Introduction
Liver cancer is the third leading cause of
cancer-related death and the sixth most commonly diagnosed cancer
worldwide (1). Hepatocellular
carcinoma (HCC) is the most common type of liver cancer. Viral
hepatitis infection is a major risk factor for hepatocellular
carcinoma in China (2,3). Despite advances in surgical
techniques, procedural interventions, and the development of
targeted drugs, the prognosis of HCC remains poor (4). Therefore, unraveling the pathogenesis
of HCC and identifying novel and effective therapeutic targets have
become the focus of current research (5–9).
FXR1 belongs to the fragile X-related (FXR) family
(10,11); previous studies have shown that
FXR1 can promote the proliferation, invasion, and migration of
colorectal cancer cells (12).
Moreover, FXR1 has been reported to play a role in the development
of oral cancer and gliomas (13,14).
However, the role and mechanism of FXR1 in HCC have not been
elucidated.
In the early stages of tumor development, TGF-β
inhibits tumor cell proliferation by inhibiting c-Myc, cyclin, and
cyclin-dependent kinase activity, preventing the transition from
the G1 to S phase. In the later stages of tumor development, TGF-β
promotes tumor progression (15–17).
The classical TGF-β pathway is mediated by SMAD proteins, which
bind to type I and II TGF-β receptor complexes (TGFBR1 and TGFBR2),
resulting in TGFBR phosphorylation and activation (11). TGFBR can further induce the
phosphorylation of SMAD2/3, and the activated SMAD2/3 can further
bind to SMAD4. Subsequently, the SMAD complex is transferred to the
nucleus to exert its effects (18,19).
One of the most critical processes that enable tumor cells to
infiltrate and metastasize is epithelial-mesenchymal transition
(EMT) (17,20). EMT causes epithelial cells to lose
their epithelial phenotypes. It also enhances tumor cell migration
and invasion, anti-apoptotic processes, and extracellular matrix
degradation (21,22).
In this study, we demonstrated that FXR1 knockdown
led to a reduction in HCC cell migration and invasion, and it
inhibited the stimulation of HCC cell migration and invasion via
modulation of TGF-β. FXR1 overexpression in HCC cells promoted cell
invasion, which was abrogated by inhibiting SMAD2/3.
Materials and methods
Cell culture
LM3 (cat. no. CL-0278), Huh7 (CL-0120), MHCC-97H
(CL-0497) and Hep3B (cat. no. CL-0102) cells were purchased from
Procell Life Science & Technology Co., Ltd. Cells were cultured
in DMEM high glucose supplemented with 10% FBS, 100 mg/ml
penicillin G, and 50 mg/ml streptomycin in a humidified incubator
at 37°C supplied with 5% CO2. MHCC-97H (CL-0497) cells
were used for western blotting.
Reagents and antibodies
TGF-β was purchased from PeproTech, Inc. FXR1 (cat.
no. 67813-1-Ig, 1:10,00 or 1:100), Ki67 (27309-1-AP; 1:100) and
β-actin (cat. no. 66009-1-Ig, 1:10,00) antibodies were purchased
from ProteinTech Group, Inc. The FXR1 antibody was diluted 1:1,000
in the western blotting assay and 1:100 in immunohistochemistry
assays. SMAD2/3 (cat. no. # 8685S, 1:1,000), N-Cadherin (cat. no. #
4061, 1:1,000), and slug (cat. no. 9585S, 1:1,000) antibodies were
purchased from Cell Signaling Technology, Inc. The secondary
antibodies used were HRP-conjugated Affinipure Goat Anti-Rabbit
(1:2,000 or 1:50; cat. no. SA00001-2) and HRP-conjugated Affinipure
Goat Anti-Mouse (1:2,000; cat. no. SA00001-1) (both from
ProteinTech Group, Inc.). HRP-conjugated Affinipure Goat
Anti-Rabbit was diluted 1:2,000 in the western blotting assay and
1:50 in immunohistochemistry assays. SMAD2/3 siRNA was purchased
from Santa Cruz Biotechnology, Inc. (cat. no. sc-37238). shRNA was
purchased from Public Protein/Plasmid Library. The sequences of the
FXR1 knockdown shRNAs were: shFXR1#1, 5′-GCTAGAGGTTTCTTGGAATTT-3′;
shFXR1#2, 5′-CGCCAGGTTCCATTTAATGAA-3′; and negative control (shNC),
5′-GTTCTCCGAACGTGTCACGTT-3′. The FXR1 expression plasmid with a
3′FLAG tag was purchased from Public Protein/Plasmid Library.
Reverse transcription-quantitative
PCR
TRIzol® reagent was used to extract total
RNA from tissues or cells, and 1 µg RNA was reverse transcribed to
cDNA using a Vazyme reverse transcription kit according to the
manufacturer's protocol (Vazyme Biotech Co., Ltd.; cat. no.
R323-01). Amplification was performed using amplification reagent
according to the manufacturer's protocol (Vazyme Biotech Co., Ltd.;
cat. no. Q711-02). The thermocycling conditions for amplification
were as follows: 95°C for 5 min, followed by 40 cycles at 95°C for
10 sec, 60°C for 30 sec, and a final step at 95°C for 15 sec, 60°C
for 60 sec and 95°C for 15 sec. β-actin was used as the
housekeeping gene. The sequences of the primers used for
amplification were: β-actin forward, 5′-CATGTACGTTGCTATCCAGGC-3′
and reverse 5′-CTCCTTAATGTCACGCACGAT-3′; SMAD2 forward,
5′-GATCCTAACAGAACTTCCGCC-3′ and reverse,
5′-CACTTGTTTCTCCATCTTCACTG-3′; and SMAD3 forward,
5′-TCCATCCCCGAAAACACTAAC-3′ and reverse,
5′-CATCTTCACTCAGGTAGCCAG-3′ (23).
The relative expression level of the target genes was calculated
using the 2−ΔΔCq method (23).
Transfection
For transient transfection of plasmids, LM3 cells
were transfected using Lipofectamine® 3000 (Thermo
Fisher Scientific, Inc.). For transfection of shRNAs (final
concentration, 30 µM), proliferating cells in a 6-well plate were
incubated in serum-free DMEM containing Lipofectamine®
3000. After 4–6 h, the cells were incubated in supplemented DMEM
for 48 h, and then treated with the newly configured puromycin (2
µg/ml) screening medium every day until no cell death was observed.
The maintenance medium contained puromycin (2 µg/ml). To transfect
siRNAs (final concentration, 20 µM), proliferating cells in a
6-well plate were incubated with serum-free DMEM supplemented with
Lipofectamine® 3000. After 4–6 h, the cells were
incubated in complete DMEM for 48 h and then selected with
puromycin (2 µg/ml) until no cellular death was observed. The
maintenance medium contained puromycin (2 µg/ml).
Western blotting
Total protein was extracted using RIPA lysis buffer
supplemented with a mixture of PMSF, aprotinin, and phosphatase
inhibitors. LM3 (cat. no. CL-0278), Huh7 (CL-0120), MHCC-97H
(CL-0497) and Hep3B (cat. no. CL-0102) cells were used for western
blotting. The protein concentration was measured using a
Bicinchoninic protein assay (Thermo Fisher Scientific, Inc.).
Proteins (30 µg per lane) were separated using SDS-PAGE on a 10%
SDS gel, transferred to PVDF membranes, and incubated with the
primary antibody, followed by incubation with the appropriate
horseradish peroxidase (HRP)-conjugated secondary antibody at room
temperature for 1 h. BeyoECL Plus (Beyotime Institute of
Biotechnology) was used for signal visualization, and images were
obtained using a Fushon Fx (Vilber Lourmat) imaging system.
Apoptosis evaluation using flow
cytometry
A Beyotime Institute of Biotechnology apoptosis kit
was used for testing (cat. no. C1069L). The cells were washed with
PBS once and detached using pancreatic enzyme cell digestion
solution, at room temperature until gentle aspiration with a
pipette was sufficient to lift cells (ensuring over-digestion was
avoided), after which the pancreatic enzyme cell digestion solution
was removed. The cells were collected, mixed gently, and
centrifuged at 1,000 × g at 4°C for 5 min, after which the cells
were collected and gently resuspend in PBS for counting. A total of
50–100 µl of the resuspended cell solution was taken, centrifuged
at 1,000 × g at 4°C for 5 min, the supernatant was discarded, 195
µl annexin V-mCherry Binding Buffer was added, and cells were
gently resuspended. A total of 5 µl annexin V-mCherry was added and
mixed gently, incubated at room temperature (20–25°C) for 10–20
min, avoiding light, followed by incubation in an ice bath.
Aluminum foil was used to protect against light. Cells were
resuspended 2–3 times during incubation to ensure appropriate and
equal staining. Subsequently, cells were analyzed using flow
cytometry (BD FACSymphony™ A3; BD Biosciences) for Annexin
V-mCherry red fluorescence (Beyotime Institute of Biotechnology
apoptosis kit; cat. no. C1069L). FlowJo™10 (BD Biosciences) was
used for analysis.
Colony formation assay
After digestion of cells using cellular pancreatic
enzyme during the logarithmic growth phase, cells were resuspended
in supplemented media and counted. A total of 400–1,000 cells/well
(generally 700 cells/well) were seeded in each experimental group
in a 6-well culture plate and cultured for 14 days, changing the
media every 3 days. After 14 days, 1 ml crystal violet dye solution
was added to each well and incubated at room temperature for 10–20
min, after which cells were washed several times, dried, and imaged
with a digital camera attached to a bright field microscope at a
magnification of ×200. Colonies consisting of ≥50 cells were
counted.
Cell migration and invasion
Cell migration was determined using a Transwell
assay. Transfected Hep3B or LM3 cells (5×104 per well)
or cells treated with 20 ng/ml TGF-β were resuspended in 500 µl
serum-free medium containing 2 µg/ml mitomycin C and added to the
upper chamber to inhibit cell proliferation, and 500 µl
supplemented media was added to the lower chamber. After incubation
at 37°C for 48 h, the cells on the upper surface of the filter
membrane were removed using a swab and the filter membrane was
incubated in 100% methanol at room temperature for 2 min. The cells
that had migrated to the underside of the filter were stained with
0.5% crystal violet at room temperature for 20 min, and images were
obtained using a bright field microscope. The number of cells that
had migrated was counted using Image Pro Plus 6.0 (Media
Cybernetics, Inc.). Cell invasion was determined using the same
method as that for cell migration except cells were seeded in a
Transwell chamber covered with Matrigel (BD Biosciences).
Mouse xenograft model
A total of 20 BALB/c nude mice were purchased from
Beijing Vital River Laboratory Animal Technology Co., Ltd. Mice
were maintained with a 12-h light/dark cycle under specific
pathogen-free conditions. All animal experiments were approved by
the Ethics Committee of the First Affiliated Hospital of Zhengzhou
University (approval no. 2019-KY-21) and in accordance with the
guidelines of the Office of Laboratory Animal Welfare. Mice were
monitored daily and euthanized according to NIH-approved criteria
for institutional humane endpoints if they failed to demonstrate a
corrective response. For tumor growth experiments, stably
transfected LM3 cells (1×105) were injected into mice by
subcutaneous injection in the abdomen. The volume of the tumor was
measured once every 2 days as follows: Volume=0.5× length × width ×
height; the total volume of each tumor did not exceed 4,400
mm3. When the tumor size reached 12–20 mm the mice were
euthanized via cervical dislocation; death was confirmed by lack of
breathing and heartbeat.
Patients
The study included 88 patients with HCC (74 male and
14 female patients) treated at the First Affiliated Hospital of
Zhengzhou University. HCC (2–3 cm) and normal tissue samples were
obtained after surgery between January 2020 and December 2021.
Patients were aged 30–76 years (median age, 56 years). Complete
clinical information was available for included patients, and none
of the patients had other malignancies or a history of chemotherapy
or radiation therapy.
Immunohistochemical analysis
A total of 88 pairs of HCC and normal tissues (74
males and 14 female patients) were obtained from the First
Affiliated Hospital of Zhengzhou University (Zhengzhou, China). The
tumors of nude mice were tested for Ki67 after removal.
For immunohistochemistry, tissue sections were
mounted on a glass slide, deparaffinized, and rehydrated. Samples
were soaked in 100% alcohol, 100% alcohol, 95% alcohol, 75% alcohol
and water for 5 min each for hydration. Heat-induced antigen
retrieval was subsequently performed using 10 nmol/l citrate buffer
(pH=6.0) for 10 min in a microwave oven (high heat). The slides
were incubated with FXR1 (cat. no. 67813-1-Ig; 1:100) or Ki67
(27309-1-AP; 1:100) antibody overnight at 4°C, followed by
incubation with a HRP labeled secondary antibody (cat. no.
SA00001-2; 1:50) at room temperature for 15 min. The slides were
further stained with DAB developer and counterstained with 5%
hematoxylin at room temperature for 3 min. Positive cells were
stained brown.
The expression levels were scored based on the
percentage of positive cells and the intensity of staining. The
score was calculated as follows: 0, <5% positively stained
cells; 1, 5–25% positively stained cells; 2, 25–50% positively
stained cells; 3, 50–75% positively stained cells; and 4, >75%
positively stained cells. The intensity was scored as follows: 0,
negative staining; 1, light yellow staining; 2, brownish yellow
staining; and 3, chocolate-brown staining. The final score was
calculated by multiplying the percentage score by the intensity
score. A score <8 was considered low, and a score ≥8, was
considered high.
Bioinformatics analysis
The Oncomine online database (http://www.oncomine.org) was used to analyze FXR1
transcripts in HCC from the Rossher liver and Rossher liver 2
datasets. UALCAN (http://ualcan.path.uab.edu) was used to analyze the
expression of FXR1 in HCC data from TCGA. GEPIA (http://gepia.cancer-pku.cn/) was used to analyze the
correlations between i) FXR1 and SMAD2/3/4 mRNA levels and ii) FXR1
expression in TCGA. The threshold for mRNA levels was customized to
allow for the stratification of subjects with low or high FXR1 mRNA
levels. TCGA raw data, such as gene expression and
clinicopathological data, were included in the UALCAN and
cBioPortal analyses (http://www.cbioportal.org). Gene Set Enrichment
Analysis (GSEA) was performed to explore potential biological
pathways based on Kyoto Encyclopedia of Genes and Genomes (KEGG)
gene sets using R software (version 4.1.2; http://www.r-project.org/). For KEGG enrichment
analysis, the ‘clusterProfiler’ R package was used to decode
potential targets downstream of FXR1, the gseKEGG function was used
to explore the underlying biological significance. Based on gene
count thresholds generated from the median value of FXR1 expression
(TPM value=4.16), patients were classified into low and high
groups.
Statistical analysis
SPSS software (version 22.0; IBM Corp.) was used for
statistical analyses. A one-way ANOVA followed by a post hoc
Bonferroni correction was used for the statistical analysis of mRNA
expression, cell migration, and invasion. A Student's t-test was
used to compare the differences between two groups. A Pearson's
correlation test was used to assess correlations. Paired samples
were tested using a paired t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
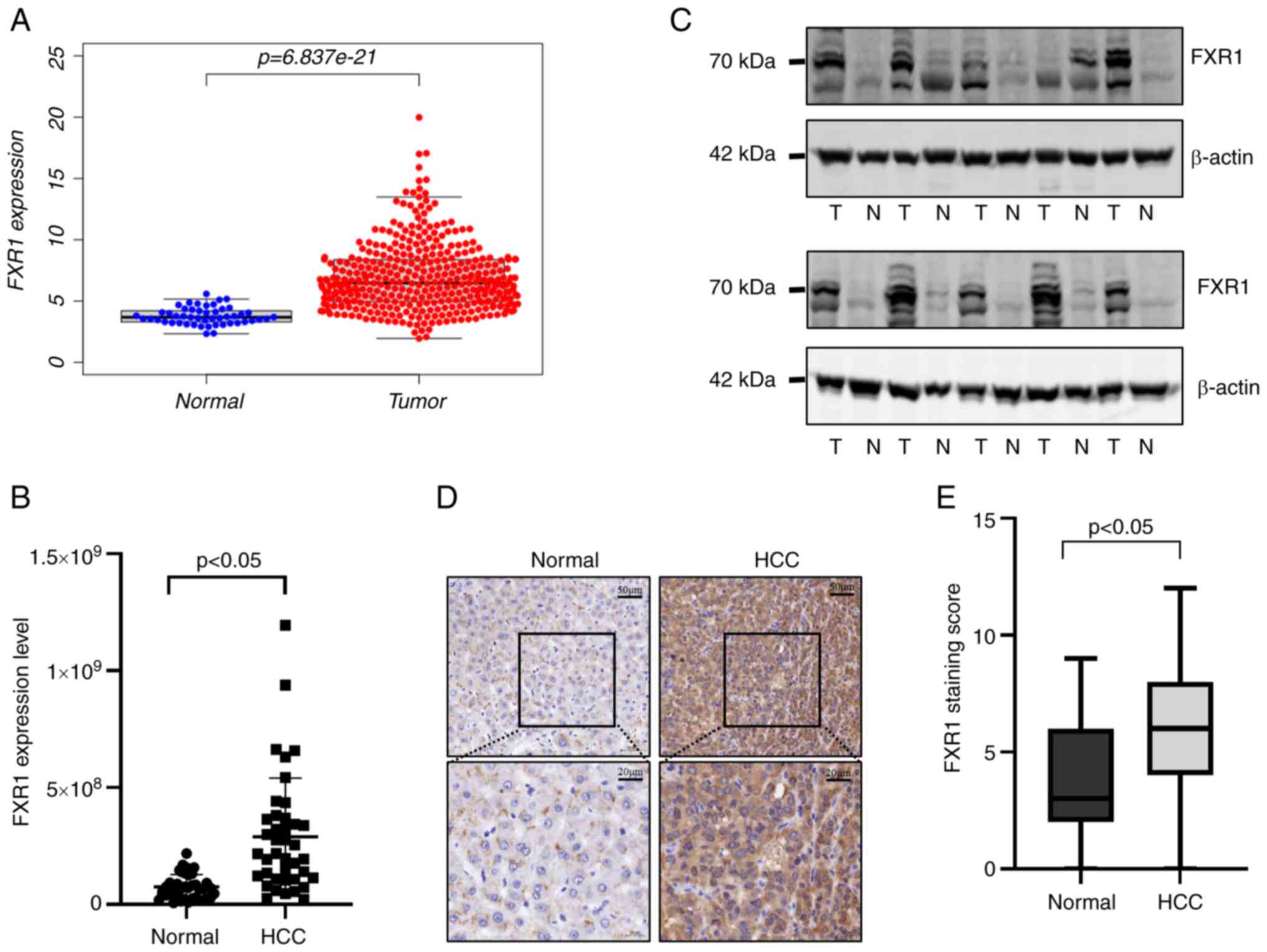
FXR1 expression is upregulated in
HCC
To investigate FXR1 expression in HCC, data from the
HCC cohort in TCGA was used; FXR1 was upregulated in HCC (Fig. 1A). Validation of FXR1 expression
was also performed using HCC and normal tissues, and the results
showed that FXR1 was highly expressed in HCC at the protein level
(Fig. 1B and C). Next, FXR1
expression was assessed in HCC and normal tissues using
immunohistochemical microarrays, and similar results were observed;
FXR1 expression was higher in HCC tissues than in normal tissues
(Fig. 1D and E).
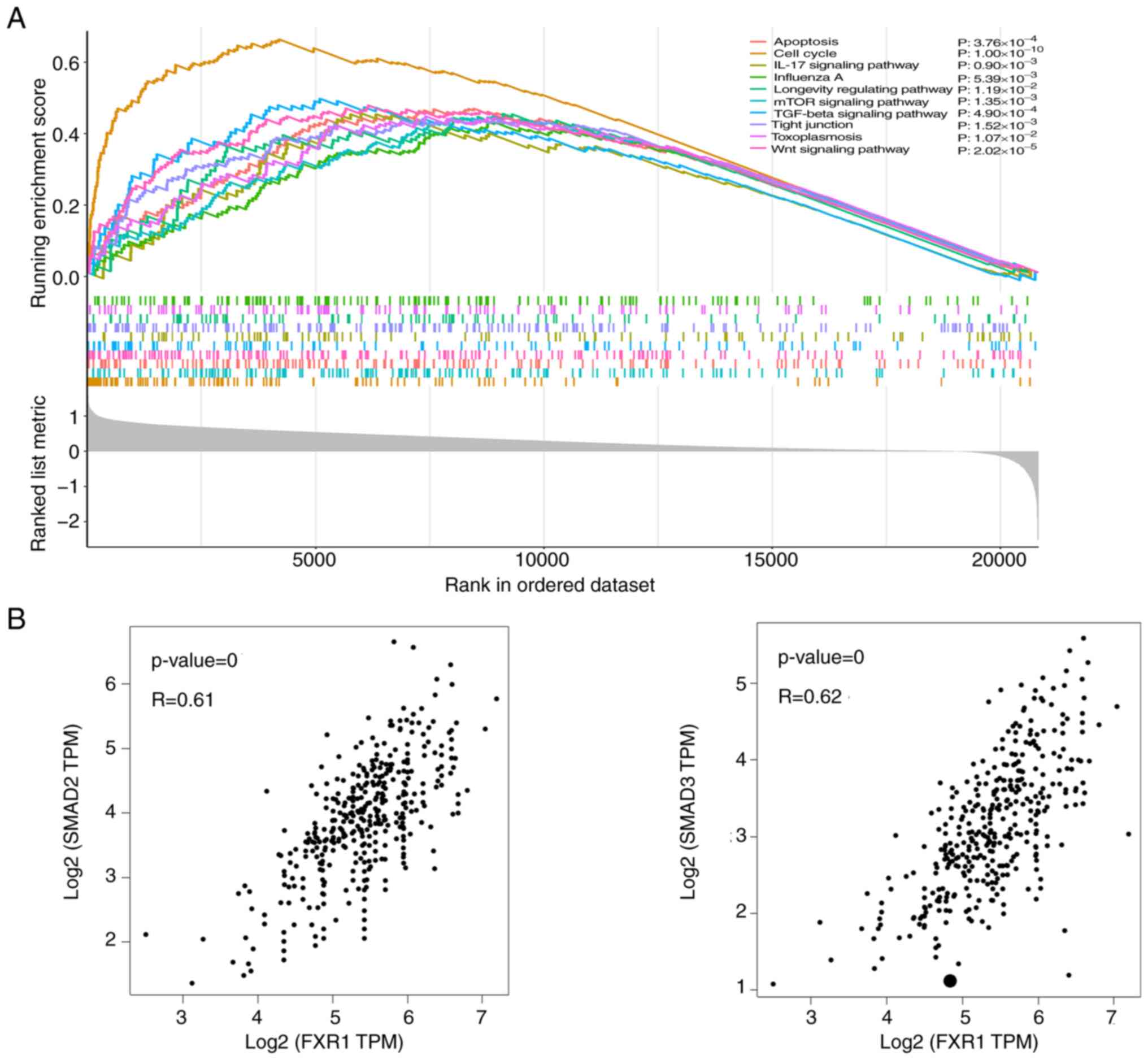
FXR1 is positively associated with
SMAD2/3/ in HCC cells
To explore potential targets downstream of FXR1,
GSEA was used to identify the latent biological pathways based on
KEGG gene sets; the top 10 corresponding genes are shown in
Fig. 2A. Bioinformatics analysis
was used to assess the correlation between FXR1 and genes from
TCGA. The mRNA expression levels of FXR1 were positively correlated
with the mRNA expression levels of SMAD2/3 (Fig. 2B).
FXR1 promotes slug/N-cadherin
expression and TGF-β-induced HCC cell migration and invasion
We further investigated the effects of FXR1 on the
migratory and invasive effects of HCC and the involvement of
SMAD2/3. The protein expression levels of FXR1 in five different
HCC cell lines were examined, and the results showed that FXR1 was
relatively highly expressed in LM3 and Hep3B cells (Fig. 3A). shRNAs were used to knock down
FXR1 expression in LM3 and Hep3B cells, and knockdown was confirmed
using western blotting (Fig. 3B).
Western blotting also showed a decrease in the expression of
N-cadherin and Slug in FXR1 knockdown cells (Fig. 3B). These results suggest that FXR1
affects the expression of EMT-related proteins. In addition, the
expression levels of SMAD2/3 decreased upon FXR1 knockdown
(Fig. 3B).
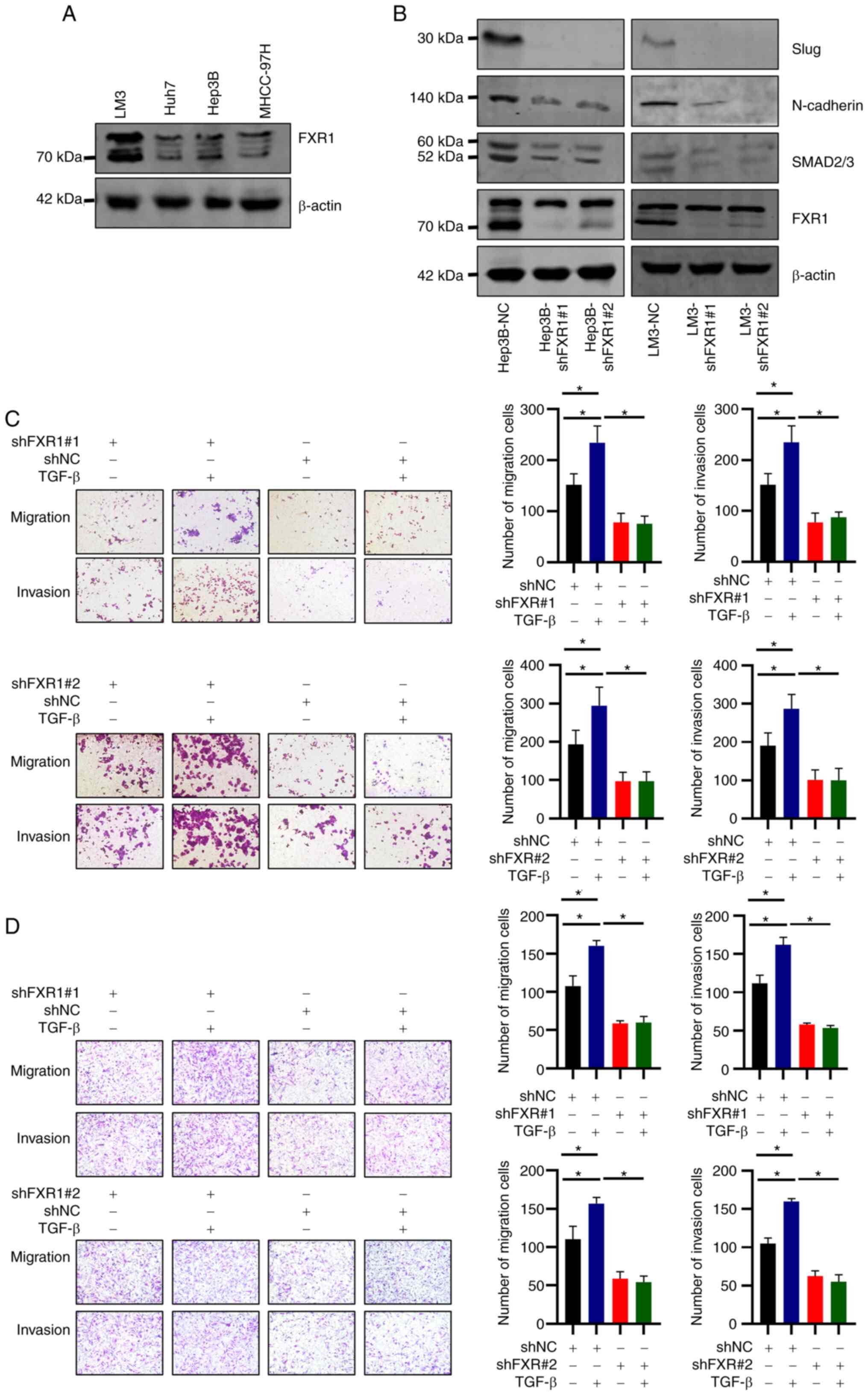 | Figure 3.FXR1 promotes slug/N-cadherin
expression and TGF-β-induced HCC cell migration and invasion. (A)
FXR1 expression in HCC cells. (B) Hep3B and LM3 cells were
transfected with shNC, shFXR1#1, or shFXR1#2, and western blotting
was used to determine knockdown efficiency. (C) Hep3B and (D) LM3
cells were transfected with shNC, shFXR1#1, or shFXR1#2, and
treated with or without TGF-β, followed by cell migration and
invasion assays. The cells were then observed microscopically
(magnification, ×200). The relative cell migration or invasion was
plotted. Cell migration or invasion was normalized to the
shNC-transfected and TGF-β-untreated group as appropriate. Note:
The difference in cell status and the time of spreading the plates
resulted in inconsistencies in the number of cells in the control
group at different times. Data are presented as the mean ± SD of
three repeats. *P<0.05. FXR1, fragile X-related 1; HCC,
hepatocellular carcinoma; sh, short hairpin; NC, negative
control. |
TGF-β promotes tumor migration and invasion through
EMT (24,25). The effect of FXR1 on the
TGF-β-induced migration and invasion in HCC cells was thus next
assessed. FXR1 knockdown cells transfected with the two different
shRNAs consistently inhibited the migration and invasion of LM3 and
Hep3B cells (Fig. 3C and D).
Additionally, shRNA-induced FXR1 knockdown inhibited the
TGF-β-induced increase in HCC cell migration and invasion when
compared with cells treated with TGF-β and transfected with shNC
(Fig. 3C and D).
Knockdown of FXR1 inhibits the
proliferation of HCC cells and promotes early apoptosis of HCC
cells
To validate the effect of FXR1 on apoptosis in HCC
cells, two different shRNAs were used to knockdown FXR1 in Hep3B
and LM3 cells, and apoptosis was assessed using flow cytometry. The
results showed that FXR1 knockdown enhanced early apoptosis in HCC
cells (Fig. 4A and B).
Additionally, the knockdown of FXR1 inhibited the proliferation of
HCC cells in the colony formation assay. These results were
validated in both Hep3B and LM3 cells (Fig. 4C and D). These results suggest that
knockdown of FXR1 inhibited the proliferation of HCC cells and
promoted early apoptosis of HCC cells.
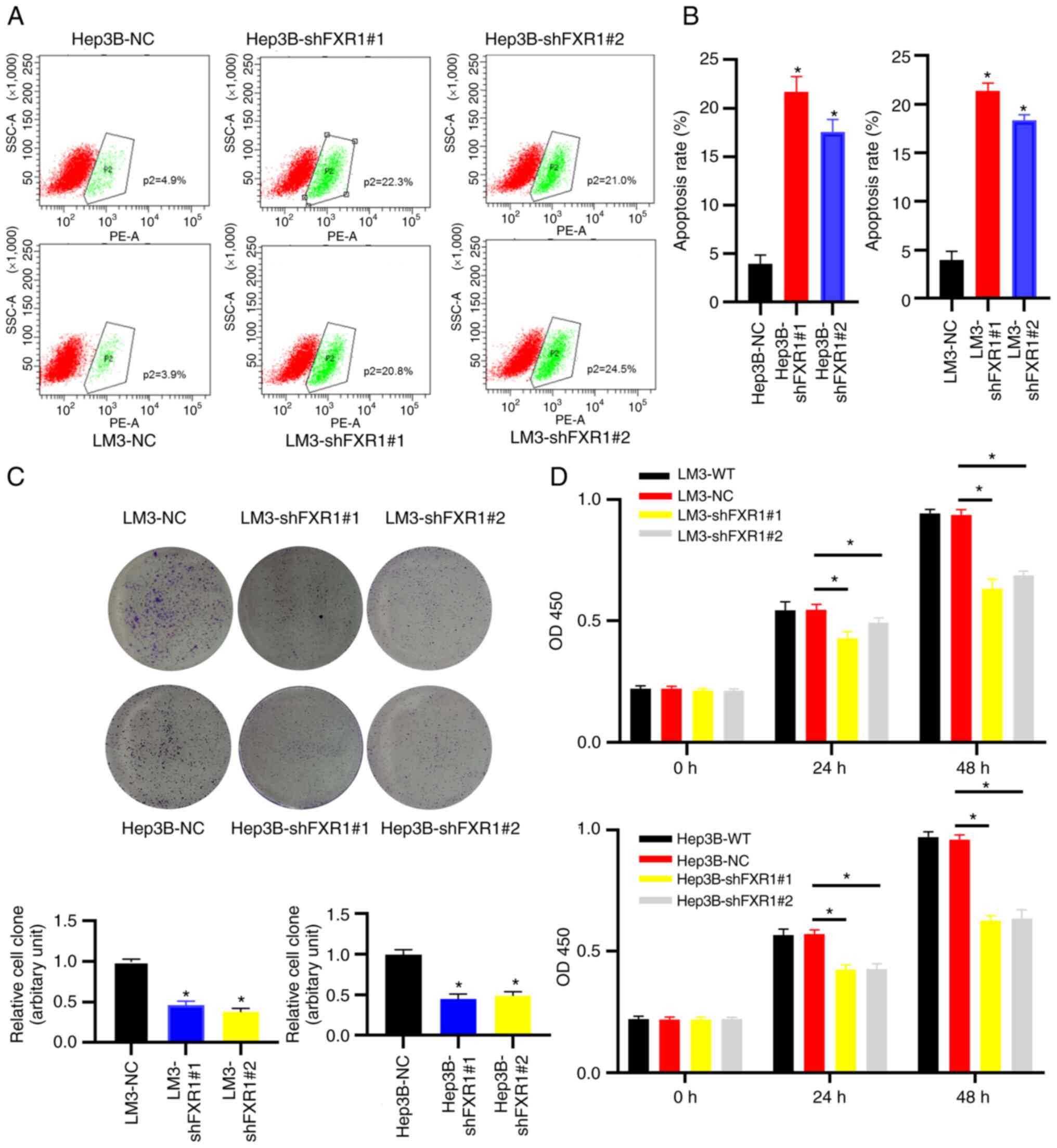 | Figure 4.Knockdown of FXR1 inhibits the
proliferation of HCC cells and promotes early apoptosis of HCC
cells. (A) Hep3B and LM3 cells were transfected with shNC, shFXR1#1
or shFXR1#2, followed by flow cytometry analysis. (B) Flow
cytometry analysis of apoptosis. (C) Hep3B and LM3 cells were
transfected with shNC, shFXR1#1 or shFXR1#2, followed by colony
formation assays and imaging with a digital camera attached to a
bright field microscope at a magnification of ×200. (D) Hep3B and
LM3 cells were transfected with shNC, shFXR1#1, or shFXR1#2,
followed by CCK-8 assays. *P<0.05. FXR1, fragile X-related 1;
NC, negative control; WT, wild-type. |
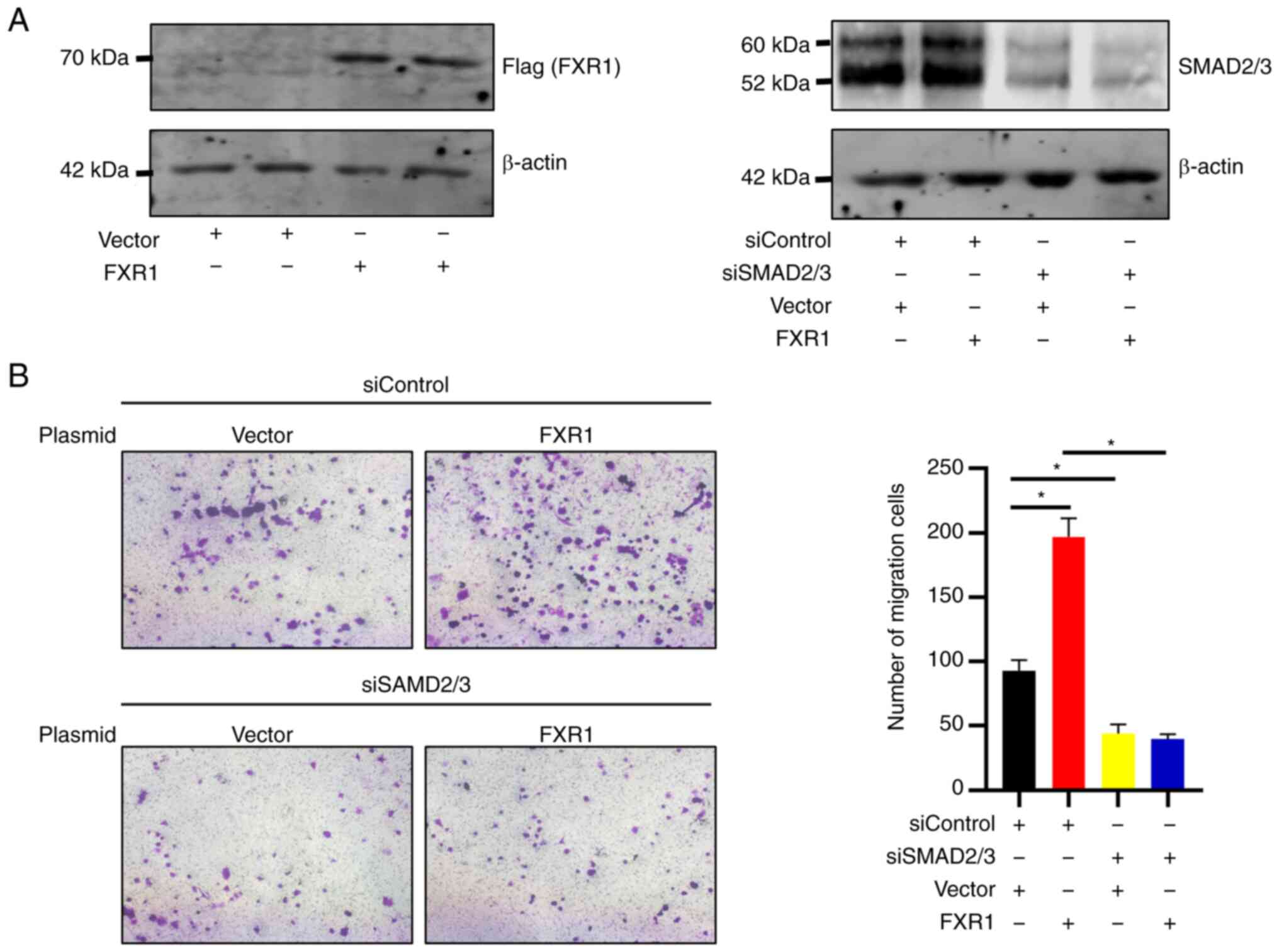
Knockdown of SMAD2/3 inhibits the
FXR1-induced increase in LM3 cell invasion
FXR1 overexpressing LM3 cell lines with or without
SMAD2/3 knockdown were established. RT-qPCR was used to verify the
knockdown efficiency of siSMAD2/3 (Fig. S1). The results showed that FXR1
overexpression enhanced LM3 cell invasion. Knockdown of SMAD2/3
inhibited the FXR1 overexpression-induced increase in the invasion
of LM3 cells (Fig. 5B). The
transfection efficiency of FXR1 and SMAD2/3 was verified using
western blotting (Fig. 5A). The
results of this experiment validated the role of SMAD2/3 in the
FXR1-induced increase in HCC invasion.
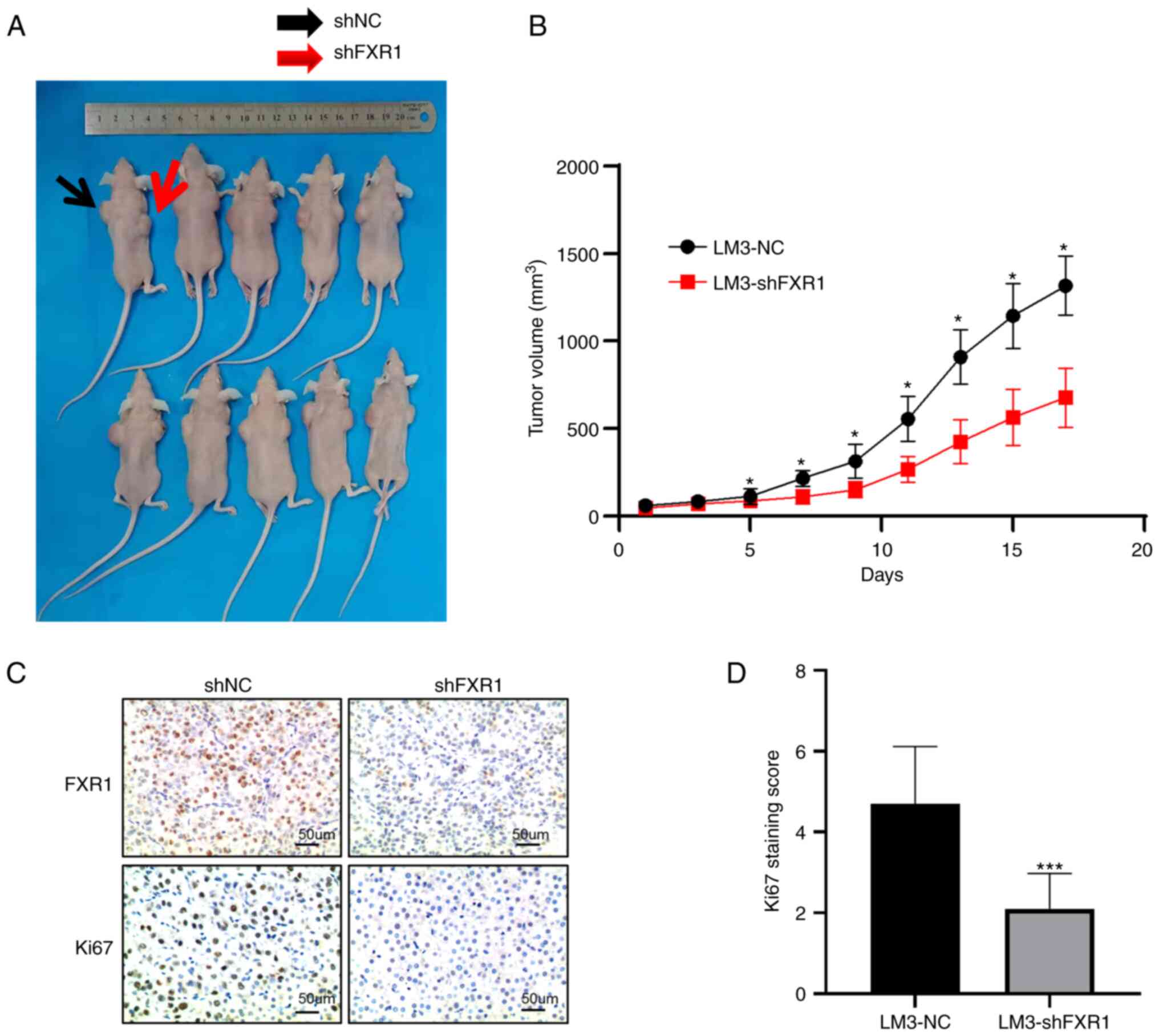
FXR1 knockdown inhibits the growth of
LM3 cells in vivo
To verify the role of FXR1 in vivo, mice were
injected with shNC-transfected LM3 cells or shFXR1-transfected LM3
cells. By measuring the volume of the tumors every 2 days, the
results showed that tumors grew slower in FXR1 knockdown cells
(Fig. 6A and B). The average
diameter of the NC tumors was 14.5 mm (range, 12.66-16.84 mm) and
the mean diameter of the shFXR1 tumors was 11.9 mm (range,
9.15-14.70 mm); the difference was statistically significant. The
expression of Ki67 in the tumors was assessed using
immunohistochemistry, which showed a significant decrease in Ki67
expression in the tumors after FXR1 knockdown (Fig. 6C and D). These data indicate that
FXR1 knockdown inhibits HCC proliferation in vivo.
Discussion
FXR1 belongs to the FXR protein family. The results
of the present study indicated that FXR1 expression was upregulated
in HCC tissues. Knockdown of FXR1 inhibited the malignant behavior
of HCC cells. Specifically, FXR1 knockdown effectively inhibited
the migratory and invasive abilities of HCC cells. FXR1 knockdown
also inhibited the proliferation of HCC cells. Furthermore, FXR1
knockdown increased early apoptosis of HCC cells. Using
bioinformatic analysis, it was shown that patients with high FXR1
expression had a worse prognosis. These results suggested that FXR1
functions as an oncogene in HCC. This finding is consistent with
the role of FXR1 in other tumors. In prostate cancer, FXR1
downregulation is associated with the inhibition of cell
proliferation, decreased cell viability, and impaired migration and
invasion of prostate cancer cells (26). FXR1 is highly expressed in gliomas,
and the knockdown of FXR1 leads to the inhibition of the malignant
biological behaviors of glioma cells (9). In colorectal cancer, FXR1 acts as an
oncogene that increases the proliferation, migration, and invasion
of cancer cells (12).
Bioinformatics is an important tool that is
increasingly being used as an initial approach to identify relevant
target proteins (27–29). Bioinformatics analysis was used in
the present study to screen SMAD2/3 as a target of the downstream
action of FXR1. TGF-β-SMAD signaling promotes the development of
EMT and the metastasis of HCC (30,31).
The best-known SMAD3 targets are EMT-related genes such as slug
(32). EMT is closely associated
with cancer metastasis (33–35),
and can be induced by environmental stresses (such as inflammation,
reactive oxygen species, hypoxia, hypoxia/reoxygenation), and
certain extracellular mediators (including TGF-β, fibroblast growth
factor-2, and epidermal growth factor) (36). The EMT-related proteins slug and
N-cadherin, play a significant role in invasive metastasis. In the
present study, western blotting was used to preliminarily validate
the expression of proteins downstream of FXR1. The results showed
that the knockdown of FXR1 inhibited the expression of SMAD2/3,
whereas knockdown of SMAD2/3 suppressed the expression of
EMT-related proteins.
TGF-β was selected for analysis in this preliminary
study as it is a well-established pathway; additional pathways will
be investigated in future research. The Smad pathway is known to be
a major transducer of TGF-β signaling and is important in
TGF-β-induced EMT (37). TGF-β
plays a complex double-edged role in tumors. Early in
tumorigenesis, TGF-β acts as a tumor suppressor through broad
multicellular inhibition; however, after tumor formation, it acts
as a pro-proliferative agent (38,39).
In HCC, TGF-β plays a key role in coordinating and regulating the
corresponding phenotype in HCC (40). GSEA showed the latent biological
pathways based on KEGG gene sets. The TGF-β pathway was thus chosen
as the primary research target in this study. TGF-β signaling in
hepatocytes is associated with liver fibrosis and carcinogenesis
(41). SMAD2/3 is a key molecule
in the TGF-β pathway. As a transcription factor, SMAD2/3 can
positively or negatively regulate the expression of several genes
(42,43). The promotion of HCC cell migration
and invasion by TGF-β was confirmed in this study. FXR1 knockdown
eliminated TGF-β-induced migration and invasion of HCC cells.
Conversely, the promotion of HCC cell invasion by FXR1
overexpression could be inhibited by SMAD2/3 knockdown. These data
suggest that SMAD2/3 is at least partially involved in the
FXR1-mediated increase in HCC cell invasion. However, the specific
regulatory mechanism by which FXR1 modulates SMAD2/3 mRNA
expression is unknown.
A nude mouse xenograft model was used to explore the
effects of FXR1 in vivo. Tumor Ki-67 protein expression is
associated with a poor prognosis (44,45).
The results of the present study showed that FXR1 knockdown
inhibited tumor growth in nude mice. Moreover, immunohistochemistry
analysis showed a significant decrease in Ki67 expression in tumors
following FXR1 knockdown. This finding suggests that FXR1
influences HCC cell proliferation in vivo. In addition to
classical SMAD signaling, TGF-β can also trigger non-classical
kinase cascades, leading to the activation of other signaling
pathways, such as PI3K, MAPK, and mTOR, which play an important
role in tumorigenesis (46–48).
Other mechanisms by which FXR1 promotes malignant behaviors in HCC
need to be further explored. Further studies are required to verify
whether FXR1 is a potential target for HCC therapy.
Since the present study did not directly confirm the
relationship between FXR1 and the SMAD pathway, instead only
showing an indirect relationship, further studies are required to
assess this.
In conclusion, the present study is the first to
show that aberrant FXR1 expression in HCC may promote the malignant
biological behavior of HCC cells. Upregulated FXR1 expression is
indicative of a poorer prognosis in HCC. In addition, SMAD2/3 was
at least partially involved in the FXR1-mediated increase in HCC
cell invasion. Thus, FXR1 may serve as a novel therapeutic target
for the management of HCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was funded by Henan Provincial Ministry of Medical
Science and Technology Research Youth Project (grant no.
SBGJ202103061) and the National Natural Science Foundation of China
(grant no. 82103282).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All of the authors have seen and confirmed the
authenticity of the raw data generated during the study. KZ, JG,
JS, CS, CP, JL, WG and SZ conceived and designed the study. KZ, JG
and JS performed the experiments and analyzed the data. JG and SZ
revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from patients
(or their parents/guardians) prior to enrollment in this study.
Ethical approval for the use of human/human tissues was obtained
from the Ethics Committee of the First Affiliated Hospital of
Zhengzhou University where the experiments were conducted prior to
the commencement of the study. This study was approved by the
Ethics Committee of the First Affiliated Hospital of Zhengzhou
University (approval no. 2019-KY-21).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
FXR
|
fragile X-related
|
|
GEPIA
|
gene expression profile interactive
analysis platform
|
|
HCC
|
hepatocellular carcinoma
|
|
HRP
|
horseradish peroxidase
|
|
PPL
|
Public Protein/Plasmid Library
|
|
TGCA
|
The Cancer Genome Atlas
|
|
TGFBR1
|
TGF-β receptor complex type I
|
|
TGFBR2
|
TGF-β receptor complex type II
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xie Y: Hepatitis B virus-associated
hepatocellular carcinoma. Adv Exp Med Biol. 1018:11–21. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang Y, Han QJ and Zhang J:
Hepatocellular carcinoma: Mechanisms of progression and
immunotherapy. World J Gastroenterol. 25:3151–3167. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Piñero F, Dirchwolf M and Pessôa MG:
Biomarkers in hepatocellular carcinoma: Diagnosis, prognosis and
treatment response assessment. Cells. 9:13702020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dimri M and Satyanarayana A: Molecular
signaling pathways and therapeutic targets in hepatocellular
carcinoma. Cancers (Basel). 12:4912020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akula SM, Abrams SL, Steelman LS, Emma MR,
Augello G, Cusimano A, Azzolina A, Montalto G, Cervello M and
McCubrey JA: RAS/RAF/MEK/ERK, PI3K/PTEN/AKT/mTORC1 and TP53
pathways and regulatory miRs as therapeutic targets in
hepatocellular carcinoma. Expert Opin Ther Targets. 23:915–929.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fujiwara N, Friedman SL, Goossens N and
Hoshida Y: Risk factors and prevention of hepatocellular carcinoma
in the era of precision medicine. J Hepatol. 68:526–549. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xing R, Gao J, Cui Q and Wang Q:
Strategies to improve the antitumor effect of immunotherapy for
hepatocellular carcinoma. Front Immunol. 12:7832362021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J,
Xing T, Ju G, Song G, Lou J and Liang J: CRISPR screens uncover
protective effect of PSTK as a regulator of chemotherapy-induced
ferroptosis in hepatocellular carcinoma. Mol Cancer. 21:112022.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoogeveen AT, Willemsen R and Oostra BA:
Fragile X syndrome, the Fragile X related proteins, and animal
models. Micros Res Tech. 57:148–155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Majumder M, Johnson RH and Palanisamy V:
Fragile X-related protein family: A double-edged sword in
neurodevelopmental disorders and cancer. Crit Rev Biochem Mol Biol.
55:409–424. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin X, Zhai B, Fang T, Guo X and Xu L:
FXR1 is elevated in colorectal cancer and acts as an oncogene.
Tumour Biol. 37:2683–2690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Majumder M and Palanisamy V: RNA binding
protein FXR1-miR301a-3p axis contributes to p21WAF1 degradation in
oral cancer. PLoS Genet. 16:e10085802020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao S, Zheng J, Liu X, Liu Y, Ruan X, Ma
J, Liu L, Wang D, Yang C, Cai H, et al: FXR1 promotes the malignant
biological behavior of glioma cells via stabilizing MIR17HG. J Exp
Clin Cancer Res. 38:372019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao M, Mishra L and Deng CX: The role of
TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. 14:111–123. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colak S and Ten Dijke P: Targeting TGF-β
signaling in cancer. Trends Cancer. 3:56–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hao Y, Baker D and Ten Dijke P:
TGF-β-mediated epithelial-mesenchymal transition and cancer
metastasis. Int J Mol Sci. 20:27672019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu HH, Chen DQ, Wang YN, Feng YL, Cao G,
Vaziri ND and Zhao YY: New insights into TGF-β/Smad signaling in
tissue fibrosis. Chem Biol Interact. 292:76–83. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aashaq S, Batool A, Mir SA, Beigh MA,
Andrabi KI and Shah ZA: TGF-β signaling: A recap of
SMAD-independent and SMAD-dependent pathways. J Cell Physiol.
237:59–85. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pallasch FB and Schumacher U: Angiotensin
Inhibition, TGF-β and EMT in Cancer. Cancers (Basel). 12:27852020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suarez-Carmona M, Lesage J, Cataldo D and
Gilles C: EMT and inflammation: Inseparable actors of cancer
progression. Mol Oncol. 11:805–823. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Babaei G, Aziz SG and Jaghi NZZ: EMT,
cancer stem cells and autophagy; The three main axes of metastasis.
Biomed Pharmacother. 133:1109092021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Syed V: TGF-β signaling in cancer. J Cell
Biochem. 117:1279–1287. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang K, Fang T, Shao Y and Wu Y:
TGF-β-MTA1-SMAD7-SMAD3-SOX4-EZH2 signaling axis promotes viability,
migration, invasion and EMT of hepatocellular carcinoma cells.
Cancer Manag Res. 13:7087–7099. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao H, Gao R, Yu C, Chen L and Feng Y: The
RNA-binding protein FXR1 modulates prostate cancer progression by
regulating FBXO4. Funct Integr Genomics. 19:487–496. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li K, Du Y, Li L and Wei DQ:
Bioinformatics approaches for anti-cancer drug discovery. Curr Drug
Targets. 21:3–17. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anashkina AA, Leberfarb EY and Orlov YL:
Recent trends in cancer genomics and bioinformatics tools
development. Int J Mol Sci. 22:121462021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsimberidou AM: Targeted therapy in
cancer. Cancer Chemother Pharmacol. 76:1113–1132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshida K, Murata M, Yamaguchi T,
Matsuzaki K and Okazaki K: Reversible human TGF-β signal shifting
between tumor suppression and fibro-carcinogenesis: Implications of
smad phospho-isoforms for hepatic epithelial-mesenchymal
transitions. J Clin Med. 5:72016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fransvea E, Angelotti U, Antonaci S and
Giannelli G: Blocking transforming growth factor-beta up-regulates
E-cadherin and reduces migration and invasion of hepatocellular
carcinoma cells. Hepatology. 47:1557–1566. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bai X, Yi M, Jiao Y, Chu Q and Wu K:
Blocking TGF-β signaling to enhance the efficacy of immune
checkpoint inhibitor. Onco Targets Ther. 12:9527–9538. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bakir B, Chiarella AM, Pitarresi JR and
Rustgi AK: EMT, MET, plasticity, and tumor metastasis. Trends Cell
Biol. 30:764–776. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu W and Kang Y: Epithelial-mesenchymal
plasticity in cancer progression and metastasis. Dev Cell.
49:361–374. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan GJ, Li QW, Shan SL, Wang WM, Jiang S
and Xu XM: Hyperthermia inhibits hypoxia-induced
epithelial-mesenchymal transition in HepG2 hepatocellular carcinoma
cells. World J Gastroenterol. 18:4781–4786. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kimura-Tsuchiya R, Ishikawa T, Kokura S,
Mizushima K, Adachi S, Okajima M, Matsuyama T, Okayama T, Sakamoto
N, Katada K, et al: The inhibitory effect of heat treatment against
epithelial-mesenchymal transition (EMT) in human pancreatic
adenocarcinoma cell lines. J Clin Biochem Nutr. 55:56–61. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang J, Xu Z, Wang Z, Du G and Lun L:
TGF-beta signaling in cancer radiotherapy. Cytokine.
148:1557092021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang M, Zhang YY, Chen Y, Wang J, Wang Q
and Lu H: TGF-β signaling and resistance to cancer therapy. Front
Cell Dev Biol. 9:7867282021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li H, He G, Yao H, Song L, Zeng L, Peng X,
Rosol TJ and Deng X: TGF-β induces degradation of PTHrP through
ubiquitin-proteasome system in hepatocellular carcinoma. J Cancer.
6:511–518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Suwa K, Yamaguchi T, Yoshida K, Murata M,
Ichimura M, Tsuneyama K, Seki T and Okazaki K: Smad
phospho-isoforms for hepatocellular carcinoma risk assessment in
patients with nonalcoholic steatohepatitis. Cancers (Basel).
12:2862020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yoshida K, Matsuzaki K, Murata M,
Yamaguchi T, Suwa K and Okazaki K: Clinico-pathological importance
of TGF-β/phospho-smad signaling during human hepatic
fibrocarcinogenesis. Cancers (Basel). 10:1832018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaminska B, Wesolowska A and Danilkiewicz
M: TGF beta signalling and its role in tumour pathogenesis. Acta
Biochim Pol. 52:329–337. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Burkhart RA, Ronnekleiv-Kelly SM and
Pawlik TM: Personalized therapy in hepatocellular carcinoma:
Molecular markers of prognosis and therapeutic response. Surg
Oncol. 26:138–145. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin LX and Tang ZY: The prognostic
molecular markers in hepatocellular carcinoma. World J
Gastroenterol. 8:385–392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stefani C, Miricescu D, Stanescu S II,
Nica RI, Greabu M, Totan AR and Jinga M: Growth factors,
PI3K/AKT/mTOR and MAPK signaling pathways in colorectal cancer
pathogenesis: Where are we now? Int J Mol Sci. 22:102602021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shorning BY, Dass MS, Smalley MJ and
Pearson HB: The PI3K-AKT-mTOR pathway and prostate cancer: At the
crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci.
21:45072020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kumari N, Reabroi S and North BJ:
Unraveling the molecular nexus between GPCRs, ERS, and EMT.
Mediators Inflamm. 2021:66554172021. View Article : Google Scholar : PubMed/NCBI
|