Introduction
During epithelial-mesenchymal transition (EMT),
which was first discovered in embryonic developmental studies,
epithelial cells undergo notable changes in morphology, as
characterized by polarized and tightly connected to adjacent cells
to being less polarized and loosely connected, with spindle-like
morphology and acquire functional properties of mesenchymal cells,
such as a motile and invasive phenotype (1–3).
Cancer cells acquires invasiveness and metastatic potential, by
exploiting the EMT machinery that endows the cells with a motile
and invasive phenotype (4).
EMT is regulated by several EMT-inducing
transcription factors that serve as master regulators of EMT
(3,4). By contrast, a number of EMT-repressing
transcription factors have also been discovered (3). Among them, Grainyhead-like 2
(GRHL2) encodes a transcription factor that has been
reported to regulate wound healing, tubulogenesis and cancer
(5,6). However, GRHL2 can exert both
tumor-promoting and -suppressive functions depending on the human
cancer involved (7–21). In pancreatic cancer, GRHL2
functions as a tumor-promoting gene by conferring metastatic
potential to cancer cells, by retaining epithelial but acquiring
stem cell properties (11). By
contrast, in breast cancer GRHL2 inhibits
anchorage-independent growth, where the loss of GRHL2 expression
has been found to be associated with higher tumor stages (14). This suggests the tumor-suppressive
role of GRHL2 in breast cancer (14). In addition, another study has
previously reported tumor-suppressive roles of GRHL2 in
breast cancer, by demonstrating that suppression of EMT by
GRHL2 increased sensitivity to anoikis (10).
To the best of our knowledge, few reports have
investigated the roles of GRHL2 in lung cancer (12). GRHL2 is overexpressed in lung
cancer compared with that in normal cells, where it was associated
with inferior prognosis. GRHL2 silencing was reported to
suppress proliferation and colony formation whilst enhancing
migration and invasion. These data are difficult to interpret,
because the results of proliferation and colony formation, together
with increased GRHL2 expression, suggested that GRHL2 serves
oncogenic roles, but those from migration and invasion assays
suggested a tumor-suppressive role. Furthermore, GRHL2 has
been demonstrated to exert oncogenic roles by stabilizing a ‘hybrid
epithelial/mesenchymal phenotype’ in H1975 lung cancer cells
(22). In that particular study,
this ‘hybrid epithelial/mesenchymal phenotype’ was considered to be
a promoter of cancer metastasis. These findings are contradictory
to some extent and suggest a highly complex role of GRHL2 in
lung cancer. This justifies further investigations into the roles
of GRHL2 in lung cancer pathophysiology.
Therefore, in the present study, the role of
GRHL2 in lung cancer was investigated using the online
bioinformatic tool, Lung Cancer Explorer (LCE) and preclinical
models, including hTERT/Cdk4-immortalized normal lung
epithelial cells and lung cancer cell lines (23–26).
Materials and methods
Cell culture
All 10 of lung cancer cell lines (HCC827, HCC4006,
HCC4017, A549, H460, H441, H661, H1792, H1975 and H2009) used in
the present study and the hTERT/Cdk4-immortalized
normal human bronchial epithelial cell lines HBEC3KT and HBEC4KT
were purchased from American Type Culture Collection and were
derived from the Hamon Center Collection University of Texas
Southwestern Medical Center (Dallas, USA) (27,28).
The lung cancer cell lines were cultured in RPMI-1640 (cat. no.
189-02025; FUJIFILM Wako Pure Chemical Corporation) supplemented
with 10% FBS (cat. no 35-079-CV; Corning, Inc.). HBEC3KT and
HBEC4KT cells were cultured in keratinocyte-serum free medium (cat.
no. 10724-011; Thermo Fisher Scientific, Inc.) supplemented with 5
ng/ml epidermal growth factor (cat. no. PHG0314; Thermo Fisher
Scientific, Inc.) and 50 ng/ml bovine pituitary extract (cat. no.
13028-014; Thermo Fisher Scientific, Inc.). All cells were cultured
at 37°C in a humidified 5% CO2 incubator. The provenance
of H1975, H2009 and H441 was confirmed by DNA fingerprinting short
tandem repeat analysis. Negativity for mycoplasma contamination in
the cultured cell lines was confirmed using the VenorGeM Onestep
kit (cat. no. 11-8050) according to the manufacturer's protocol
(Minerva Biolabs GmbH) or by mycoplasma group-specific PCR
(29).
Small-interfering RNA (siRNA)
transfection
A total of 3×105 cells (HBEC4KT, H1975,
H2009 and H441) were transfected with predesigned siRNA (Silencer
Select RNAi; cat. no. 4427037, Thermo Fisher Scientific, Inc.) to a
final concentration of 6.25 nM using Lipofectamine®
RNAiMAX (cat. no. 13778-150; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Knockdown of GRHL2
was performed using two siRNA oligos, si-GRHL2#1 (cat. no.
4427037; ID, s36755; Thermo Fisher Scientific, Inc.) and
si-GRHL2#2 (cat. no. 4427037, ID, s36756; Thermo Fisher
Scientific, Inc.). The negative control used was Silencer™ Select
Negative Control No. 1 siRNA (siNC; cat. no. 4390844; Thermo Fisher
Scientific, Inc.). All transfections were performed at 37°C in a
humidified 5% CO2 incubator. Durations of transfection
were two days for cell proliferation and colony formation assays
and four days for harvesting cells for RNA and protein extractions.
Subsequent experimental procedures were performed immediately after
completion of the transfections.
GRHL2 overexpression
A custom-ordered GRHL2 expression vector,
pLenti6/V5-GW/GRHL2, was constructed by replacing the coding
sequence of LacZ in a pLenti6/V5-GW/lacZ vector (cat. no.
K4955-10; Thermo Fisher Scientific, Inc.) with the GRHL2
reference sequence (accession no. NM_024915.4), purchased from
GenScript. 2×105 of A549 cells plated in 6 cm dishes
were transiently transfected with 1.5 ug of either the
pLenti6/V5-GW/GRHL2 or the pLenti6/V5-GW/lacZ vector used as a
control using Fugene®4K transfection reagent (cat. no.
E5911; Promega Corporation) as aforementioned. Subsequently, 2 days
of culture at 37°C in a humidified 5% CO2 incubator
after the transfection, cells were harvested for protein or RNA
extraction, or they were re-plated for proliferation or colony
formation assays. Transfection efficiency was evaluated in
1×105 A549 cells plated in 6-well plates by
co-transfecting with 0.32 µg EGFP-expressing vector pEGFP-N3 (cat.
no. 6080; Clontech.) at the ratio of 1:1 to the pLenti6/V5-GW/GRHL2
or pLenti6/V5-GW/lacZ vector (cat. no. K4955-10, Thermo Fisher
Scientific, Inc.).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Four days after transfection, total RNA was
extracted from the cells (HBEC4KT, H1975, H2009 and H441) using the
RNeasy mini kit (cat. no. 74106; Qiagen GmbH) following the
manufacturer's protocol. Reverse transcription was completed using
the SuperScript IV First-Strand Synthesis System with a random
primer system (cat. no. 18091050; Thermo Fisher Scientific, Inc.)
Using the PowerUp™ SYBR™ Green Master Mix (cat. no. A25742; Thermo
Fisher Scientific, Inc.), qPCR was performed for E-cadherin,
VIMENTIN, TWIST1, Zinc finger E-box-binding homeobox 1 (ZEB1),
snail homolog (SNAI)1 and SNAI2. The
thermocycling conditions were follows: 95°C for 20 sec, 40 cycles
of 95°C for 3 sec and 60°C for 30 sec. The melt condition was
follows; 95°C for 15 sec, 60°C for 1 min and 95°C for 15 sec.
GAPDH was used as an internal control, and the relative
expression level was calculated by the ΔΔCq method (30). The primer sequences are shown in
Table SI.
Cell proliferation assays
In total, 2 days after the transfection of siRNA,
the cells (HBEC4KT, H1975, H2009, or H441) were plated into 96-well
plates at 5,000 cells/well. Subsequently, 2 days later, a
colorimetric proliferation assay was performed using the Cell
Counting Kit-8 (CCK-8: cat. no. CK04; Dojindo Molecular
Technologies, Inc.) according to the manufacturer's protocol. Cells
were incubated at 37°C, 5% CO2 for 2 h, measured
absorbance at 450 nm and at 600 nm as a reference wavelength.
Colony formation assay
In total, 2 days after siRNA transfection, the cells
(H441, H1975, H2009, and HBEC4KT) were replated into six-well
plates at 1,000 cells/well for GRHL2 silencing experiments.
After 10 days of culture at 37°C, 5% CO2, the colonies
were stained with a solution containing 0.5% methylene blue
tetrahydrate (cat. no. 137-06982; FUJIFILM Wako Pure Chemical
Corporation) and 50% ethanol for fixation) at room temperature for
30 min. Colonies >1 mm in diameter were counted manually.
Western blot analysis
Western blotting was conducted as described
previously using whole cell lysates (31). Lysate of transfected cells was
collected 4 days after siRNA transfection and 2 days after
GRHL2 overexpression. Cell lysate of untransfected normal
and lung cancer cell lines was collected at ~80% confluency. The
primary antibodies used were rabbit anti-GRHL2 (cat. no. HPA062839;
Merck KGaA; 1:500), mouse anti-E-cadherin (cat. no. 610181; BD
Biosciences; 1:1,000), mouse anti-vimentin (cat. no. HPA001762;
Merck KGaA; 1:500), rabbit anti-AKT (pan; cat. no. 4691; Cell
Signaling Technology, Inc.; 1:1,000), rabbit anti-phosphorylated
(p-)-AKT (ser473; cat. no. 4060; Cell Signaling Technology, Inc.;
1:1,000), rabbit anti-p44/42MAPK (Erk1/2; cat. no. 9102; Cell
Signaling Technology, Inc.; 1:1,000), rabbit anti-p-p44/42MAPK
(Erk1/2; cat. no. 4370; Cell Signaling Technology, Inc.; 1:1,000)
and mouse anti-β-actin (cat. no. A2228; Merck KGaA; 1:2,000). The
β-actin protein level was used as a control for protein loading.
The primary antibodies were incubated at 4°C overnight. The
secondary antibodies conjugated with horseradish peroxidase (HRP)
were anti-rabbit (cat. no. NA934-1ML; Cytiva) and anti-mouse
antibodies (cat. no. NA931-1ML; Cytiva) at a 1:2,000 dilution and
incubated at room temperature for 1 h. Primary and secondary
antibodies of GRHL2 were diluted by Can Get Signal™ (cat. no.
NKB-101; Toyobo Life Science). The intensities of the bands were
quantified with Fiji (v.2.9.0/1.53t) (https://imagej.net/software/fiji/) (32) or ImageJ (v.1.53k) (https://imagej.nih.gov/ij/index.html)
(33), where values of intensities
normalized to β-actin, AKT or ERK were shown below the
corresponding band images in the figures.
DNA copy number analysis
To perform a DNA copy number analysis, whole-exome
sequencing (dbGaP Study Accession: phs001823.v1.p1; http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001823.v1.p1)
(34). A total of 68 lung
adenocarcinoma, 20 lung squamous cell carcinoma and 63 non-small
cell lung cancer (NSCLC)-histology not otherwise specified lung
cancer cell lines were used (Table
SII). Copy number variation (CNV) was determined using the R
package ‘DNAcopy’ (R version 3.6, r-project.org/; DNAcopy version
1.36, https://bioconductor.org/packages/release/bioc/html/DNAcopy.html)
with several modifications.
Choice of diploid controls
Because of differences in target capture enrichment
during whole-exome sequencing, diploid controls were selected from
the same batch as tumor samples. These batches were determined
computationally by estimating the amount of noise (deviation from
copy number segments) for each pair of tumor and control samples,
followed by the hierarchical clustering of these noise values.
Typically, tumor-control pairs from the same batch will have
noticeably lower noise values. Next diploid controls were compared
to other controls (from the same batch) to generate CNV
segmentation plots. This allowed a further selection of controls
with no or few CNV.
Recalibration
Because tumor samples are not diploid and frequently
do not have an equal amount of copy number gains and losses, copy
numbers were recalibrated by the visual inspection of each plot. In
general, the lowest segments were set to one copy
[log2(tumor/control)=−1], which shifted the subsequent
segments so that log2(tumor/control) can be 0 (2
copies), 0.58 (3 copies), 1 (4 copies), and so on.
Statistical analysis
The Lung Cancer Explorer (LCE; http://lce.biohpc.swmed.edu/lungcancer/)
was used to analyze the data from The Cancer Genome Atlas (TCGA;
http://portal.gdc.cancer.gov/) and to
perform meta-analyses on expression levels of GRHL2 in tumor
vs. normal tissues and impact of GRHL2 expression on
survival in patients with lung cancer. Univariate and multivariate
analyses were performed using EZR (v. 1.55;
jichi.ac.jp/saitama-sct/SaitamaHP.files/statmedEN.html) (35). Pearson's correlation coefficients
with associated P-values were calculated for correlation analyses
by using EZR (v. 1.55). Statistically significant differences
between two comparisons (control and one of the investigated
groups) or >2 groups (control and multiple of the investigated
groups) were analyzed by unpaired t-test or one-way ANOVA with
Dunnett's post hoc test respectively, using the SPSS software
(v.28; IBM Corp.). P<0.05 were considered to be statistically
significant difference.
Results
GRHL2 expression is upregulated in
lung cancer but is not associated with patient survival
To investigate the GRHL2 expression profile
in lung cancer and its impact on patient survival, statistical
analyses were performed using LCE (23), with focus on a dataset of TCGA
(cancer. gov/ccg/research/genome-sequencing/tcga). GRHL2
expression was revealed to be significantly upregulated in both
lung adenocarcinoma and squamous cell carcinoma tissues compared
with that in normal lung tissues (Fig.
1A) in TCGA dataset. This result was confirmed by a
meta-analysis of six datasets, including TCGA. GRHL2
expression in adenocarcinoma and squamous cell carcinoma tissues
was 1.31 and 1.61-fold higher compared with that in normal tissue,
respectively (Fig. 1B). In
addition, copy number analysis by whole-exome sequencing, carried
out as part of a different study (34), revealed increases in GRHL2
copy numbers. Specifically, 51/68 (75.0%) adenocarcinoma, 14/20
(70.0%) squamous cell carcinoma and 40/63 (63.5%) NSCLC-not
otherwise specified cell lines had > three GRHL2 copies
(mean ± SD; 3.35±1.25 in adenocarcinoma, 3.35±1.31 in squamous cell
carcinoma and 2.94±0.97 in NSCLC-otherwise specified cell lines;
Fig. 1C). These data suggest that
the increased GRHL2 expression may have resulted from gene
amplification. Subsequently, it was investigated whether
GRHL2 expression was associated with patient survival in
patients with adenocarcinoma or squamous cell carcinoma.
Kaplan-Meier analysis in TCGA dataset revealed that overall
survival was not significantly different in the GRHL2 high-
and low-expression adenocarcinoma and squamous cell carcinoma
groups, following division using the median expression value
(Fig. 1D). Univariate and
multivariate Cox regression analysis in TCGA dataset showed that
GRHL2 expression was not associated with survival in
patients with either adenocarcinoma (Table I) or squamous cell carcinoma
(Table II). In addition, the
absence of correlation between GRHL2 expression between
patient survival in lung cancer was also confirmed by a metanalysis
using LCE (Fig. 1E). These data
suggest that the expression of GRHL2 was upregulated in both
adenocarcinoma and squamous cell carcinoma, but this phenomenon was
not associated with patient survival.
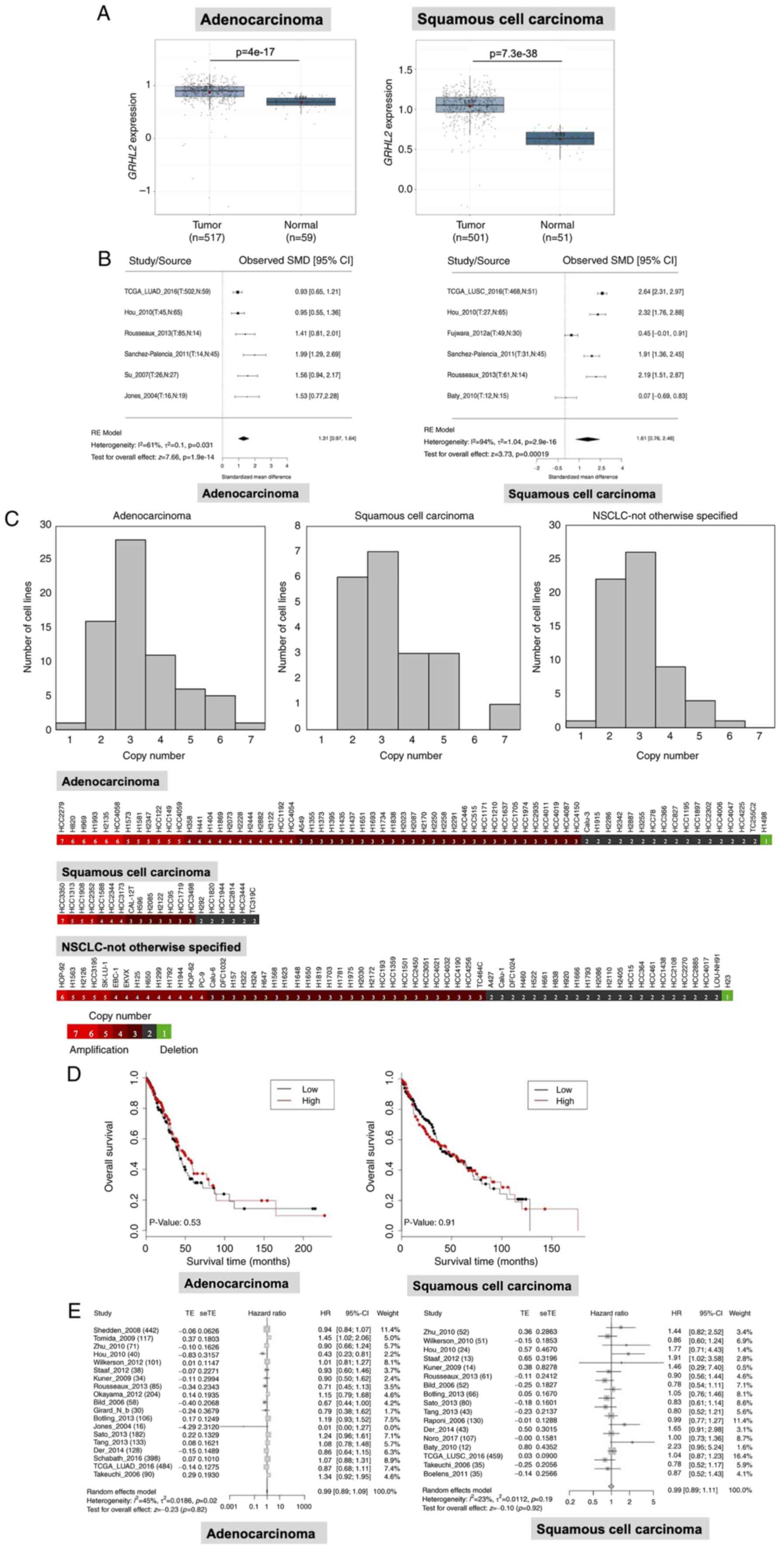 | Figure 1.GRHL2 is expressed at high levels in
lung cancer. (A) Expression analysis of GRHL2 in lung
adenocarcinoma and squamous cell carcinoma in TCGA dataset. (B)
Meta-analysis of GRHL2 expression in lung adenocarcinoma and
squamous cell carcinoma from six studies. T, tumor, N, normal, RE,
random effects. (C) DNA copy number analysis of GRHL2 showing
frequent increases in the copy number of GRHL2 in NSCLC cell lines.
(D) Kaplan-Meier survival curves for patients with high or low
GRHL2 expression (the cut-off value is the median expression
level). (E) Survival meta-analysis for GRHL2 in adenocarcinoma
(left) and squamous cell carcinoma (right). In each forest plot,
the name of each study is followed by the number of total tumor
samples. GRHL2, Grainyhead-like 2; SMD, standardized mean
difference; TE, estimated treatment effect; seTE, standard error of
treatment effect; HR, hazard ratio; CI, confidence interval, TCGA,
The Cancer Genome Atlas; NSCLC, non-small cell lung cancer. |
 | Table I.Univariate and multivariate analyses
of cox multivariate regression analysis in adenocarcinoma in TCGA
cohort. |
Table I.
Univariate and multivariate analyses
of cox multivariate regression analysis in adenocarcinoma in TCGA
cohort.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Sex |
|
|
|
|
|
|
|
Female | Reference |
|
| Reference |
|
|
|
Male | 0.9058 | 0.632–1.298 | 0.5903 | 0.8083 | 0.5362–1.219 | 0.3097 |
| Age, years |
|
|
|
|
|
|
|
>65 | Reference |
|
| Reference |
|
|
|
≤65 | 0.7696 | 0.5353–1.107 | 0.1575 | 0.6787 | 0.4589–1.004 | 0.0522 |
| Smoking status |
|
|
|
|
|
|
|
Never | Reference |
|
| Reference |
|
|
|
Former | 0.9214 | 0.5095–1.666 | 0.7864 | 1.2300 | 0.6490–2.332 | 0.5255 |
|
Current | 0.7176 | 0.3668–1.404 | 0.3324 | 0.7643 | 0.3741–1.561 | 0.4609 |
| Stage |
|
|
|
|
|
|
| I | Reference |
|
| Reference |
|
|
| II | 2.516 | 1.582–4.002 |
<0.0001a | 3.0790 | 1.8620–5.091 |
<0.0001a |
|
III | 4.426 | 2.774–7.062 |
<0.0001a | 4.8250 | 2.9390–7.922 |
<0.0001a |
| IV | 3.266 | 1.665–6.407 | 0.0006a | 4.4210 | 2.1770–8.981 |
<0.0001a |
| GRHL2 mRNA |
|
|
|
|
|
|
|
Low | Reference |
|
| Reference |
|
|
|
High | 0.9162 | 0.6413–1.309 | 0.1575 | 0.9025 | 0.6140–1.327 | 0.6018 |
| Table II.Univariate and multivariate analyses
of cox multivariate regression analysis in squamous cell carcinoma
in TCGA cohort. |
Table II.
Univariate and multivariate analyses
of cox multivariate regression analysis in squamous cell carcinoma
in TCGA cohort.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Sex |
|
|
|
|
|
|
|
Female | Reference |
|
| Reference |
|
|
|
Male | 1.038 | 0.7188–1.498 | 0.8434 | 1.0120 | 0.6924–1.478 | 0.9528 |
| Age, years |
|
|
|
|
|
|
| >65
years | Reference |
|
| Reference |
|
|
| ≤65
years | 0.767 | 0.5447–1.080 | 0.1286 | 0.6727 | 0.4708–0.9611 | 0.0294 |
| Smoking status |
|
|
|
|
|
|
|
Never | Reference |
|
| Reference |
|
|
|
Former | 0.4628 | 0.1695–1.264 | 0.1327 | 0.4100 | 0.1462–1.150 | 0.0903 |
|
Current | 0.5929 | 0.2114–1.663 | 0.3205 | 0.5453 | 0.1894–1.570 | 0.2611 |
| Stage |
|
|
|
|
|
|
| I | Reference |
|
| Reference |
|
|
| II | 1.202 | 0.8161–1.771 | 0.3514 | 1.1420 | 0.7654–1.704 | 0.5150 |
|
III | 1.499 | 0.9979–2.251 | 0.0512 | 1.6030 | 1.060–2.424 | 0.0254 |
| IV | 2.042 | 0.6373–6.542 | 0.2295 | 1.8550 | 0.5745–5.992 | 0.3014 |
| GRHL2 mRNA |
|
|
|
|
|
|
|
Low | Reference |
|
| Reference |
|
|
|
High | 1.008 | 0.7317–1.387 | 0.963 | 0.9540 | 0.6845–1.330 | 0.7812 |
GRHL2 expression is associated with
the epithelial phenotype in lung cancer
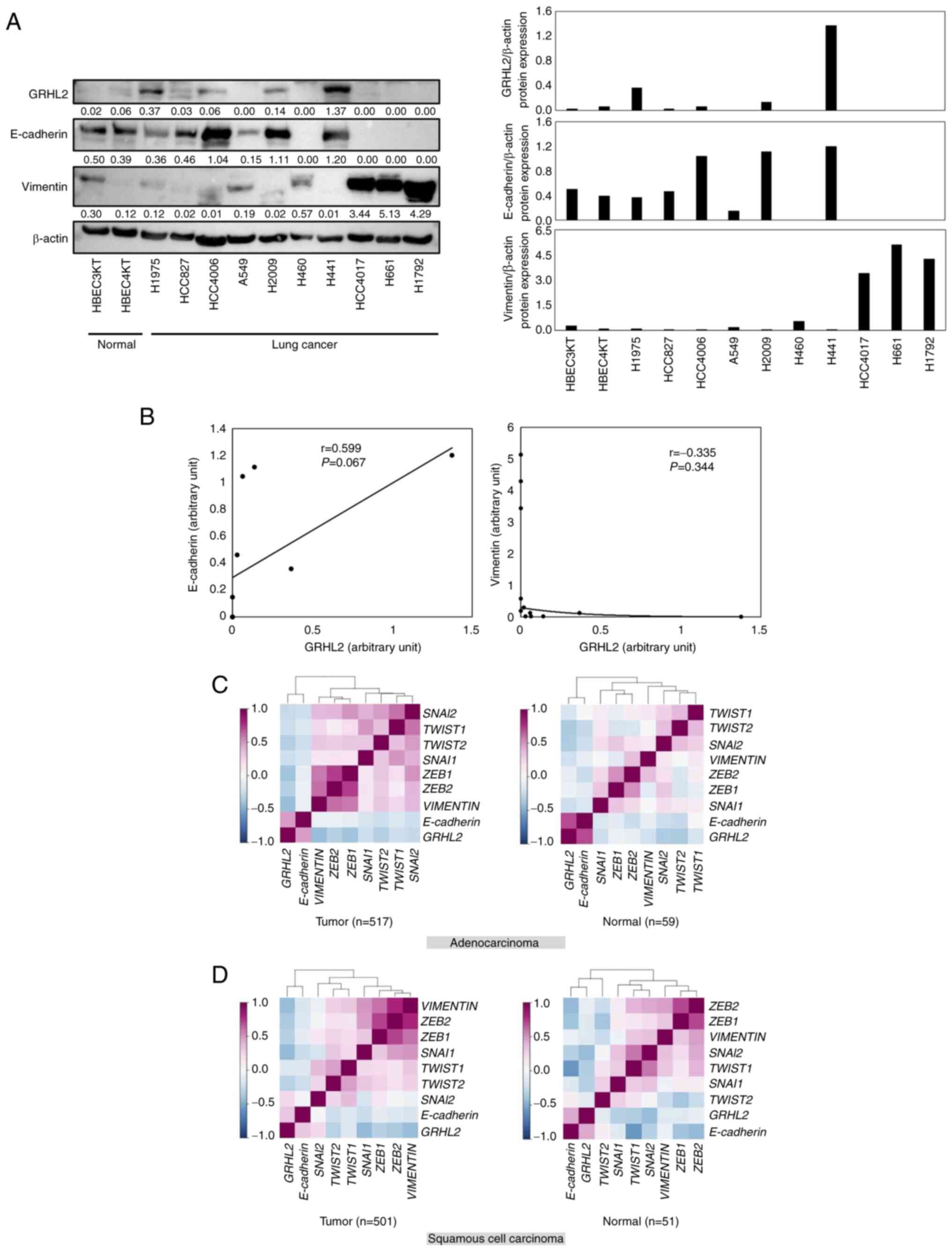
The association between GRHL2 protein expression and
the EMT status in lung cancer was next investigated by evaluating
the expression levels of GRHL2, E-cadherin (an epithelial marker)
and vimentin (a mesenchymal marker) in a panel of lung cancer cell
lines (HCC827, HCC4006, HCC4017, A549, H460, H441, H661, H1792,
H1975, and H2009). As shown in Fig. 2A
and B, expression levels of GRHL2 were not significantly
positively correlated with E-cadherin (Pearson's correlation
coefficient=0.059; P=0.067) or negatively correlated with Vimentin
(Pearson's correlation coefficient=0.335; P=0.344). The correlation
among the expression levels of GRHL2, E-cadherin, vimentin
and master EMT transcription factors ZEB1, ZEB2, SNAI1, SNAI2,
TWIST1 and TWIST2, was then investigated using LCE. It
was shown that GRHL2 expression was positively and
negatively correlated with E-cadherin and vimentin,
respectively, in both patients with adenocarcinoma and patients
with squamous cell carcinoma (Fig. 2C
and D; Table SIII). This
suggests that GRHL2 expression is associated with the
epithelial phenotype in lung cancer. In addition, GRHL2
expression was found to be negatively correlated with five of the
six (except SNAI2) of the master EMT transcription factors
(Fig. 2C and D; Table SIII).
GRHL2 silencing induces a partial EMT
in an hTERT/Cdk4-immortalized normal lung epithelial cell line
without affecting growth
Next, to evaluate the impact of GRHL2
silencing on EMT in normal lung epithelial cells, knockdown
experiments were performed using the hTERT/Cdk4
immortalized normal lung epithelial cell line HBEC4KT (28). This cell line does not harbor
genetic alterations that may affect EMT phenotypes (28). GRHL2 silencing was found to
decrease E-cadherin whilst increasing vimentin expression, with
upregulation in expression also observed in master EMT
transcription factors TWSIT1 (statistically significant) and
ZEB1 (not statistically significant; Fig. 3A and B). These suggest that partial
EMT, characterized by decreased E-cadherin and increased Vimentin,
albeit without completely switching their expression, has occurred
(36). To evaluate the effects of
this partial EMT on cell proliferation and colony formation, CCK-8
and colony formation assays were performed but no significant
differences could be identified except for increased viability in
si-GRHL2#2-treated cells (Fig.
3C and D). Therefore, these data suggest that GRHL2
silencing promoted partial EMT in this cell line, but it did not
significantly affect cell proliferation.
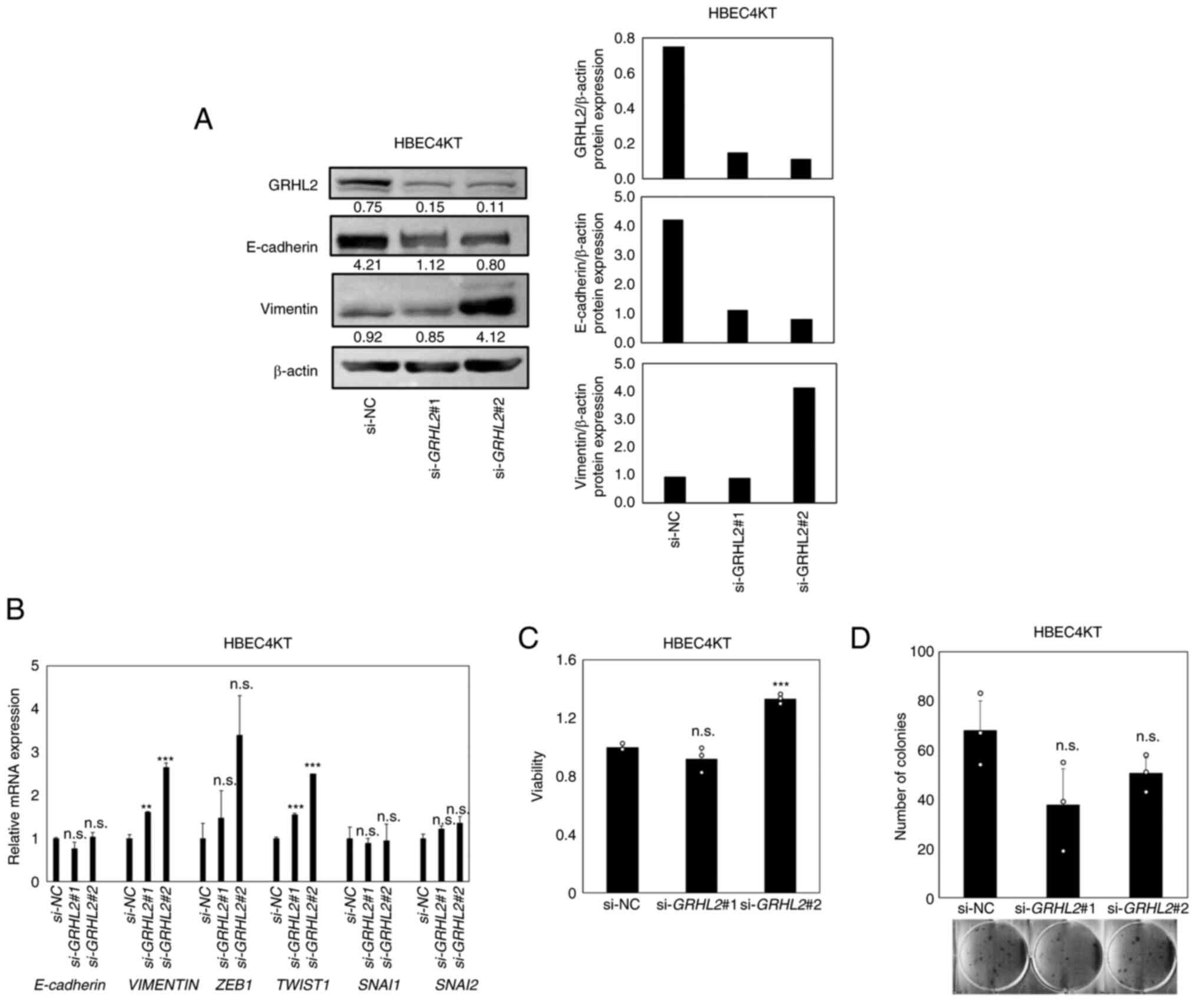 | Figure 3.GRHL2 silencing induces a partial EMT
in hTERT/Cdk4-immortalized normal lung epithelial cell line
(HBEC4KT) without affecting its growth. (A) Western blotting of
GRHL2, E-cadherin and Vimentin in the immortalized normal bronchial
epithelial cell line HBEC4KT transfected with either control or
GRHL2 siRNA. Values below bands indicate quantitation of band
intensities normalized to β-actin. Quantification of band
intensities is shown in the right graph. (B) Reverse
transcription-quantitative PCR of E-cadherin, VIMENTIN and four
master EMT regulators in HBEC4KT cells, transfected with either
control (si-NC) or GRHL2 siRNA. (C) Proliferation and (D) colony
formation assays for HBEC4KT cells transfected with either control
or GRHL2 siRNA. Results are shown as the mean ± SD (n=3), where
comparisons were performed with one-way ANOVA followed by the
Dunnett test. **P<0.01 and ***P<0.001 vs. si-NC. GRHL2,
Grainyhead-like 2, siRNA, small-interfering RNA; NC, negative
control; n.s., non-significant; EMT, epithelial-to-mesenchymal
transition; ZEB, Zinc finger E-box-binding homeobox; SNAI, snail
homolog. |
GRHL2 silencing induces partial EMT in
lung cancer with cell type-dependent effects on growth
Next, three lung adenocarcinoma cancer cell lines
H1975, H2009 and H441 were selected for evaluating the effects of
GRHL2 silencing on EMT status and growth. H1975 [TP53
(Arg273His) and EGFR (Thr790Met, Leu858Arg)
mutant) and H2009 (TP53 (Arg273Leu) and KRAS
(Gly12Ala) mutant] were selected due to the expression of
significant levels of GRHL2 protein (Fig. 2A) and due to the possession of two
major representative lung adenocarcinoma driver mutations (37,38).
In addition, H441 [TP53 (Arg158Leu) and KRAS
(Gly12Val) mutant] was selected for experimentation because
it expressed the highest levels of GRHL2 protein among the lung
cancer cell line panel tested (Fig.
2A). GRHL2 silencing did not alter E-cadherin protein or
mRNA expression in any of the three aforementioned cell lines
(Fig. 4A). By contrast, increased
vimentin protein and mRNA expression levels in H1975 and H2009
cells were observed. However, GRHL2 silencing significantly
increased vimentin mRNA expression but did not affect protein
expression in H441 cells (Fig. 4A and
B). GRHL2 silencing resulted in the upregulation of
ZEB1 expression in H1975, TWIST1 expression in H2009
and SNAI2 in H2009 and H441, suggesting a partial EMT and
variable response to this silencing (Fig. 4A and B). GRHL2 silencing also
increased cell viability in H1975 cells but not in either H2009 or
H441 cells (Fig. 4C). The colony
formation assay revealed a significant decrease in colony formation
by H2009 cells, but no change was observed in H1975 cells (Fig. 4D). H441 cells did not form colonies
and therefore could not be tested (Fig.
4D). These results suggest that GRHL2 silencing induced
EMT in the lung cancer cell lines, but its effects on proliferation
and colony formation were cell type-dependent and varied greatly
among the lung adenocarcinoma cell lines.
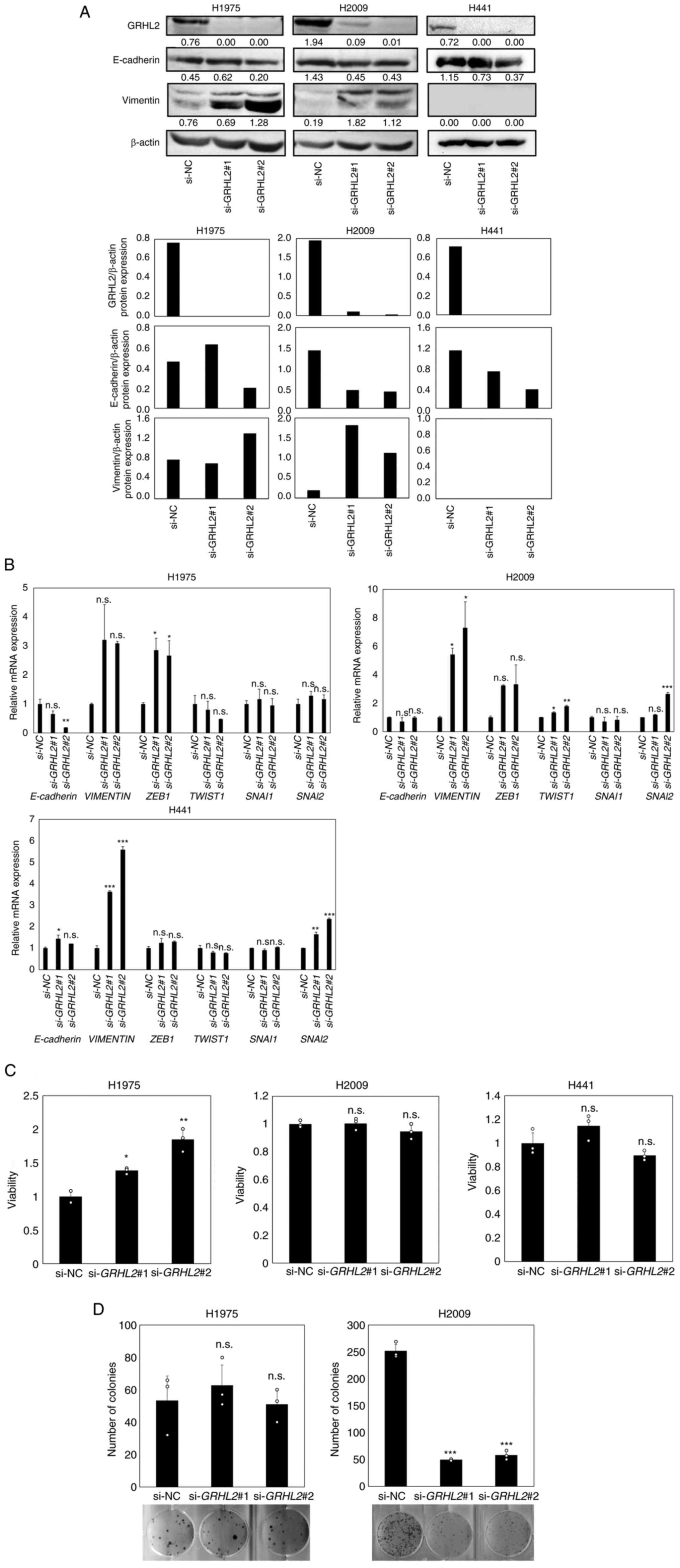 | Figure 4.GRHL2 silencing induces a partial EMT
in lung cancer but its effects on growth are cell type-dependent.
(A) Western blotting of GRHL2, E-cadherin and vimentin in the lung
cancer cell lines H1975 and H2009 transfected with control or GRHL2
siRNA. Values below bands indicate quantitation of band intensities
normalized to β-actin. Quantification of band intensities is shown
in the bottom graph. (B) Reverse transcription-quantitative PCR of
E-cadherin, vimentin and four master EMT regulator genes in H1975
and H2009 cells transfected with control (si-NC) or GRHL2 siRNA.
(C) Proliferation and (D) colony formation assays of H1975 and
H2009 cells transfected with either control or GRHL2 siRNA. Results
are shown as the mean ± SD, assessed by one-way ANOVA followed by
the Dunnett test. *P<0.05, **P<0.01 and ***P<0.001 vs.
si-NC. GRHL2, Grainyhead-like 2; siRNA, small-interfering RNA; NC,
negative control; n.s., non-significant; EMT,
epithelial-to-mesenchymal transition; ZEB, Zinc finger
E-box-binding homeobox; SNAI, snail homolog. |
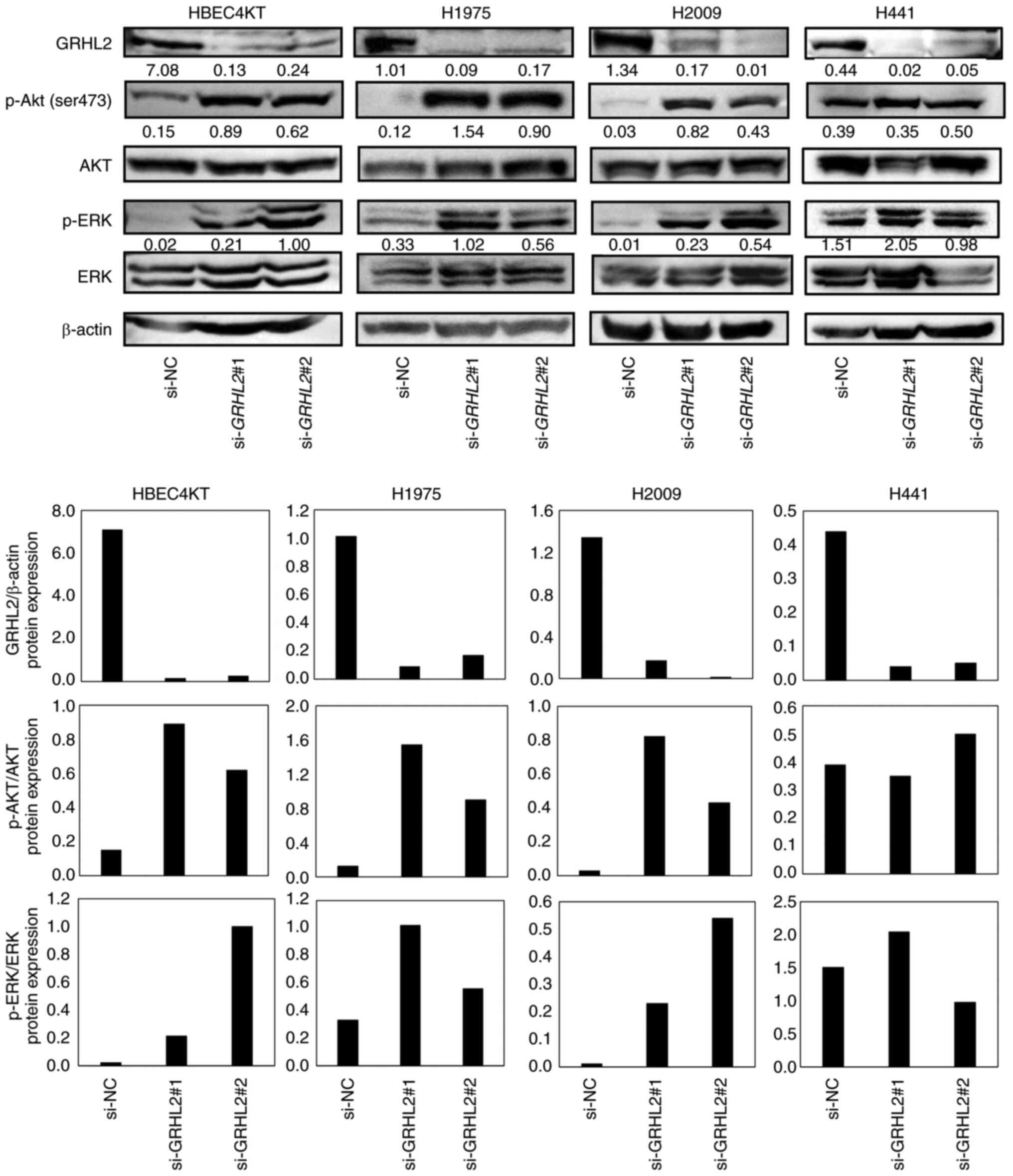
GRHL2 silencing promotes the
phosphorylation of AKT and ERK
Compared with the present study, numerous studies
have reported that EMT can promote cancer cell proliferation
(39–41). Additionally, several studies have
found oncogenic roles in GRHL2 by showing growth suppression
in several cancer cell types, including breast, pancreatic and
colorectal cancers, after GRHL2 silencing (5,6,11,16,18,20).
Growth suppression induced by GRHL2 silencing occurred
through inhibition of the AKT or ERK pathways (18,20).
This finding that GRHL2 silencing causes the inhibition of
AKT and ERK is inconsistent with the function of GRHL2 as an
EMT suppressor because the AKT and ERK pathways are associated with
EMT in various types of canceer including lung and colorectal
cancers (42–44). It was therefore hypothesized that
the differential effects of GRHL2 silencing on growth may be
due to whether the AKT or ERK pathway was induced or suppressed by
GRHL2 silencing. Therefore, the effect of GRHL2
silencing on the phosphorylation of AKT and ERK was next assessed
in HBEC4KT, H1975, H2009 and H441 cell lines by western blotting.
GRHL2 silencing enhanced the phosphorylation of both AKT and
ERK in HBEC4KT, H1975 and H2009 cell lines, but it did not affect
either AKT or ERK in H441 (Fig. 5).
The finding that both p-AKT and p-ERK were equally upregulated in
HBEC4KT, H1975 and H2009 cells was unexpected, because the effects
of GRHL2 silencing in these cells further downstream
differed substantially. This suggests that the differential effects
of GRHL2 silencing on cell proliferation among normal
bronchial epithelial and lung cancer cells do not occur through the
differences in the phosphorylation of the AKT or ERK pathways.
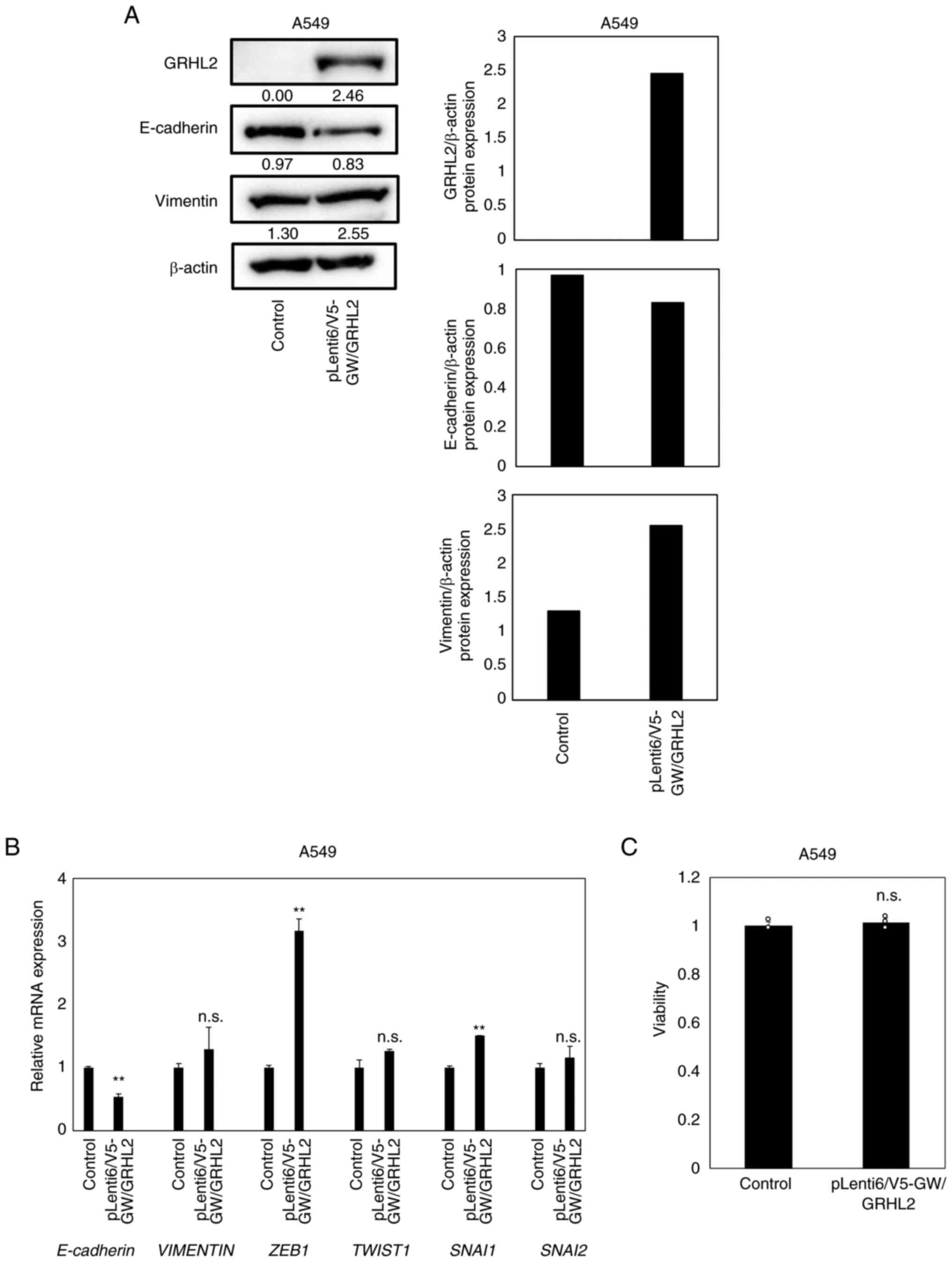
GRHL2 overexpression does not affect
the proliferation in A549 cells
To evaluate the effects of GRHL2
overexpression in lung cancer cells, GRHL2 overexpression
experiments were performed using A549 cells [TP53 wild-type,
KRAS(Gly12Ser) and liver kinase B1
(Gln37Ter) mutant], which lacked detectable expression
levels of the GRHL2 protein (Fig.
2A). In total, ~50% transfection efficiency was confirmed by
co-transfection with the EGFP-expressing vector pEGFP-N3
(Fig. S1). Overexpression of the
GRHL2 protein was achieved in the A549 cells transfected with the
GRHL2-expressing vector (Fig.
6A). GRHL2 overexpression reduced the protein expression
of E-cadherin, whilst the protein expression of vimentin remained
unaltered (Fig. 6A). GRHL2
overexpression decreased the mRNA expression of E-cadherin
and the increased mRNA expression of ZEB1 and SNAI1
without affecting TWIST1 or SNAI2 expression
(Fig. 6B). However, GRHL2
overexpression did not affect A549 proliferation (Fig. 6C).
Discussion
Both oncogenic and tumor suppressive roles of
GRHL2 have been reported in various malignancies (7–21). A
study previously showed that GRHL2 is overexpressed in lung
cancer, which is in turn associated with poor prognosis. In
addition, silencing GRHL2 expression was found to suppress
proliferation and colony formation, but it also promoted migration
and invasion in lung cancer cell lines (12). Based on the finding that
GRHL2 silencing promoted migration and invasion in a lung
cancer cell line H1299, this previous study concluded that
GRHL2 primarily functions as a tumor suppressor by
suppressing metastasis (12).
However, this previous study also presented data suggesting
tumor-promoting roles of GRHL2, including that higher
expression levels of GRHL2 in lung cancer samples are associated
with poorer patient prognosis. In addition, suppressed
proliferation and colony formation resulted from GRHL2 silencing
(12). Therefore, the roles of
GRHL2 in lung cancer remain unclear. In the present study,
increased expression of GRHL2 was observed in lung cancer
tissues without association with patient survival. Expression data
from a public database together with the experimental results of
the present study indicated that GRHL2 likely serves an
important role in maintaining an epithelial phenotype in lung
cancer. Nevertheless, despite its EMT-suppressing function in lung
cancer, it was shown that GRHL2 silencing resulted in either
growth-promoting or -suppressing effects in lung cancer cell
lines.
Consistent with a previous study (12), it was shown that GRHL2 is
overexpressed in lung cancer in a meta-analysis of gene expression
datasets. In addition, this previous study also showed that
patients with lung cancer and higher protein expression of GRHL2
had significantly shorter survival (12). However, the meta-analysis in the
present study revealed the absence of association between
GRHL2 expression and patient survival in lung cancer,
suggesting that GRHL2 expression is not associated with
survival in patients with lung cancer. This was unexpected because
of the apparent EMT-suppressing function of GRHL2. Bi-directional
effects of GRHL2 on the growth of lung cancer cells were
observed, which likely explains the reason underlying this
ambiguous result.
GRHL2 is one of the several transcription
factors that can suppress EMT (3,6).
However, the predominant roles that GRHL2 plays in
maintaining the epithelial phenotypes in lung cancer remain to be
clarified. The present study suggested an association between
GRHL2 expression and epithelial phenotypes was identified in
a panel of lung cancer cell lines and clinical tumors tissues.
Additionally, GRHL2 silencing induced partial EMT in lung
cancer cell lines and an immortalized normal bronchial epithelial
cell line. These results suggest an important role of GRHL2
in maintaining an epithelial phenotype in lung cancer.
Despite the partial EMT induced by GRHL2
silencing in the lung cancer cell lines, its impact on growth
differed substantially among the cell lines tested, as shown by
enhanced viability in H1975 and inhibited clonogenic growth in
H2009. To gain insights into the interpretation of these results,
the phosphorylation of AKT and ERK was investigated because these
growth-promoting proteins were previously found to be suppressed by
GRHL2 silencing (18,20).
GRHL2 silencing resulted in increased levels of p-AKT and
p-ERK in all cell lines studied, suggesting that the differential
activation of either AKT or ERK may not be attributable to the
observed differences in the effects of GRHL2 silencing on
cell proliferation. Notably, one previous study (20) reported that GRHL2 silencing
suppressed the proliferation of two colorectal cancer cell lines
through decreased AKT phosphorylation. However, in the present
study, it was shown that GRHL2 silencing resulted in the
increased activation of AKT even in the H2009 lung cancer cell
line, which in turn inhibited colony formation (Fig. 4D). These findings suggested that the
effects of GRHL2 on the AKT or ERK pathways markedly vary
among cancer types. Therefore, the effects of GRHL2
silencing on the phosphorylation status of AKT and ERK should be
evaluated in a variety of human cancer cell lines, such as breast
and pancreatic cancer. ERK can serve a pro-apoptotic function when
it is translocated into the mitochondria instead of the nucleus
(45). Therefore, it remains
possible that upregulated ERK activity may not promote, but rather
suppress, cell proliferation in the experiments performed in the
present study.
A discrepancy was observed in the results of cell
viability and colony formation assays in H2009 cells, whereby
GRHL2 silencing did not affect proliferation but inhibited
colony formation. It was hypothesized that these contradictory
results could be explained by the differences in the properties of
cancer cells these two assays evaluate. The viability assay
evaluates cell proliferation, whereas colony formation assays
measure the ability of individual cells to survive and propagate
over long periods of time, typically over 10–14 days (46,47).
Notably, a previous study (46)
described that colony formation assays can be used to evaluate
self-renewing capacity in vitro. Relevant roles of
GRHL2 in self-renewal have been reported (48,49).
The growth suppression induced by GRHL2 silencing in the
present study was only observed in colony formation but not in
viability assays in H2009, suggesting the involvement of
GRHL2 in cell stemness. Collectively, results of the present
study in H2009 cells suggest that GRHL2 may confer
anti-apoptotic and/or self-renewing abilities. Therefore, this
hypothesis warrants further investigation in future studies.
GRHL2 overexpression did not affect
proliferation in A549 cells, which lacked detectable expression
levels of the endogenous GRHL2 protein. Both growth-promoting and
-suppressive effects of GRHL2 overexpression have been
reported in numerous types of different cell models (8,10,13–16,20).
To further evaluate the effects of GRHL2 overexpression on
proliferation and clonogenic growth in lung cells, GRHL2
overexpression experiments using additional lung cancer cell lines
and immortalized normal lung epithelial cells are planned as future
experiments. In addition, it was an unexpected observation that
GRHL2 overexpression decreased the expression of E-cadherin
on both mRNA and protein levels because of its function of positive
transcription factor for E-cadherin (3). One possible explanation for this
result is the increased ZEB1 expression in A549 cell, which
was also induced in response to GRHL2 overexpression. It was
possible that ZEB1 counteracted the function of GRHL2 as a
positive regulator of E-cadherin expression, leading to decreased
levels of E-cadherin expression. To test this hypothesis,
luciferase reporter assay is required for evaluating
transcriptional activity of E-cadherin in
GRHL2-overexpressing cells with or without ZEB1
silencing.
Altogether, the present study revealed that
GRHL2 expression was associated with an epithelial
phenotype, but its expression was not associated with prognosis in
lung cancer. Gene silencing experiments suggested that GRHL2
is not likely to be a definitive tumor suppressor or an oncogene,
instead acting as either of them depending on the cell context.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported, in part, by the Grant-in-Aid
for Exploratory Research (grant no. 19K22617), Grant-in-Aid for
Scientific Research (grant no. 21H02924), the 45th (2020) Aichi
Cancer Research Foundation to M. Sato, Nagoya University CIBoG
program from MEXT WISE program and National Cancer Institute,
SPORE: Developing New Rationale, Personalized Medicine for Lung
Cancer (grant no.,P50 CA070907).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
NK and MS conceived and designed the study. NK, KM,
KK, NM, MT, YT, YI, MY, NS and MS acquired data. NK, KM, LG, LC,
YX, IT, MM, LG, JDM and MS analyzed and interpreted the data. LG
and JDM provided technical or material support. NK, NS, LG, JDM and
MS wrote, reviewed and revised the manuscript. All authors have
read and approved the final version of the manuscript. NK and MS
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
JM receives royalties from the NIH and University
of Texas Southwestern for distribution of human cell lines. The
remaining authors declare that they have no competing
interests.
References
|
1
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lim J and Thiery JP:
Epithelial-mesenchymal transitions: Insights from development.
Development. 139:3471–3486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frisch SM, Farris JC and Pifer PM: Roles
of grainyhead-like transcription factors in cancer. Oncogene.
36:6067–6073. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He J, Feng C, Zhu H, Wu S, Jin P and Xu T:
Grainyhead-like 2 as a double-edged sword in development and
cancer. Am J Transl Res. 12:310–331. 2020.PubMed/NCBI
|
|
7
|
Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT,
Yang Z, Ye J, Muller J, Koh CM, Guccione E, et al:
GRHL2-miR-200-ZEB1 maintains the epithelial status of
ovarian cancer through transcriptional regulation and histone
modification. Sci Rep. 6:199432016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cieply B, Farris J, Denvir J, Ford HL and
Frisch SM: Epithelial-mesenchymal transition and tumor suppression
are controlled by a reciprocal feedback loop between ZEB1 and
grainyhead-like-2. Cancer Res. 73:6299–6309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cieply B, Riley P IV, Pifer PM, Widmeyer
J, Addison JB, Ivanov AV, Denvir J and Frisch SM: Suppression of
the epithelial-mesenchymal transition by grainyhead-like-2. Cancer
Res. 72:2440–2453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Farris JC, Pifer PM, Zheng L, Gottlieb E,
Denvir J and Frisch SM: Grainyhead-like 2 reverses the metabolic
changes induced by the oncogenic epithelial-mesenchymal transition:
Effects on anoikis. Mol Cancer Res. 14:528–538. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishino H, Takano S, Yoshitomi H, Suzuki
K, Kagawa S, Shimazaki R, Shimizu H, Furukawa K, Miyazaki M and
Ohtsuka M: Grainyhead-like 2 (GRHL2) regulates epithelial
plasticity in pancreatic cancer progression. Cancer Med.
6:2686–2696. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pan X, Zhang R, Xie C, Gan M, Yao S, Yao
Y, Jin J, Han T, Huang Y, Gong Y, et al: GRHL2 suppresses
tumor metastasis via regulation of transcriptional activity of RhoG
in non-small cell lung cancer. Am J Transl Res. 9:4217–4226.
2017.PubMed/NCBI
|
|
13
|
Shen J, Lv X and Zhang L: GRHL2
acts as an anti-oncogene in bladder cancer by regulating ZEB1 in
epithelial-mesenchymal transition (EMT) process. Onco Targets Ther.
13:2511–2522. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Werner S, Frey S, Riethdorf S, Schulze C,
Alawi M, Kling L, Vafaizadeh V, Sauter G, Terracciano L, Schumacher
U, et al: Dual roles of the transcription factor grainyhead-like 2
(GRHL2) in breast cancer. J Biol Chem. 288:22993–23008.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiang J, Fu X, Ran W and Wang Z:
Grhl2 reduces invasion and migration through inhibition of
TGFβ-induced EMT in gastric cancer. Oncogenesis. 6:e2842017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiang X, Deng Z, Zhuang X, Ju S, Mu J,
Jiang H, Zhang L, Yan J, Miller D and Zhang HG: Grhl2
determines the epithelial phenotype of breast cancers and promotes
tumor progression. PLoS One. 7:e507812012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Z, Wu D, Chen Y, Min Z and Quan Y:
GRHL2 inhibits colorectal cancer progression and metastasis
via oppressing epithelial-mesenchymal transition. Cancer Biol Ther.
20:1195–1205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen W, Kang KL, Alshaikh A, Varma S, Lin
YL, Shin KH, Kim R, Wang CY, Park NH, Walentin K, et al:
Grainyhead-like 2 (GRHL2) knockout abolishes oral cancer
development through reciprocal regulation of the MAP kinase and
TGF-β signaling pathways. Oncogenesis. 7:382018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Faddaoui A, Sheta R, Bachvarova M, Plante
M, Gregoire J, Renaud MC, Sebastianelli A, Gobeil S, Morin C, Ghani
K and Bachvarov D: Suppression of the grainyhead transcription
factor 2 gene (GRHL2) inhibits the proliferation, migration,
invasion and mediates cell cycle arrest of ovarian cancer cells.
Cell Cycle. 16:693–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu F, He Z, Sun C and Rong D: Knockdown of
GRHL2 inhibited proliferation and induced apoptosis of
colorectal cancer by suppressing the PI3K/Akt pathway. Gene.
700:96–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quan Y, Jin R, Huang A, Zhao H, Feng B,
Zang L and Zheng M: Downregulation of GRHL2 inhibits the
proliferation of colorectal cancer cells by targeting ZEB1. Cancer
Biol Ther. 15:878–887. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jolly MK, Tripathi SC, Jia D, Mooney SM,
Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E and Levine
H: Stability of the hybrid epithelial/mesenchymal phenotype.
Oncotarget. 7:27067–27084. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai L, Lin S, Girard L, Zhou Y, Yang L, Ci
B, Zhou Q, Luo D, Yao B, Tang H, et al: LCE: An open web portal to
explore gene expression and clinical associations in lung cancer.
Oncogene. 38:2551–2564. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sato M, Shay JW and Minna JD: Immortalized
normal human lung epithelial cell models for studying lung cancer
biology. Respir Investig. 58:344–354. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sato M, Vaughan MB, Girard L, Peyton M,
Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW and
Minna JD: Multiple oncogenic changes (K-RAS(V12), p53 knockdown,
mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer
a full malignant phenotype on human bronchial epithelial cells.
Cancer Res. 66:2116–2128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Phelps RM, Johnson BE, Ihde DC, Gazdar AF,
Carbone DP, McClintock PR, Linnoila RI, Matthews MJ, Bunn PA Jr,
Carney D, et al: NCI-navy medical oncology branch cell line data
base. J Cell Biochem Suppl. 24:32–91. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ramirez RD, Sheridan S, Girard L, Sato M,
Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, et al:
Immortalization of human bronchial epithelial cells in the absence
of viral oncoproteins. Cancer Res. 64:9027–9034. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van Kuppeveld FJ, Johansson KE, Galama JM,
Kissing J, Bölske G, van der Logt JT and Melchers WJ: Detection of
mycoplasma contamination in cell cultures by a mycoplasma
group-specific PCR. Appl Environ Microbiol. 60:149–152. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato M, Girard L, Sekine I, Sunaga N,
Ramirez RD, Kamibayashi C and Minna JD: Increased expression and no
mutation of the Flap endonuclease (FEN1) gene in human lung cancer.
Oncogene. 22:7243–7246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schindelin J, Arganda-Carreras I, Frise E,
Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S,
Schmid B, et al: Fiji: An open-source platform for biological-image
analysis. Nat Methods. 9:676–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH image to imageJ: 25 Years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McMillan EA, Ryu MJ, Diep CH, Mendiratta
S, Clemenceau JR, Vaden RM, Kim JH, Motoyaji T, Covington KR,
Peyton M, et al: Chemistry-first approach for nomination of
personalized treatment in lung cancer. Cell. 173:864–878.e29. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saitoh M: Involvement of partial EMT in
cancer progression. J Biochem. 164:257–264. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Das AK, Sato M, Story MD, Peyton M, Graves
R, Redpath S, Girard L, Gazdar AF, Shay JW, Minna JD and Nirodi CS:
Non-small-cell lung cancers with kinase domain mutations in the
epidermal growth factor receptor are sensitive to ionizing
radiation. Cancer Res. 66:9601–9608. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sunaga N, Imai H, Shimizu K, Shames DS,
Kakegawa S, Girard L, Sato M, Kaira K, Ishizuka T, Gazdar AF, et
al: Oncogenic KRAS-induced interleukin-8 overexpression promotes
cell growth and migration and contributes to aggressive phenotypes
of non-small cell lung cancer. Int J Cancer. 130:1733–1744. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen L, Liu H, Ji Y, Ma Z, Shen K,
Shangguan X, Qian H, Zhao Y, Pan CW and Xue W: Downregulation of
SHMT2 promotes the prostate cancer proliferation and metastasis by
inducing epithelial-mesenchymal transition. Exp Cell Res.
415:1131382022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He F, Feng G, Ma N, Midorikawa K, Oikawa
S, Kobayashi H, Zhang Z, Huang G, Takeuchi K and Murata M: GDF10
inhibits cell proliferation and epithelial-mesenchymal transition
in nasopharyngeal carcinoma by the transforming growth
factor-β/Smad and NF-κB pathways. Carcinogenesis. 43:94–103.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang W, Huang H, Xiao Y, Wang L, Zhang T,
Fang X and Xia X: UBE2T is upregulated, predicts poor prognosis,
and promotes cell proliferation and invasion by promoting
epithelial-mesenchymal transition via inhibiting autophagy in an
AKT/mTOR dependent manner in ovarian cancer. Cell Cycle.
21:780–791. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ding C, Luo J, Li L, Li S, Yang L, Pan H,
Liu Q, Qin H, Chen C and Feng J: Gab2 facilitates
epithelial-to-mesenchymal transition via the MEK/ERK/MMP signaling
in colorectal cancer. J Exp Clin Cancer Res. 35:52016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Karimi Roshan M, Soltani A, Soleimani A,
Rezaie Kahkhaie K, Afshari AR and Soukhtanloo M: Role of AKT and
mTOR signaling pathways in the induction of epithelial-mesenchymal
transition (EMT) process. Biochimie. 165:229–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu M, Cao FL, Li N, Gao X, Su X and Jiang
X: Leptin induces epithelial-to-mesenchymal transition via
activation of the ERK signaling pathway in lung cancer cells. Oncol
Lett. 16:4782–4788. 2018.PubMed/NCBI
|
|
45
|
Cook SJ, Stuart K, Gilley R and Sale MJ:
Control of cell death and mitochondrial fission by ERK1/2 MAP
kinase signalling. FEBS J. 284:4177–4195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brix N, Samaga D, Belka C, Zitzelsberger H
and Lauber K: Analysis of clonogenic growth in vitro. Nat Protoc.
16:4963–4991. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Franken NAP, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen AF, Liu AJ, Krishnakumar R, Freimer
JW, DeVeale B and Blelloch R: GRHL2-Dependent enhancer
switching maintains a pluripotent stem cell transcriptional
subnetwork after exit from naive pluripotency. Cell Stem Cell.
23:226–238.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gao X, Bali AS, Randell SH and Hogan BL:
GRHL2 coordinates regeneration of a polarized mucociliary
epithelium from basal stem cells. J Cell Biol. 211:669–682. 2015.
View Article : Google Scholar : PubMed/NCBI
|