Introduction
Neuroendocrine tumors (NETs) comprise a tumor class
originating from neuroendocrine cells and peptidergic neurons, and
are common in the respiratory and gastrointestinal tracts.
Well-differentiated NETs (WDNETs) are a rare type of NET,
comprising <1% of all cases (1,2).
WDNETs have a better prognosis than neuroendocrine carcinoma,
therefore, for patients with WDNETs and without metastases,
surgical resection is the preferred approach. In the present study,
a patient with a WDNET, also known as primary renal carcinoid
tumor, was recently admitted to the Department of Urology, The
Affiliated Hospital of Zunyi Medical University (Zunyi, China).
Postoperative follow-up for 1 year indicated no tumor recurrence or
metastasis. The present study retrospectively analyzed the
patient's medical records, diagnosis and treatment process. This
information was combined with an analysis of the literature, aiming
to share the experience of diagnosis and treatment of WDNETs, and
provide new strategies for patient follow-up.
Case report
A 45-year-old female patient was admitted to The
Affiliated Hospital of Zunyi Medical University with right-sided
lumbago in November 2021. By analyzing the patient's history, it
was discovered that occasional right lumbago, accompanied by
general fatigue, has been experienced for several months. There was
no record of fever, bellyache, hematuria, frequent urination,
hesitancy or loss of weight. The patient had previously undergone a
total hysterectomy 5 years prior to admission and had no history of
hypertension, diabetes, cancer or other relevant familial diseases.
Following a physical examination, no abnormal signs were noted. No
significant abnormalities were noted in the expression levels of
chromogranin A (CGA) and in other biological functions on blood
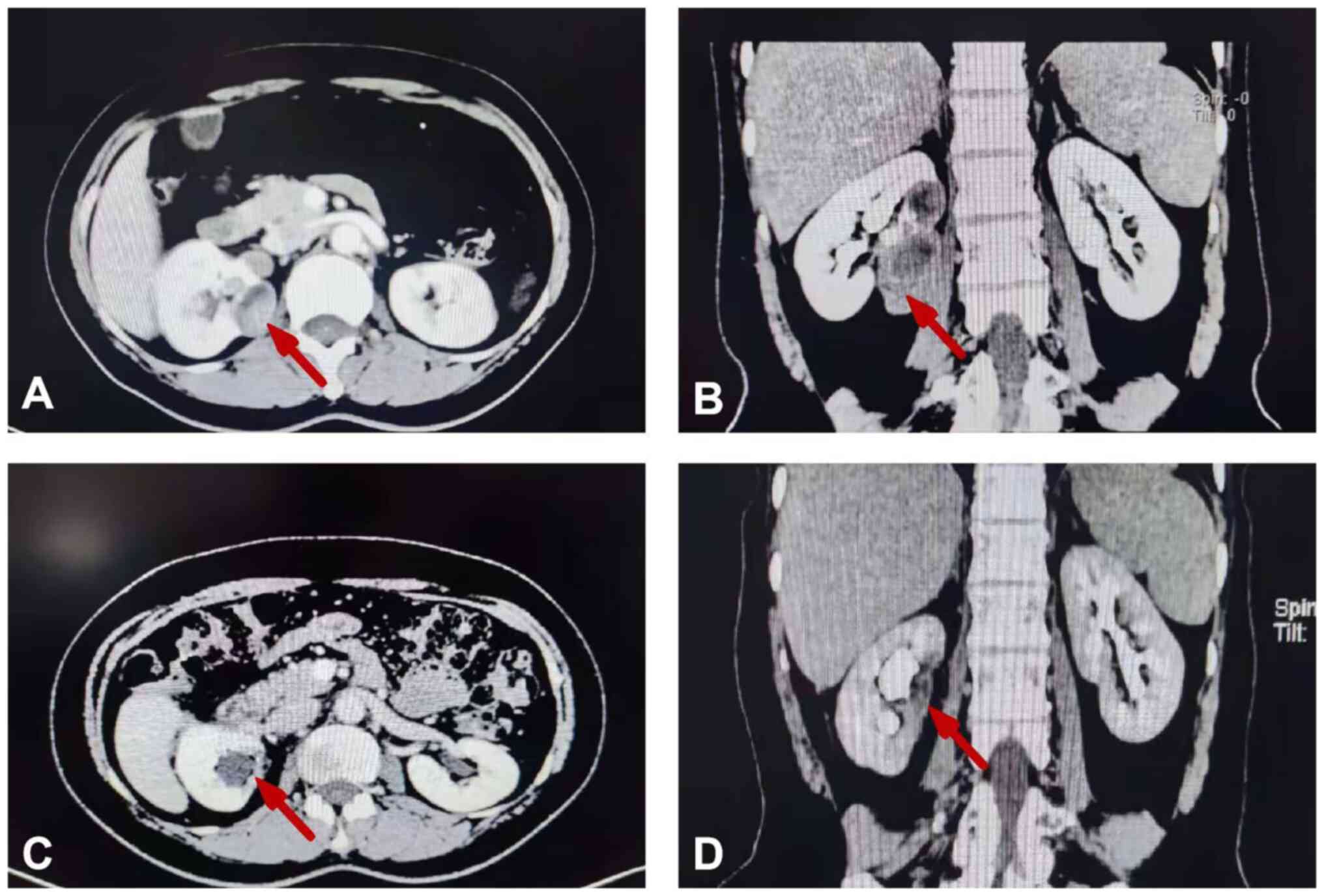
tests. Abdominal computed tomography (CT) in the venous phase
demonstrated a well-circumscribed, slightly enhanced mass with an
approximate size of 44×34×70 mm and a solid component; this mass
was accompanied by sporadic fatty and calcified components, and was
located in the right renal area. The left kidney function was
normal (Fig. 1A and B). No evidence
of distant or nodal metastases was present. Vascular examination
indicated that the right renal arteries and the branch arteries
were normal in shape without filling defects; the right renal vein
and inferior vena cava were also normal. Additional examinations
did not show any abnormalities. The clinical diagnosis was
initially of a right renal paraganglioma (PPGL). A laparoscopic
partial nephrectomy of the right kidney was performed under general
anesthesia. During the operation, the tumor was discovered in the
renal sinus, between the renal vein, renal artery and renal pelvis,
and there was no vascular or lymphatic invasion. The renal artery
was blocked with vascular blocking clips and the tumor was removed
completely. Intraoperative blood pressure was within the normal
range without significant fluctuation. After the operation, the
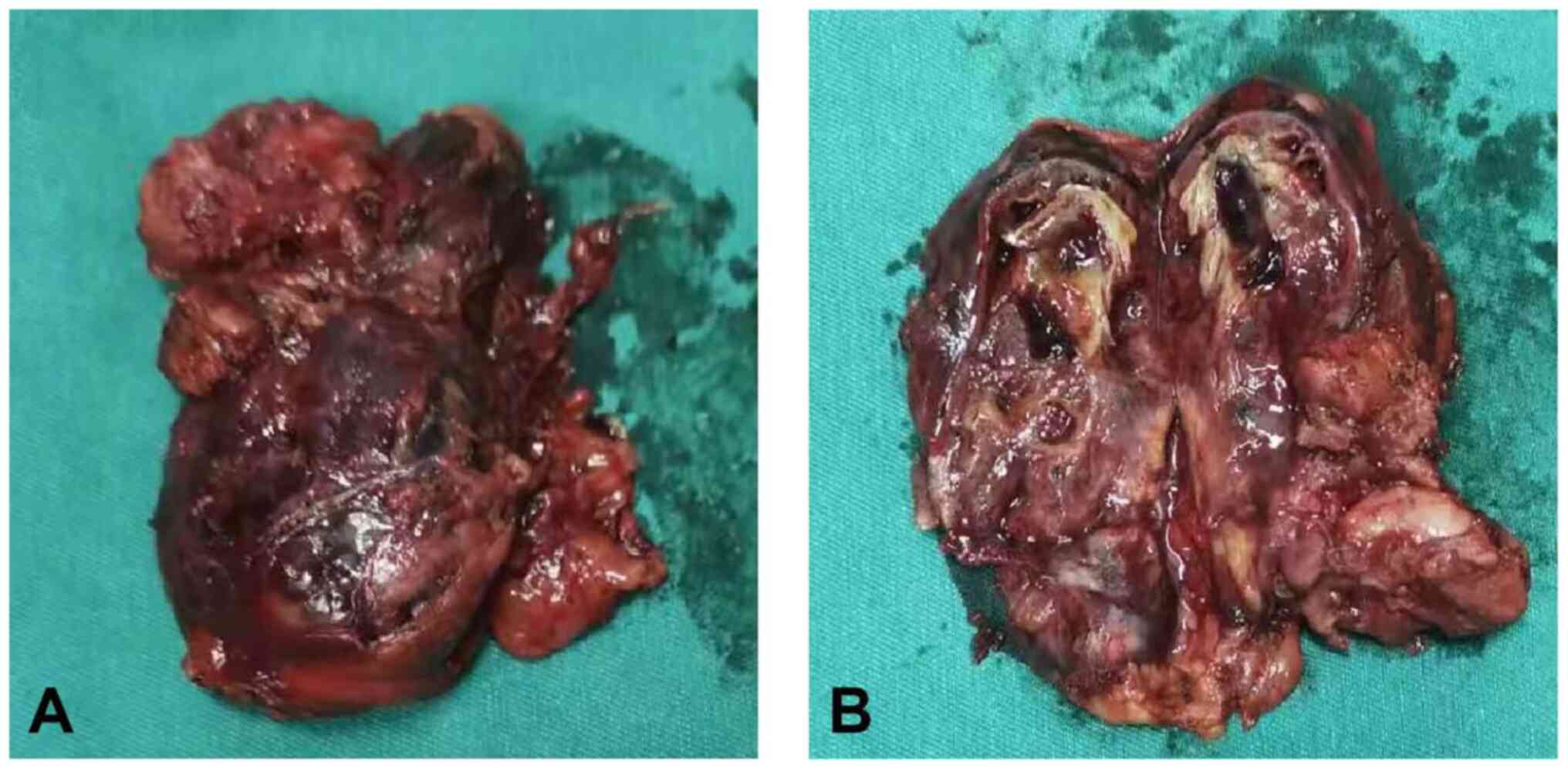
tumor specimen was sent to the Department of Pathology. Gross
examination of the tumor specimen indicated a solid mass of
40×30×70 mm, and the cut surfaces were gray with a complete capsule
(Fig. 2). The tissue samples were
fixed with 4% formaldehyde for 24 h at room temperature. After
paraffin embedding, the tissue samples were prepared into 5 µm
sections and stained with hematoxylin and eosin for 5 and 2 min,
respectively. At room temperature, the tissue samples were fixed
with 4% formaldehyde for 24 h. After paraffin embedding, the tissue
samples were prepared into 5-µm sections, and stained with
hematoxylin and eosin for 5 and 2 min, respectively. Examination
under a light microscope revealed that the presence of normal
kidney tissue and tumor tissue (Fig.
3A) and the tumors were composed of a single population of
small, round- to oval-shaped cells with inconspicuous nucleoli,
scant cytoplasm and a distinctive ‘salt and pepper’ chromatin
pattern (Fig. 3B). The tumor cells
were arranged in cords (Fig. 3C),
flakes and even nest-like structures (Fig. 3D). The mitotic rate was <4/10
high-power field. The results of immunohistochemical staining
indicated that these tumor cells were diffusely positive for the
expression of CGA (Fig. 3E),
synaptophysin (Syn) (Fig. 3F) and
cluster of differentiation (CD)56 (Fig.
3G). The Ki-67 index was <10% (Fig. 3H). Therefore, a diagnosis of a
right-sided renal WDNET was confirmed. At 1-year post-surgery,
computed tomography indicated the absence of any residual tumor and
mild hydronephrosis in the right kidney following partial
nephrectomy, which indicated a good prognosis for the patient
(Fig. 1C and D). Patient was
followed up every 3–12 months.
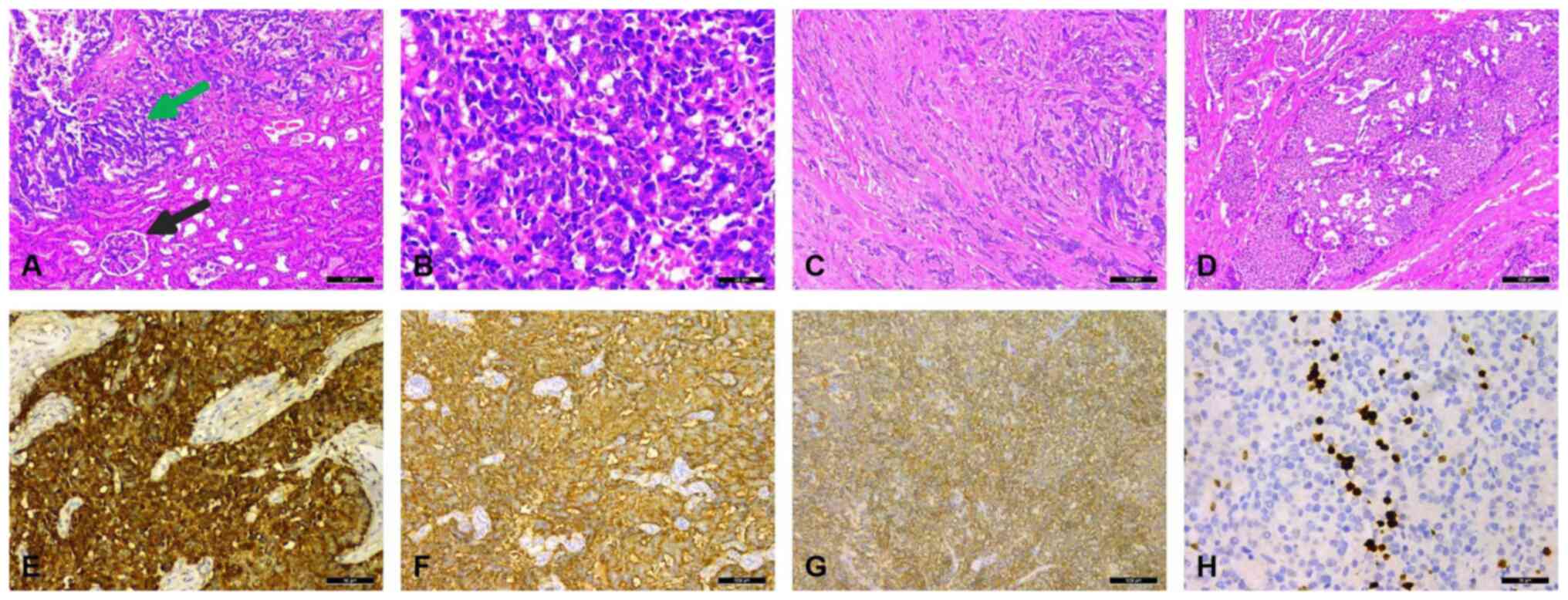 | Figure 3.Pathological features of the mass. (A)
The presence of normal kidney tissue and tumor tissue (the black
arrow indicates the glomerulus and the green arrow indicates the
tumor tissue) (H&E staining; magnification, ×100). (B) The
tumors were composed of a single population of small, round- to
oval-shaped cells with inconspicuous nucleoli, scant cytoplasm, and
a distinctive ‘salt and pepper’ chromatin pattern (H&E
staining; ×200 magnification). (C) The tumor cells were arranged in
cords (H&E staining; magnification, ×100). (D) Nest-like
structures were present (H&E staining; magnification, ×100).
(E) Positive immunostaining of the tumor cells for chromogranin A
expression (magnification, ×100). (F) Intense positive
immunostaining for synaptophysin expression in the tumor cells
(magnification, ×100). (G) Positive immunostaining for cluster of
differentiation 56 in the tumor cells (magnification, ×100). (H)
The Ki-67 index was <10%. H&E, hematoxylin and eosin. |
Discussion
NETs are relatively rare. According to a study
performed in the United States, the incidence rate is ~7
cases/100,000 individuals (3). The
majority of NETs occur in the gastrointestinal tract and lung, and
WDNETs account for 0.3-0.4% of all NETs (4,5). A
study by McGarrah et al (6)
indicated that the incidence of WDNETs was only 0.13 cases per 1
million subjects. WDNETs tend to occur between the ages of 40 and
70 years. No significant difference has been noted in the incidence
of WDNETs between male and female patients. It has been shown that
the incidence on the right side is 53.6% higher than that on the
left side. The incidence in the renal parenchyma (92.8%) is
significantly higher compared with that in the renal pelvis (7.2%)
(7).
No neuroendocrine cells have been found in the
kidney; therefore the tissue source of WDNETs is not clear, but may
be related to the neural crest or pancreatic tissue that is
mislocated to the kidney during embryogenesis. Multipotent stem
cells differentiate into neuroendocrine cells. Chronic inflammation
causes metaplasia of the pelvicalyceal urothelium. Certain
seemingly primary renal carcinoids can constitute a metastatic
focus from an undiscovered primary lesion (8–11).
Previous studies support a hypothesis, also known as the
co-existence hypothesis, which proposes that the proliferation of
scattered neuroendocrine cells originates from the epithelium of a
horseshoe or polycystic kidney (12,13).
It has been found that the risk of developing carcinoids in the
horseshoe kidney is 62- to 82-fold higher than that noted in the
normal kidney. Furthermore, in a study of renal carcinoid tumors,
~15.6% of patients had horseshoe kidneys (11).
WDNETs lack characteristic clinical manifestations,
with the main reported symptoms being lumbago, abdominal swelling,
an abdominal mass and hematuria. Due to the secretion of vasoactive
substances by certain tumors, 10–15% of patients may have carcinoid
syndromes, such as diarrhea, facial flushing and dyspnea (14,15). A
total of 20–25% of patients do not present with any clinical
symptoms, with the WDNET only found during routine clinical
examinations (16). Conventional
complementary examinations, such as CT and magnetic resonance
imaging (MRI), do not distinguish WDNETs from other renal tumors.
WDNETs generally present with a well-circumscribed, non-enhanced or
slightly enhanced mass on CT, with a solid component; however, they
are sporadically accompanied by cystic and calcified components. On
MRI, WDNETs mostly demonstrate a heterogeneous signal intensity on
both T1 and T2-weighted images (17). However, the signal characteristics
of WDNETs are varied. The presence of hemorrhagic necrosis in the
tumor directly affects the MRI signal characteristics and is the
main factor leading to signal inconsistency (17). A study has shown that gallium-68
positron emission tomography (PET)/CT is more sensitive than CT,
MRI or [18F]fluorodeoxyglucose-PET/CT for detecting
WDNETs (18). In addition, RNA
sequencing is useful for improving diagnostic accuracy; however,
the patient in the present case refused to undergo this process. A
diagnosis of WDNET requires pathological and immunohistochemical
analyses. The majority of WDNETs are solid masses with clear
boundaries and often present with grayish white or grayish brown
sections (19). Hemorrhage and
necrosis are rare (17). In the
current study, the tumor cells were arranged into cords, ribbons
and trabecular structures with abundant sinusoids. They were round
or polygonal, with eosinophilic cytoplasm and unclear boundaries,
round basophilic nuclei, uneven granular nuclear chromatin, mitotic
appearance and rare necrosis (20).
WDNETs specifically express neuroendocrine markers, such as Syn,
CD56 and CGA. The specificity and sensitivity of these markers for
the diagnosis of WDNETs is high. Among them, NSE exhibits high
sensitivity and poor specificity, whereas CGA exhibits optimal
specificity and poor sensitivity, therefore multiple markers need
to be tested simultaneously for a successful clinical diagnosis
(10). Previous studies have shown
that insulinoma-associated protein 1 has a significant advantage
with regard to the sensitivity and specificity for the diagnosis of
NETs (21–23). However, prior to the diagnosis and
treatment of primary renal highly differentiated endocrine tumors,
metastasis to other sites should be excluded. WDNETs have a low
degree of malignancy, slow growth and optimal prognosis. Surgical
resection is the preferred approach for patients without
metastases; however in the presence of metastasis, either pre- or
post-operatively, systemic therapies for renal NETs have been
directed by the treatment of non-renal NETs (24). Romero et al (10) indicated that ~50% of patients with
WDNETs undergoing radical nephrectomy demonstrated no recurrence or
metastasis following an average follow-up time of 43 months.
However, it has been shown that certain cases can develop systemic
multiple metastases several years after nephrectomy; therefore,
patients still need to be followed up every 3 months for life even
if the tumor cells are well differentiated, low grade or at an
early clinical stage (25). In the
present study, the postoperative pathological diagnosis
demonstrated a highly differentiated NET. However, both WDNETS and
renal PPGL are NETs with various similarities in their biological
behavior and disease prognosis. It remains unknown whether they can
be managed as a single disease. For patients from urban areas and
those with a high financial status, the follow-up process should
adhere to the follow-up standards for NETs, which is assessment
every 3–12 months for life; however, for patients from remote rural
areas and for economically disadvantaged patients, the follow-up
process should adhere to the follow-up standards of non-high-risk
benign PPGL, which requires only annual follow-up assessments for
10 years after surgery (26). This
measure can increase the available options for patient follow-up,
reduce the financial burden and psychological pressure on patients,
and improve their compliance for follow-up, which in turn may be
more beneficial to these patients.
In summary, WDNETs are rare, their clinical and
imaging manifestations are non-specific, and the confirmation of
their diagnosis depends on immunohistochemical analysis. These
tumors have a low degree of malignancy and optimal prognosis, and
for patients without metastases, surgical resection is the
preferred approach; however, if metastases develop preoperatively
or postoperatively, the systemic treatment approach for renal NETs
is guided by the treatment of non-renal NETs. Certain patients can
still develop recurrence and metastasis following surgery, and the
postoperative follow-up needs to be performed strictly with
reference to renal NETs. If patients find regular follow-up
inconvenient and have financial difficulties, their follow-up can
be performed according to that of non-high-risk benign PGL, which
can improve patient compliance for follow-up and may be more
beneficial to them.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Science and Technology
Project of Zunyi City, China (grant no. HZ-2021-11) and Science and
Technology Foundation of Guizhou Province, China [grant no.
ZK(2022)665].
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DY and WT were the main contributors to analyzing
patient data and the writing and revision of this manuscript. TW
provided the initial idea and medical images. GL and ZZ provided
important suggestions for the patient treatment. DY and TW confirm
the authenticity of all the raw data. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the Affiliated Hospital of Zunyi Medical
University. Ethical review approval number
KLL-2021-132/KLL-2021-331. Written informed consent was obtained
from the patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sun K, You Q, Zhao M, Yao H, Xiang H and
Wang L: Concurrent primary carcinoid tumor arising within mature
teratoma and clear cell renal cell carcinoma in the horseshoe
kidney: Report of a rare case and review of the literature. Int J
Clin Exp Pathol. 6:2578–2584. 2013.PubMed/NCBI
|
|
2
|
Seker KG, Sam E, Sahin S, Yenice MG, Aktas
AG, Simsek A and Tugcu V: Partial nephrectomy in horseshoe kidney:
Primary carcinoid tumor. Arch Ital Urol Androl. 89:316–318. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dasari A, Shen C, Halperin D, Zhao B, Zhou
S, Xu Y, Shih T and Yao JC: Trends in the incidence, prevalence,
and survival outcomes in patients with neuroendocrine tumors in the
United States. JAMA Oncol. 3:1335–1342. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Darbà J and Marsà A: Exploring the current
status of neuroendocrine tumours: A population-based analysis of
epidemiology, management and use of resources. BMC Cancer.
19:12262019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lane BR, Jour G and Zhou M: Renal
neuroendocrine tumors. Indian J Urol. 25:155–160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McGarrah PW, Westin G, Hobday TJ, Scales
JA, Ingimarsson JP, Leibovich BC and Halfdanarson TR: Renal
neuroendocrine neoplasms: A Single-center experience. Clin
Genitourin Cancer. 18:e343–e349. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen AH, O'Leary MP, De Andrade JP,
Ituarte PHG, Kessler J, Li D, Singh G and Chang S: Natural history
of renal neuroendocrine neoplasms: A NET by any other name. Front
Endocrinol (Lausanne). 11:6242512021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Priemer DS, Montironi R, Wang L,
Williamson SR, Lopez-Beltran A and Cheng L: Neuroendocrine tumors
of the prostate: Emerging insights from molecular data and updates
to the 2016 World health organization classification. Endocr
Pathol. 27:123–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamada Y and Beltran H: Clinical and
biological features of neuroendocrine prostate cancer. Curr Oncol
Rep. 23:152021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Romero FR, Rais-Bahrami S, Permpongkosol
S, Fine SW, Kohanim S and Jarrett TW: Primary carcinoid tumors of
the kidney. J Urol. 176:2359–2366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krishnan B, Truong LD, Saleh G, Sirbasku
DM and Slawin KM: Horseshoe kidney is associated with an increased
relative risk of primary renal carcinoid tumor. J Urol.
157:2059–2066. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Y, Shu Y, He L and Wu K: Primary
renal carcinoid tumors: Three case reports. Medicine (Baltimore).
100:e247142021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang H and Zhang H: Clinical and
pathological features of primary renal well-differentiated
neuroendocrine tumor. Onco Targets Ther. 15:587–596. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosenberg JE, Albersheim JA, Sathianathen
NJ, Murugan P and Weight CJ: Five new cases of primary renal
carcinoid tumor: Case reports and literature review. Pathol Oncol
Res. 26:341–346. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jabaji R, Kern T, Shen D, Chu W and
Merchant M: Primary renal carcinoid tumor: Report of two cases.
Perm J. 24:1972020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Omiyale AO and Venyo AK: Primary carcinoid
tumour of the kidney: A review of the literature. Adv Urol.
2013:5793962013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoon JH: Primary renal carcinoid tumor: A
rare cystic renal neoplasm. World J Radiol. 5:328–333. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mufarrij P, Varkarakis IM, Studeman KD and
Jarrett TW: Primary renal carcinoid tumor with liver metastases
detected with somatostatin receptor imaging. Urology. 65:10022005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang XH, Lu X, He B, Jiang YX, Yu WJ, Wang
H, Zhang W and Li YJ: Clinicopathologic features of primary renal
neuroendocrine carcinoma. Zhonghua Bing Li Xue Za Zhi. 47:851–856.
2018.(In Chinese). PubMed/NCBI
|
|
20
|
Gu X, Cheng M and Herrera GA: Kidney
carcinoid tumor: Histological, immunohistochemical and
ultrastructural features. Ultrastruct Pathol. 42:18–22. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rindi G, Mete O, Uccella S, Basturk O, La
Rosa S, Brosens LAA, Ezzat S, de Herder WW, Klimstra DS, Papotti M
and Asa SL: Overview of the 2022 WHO classification of
neuroendocrine neoplasms. Endocr Pathol. 33:115–154. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maleki Z, Nadella A, Nadella M, Patel G,
Patel S and Kholová I: INSM1, a novel biomarker for detection of
neuroendocrine neoplasms: Cytopathologists View. Diagnostics
(Basel). 11:21722021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sappenfield R, Gonzalez IA, Cao D and
Chatterjee D: Well-differentiated rectal neuroendocrine tumors:
Analysis of histology, including insulinoma-associated protein 1
expression, and biologic behavior, involving a large cohort of 94
cases. Hum Pathol. 104:66–72. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deacon MJ, Harvey H, Shah C and Khan A: A
rare case of a large primary renal neuroendocrine tumour: A case
report and brief review of literature. Cureus.
13:e197432021.PubMed/NCBI
|
|
25
|
Moch H, Cubilla AL, Humphrey PA, Reuter VE
and Ulbright TM: The 2016 WHO classification of tumours of the
urinary system and male genital organs-part A: Renal, penile, and
testicular tumours. Eur Urol. 70:93–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Plouin PF, Amar L, Dekkers OM, Fassnacht
M, Gimenez-Roqueplo AP, Lenders JW, Lussey-Lepoutre C and Steichen
O; Guideline Working Group, : European Society of Endocrinology
Clinical Practice Guideline for long-term follow-up of patients
operated on for a phaeochromocytoma or a paraganglioma. Eur J
Endocrinol. 174:G1–G10. 2016. View Article : Google Scholar : PubMed/NCBI
|