinical stage | International
Prognostic Index | Treatment | Treatment
response | Outcome from
remission until publication follow-up | (Refs.) |
|---|
| Malkan et
al, 2021 | Turkey | 32 | Male | Vomiting, nausea
and diarrhea, anuria, fever and hepatosplenomegaly | Pancytopenia,
hypertriglyceridemia, elevated transaminases and CRP, high level of
LDH and fer-ritin, hypercalcemia, hemophagocytosis | NA | NA | NA | HLH-2004,
R-EPOCH | CR | Survival | (23) |
| Desai et al,
2020 | USA | 73 | Female | Fever,
splenomegaly | Cytopenia,
hypertriglyceridemia, hyperferritinemia, hemophagocytosis | NA | IV | NA | HLH-1994,
R-CHOP | ND | Death | (27) |
| Tang et al,
2020 | China | 64 | Female | Upper abdominal
pain and continuous high-grade fever | Anemia, elevated
CRP and procalcitonin, liver dysfunction, lymphadenopathy | GC | NA | NA | R-CHOP | ND | Death | (59) |
| Wu et al,
2020 | China | 66 | Male | Continuous
high-grade fever with no obvious cause | Bicytopenia,
increased level of ferritin, sIL-2R and triglycerides,
hemophagocytosis, lymphadenopathy, splenomegaly | nGC | III B | High risk | R+ECHOP,
intrathecal chemotherapy (methotrexate, cytarabine,
dexamethasone) | CR | Survival | (60) |
| Kim et al,
2017 | Korea | 57 | Male | Fever, weight
loss | Bicytopenia,
increased levels of ferritin, soluble CD25, LDH and
β2-microglobulin, splenomegaly, hyperdiploidy and complex
abnormalities, clonal rearrangement of the IGH gene | NA | NA | NA | R-CHOP | CR | Survival | (24) |
| Patel et al,
2017 | USA | 57 | Male | Weakness, fatigue,
confusion, dizziness and mild jaundice | Bicytopenia,
elevated creatinine, LDH, ferritin, triglycerides, liver
dysfunction, coagulopathy, splenomegaly, lymphadenopathy | NA | NA | NA | Dexamethasone,
ifosfamide, carboplatin and etoposide | ND | Death | (61) |
| Patel et al,
2017 | USA | 70 | Female | Left facial
weakness, fatigue, night sweats, fever, decreased appetite and
weight loss | Pancytopenia,
elevated LDH, ferritin, coagulopathy, hypertriglyceridemia,
sCD-25 | NA | IV B | NA | R-CHOP | ND | Survival | (62) |
| Entezari et
al, 2015 | USA | 66 | Male | Fevers and altered
mental status without focal neurological deficits | Bicytopenia,
splenomegaly elevated LDH, serum ferritin, fibrinogen, triglyceride
and sCD25, low NK cell levels, hemophagocytosis | nGC | NA | NA | High-dose
dexamethasone, cyclosporine, R-CHOP | ND | Survival | (63) |
| Li et al,
2014 | China | 48 | Male | High-grade fever,
cough, weight loss, hepatosplenomegaly, lymphadenopathy | Pancytopenia, liver
dysfunction, elevation of triglyceride, LDH and serum ferritin,
hemophagocytosis LDH, serum ferritin | GC | IV B | High risk | High-dose
methylprednisolone, cyclophosphamide, vincristine, doxorubicin and
etoposide, R+ECHOP | At first CR,
relapsed after 8 cycles of chemotherapy | Survival | (25) |
| Kuo et al,
2014 | China (Taiwan) | 51 | Female | Intermit-tent
fever | Anemia, elevated
CRP, hemophagocytosis | NA | NA | NA | E-POCH | ND | Survival | (64) |
| Wang et al,
2014 | China | 46 | Male | Persistent fever,
vibration white fingers, discontinuous arthralgia, jaundice,
abdominal distention, anorexia, hepatosplenomegaly, scrotal edema
and loss of body weight | Pancytopenia,
coagulopathy, splenohepatomegaly, hemophagocytosis | NA | I B | NA | HLH-2004,
R-CHOP | ND | Survival | (65) |
| Davidson-Moncada
et al, 2013 | Greek | 74 | Male | Increasing
abdominal pain, nausea, vomiting, splenomegaly,
lymphadenopathy | Anemia, liver
dysfunction, coagulopathy, lymphadenopathy, elevated fibrinogen
levels, serum ferritin and LDH, hemophagocytosis | NA | NA | NA | High-dose
corticosteroids with rituximab, cyclophosphamide | ND | Death | (26) |
| Sano et al,
2007 | Japan | 63 | Male | Fever and upper
abdominal discomfort, hepatosplenomegaly | Pancytopenia, liver
dysfunction, elevation of LDH, serum ferritin, sIL-2R, CRP,
lymphadenopathy; | NA | IV | High risk | High-dose
methylprednisolone, R-CHOP | CR | Survival | (66) |
The current case presents several intriguing
findings. First, the most plausible diagnosis was DLBCL with
plasmacytic differentiation. From the imaging results, lymphoma was
identified. However, surgeons found that neither the adrenal gland
nor the palpable lymph nodes were palpable for excision biopsy.
Finally, the diagnosis of ABC DLBCL was established through a bone
biopsy. A previous study revealed that different diagnoses can be
made by examining lymph node biopsies and other sites of
involvement, and the diagnosis concordance rate between bone marrow
and lymph nodes was 84.85% (28).
Most patients with discordant bone marrow involvement (BMI)
progress to invasive lymphoma due to transformation of the original
indolent neoplasm or benign lymphoid aggregation, and downgrading
from high- to low-grade malignancy has also been observed. It is
well known that BMI occurs in 10–30% of DLBCL cases. Since BMI in
DLBCL is generally recognized as a high-risk advanced disease, it
contributes to a higher IPI score. Unexpectedly, a previous study
showed that patients with discordant BMI exhibited characteristics
similar to patients without BMI (29). Patients with a concordant BMI
exhibited a negative prognosis with a lower CR rate. A
retrospective analysis suggested that discordant BMI might be
overestimated, as some subsets of cases may be partially due to
considerable morphological and immunohistological overlap, relying
on individual interpretations or different bone marrow
microenvironments (30). In the
present study, IgM κ chain was detected in both serum and urine
electrophoresis, and sκ was positive in the initial flow cytometry
analysis. In addition, following the first E-POCH-R chemotherapy,
flow cytometry showed that there were plasma cells expressing CD38.
Although plasmacytic differentiation is unusual in DLBCL, it has
been established that ABC-DLBCL is closely associated with
plasmablasts that have passed through the germinal center reaction
and express IgM (31). Caution is
advised, and the diagnosis of DLBCL with plasmacytic
differentiation must be distinguished from the transformation from
lymphoplasmacytic lymphoma (LPL). However, LPL is indolent, and
transformation may occur only at a frequency of 5–13% (32). Furthermore, the prognosis of DLBCL
transformed from LPL appears to be worse than that of de
novo DLBCL, with a survival time of ~2 years after
transformation (33). Although the
most frequent somatic mutation in LPL is MYD88 L265P, this mutation
is also common in ABC DLBCL. More importantly, patients with ABC
DLBCL frequently express IgM (34). However, DLBCL transformation from
LPL needs to be confirmed by the pathology results of lymph node
biopsies before and after transformation. As the patient in the
present study did not present with any manifestation of LPL before
the onset of disease, the patient was diagnosed with DLBCL with
plasmacytic differentiation.
The second item of note in the present study was
that it was hypothesized that the NF-κB signaling pathway was
activated. In HLH, disruption of granule-mediated cytotoxicity
impairs apoptosis in target cells and causes uncontrolled
activation of T cells, which release proinflammatory cytokines
(1). Moreover, previous data have
demonstrated that high levels of serum TNF-α, IL-6 and IL-10 in
B-LAHS may be due to the production of neoplastic B cells (35). There is accumulating evidence that
TNF-α, a critical mediator of inflammatory immune responses, can be
produced by B cells in an autocrine manner (36). The TNF-α gene is well known as an
activator of NF-κB and is an NF-κB-regulated gene. The mechanism by
which TNF-α mRNA translation is increased may involve strong
activation of NF-κB via MYD88 in the early phase (37). It has been revealed that
leukemia-initiating cells retain p65 (component of NF-κB) nuclear
translocation even after serum-free culture, suggesting that
constitutive NF-κB activity in cells is maintained via autonomous
TNF-α secretion (38). This
observation suggests that NF-κB is spontaneously activated via
autocrine/paracrine TNF-α, forming an autocrine positive feedback
loop between TNF-α and NF-κB activation (37,39,40).
A similar situation may exist between IL-6 and NF-κB activation.
Hashwah et al (41)
reported that ABC DLBCL cells had developed complementary
mechanisms that ensured constitutively active IL-6 signaling.
Collectively, this evidence supports the hypothesis that NF-κB is
constitutively activated by neoplastic B cells and generates an
intense hyperimmune response. Above all, it is considered that
tumor B cells secreted the inflammatory cytokines and activated the
NF-κB consistently in the present patient.
Third, ER signaling pathway activation was mediated
by lymphoma cells with plasmacytic differentiation. ER stress is
obligatory in the modification and trafficking of proteins
(42). In the presence of the
triggers, misfolded proteins exceed the rate of protein refolding
or degradation, accumulate and induce ER stress (43). During DLBCL-associated HLH, IL-6
could induce the differentiation of activated B cells into
antibody-producing plasma cells (44). B-cell-specific Bcl-2 overexpression
can also increase the antibody-producing cell population (45). In addition, MYD88 L265P mutations
were discovered to be strongly associated with IgM secretion in
LPL/IgM monoclonal gammopathy of undetermined significance
(34,46). In plasma B cells, excessive
production of monoclonal immunoglobulin induced ER stress. In
response to ER stress, cells activate compensatory mechanisms, such
as the unfolded protein response (UPR), eventually leading to ER
stress-related apoptosis. If ER stress cannot be resolved,
prolonged ER stress may induce the activation of the inflammatory
signaling pathways, including that of NF-κB (47). Overall, constitutive NF-κB activity
in tumor cells could increase tumor cell survival and resistance to
cytotoxic agents (48). These data
suggested that the large amount of immunoglobulin production by
neoplastic B cells was also a possible explanation for the
constitutive activation of ER stress and the NF-κB pathway. In the
present case, there is a great possibility that the overproduction
of immunoglobulin secreted by lymphoma cells with plasmacytic
differentiation activated ER stress and the NF-κB pathway.
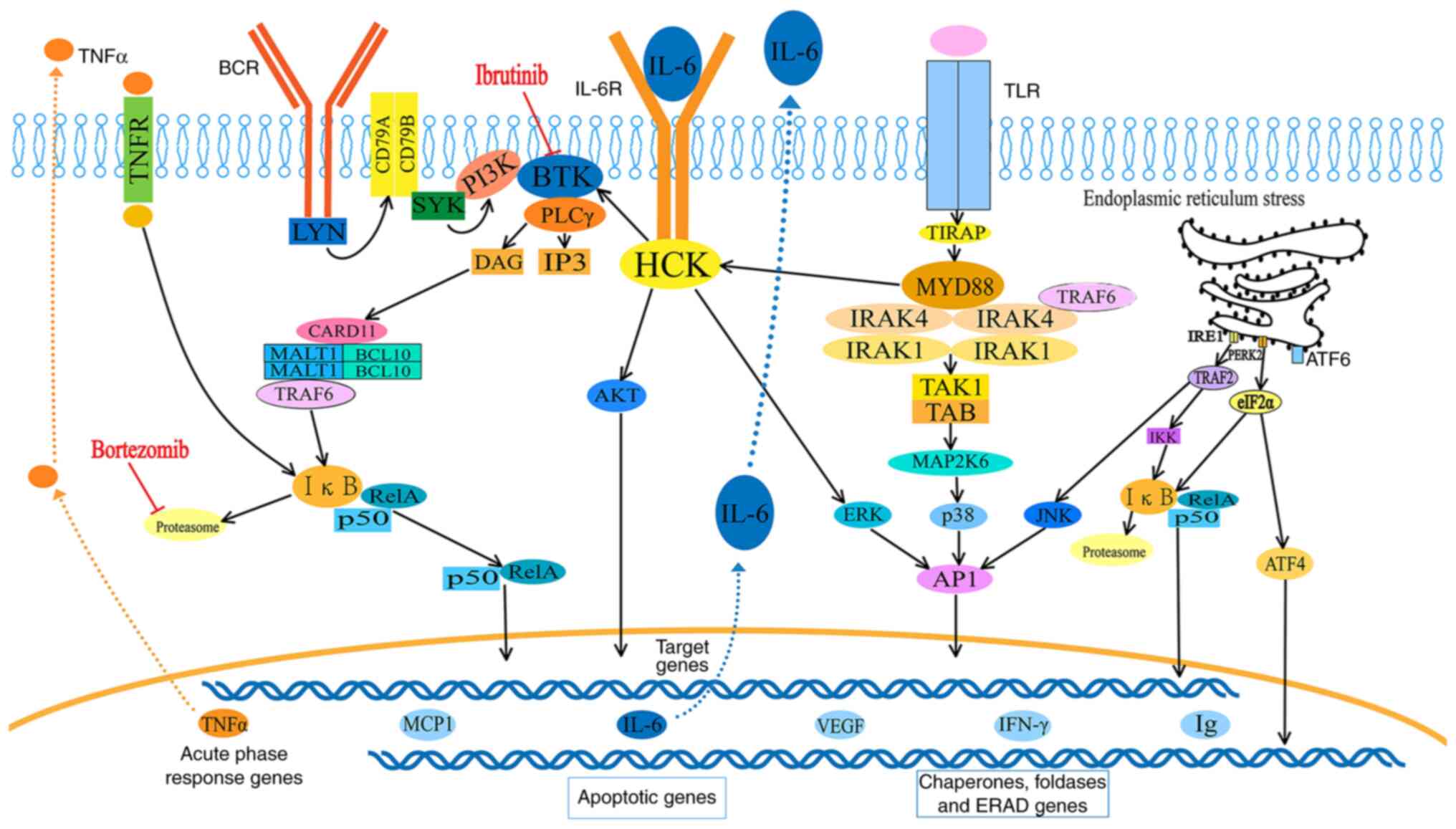
The effects of MYD88 on the NF-κB and ER signaling
pathways are presented in Fig. 6.
After stimulation of Toll-like receptors (TLRs) by
pathogen-associated molecular patterns, MYD88 is recruited to the
activated receptor via TIRAP and forms complexes with interleukin-1
receptor-associated kinase (IRAK)1 and 4. IRAK1 is then
phosphorylated by IRAK4, separates from MYD88, and interacts with
the E3 ubiquitin ligase TNF receptor-associated factor 6 (TRAF6).
On the one hand, active TRAF6 recruits and activates TAK1, which
mediates phosphorylation of the IκB kinase (IKK) complex. The
activated IKK complex then marks IκB for ubiquitination and
proteasomal degradation, consequently releasing NF-κB dimers
(49). On the other hand, active
TRAF6 phosphorylates mitogen-activated protein kinase 6 (MAP2K6),
and MAP2K6 activates MAPKs, including c-Jun N-terminal kinase
(JNK), p38 and extracellular signal-regulated kinase (ERK), to
stimulate activating protein-1 (AP-1) activity. The free NF-κB
dimers and active AP-1 translocate into the nucleus to activate the
expression of target genes, including IgM, TNF-α, IFN-γ, IL-6,
IL-10, monocyte chemoattractive protein-1 and VEGF. Mutated MYD88
also transcriptionally upregulates and transactivates the SRC
family member hematopoietic cell kinase through IL-6, which
triggers the activation of Bruton's tyrosine kinase (BTK).
In response to many paraproteins, ER stress induces
the UPR, which includes three pathways, the PERK-eIF2α-ATF4,
IRE1-XBP1 and the ATF6 axes, which contributes to avoiding fatal ER
stress through the activation of the ER chaperon and ER-associated
degradation (47). Activation of
the PERK/eIF-2α axis, on the one hand, results in NF-κB activation
by reducing IκB protein. On the other hand, the above course
increases ATF-4, which can negatively regulate the activity of
NF-κB and JNK, thus increasing AP-1 (50). The IRE-1α activates TRAF-2
activates IKK or JNK, both of which activate the transcription of
inflammatory genes.
It is generally agreed that ABC DLBCL presents with
significantly inferior progression-free survival (PFS) and overall
survival (OS) times compared with germinal center B-cell (GCB)
DLBCL (51). Up to 29% of ABC
DLBCL cases were originally discovered to have the MYD88 L265P
mutation (52). The
lymphomagenesis mechanism of L265P might facilitate its ability to
drive and increase the activation of NF-κB (49). In addition to TLR-related NF-κB
activity, B-cell antigen receptor (BCR)-dependent NF-κB activation
is of critical importance. This constitutive NF-κB activation,
which has been shown to increase lymphoma cell survival and repress
chemotherapy-induced cytotoxicity, might partly explain the
enhanced resistance of ABC DLBCL toward frontline chemotherapy
(45). Consequently, the
inhibition of NF-κB activation represents an attractive therapeutic
strategy in ABC DLBCL.
Lymphoma cells in ABC tumors take advantage of BCR
expression, which associates with increased tyrosine
phosphorylation levels of CD79 complexes and induces sustained BCR
signaling that is activated through the NF-κB pathway to sustain
neoplastic proliferation (53).
Hence, ibrutinib, an inhibitor of BTK, is suggested to be a
treatment of ABC DLBCL (7). A
recent double-blind phase III study evaluated the ibrutinib plus
R-CHOP regimen in untreated ABC DLBCL, and the results showed
improvements in event-free survival, PFS, and OS times, with
manageable safety only in patients aged <60 years (9). In patients aged ≥60 years, ibrutinib
plus R-CHOP was associated with worse outcomes and increased
toxicity. A previous phase I/II trial involving patients with RR
DLBCL reported that ABC tumors with BCR mutations and concomitant
MYD88 mutations frequently responded to ibrutinib (4/5; 80%)
(7). Notably, DLBCL with mutated
MYD88 but without BCR mutations (CD79B) was unresponsive. This
outcome might be associated with abnormal MYD88-activated chronic
BCR signaling through a BCR-independent mechanism or some
non-genetic mechanism. However, patients who exhibited mutations in
CD79B and MYD88 were associated with an increased risk of relapse
and progression (54).
Lenalidomide can impact NF-κB signaling and type I
interferon to improve the efficacy of ABC DLBCL. In the first phase
III trial (REMARC study) comparison of lenalidomide with placebo in
patients with DLBCL treated with R-CHOP, the 2-year PFS rate
improved from 75 to 80% in the lenalidomide group. It is noteworthy
that the patients were 60 to 80 years old, and further clinical
trials need to define the effect of this agent on other age groups
(8). Thereafter, a subpopulation
analysis was performed to assess the impact of lenalidomide
maintenance therapy (55). The
results showed a trend toward improved PFS in patients undergoing
dose reductions, which might be associated with bone marrow
reserves.
BTZ, a proteasome inhibitor, could prevent the
degradation of proapoptotic proteins in HLH and increase the
protein load within the cell, consistently stimulating ER stress,
and finally inducing apoptosis in cells. Furthermore, BTZ inhibited
the activation of the 26S proteasome complex, blocking the
degradation of IκBα. Consequently, NF-κB was unable to mediate
transcription, and signaling activity was suppressed. BTZ is now
widely used in the treatment of MM as an initial therapy (56). In 2009, Dunleavy et al
(57) reported that BTZ was an
effective regimen to treat DLBCL. When combined with chemotherapy,
BTZ resulted in a notably higher response and median OS time in ABC
than in GCB DLBCL. Notably, in an open-label, randomized
controlled, phase III trial evaluating DLBCL with BTZ, there was no
difference in PFS time between the R-CHOP and BTZ+R-CHOP (B+R-CHOP)
groups in ABC or GCB DLBCL populations (58). Among patients with mutations that
are known to be associated with the NF-κB pathway (MYD88, CD79A and
CD79B), the addition of BTZ had no significant effect on their
survival, although the regimen was well tolerated. However, these
results might be influenced by the restricted populations and
insufficient doses of BTZ. To gain insight into patient outcomes
due to the extended regimen, a multicenter, open label phase I/II
study for newly diagnosed CD20+ DLBCL and a higher risk
profile (IPI ≥2) aimed at the use of R-CHOP and a biological agent,
as well as a signaling inhibitor (ibrutinib), is underway (10). This trial may reveal the
responsiveness to innovative multimodality
immune-signaling-biological-chemotherapy regimens in patients
stratified by molecular classifiers.
In summary, the current study presented the case of
a patient with HLH triggered by ABC DLBCL exhibiting plasmacytic
differentiation and the MYD88 L265P mutation. Prompt recognition of
HLH and an accurate diagnosis of underlying disease are obligatory
and vital. Treatments not only control cytokine release but also
target the underlying disorder and significantly affect the
prognosis of patients. Therefore, a reliable diagnosis of lymphoma
and precision therapy pointing to gene aberrations need to be
examined in further research. As suggested by the present case, the
application of B+R-CHOP may be successful in the treatment of the
subtype of ABC DLBCL characterized by NF-κB and ER stress pathway
activity.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC contributed to the analysis and interpretation of
the data, as well as the drafting of the manuscript. LZ and HZ
contributed to data analysis and manuscript preparation. GF
contributed to the conception of the manuscript. XZ helped perform
the analysis, with constructive discussions. All authors read and
approved the final version of the manuscript. LZ and GF confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The patient provided written informed consent for
the present study.
Patient consent for publication
The patient provided written informed consent for
the publication of all the data and associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Al-Samkari H and Berliner N:
Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 13:27–49.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yildiz H, Van Den Neste E, Defour JP,
Danse E and Yombi JC: Adult haemophagocytic lymphohistiocytosis: A
review. QJM. Jan 14–2020.(Epub ahead of print). PubMed/NCBI
|
|
3
|
Jordan MB, Allen CE, Greenberg J, Henry M,
Hermiston ML, Kumar A, Hines M, Eckstein O, Ladisch S, Nichols KE,
et al: Challenges in the diagnosis of hemophagocytic
lymphohistiocytosis: Recommendations from the North American
consortium for histiocytosis (NACHO). Pediatr Blood Cancer.
66:e279292019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Henter JI, Horne A, Aricó M, Egeler RM,
Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski
J and Janka G: HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer.
48:124–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang J, Huang J and Zeng Q: Network
meta-analysis of targeted therapies for diffuse large B cell
lymphoma. BMC Cancer. 20:12182020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sarkozy C and Sehn LH: Management of
relapsed/refractory DLBCL. Best Pract Res Clin Haematol.
31:209–216. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson WH, Young RM, Schmitz R, Yang Y,
Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano
J, et al: Targeting B cell receptor signaling with ibrutinib in
diffuse large B cell lymphoma. Nat Med. 21:922–926. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thieblemont C, Tilly H, Gomes da Silva M,
Casasnovas RO, Fruchart C, Morschhauser F, Haioun C, Lazarovici J,
Grosicka A, Perrot A, et al: Lenalidomide maintenance compared with
placebo in responding elderly patients with diffuse large b-cell
lymphoma treated with first-line rituximab plus cyclophosphamide,
doxorubicin, vincristine, and prednisone. J Clin Oncol.
35:2473–2481. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Younes A, Sehn LH, Johnson P, Zinzani PL,
Hong X, Zhu J, Patti C, Belada D, Samoilova O, Suh C, et al:
Randomized phase III trial of ibrutinib and rituximab plus
cyclophosphamide, doxorubicin, vincristine, and prednisone in
non-germinal center B-cell diffuse large B-cell lymphoma. J Clin
Oncol. 37:1285–1295. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Denker S, Bittner A, Na IK, Kase J, Frick
M, Anagnostopoulos I, Hummel M and Schmitt CA: A phase I/II
first-line study of R-CHOP plus B-cell
receptor/NF-κB-double-targeting to molecularly assess therapy
response. Int J Hematol Oncol. 8:IJH202019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilson WH, Wright GW, Huang DW, Hodkinson
B, Balasubramanian S, Fan Y, Vermeulen J, Shreeve M and Staudt LM:
Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of
DLBCL. Cancer Cell. 39:1643–1653.e3. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Henter JI, Aricò M, Egeler RM, Elinder G,
Favara BE, Filipovich AH, Gadner H, Imashuku S, Janka-Schaub G,
Komp D, et al: HLH-94: A treatment protocol for hemophagocytic
lymphohistiocytosis. HLH study GRoup of the histiocyte society. Med
Pediatr Oncol. 28:342–347. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hans CP, Weisenburger DD, Greiner TC,
Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E,
Braziel RM, Jaffe ES, et al: Confirmation of the molecular
classification of diffuse large B-cell lymphoma by
immunohistochemistry using a tissue microarray. Blood. 103:275–282.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fonseca R, Bergsagel PL, Drach J,
Shaughnessy J, Gutierrez N, Stewart AK, Morgan G, Van Ness B, Chesi
M, Minvielle S, et al: International myeloma working group
molecular classification of multiple myeloma: Spotlight review.
Leukemia. 23:2210–2221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yohe S and Thyagarajan B: Review of
clinical next-generation sequencing. Arch Pathol Lab Med.
141:1544–1557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bartlett NL, Wilson WH, Jung SH, His ED,
Maurer MJ, Pederson LD, Polley MC, Pitcher BN, Cheson BD, Kahl BS,
et al: Dose-adjusted EPOCH-R compared with R-CHOP as frontline
therapy for diffuse large B-cell lymphoma: Clinical outcomes of the
phase III intergroup trial alliance/CALGB 50303. J Clin Oncol.
37:1790–1799. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Daudignon A, Quilichini B, Ameye G, Poirel
H, Bastard C and Terré C: Cytogenetics in the management of
multiple myeloma: An update by the groupe francophone de
cytogénétique hématologique (GFCH). Ann Biol Clin (Paris).
74:588–595. 2016.PubMed/NCBI
|
|
18
|
Tian E: Fluorescence in situ hybridization
(FISH) in multiple myeloma. Methods Mol Biol. 1792:55–69. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Campo M and Berliner N: Hemophagocytic
lymphohistiocytosis in adults. Hematol Oncol Clin North Am.
29:915–925. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
La Rosee P, Horne A, Hines M, von Bahr
Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J,
Girschikofsky M, Jordan MB, et al: Recommendations for the
management of hemophagocytic lymphohistiocytosis in adults. Blood.
133:2465–2477. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang Y, Cui M, Fu X, Han L, Zhang L, Li
L, Li X, Sun Z, Wu J, Zhang X, et al: Lymphoma associated
hemophagocytic syndrome: A single-center retrospective study. Oncol
Lett. 16:1275–1284. 2018.PubMed/NCBI
|
|
22
|
Yu JT, Wang CY, Yang Y, Wang RC, Chang KH,
Hwang WL and Teng CL: Lymphoma-associated hemophagocytic
lymphohistiocytosis: Experience in adults from a single
institution. Ann Hematol. 92:1529–1536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malkan UY, Albayrak M, Yildiz A, Maral S,
Afacan Ozturk HB and Comert P: A rare case of diffuse large B-cell
lymphoma-associated hemophagocytic lymphohistiocytosis. J Oncol
Pharm Pract. 27:250–252. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim MS, Cho YU, Jang S, Seo EJ, Lee JH and
Park CJ: A case of primary bone marrow diffuse large B-cell
lymphoma presenting with fibrillar projections and hemophagocytic
lymphohistiocytosis. Ann Lab Med. 37:544–546. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li X, He Y, Wang D, Hu Y, Wang W and Huang
R: A case of diffuse large B-cell lymphoma complicated by
hemophagocytic syndrome. Clin Lab. 60:681–683. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davidson-Moncada JK, McDuffee E and
Roschewski M: CD5+ diffuse large B-cell lymphoma with
hemophagocytosis. J Clin Oncol. 31:e76–e79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Desai A, Saul EE, Chapman JR, Lekakis L
and Pimentel A: Adult lymphoma-associated hemophagocytic
lymphohistiocytosis: A clinical case series in a predominantly
hispanic cohort. J Med Cases. 11:256–261. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kwoun WJ, Ahn JY, Park PW, Seo YH, Kim KH,
Seo JY, Lee HT and Yoo KH: How useful is bone marrow study as an
initial investigative tool without lymph node biopsy in malignant
lymphoma?: Eleven years of experience at a single institution. J
Clin Lab Anal. 33:e228412019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chigrinova E, Mian M, Scandurra M, Greiner
TC, Chan WC, Vose JM, Inghirami G, Chiappella A, Baldini L, Ponzoni
M, et al: Diffuse large B-cell lymphoma with concordant bone marrow
involvement has peculiar genomic profile and poor clinical outcome.
Hematol Oncol. 29:38–41. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pezzella F, Munson PJ, Miller KD,
Goldstone AH and Gatter KC: The diagnosis of low-grade peripheral
B-cell neoplasms in bone marrow trephines. Br J Haematol.
108:369–376. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Leval L and Harris NL: Variability in
immunophenotype in diffuse large B-cell lymphoma and its clinical
relevance. Histopathology. 43:509–528. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin P, Molina TJ, Cook JR and Swerdlow SH:
Lymphoplasmacytic lymphoma and other non-marginal zone lymphomas
with plasmacytic differentiation. Am J Clin Pathol. 136:195–210.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boiza-Sánchez M, Manso R, Balagué O,
Chamizo C, Askari E, Salgado RN, Blas-López C, Aguirregoicoa-García
E, Menárguez J, Santonja C, et al: Lymphoplasmacytic lymphoma
associated with diffuse large B-cell lymphoma: Progression or
divergent evolution? PLoS One. 15:e02416342020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang W and Lin P: Lymphoplasmacytic
lymphoma and Waldenström macroglobulinaemia: Clinicopathological
features and differential diagnosis. Pathology. 52:6–14. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ohno T, Ueda Y, Nagai K, Takahashi T,
Konaka Y, Takamatsu T, Suzuki T, Sasada M and Uchiyama T; Kyoto
University Hematology/Oncology Study Group, : The serum cytokine
profiles of lymphoma-associated hemophagocytic syndrome: A
comparative analysis of B-cell and T-cell/natural killer cell
lymphomas. Int J Hematol. 77:286–294. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aggarwal BB: Signalling pathways of the
TNF superfamily: A double-edged sword. Nat Rev Immunol. 3:745–756.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pękalski J, Zuk PJ, Kochańczyk M, Junkin
M, Kellogg R, Tay S and Lipniacki T: Spontaneous NF-κB activation
by autocrine TNFα signaling: A computational analysis. PLoS One.
8:e788872013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kagoya Y, Yoshimi A, Kataoka K, Nakagawa
M, Kumano K, Arai S, Kobayashi H, Saito T, Iwakura Y and Kurokawa
M: Positive feedback between NF-κB and TNF-α promotes
leukemia-initiating cell capacity. J Clin Invest. 124:528–542.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hayakawa K, Meng Y, Hiramatsu N, Kasai A,
Yao J and Kitamura M: Spontaneous activation of the NF-kappaB
signaling pathway in isolated normal glomeruli. Am J Physiol Renal
Physiol. 291:F1169–F1176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee TK, Denny EM, Sanghvi JC, Gaston JE,
Maynard ND, Hughey JJ and Covert MW: A noisy paracrine signal
determines the cellular NF-kappaB response to lipopolysaccharide.
Sci Signal. 2:ra652009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hashwah H, Bertram K, Stirm K, Stelling A,
Wu CT, Kasser S, Manz MG, Theocharides AP, Tzankov A and Müller A:
The IL-6 signaling complex is a critical driver, negative
prognostic factor, and therapeutic target in diffuse large B-cell
lymphoma. EMBO Mol Med. 11:e105762019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hasnain SZ: Endoplasmic reticulum and
oxidative stress in immunopathology: Understanding the crosstalk
between cellular stress and inflammation. Clin Transl Immunology.
7:e10352018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nikesitch N, Lee JM, Ling S and Roberts
TL: Endoplasmic reticulum stress in the development of multiple
myeloma and drug resistance. Clin Transl Immunology. 7:e10072018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang HQ, Jia L, Li YT, Farren T, Agrawal
SG and Liu FT: Increased autocrine interleukin-6 production is
significantly associated with worse clinical outcome in patients
with chronic lymphocytic leukemia. J Cell Physiol. 234:13994–14006.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Knittel G, Liedgens P, Korovkina D,
Pallasch CP and Reinhardt HC: Rewired NFκB signaling as a
potentially actionable feature of activated B-cell-like diffuse
large B-cell lymphoma. Eur J Haematol. 97:499–510. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao
Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al: MYD88
L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J
Med. 367:826–833. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Maamoun H, Abdelsalam SS, Zeidan A,
Korashy HM and Agouni A: Endoplasmic reticulum stress: A critical
molecular driver of endothelial dysfunction and cardiovascular
disturbances associated with diabetes. Int J Mol Sci. 20:16582019.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Obeng EA, Carlson LM, Gutman DM,
Harrington WJ Jr, Lee KP and Boise LH: Proteasome inhibitors induce
a terminal unfolded protein response in multiple myeloma cells.
Blood. 107:4907–4916. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Grondona P, Bucher P, Schulze-Osthoff K,
Hailfinger S and Schmitt A: NF-κB activation in lymphoid
malignancies: Genetics, signaling, and targeted therapy.
Biomedicines. 6:382018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Treon SP, Xu L, Guerrera ML, Jimenez C,
Hunter ZR, Liu X, Demos M, Gustine J, Chan G, Munshi M, et al:
Genomic landscape of Waldenström macroglobulinemia and its impact
on treatment strategies. J Clin Oncol. 38:1198–1208. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chapuy B, Stewart C, Dunford AJ, Kim J,
Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et
al: Molecular subtypes of diffuse large B cell lymphoma are
associated with distinct pathogenic mechanisms and outcomes. Nat
Med. 24:679–690. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Visco C, Tanasi I, Quaglia FM, Ferrarini
I, Fraenza C and Krampera M: Oncogenic mutations of MYD88 and CD79B
in diffuse large B-Cell lymphoma and implications for clinical
practice. Cancers. 12:29132020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Young RM and Staudt LM: Targeting
pathological B cell receptor signalling in lymphoid malignancies.
Nat Rev Drug Discov. 12:229–243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Vermaat JS, Somers SF, de Wreede LC, Kraan
W, de Groen RA, Schrader AM, Kerver ED, Scheepstra CG, Berenschot
H, Deenik W, et al: MYD88 mutations identify a molecular subgroup
of diffuse large B-cell lymphoma with an unfavorable prognosis.
Haematologica. 105:424–434. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Thieblemont C, Howlett S, Casasnovas RO,
Mounier N, Perrot A, Morschhauser F, Fruchart C, Daguindau N, van
Eygen K, Obéric L, et al: Lenalidomide maintenance for diffuse
large B-cell lymphoma patients responding to R-CHOP: Quality of
life, dosing, and safety results from the randomised controlled
REMARC study. Br J Haematol. 189:84–96. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Keats JJ, Fonseca R, Chesi M, Schop R,
Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al:
Promiscuous mutations activate the noncanonical NF-kappaB pathway
in multiple myeloma. Cancer Cell. 12:131–144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dunleavy K, Pittaluga S, Czuczman MS, Dave
SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM and
Wilson WH: Differential efficacy of bortezomib plus chemotherapy
within molecular subtypes of diffuse large B-cell lymphoma. Blood.
113:6069–6076. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Davies A, Cummin TE, Barrans S, Maishman
T, Mamot C, Novak U, Caddy J, Stanton L, Kazmi-Stokes S, McMillan
A, et al: Gene-expression profiling of bortezomib added to standard
chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An
open-label, randomised, phase 3 trial. Lancet Oncol. 20:649–662.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tang S, Zhao C and Chen W: Aggressive
diffuse large B-cell lymphoma with hemophagocytic
lymphohistiocytosis: Report of one case. Int J Clin Exp Pathol.
13:2392–2396. 2020.PubMed/NCBI
|
|
60
|
Wu R, Deng X, Hao S and Ma L: Successful
treatment of diffuse large B-cell lymphoma with secondary
hemophagocytic lymphohistiocytosis by R-CHOP-E regimen: A case
report. J Int Med Res. 48:3000605198822332020.PubMed/NCBI
|
|
61
|
Patel R, Patel H, Mulvoy W and Kapoor S:
Diffuse large B-cell lymphoma with secondary hemophagocytic
lymphohistiocytosis presenting as acute liver failure. ACG Case Rep
J. 4:e682017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Patel A, Vakiti A, Chilkulwar A and
Mewawalla P: Hemophagocytic lymphohistiocytosis secondary to bone
marrow only B-cell lymphoma: A very rare entity with an even rarer
presentation. J Hematol. 6:49–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Entezari V, Agwa E, Ruiz SJ, Lietman SA,
Silver BJ and Jegalian AG: Hemophagocytic lymphohistiocytosis
secondary to localized large B-cell lymphoma in a patient with
history of knee arthroplasty. Leuk Lymphoma. 56:1521–1523. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kuo CY, Yeh ST, Huang CT and Lin SF:
Diffuse large B-cell lymphoma presenting with type B lactic
acidosis and hemophagocytic syndrome. Kaohsiung J Med. 30:428–429.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang L, Zhang XH, Zhou YL and Yin XL:
Hemophagocyte lymphohistiocytosis secondary to bilateral epididymal
diffuse large B-cell lymphoma. Indian J Hematol Blood Transfus. 30
(Suppl 1):S93–S96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sano T, Sakai H, Takimoto K and Ohno H:
Rituximab alone was effective for the treatment of a diffuse large
B-cell lymphoma associated with hemophagocytic syndrome. Int J Clin
Oncol. 12:59–62. 2007. View Article : Google Scholar : PubMed/NCBI
|