Introduction
Low-grade gliomas (LGGs) approximately represent
10–20% of all primary brain tumours; they are more commonly
diagnosed in younger individuals with a sporadic occurrence,
although their precise aetiology is still not well understood
(1). The clinical presentation of
LGGs can vary depending on the location and size of the tumour, but
commonly, symptoms such as headaches, cognitive impairment, focal
neurological deficits and changes in behaviour or personality have
been reported (1). It is well known
that LGGs can arise in various locations within the brain,
including the cerebral hemispheres, brainstem and cerebellum
(1). Regarding the prognosis of
LGGs, it can vary depending on various factors, such as tumour
location, extent of resection, age of the patient as well as
molecular characteristics. Nevertheless, their behaviour is
generally more favourable, mainly compared to high-grade gliomas,
with median survival ranging from 4.7 to 9.8 years (1).
The paradigmatic LGG subtype is represented by the
pilocytic astrocytoma (PA), which is considered a grade 1 tumour by
the World Health Organization (WHO). Herein, we will focus on the
clinicopathological, neurosurgical and molecular features of this
rare entity through a multidisciplinary approach, also taking into
consideration its variants and their prognosis.
Characteristics of pilocytic astrocytomas
(PA)
PAs are the most common paediatric tumours of the
central nervous system (CNS) accounting for 5% of all gliomas and
15–17% of all children and adolescent brain tumours (between 0 and
19 years), whereas they are rare in adults (about 2% of all adult
brain tumours) (2). The incidence
rate is 0.91 cases per 100.000 population, being highest in young
children but decreasing with advancing age (2). PAs are well-circumscribed,
slow-growing and low-grade astrocytic neoplasms that can arise
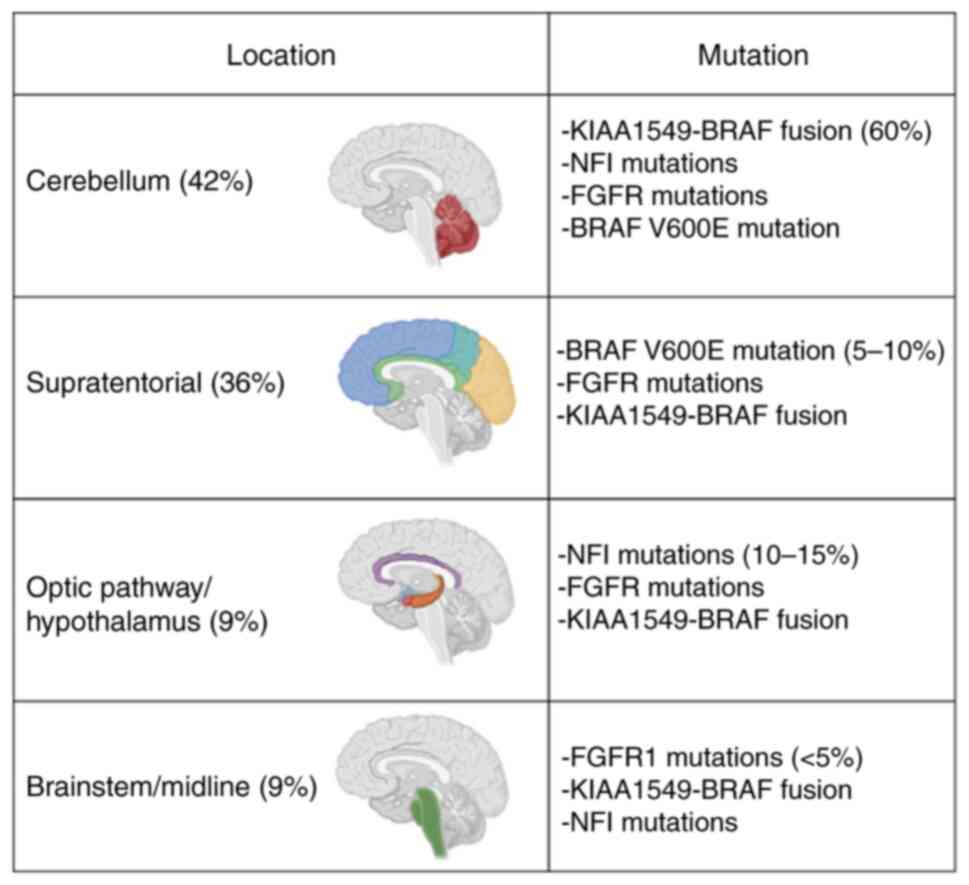
throughout the neuroaxis but are most common in the cerebellum
(42%), followed by supratentorial sites (36%), optic pathway and
hypothalamus (9%), brainstem (9%) and spinal cord (2%) (2,3).
Despite the cerebellar site being most common in the paediatric
population, there is no difference between the cerebellar and
supratentorial localisation in adults (3). The majority of PA cases are sporadic,
but they are often associated with neurodevelopmental disorders
with germline mutations in the Mitogen-activated protein kinase
(MAPK) pathway, such as Neurofibromatosis type-1 (NF1), Noonan
syndrome and encephalocraniocutaneous lipomatosis (2,4–6).
Clinical features
The signs and symptoms of PA are usually due to mass
effect and ventricular obstruction and are strictly related to
anatomical localisation. In detail, cerebellar PAs are generally
characterised by symptoms due to loss of balance and coordination
such as ataxia, dizziness and gait instability (7). Other symptoms have been reported due
to the development of hydrocephalus and increased intracranial
pressure such as headache and vomiting (7). On the other hand, PAs of the optic
pathway can cause visual loss, strabismus and protrusion of the
eyeball (7), whereas the
hypothalamic localisation can present with hypothalamic/pituitary
dysfunctions, such as obesity and diabetes insipidus (2); in addition, patients may have
associated emaciation, failure to thrive and poor clinical outcome
compared to PAs in other sites. Finally, spinal PAs are associated
with back pain, paresis and kyphoscoliosis (2).
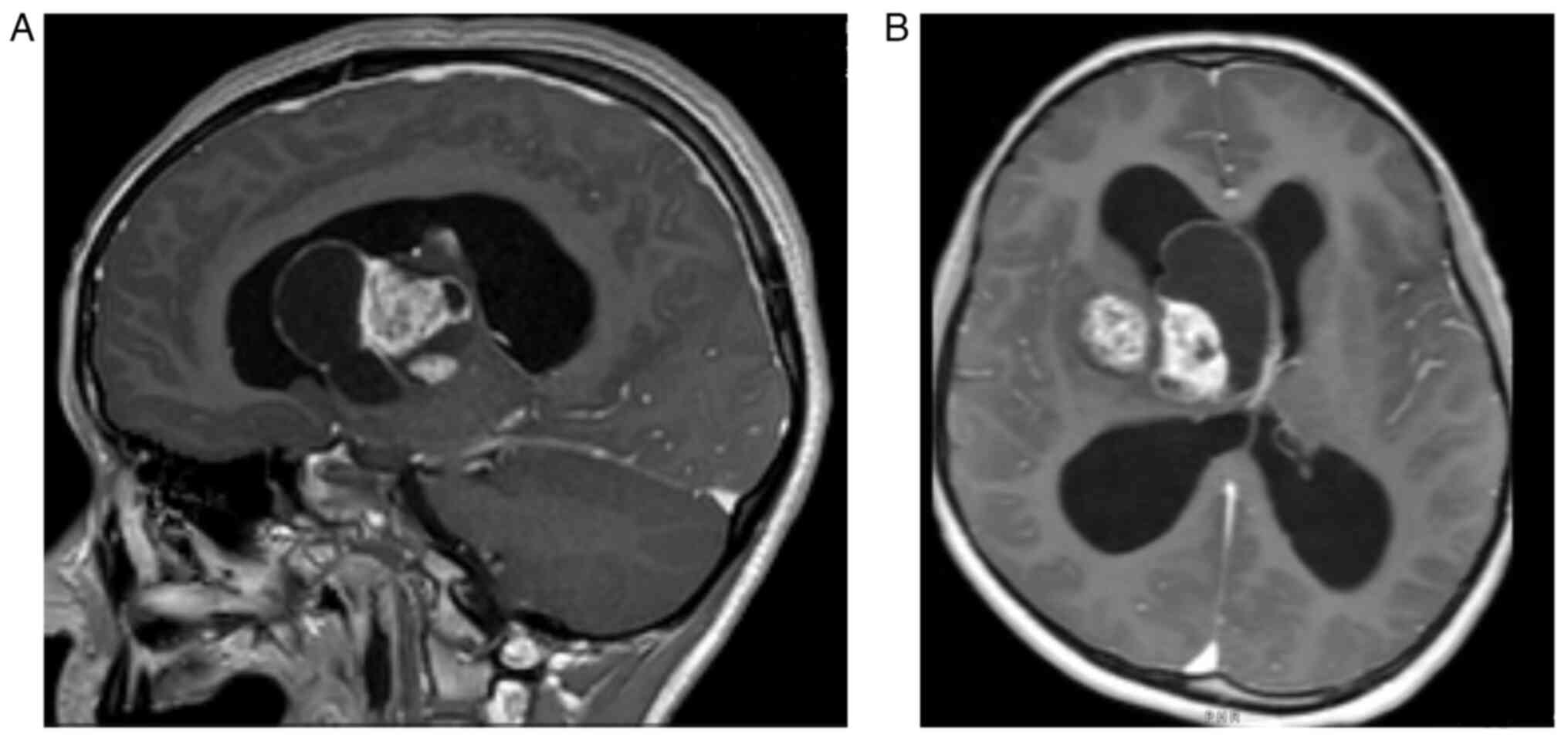
Paediatric PAs are frequently subtentorial and
affect the cerebellum with cystic, circumscribed and indolent
lesions (Fig. 1A and B). However,
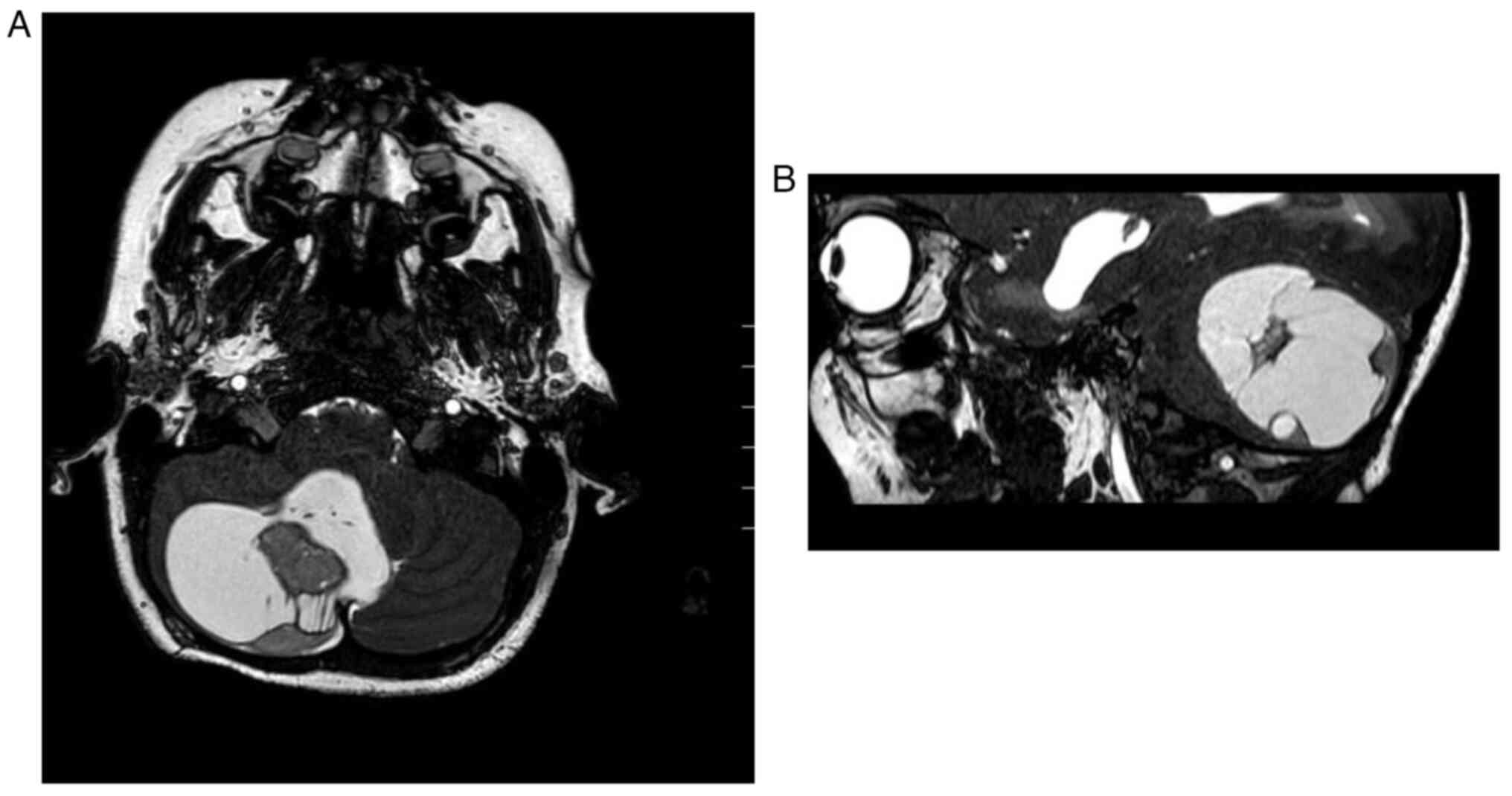
adult PAs are uncommon, but supratentorial lobar (mainly temporal
or parietal regions) are the most frequent sites (Fig. 2A and B). In addition, similar to
paediatric cases (8) in terms of
survival and neurological function, adult PAs have been reported to
have quite a benign course. Nevertheless, a recurrence incidence of
30% has been reported in adult PAs with a possible malignant
aggressive transformation (9).
Imaging
The majority of PAs can be identified on magnetic
resonance imaging (MRI) as well-circumscribed oval-shaped lesions
with cystic components and contrast-enhancing mural nodules
(Fig. 1 and Fig. 2). The solid component of PA is
typically iso to hypointense on T1 imaging and hyperintense on T2
(10). Moreover, vasogenic oedema
can be present, but it is less conspicuous than in higher-grade
tumours, as well as non-specific calcifications (7). PAs of the optic pathways usually form
fusiform masses, are accompanied by enlargement of the optic tract
and are mostly associated with NF1 patients (3,11). An
exophytic growth of PA is encountered when the tumour is localised
in the posterior fossa, especially in the brainstem (3).
Pathological findings
PAs are slow-growing soft grey tissue, that
characteristically occur in association with a cyst in the wall of
which the tumour forms a small mural nodule, typically in the
cerebellum (2). Sometimes,
associated cysts may also occur in the spinal cord, basal ganglia
or cerebral hemispheres (2). On
smears, intraoperative examination shows cells with round to
spindle nuclei with characteristic long ‘hairlike’ processes,
Rosenthal fibres and eosinophilic granular bodies. Hemosiderin
deposits, calcifications or spread in the subarachnoid space and
occasional necrosis may be commonly present (2). On histopathological examination, PAs
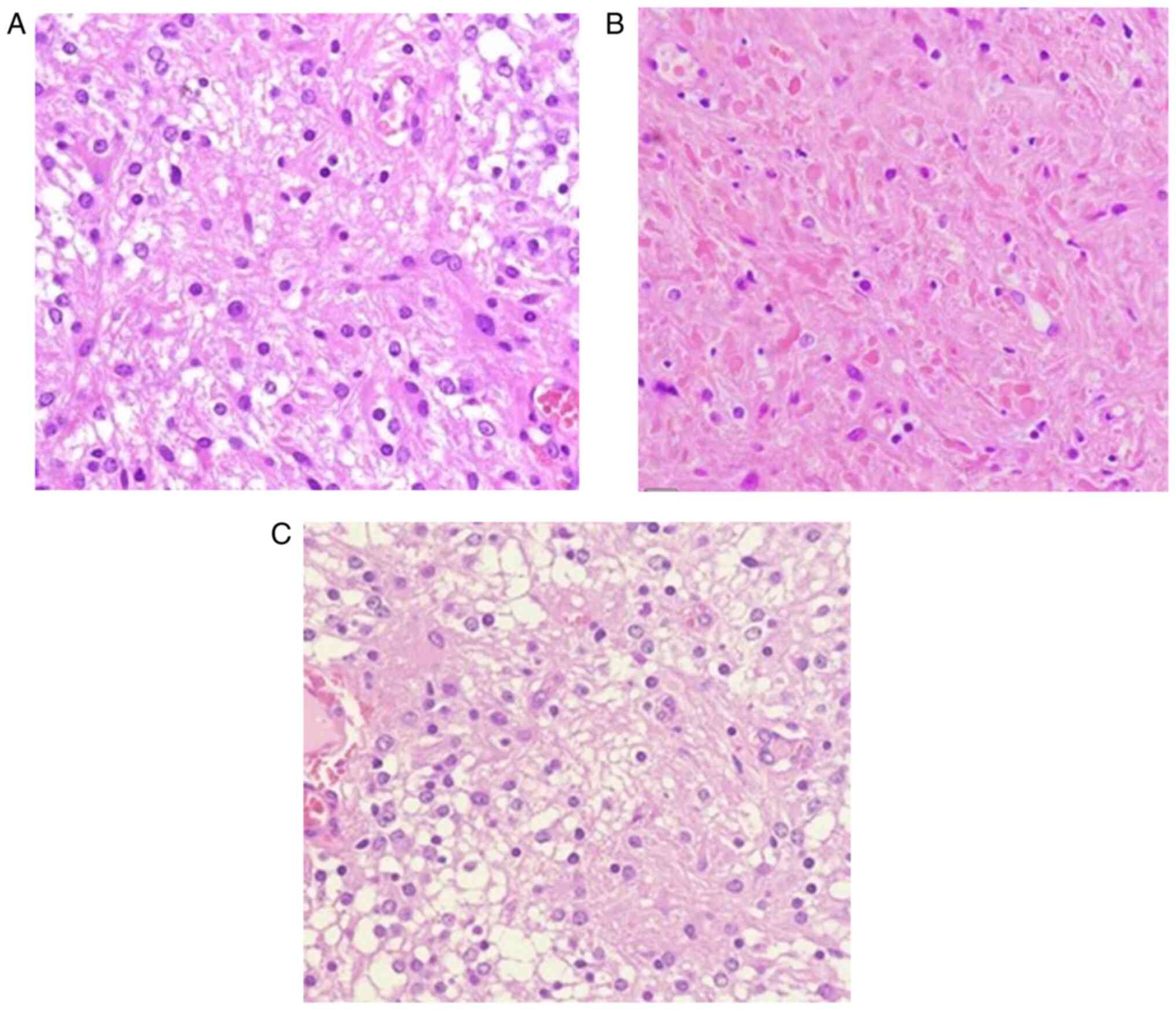
exhibit a low-to-moderate cellularity characterised by a biphasic
architectural pattern, in which compact and microcystic areas are
intermingled. Microscopically, densely fibrillated areas composed
of elongated cells (piloid cells) with bipolar ‘hairlike’ processes
(Fig. 3A) and bland nuclei rich in
Rosenthal fibres (Fig. 3B)
alternate with loose areas (Fig.
3C), with a varied degree of myxoid component composed of
multipolar oligodendrocyte-like cells with round nuclei and short
cytoplasmatic extension. Multinucleated cells with a
‘pennies-on-a-plate’ appearance may be present, as well as
eosinophilic granular bodies. Mitotic activity is usually very rare
(3,12). A variable amount of inflammatory
cells may be encountered in PAs, mainly represented by T cells,
although the role of CD4+ and CD8+ remains unclear. However, the
average ratio of CD8+/CD4+ cells in PA has been reported as
significantly higher than that in normal tissue, in which CD8+ and
CD4+ cells were equally present (13). The higher CD8+/CD4+ ratio in PA may
suggest a preferential recruitment of CD8 T cells to the tumour
microenvironment, rather than a nonspecific migration of CD8 and
CD4 T cells from adjacent normal brain (13). On the other hand,
microglia/macrophages increase in low-grade gliomas (LGG),
especially in PAs without clustering around vessels (14). Vascular proliferation with
glomeruloid features and hyalinised vessels, as well as necrosis
without pseudopalisading, can occur, but differential diagnoses
with high-grade gliomas must be ruled out (2). Three different histological patterns
of PAs can be recognised: 1) a biphasic pattern, which is the most
common; 2) a predominantly compact pattern, mainly composed of
pilocytic cells and Rosenthal fibres, more common in adult patients
and 3) a predominantly loose pattern composed by multipolar cells,
mimicking an oligodendroglioma, often associated with Fibroblast
Growth Factor Receptor-1 (FGFR1) alterations (2). Microscopic infiltration of
leptomeninges can be seen, especially in cerebellar and optic
pathways PA. In particular, in the case of PA of the optic nerve,
the tumour grows in the subarachnoid space between the nerve and
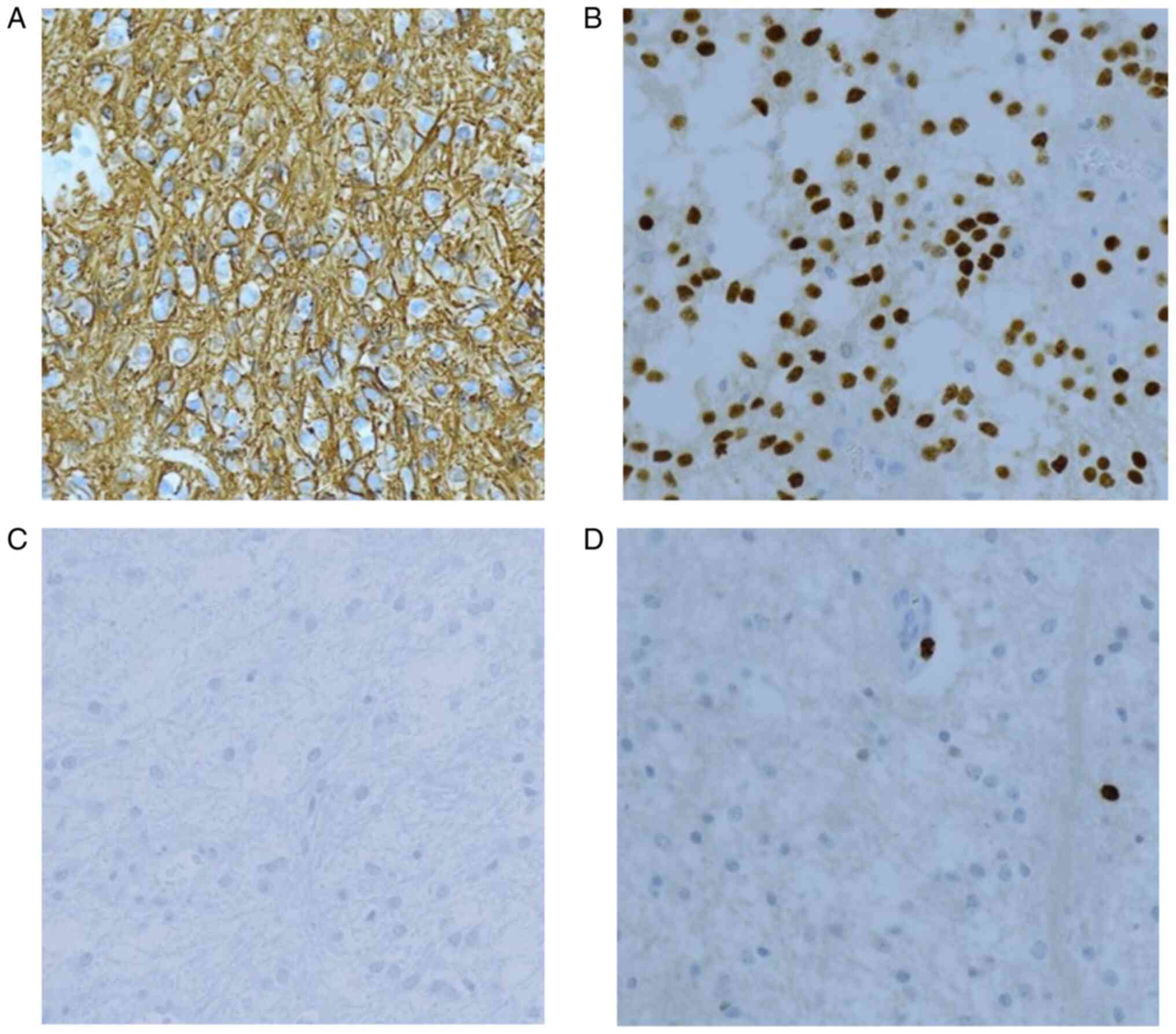
the dural sheath (2,3). Immunohistochemical analysis of PAs
show a strong and diffuse positivity for GFAP (Fig. 4A), S100, OLIG2 (Fig. 4B), SOX10 and p16, mainly in the
classical pilocytic cells, while microcystic areas are only weakly
GFAP immunoreactive (2,12). The majority of cases are positive
for synaptophysin and negative for NFP, NeuN, chromogranin and
CD34, although the expression of the last has been reported in some
hypothalamic cases. IDH1 (R132H) (Fig.
4C) and H3K27M are not expressed. The proliferative index of
Ki-67 is usually very low, less than 1% (2) (Fig.
4D).
Differential diagnosis of PA
Firstly, two different PA subtypes must be
recognised: pilomyxoid astrocytoma (PMA) and pilocytic astrocytoma
with histological features of anaplasia. PMA occurs predominantly
in children, in the hypothalamic region, the suprasellar region
being the most common location (67%) and shows an aggressive
clinical course with a high rate of recurrence, poor clinical
outcome and frequent cerebrospinal dissemination. On MRI, these
tumours appear more solid and uniformly enhancing compared with
typical PA. They are histologically characterised by increased
cellularity composed of piloid cells admixed in abundant myxoid
background; this variant typically lacks Rosenthal fibres and
eosinophilic granular bodies. Therefore, there is sufficient
evidence to support the classification of PMA as a more aggressive
and higher grade variation of PA (15); based on imaging findings, it is
really difficult to differentiate PA and PMA (16,17).
The most prominent imaging characteristic of PMA is represented by
intratumoural haemorrhage, which is much less common in PA. In
addition, PMA shows a higher recurrence rate and often prominent
cerebral-spinal fluid dissemination (18). Moreover, PA can be distinguished
from pleomorphic xanthoastrocytoma (PXA), an uncommon, benign
tumour in the brain that most likely develops from astrocytes, but
presents with pleomorphic, xanthomatous cells (2,19).
Additionally, PXA is most commonly found in the cerebral hemisphere
and the leptomeninges; it occasionally occurs in the spinal cord,
rarely evolving into a more malignant tumour (20). However, PA differs from
dysembryoplastic neuroepithelial tumours by the absence of
mucin-rich nodules and microcysts with floating neurons (2,21).
Finally, the diffuse leptomeningeal glioneuronal tumour (DLGNT), a
rare entity typically present in children, sometimes may present
with focal areas of pilocytic features (2). Nevertheless, either 1p/19q codeletion
or isolated 1p deletion with mitogen-activated protein kinase
(MAPK) activating alterations may be considered characteristics in
DLGNT (2). This latter finding
together with the absence of isocitrate dehydrogenase (IDH)
mutations may suggest a peculiar molecular profile of DLGNT
(2).
In the differential diagnoses of PA, some additional
low- and high-grade glial tumours must be considered (2,3). In
particular, low-grade diffuse gliomas, such as IDH-mutant
astrocytoma and oligodendroglioma, are diffusely infiltrative
without Rosenthal fibres and eosinophilic granular bodies (2,3). On
the other hand, the distinction from high-grade gliomas can be
challenging only when PA presents high-grade histological features,
such as vascular proliferation and/or necrosis, but a solid growth
pattern, the presence of bipolar cells and Rosenthal fibres and low
mitotic activity can be helpful in these cases (2,3)
Molecular features
PAs are correlated with a wide range of molecular
alterations, mostly affecting the MAPK pathway (Fig. 5) (22). The most common alteration is a
rearrangement of the BRAF gene, resulting in the gene fusion
KIAA1549-BRAF (22,23). Other alterations of the MAPK pathway
include NF1 mutations, BRAFV600E mutations, other types of BRAF
fusions, KRAS mutations, FGFR1 mutations of fusions, NTRK family
receptor tyrosine kinase fusions and RAF1 gene fusions. Moreover, a
recent study showed that specific miRNAs, such as miR-155, miR-34a
and miR-503, target genes that are involved in the regulation of
the MAPK pathway in PA (24).
The KIAA1549-BRAF fusion is the most common
molecular alteration, especially in cerebellar PA, accounting for
60% of all cases (2,22,23).
The fusion is characterised by the tandem duplication at location
7q34, resulting in the replacement of the N-terminal end of various
KIAA1549 protein exons with the N-terminal regulatory region of
BRAF, while the retained BRAF kinase domain becomes unregulated and
constitutively active. The most common fusion is between exon 16 of
KIAA1549 and exon 9 of BRAF, followed by fusion of 15-9 exons and
16–11 exons, and then by other rare exon combinations (22). KIAA1549-BRAF fusion is recognised as
a diagnostic marker in paediatric PA, whereas it is rare in adults;
however, the presence of this alteration has been reported in some
cases of oligodendroglioma IDH-mutated, 1p19q codeleted (25,26).
Other BRAF fusions account for less than 5% (2). Several gene partners for BRAF fusions
have been found in small numbers of cases, including FAM131B,
RNF130, CLCN6, MKRN1, GNA11, QKI, FZR1, MACF1, GTF2I and recently
GNAI3, all resulting in the loss of the N-terminal region of BRAF
and the retention of the kinase domain (27–33).
NF1 somatic mutations or loss of a single wild-type
allele accounts for 10–15% of all cases and are generally found in
optic pathways tumours in patients with NF1 syndrome (2). Up to 33% of NF1 patients have a PA
with anaplastic features and an aggressive clinical course
(34).
BRAF V600E mutation accounts for 5–10% of all cases
and is mostly found in supratentorial tumours and other glial and
glioneuronal tumours, such as gangliogliomas and pleomorphic
xanthoastrocytoma (2). It consists
of the replacement of valine with glutamic acid at position 600 of
the BRAF gene. BRAF V600E is the third most common mutation in PAs
after KIAA1549-BRAF fusion, but the two alterations are rarely
present at the same time.
FGFR1 hotspot point mutations (p.N546K, p.K656E),
FGFR1-TACC1 fusion and FGFR1-internal tandem duplication
(FGFR1-ITD) account for less than 5% of all cases and are mainly
found in midline/brainstem tumours (2,35–37).
FGFR1 mutations are also reported in up to 60% of other glial
tumours, such as dysembryoplastic neuroepithelial tumours (38). The frequency of FGFR1 mutations
increases with the age of the patients and is associated with
higher prevalence in sporadic optic pathway PAs in adults (39). Extremely rare molecular alterations
were also identified, such as NTRK fusion with several different
types of partners resulting in a constitutive dimerization and
kinase activation (36),
RAF1-STAGP3 fusion and RAF1-NFIA fusion resulting in constitutive
RAF-1 kinase activity that leads to MEK 1/2 activation and
increased cancer cell proliferation (40,41),
KRAS mutations (42) and ROS1
fusions (43). Furthermore, PAs may
also exhibit H3 p.K27M mutation as an exception to the rule; by
contrast, diffuse midline gliomas behave aggressively when the H3
K27M mutation is documented (44,45).
Therefore, taking into consideration this latter unusual molecular
finding in PA, the histone H3 K27M mutation should not be
considered an exclusive criterion for the diagnosis of high-grade
gliomas.
On a molecular level, pilomyxoid astrocytomas
harbour MAPK pathway alterations similar to typical PA, in
particular, the presence of fusion gene KIAA1549-BRAF. Furthermore,
a recent study revealed additional alterations in retinoic
acid-mediated apoptosis and MAPK signalling pathways and key hub
genes that may potentially be involved in tumour growth and
progression, including BRAF, LUC7L2, MKRN1, RICTOR, TP53, HIPK2,
HNF4A, POU5F and SOX4 (2,46). A distinct methylation signature
called ‘DNA methylation class anaplastic astrocytoma with piloid
features’ is encountered in pilocytic astrocytoma with unusual
histological anaplastic changes (47). This latter variant is characterised
by hypercellularity, cytologic atypia with or without necrosis and
brisk mitotic activity (at least 5 mitoses per 10 high power
fields) in an otherwise well-circumscribed, non-infiltrative lesion
at the initial or recurrent diagnosis (2). Molecular features of these tumours are
NF1 mutations (33%), BRAF duplications (30%), loss of ALT (69%) and
ATRX expression (57%) and an alternative lengthening of telomeres
phenotype (69%), the latter associated with worse overall survival
(13 months) together with the presence of necrosis and anaplasia
(2,29).
Treatment and prognosis
The treatment recommendations were based on outcomes
studies relating to patient age, tumour location, surgical
treatment and eventually radiation therapy. The gold standard of
treatment is represented by surgical excision with complete margin
resection, achieving minimal neurological damage. In fact, the
complete resection has been considered the curative procedure for
PA. In cystic lesions, the resection of only the nodule is
recommended, but not the cyst wall (48); nevertheless, an increased thickness
of the cystic wall suggests en-bloc removal of the neoplastic
lesion (48,49). In detail, for subtentorial
localisation of PA, the lesion should be treated through a
telovelar suboccipital approach. At surgery, a clear cleavage plane
may be identified for a favourable complete removal. Frequently, a
serum-blood thin-walled cyst causing significant compression to the
cerebellar structures may be promptly revealed. If greyish richly
vascularised mural nodules were found, they should be removed. On
the other hand, in the occurrence of supratentorial PAs, commonly a
frontal transcortical transventricular surgical approach has been
chosen. Following a frontal craniotomy, using a transnuchal
corridor, the involved frontal horn of the lateral ventricle has
been reached, where a voluminous thalamic lesion characterised by a
thin-wall, transparent, vascularized, soft, pink cystic mass is to
be removed. Conventional radiotherapy is not required; the more
appropriate treatment seems to be serial follow-up, since
radiotherapy frequently may carry significant sequelae.
Nevertheless, if the tumour cannot be completely surgically removed
due to its location, adults and older children may be subjected to
radiation therapy to destroy any remaining neoplastic elements.
Finally, following surgery, chemotherapy should be applied in
younger children to avoid the risk of long-term growth and
developmental issues. If PA recurs, a new surgical approach should
be performed. It has been reported that stereotactic radiosurgery
may achieve good results for residual and recurrent PA (50).
PAs are associated with a favourable prognosis and
overall survival (90% in 10 years for paediatric patients, 70% in
adults) (7), depending on the
tumour location and resection, clinical manifestations and age of
the patient. KIAAA1549-BRAF fusion has been associated with
improved progression-free survival (PFS) and overall better
prognosis in paediatric PA (51,52).
Hypothalamic and optic pathway location has less favourable
progression-free and overall survival due to an often incomplete
surgical resection (53). In
addition, pilomyxoid astrocytomas, pilocytic astrocytomas with
histological features of anaplasia, and PA with leptomeningeal
dissemination show aggressive behaviour and poorer overall survival
(7,29).
Total resection is associated with better overall
survival (29,54), whereas adjuvant radiotherapy,
although associated with excellent progression-free survival (PFS)
and overall survival (OS) (71–90 and 92–100% respectively, in 5
years), shows long-term side effects, such as second malignancies,
suggesting that malignant transformation of PA occurs mainly in
tumours treated with prior irradiation (54). Standard treatment with chemotherapy
has been considered in the treatment of patients with low-grade
gliomas and showed a 5-year PFS between 35 and 45% (54,55).
These protocols comprehend the association of carboplatin with
vincristine, monotherapy with vinblastine or a combination of
thioguanine, procarbazine, lomustine and vincristine (54). Temozolomide has been reported not to
be effective (29).
Novel therapies involving agents targeting MAPK
signalling pathway dysregulation are currently in development. The
most studied target agents are BRAF and MEK inhibitors (54,55). A
phase II study with a MEK-inhibitor, selumetinib, in relapsed and
refractory low-grade gliomas in paediatric patients showed benefits
in patients with hypothalamic/optic pathway gliomas with a 24%
portal response and 56% of patients with prolonged stable disease
(56). Moreover, the treatment of
progressive paediatric low-grade glioma with trametinib has
demonstrated benefits, including 100% disease control in 18
paediatric patients (57). A
combination of MEK inhibitors and chemotherapy agents such as
vinblastine is currently under evaluation. Clinical trials with
early-generation BRAF inhibitors (Vemurafenib, Dabrafenib,
Encorafenib) are evaluating their effects on paediatric low-grade
gliomas compared to standard chemotherapy treatment. Dabrafenib
plus trametinib showed clinically meaningful activity in patients
with BRAFV600E-mutated recurrent or refractory high- and low-grade
gliomas (58). Moreover, the same
combination of BRAF inhibitors showed an excellent overall response
rate (ORR) (47%) compared to carboplatin plus vincristine (11%)
(59). Other studies comparing the
effect of target therapy vs. standard treatment, such as
selumetinib vs. carboplatin plus vincristine (NCT03871257) or
trametinib vs. carboplatin plus vincristine in the Phase III LOGGIC
trial, are currently ongoing, as well as the use of next-generation
dimer inhibitors, such as PLX8394 and TAK580, that can potentially
prevent RAF dimer formation and block MAPK activation (29). Moreover, mTOR inhibitors are also
under investigation for the treatment of LGGS as monotherapy and/or
in combination with other agents. In a phase II study, Everolimus
showed a 2-year PFS of 39 and OS 93% in patients with relapsed LGGs
(60). Additionally, Bevacizumab
showed beneficial responses in a series of small studies of adult
patients with unresectable PA (61,62).
Finally, immune checkpoint blockade using PD-L1 or CTLA4 inhibitors
may actually not be considered a potential therapeutic choice for
unresectable or recurrent PA since a low positive rate, as well as
a low percentage of positive neoplastic or immune cells, has been
documented (63). However, the
amount of infiltrating T cells in PA is variable, although the
contribution of CD4+ and CD8+ T cells remains unclear. In detail,
higher levels of T cell infiltration have been encountered in LGG
in comparison to their malignant counterparts, suggesting that CD8+
T cell content is related to improved patient survival (64). Moreover, the intriguing role of T
cells in LGG growth has been also revealed by sex-specific
differences in T-cell content in supratentorial vs. infratentorial
PA (65). The additional lack of
tumoral PD-1/PD-L1 immunoexpression strongly suggests the need for
further investigations to better define immune cell-directed
targets in PA (65).
Conclusions and future perspectives
The recent updating of the WHO Classification of CNS
tumours has focused on the need to integrate molecular information
into the neuropathological profile of brain tumours, including PA.
Since molecular findings appear complex, the attempts to utilise
specific individualised treatment for PA will need the results of
ongoing trials targeting the MAP kinase pathway. Future insights
including morphological, molecular and epigenetic characteristics
of PA are mandatory to approach a personalised treatment, also
taking into consideration the low rate of PA incidence in children
and adults, which requires a multi-institution team or the
institution of a specific registry to determine the optimal
targeted agents. In this direction, the application of different
methodologies, such as fluorescent in situ hybridisation,
polymerase chain reaction (PCR) and reverse transcription-PCR, may
be helpful to better define characteristic molecular alterations in
PA, driving the choice of an individual oncological treatment, even
in the event of recurrence. In particular, molecular targets are
continually expanding, including novel BRAF and RAF gene fusions
such as FAM131-BRAF, SRGAP3-RAF1, RNF130-BRAF, CLCN6-BRAF,
MKRN1-BRAF and GNAI1-BRAF, which may represent an important area
for continued investigation to better characterise their
therapeutic and prognostic significance.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CP, AI and GT contributed to conception, design and
the draft of the manuscript. CP, VF and AG were involved in
acquisition and interpretation of data. MM, AI and GT critically
reviewed/edited the manuscript. AG and GT confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethical approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
Local Bioethical Committee at the University Hospital ‘G. Martino’
of Messina (Messina, Italy) and with the 1964 Helsinki Declaration
and its later amendments or comparable ethical standards. The
present study was submitted to The Institutional Review Board of
the University Hospital of Messina (Messina, Italy) to discuss and
approve the study (prot. 47/19; May 2, 2019). Written anonymized
informed consent was obtained from all patients; for children, the
consent was signed by the parents/guardians.
Patient consent for publication
Written informed consent for the publication of
their data was obtained from all patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ius T, Cesselli D, Isola M, Pauletto G,
Tomasino B, D'Auria S, Bagatto D, Pegolo E, Beltrami AP, Loreto CD
and Skrap M: Incidental Low-Grade Gliomas: Single-Institution
management based on clinical, surgical, and molecular data.
Neurosurgery. 86:391–399. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Collins VP, Jones DTW and Giannini C:
Pilocytic astrocytoma: Pathology, molecular mechanisms and markers.
Acta Neuropathol. 129:775–788. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lodi M, Boccuto L, Carai A, Cacchione A,
Miele E, Colafati GS, Diomedi Camassei F, De Palma L, De Benedictis
A, Ferretti E, et al: Low-Grade gliomas in patients with noonan
syndrome: Case-Based review of the literature. Diagnostics (Basel).
10:5822020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gutmann DH, McLellan MD, Hussain I, Wallis
JW, Fulton LL, Fulton RS, Magrini V, Demeter R, Wylie T, Kandoth C,
et al: Somatic neurofibromatosis type 1 (NF1) inactivation
characterizes NF1-associated pilocytic astrocytoma. Genome Res.
23:431–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valera ET, McConechy MK, Gayden T, Rivera
B, Jones DTW, Wittmann A, Han H, Bareke E, Nikbakht H, Mikael L, et
al: Methylome analysis and whole-exome sequencing reveal that brain
tumors associated with encephalocraniocutaneous lipomatosis are
midline pilocytic astrocytomas. Acta Neuropathol. 136:657–660.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salles D, Laviola G, Malinverni ACM and
Stávale JN: Pilocytic Astrocytoma: A review of general, clinical,
and molecular characteristics. J Child Neurol. 35:852–858. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bell D, Chitnavis BP, Al-Sarraj S, Connor
S, Sharr MM and Gullan RW: Pilocytic astrocytoma of the
adult-clinical features, radiological features and management. Br J
Neurosurg. 18:613–616. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ryu HH, Jung TY, Lee GJ, Lee KH, Jung SH,
Jung S and Baek HJ: Differences in the clinical courses of
pediatric and adult pilocytic astrocytomas with progression: A
single-institution study. Child's Nerv Syst. 31:2063–2069. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Opancina V, Esposito S, Di Meco F, Bruno
E, Moscatelli M, Vetrano IG, Chiapparini L, Opancina M, Farinotti
M, Zdravkovic N, et al: Magnetic resonance imaging characteristics
of pediatric pilocytic astrocytoma. Neurol Sci. 44:4033–4040. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ajithkumar T, Taylor R and Kortmann RD:
Radiotherapy in the management of paediatric low-grade gliomas.
Clin Oncol (R Coll Radiol). 31:151–161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Santino SF, Salles D, Stávale JN and
Malinverni ACM: Pathophysiological evaluation of pilocytic
astrocytoma in adults: Histopathological and immunohistochemical
analysis. Pathol Res Pract. 248:1545932023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Griesinger AM, Birks DK, Donson AM, Amani
V, Hoffman LM, Waziri A, Wang M, Handler MH and Foreman NK:
Characterization of distinct immunophenotypes across pediatric
brain tumor types. J Immunol. 191:4880–4888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Annovazzi L, Mellai M, Bovio E, Mazzetti
S, Pollo B and Schiffer D: Microglia immunophenotyping in gliomas.
Oncol Lett. 15:998–1006. 2018.PubMed/NCBI
|
|
15
|
Komotar RJ, Mocco J, Carson BS, Sughrue
ME, Zacharia BE, Sisti AC, Canoll PD, Khandji AG, Tihan T, Burger
PC and Bruce JN: Pilomyxoid astrocytoma: A review. MedGenMed.
6:422004.PubMed/NCBI
|
|
16
|
Lee IH, Kim JH, Suh YL, Eo H, Shin HJ, Yoo
SY and Lee KS: Imaging characteristics of pilomyxoid astrocytomas
in comparison with pilocytic astrocytomas. Eur J Radiol.
79:311–316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alkonyi B, Nowak J, Gnekow AK, Pietsch T
and Warmuth-Metz M: Differential imaging characteristics and
dissemination potential of pilomyxoid astrocytomas versus pilocytic
astrocytomas. Neuroradiology. 57:625–638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chaulagain D, Smolanka V and Smolanka A:
Pilocytic astrocytoma: A literature review. Int Neurol J. 18:39–45.
2022.
|
|
19
|
She D, Liu J, Zeng Z, Xing Z and Cao D:
Diagnostic accuracy of diffusion weighted imaging for
differentiation of supratentorial pilocytic astrocytoma and
pleomorphic xanthoastrocytoma. Neuroradiology. 60:725–733. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shaikh N, Brahmbhatt N, Kruser TJ, Kam KL,
Appin CL, Wadhwani N, Chandler J, Kumthekar P and Lukas RV:
Pleomorphic xanthoastrocytoma: A brief review. CNS Oncol.
8:CNS392019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rahim S, Ud Din N, Abdul-Ghafar J,
Chundriger Q, Khan P and Ahmad Z: Clinicopathological features of
dysembryoplastic neuroepithelial tumor: A case series. J Med Case
Rep. 17:3272023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Salles D, Santino SF, Ribeiro DA,
Malinverni ACM and Stávale JN: The involvement of the MAPK pathway
in pilocytic astrocytomas. Pathol Res Pract. 232:1538212022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cler SJ, Skidmore A, Yahanda AT, Mackey K,
Rubin JB, Cluster A, Perkins S, Gauvain K, King AA, Limbrick DD, et
al: Genetic and histopathological associations with outcome in
pediatric pilocytic astrocytoma. J Neurosurg Pediatr. 29:504–512.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones TA, Jeyapalan JN, Forshew T,
Tatevossian RG, Lawson AR, Patel SN, Doctor GT, Mumin MA, Picker
SR, Phipps KP, et al: Molecular analysis of pediatric brain tumors
identifies microRNAs in pilocytic astrocytomas that target the MAPK
and NF-κB pathways. Acta Neuropathol Commun. 3:862015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rodriguez FJ, Schniederjan MJ, Nicolaides
T, Tihan T, Burger PC and Perry A: High rate of concurrent
BRAF-KIAA1549 gene fusion and 1p deletion in disseminated
oligodendroglioma-like leptomeningeal neoplasms (DOLN). Acta
Neuropathol. 129:609–610. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kumar A, Pathak P, Purkait S, Faruq M, Jha
P, Mallick S, Suri V, Sharma MC, Suri A and Sarkar C: Oncogenic
KIAA1549-BRAF fusion with activation of the MAPK/ERK pathway in
pediatric oligodendrogliomas. Cancer Genet. 208:91–95. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roth JJ, Santi M, Pollock AN, Harding BN,
Rorke-Adams LB, Tooke LS and Biegel JA: Chromosome Band 7q34
Deletions Resulting in KIAA 1549-BRAF and FAM 131 B-BRAF fusions in
pediatric low-grade gliomas. Brain Pathol. 25:182–192. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ryall S, Arnoldo A, Krishnatry R, Mistry
M, Khor K, Sheth J, Ling C, Leung S, Zapotocky M, Guerreiro
Stucklin A, et al: Multiplex detection of pediatric low-grade
glioma signature fusion transcripts and duplications using the
NanoString nCounter system. J Neuropathol Exp Neurol. 76:562–570.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gregory TA, Chumbley LB, Henson JW and
Theeler BJ: Adult pilocytic astrocytoma in the molecular era: A
comprehensive review. CNS Oncol. 10:CNS682021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin R, Kenyon A, Wang ZX, Cai J, Iacovitti
L and Kenyon LC: Pilocytic astrocytoma harboring a novel GNAI3-BRAF
fusion. Neuropathology. 43:391–395. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Laviv Y, Toledano H, Michowiz S,
Dratviman-Storobinsky O, Turm Y, Fichman-Horn S, Kagnovski E and
Goldenberg-Cohen N: BRAF, GNAQ, and GNA11 mutations and copy number
in pediatric low-grade glioma. FEBS Open Bio. 2:129–134. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zwaig M, Baguette A, Hu B, Johnston M,
Lakkis H, Nakada EM, Faury D, Juretic N, Ellezam B, Weil AG, et al:
Detection and genomic analysis of BRAF fusions in Juvenile
Pilocytic Astrocytoma through the combination and integration of
multi-omic data. BMC Cancer. 22:12972022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tomić TT, Olausson J, Wilzén A, Sabel M,
Truvé K, Sjögren H, Dósa S, Tisell M, Lannering B, Enlund F, et al:
A new GTF2I-BRAF fusion mediating MAPK pathway activation in
pilocytic astrocytoma. PLoS One. 12:e01756382017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rodriguez EF, Scheithauer BW, Giannini C,
Rynearson A, Cen L, Hoesley B, Gilmer-Flynn H, Sarkaria JN, Jenkins
S, Long J and Rodriguez FJ: PI3K/AKT pathway alterations are
associated with clinically aggressive and histologically anaplastic
subsets of pilocytic astrocytoma. Acta Neuropathol. 121:407–420.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Engelhardt S, Behling F, Beschorner R,
Eckert F, Kohlhof P, Tatagiba M, Tabatabai G, Schuhmann MU, Ebinger
M and Schittenhelm J: Frequent FGFR1 hotspot alterations in
driver-unknown low-grade glioma and mixed neuronal-glial tumors. J
Cancer Res Clin Oncol. 148:857–866. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jones DT, Hutter B, Jäger N, Korshunov A,
Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, et
al: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic
astrocytoma. Nat Genet. 45:927–932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Asuzu DT, Desai B, Maggio D, Mandell J,
Ray-Chaudhury A, Abdullaev Z, Aldape K, Heiss J and Buchholz AL:
FGFR1-TACC1 fusion associated with malignant transformation in a
primary spinal cord glioma: A case report. J Spine Surg. 7:434–438.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rivera B, Gayden T, Carrot-Zhang J, Nadaf
J, Boshari T, Faury D, Zeinieh M, Blanc R, Burk DL, Fahiminiya S,
et al: Germline and somatic FGFR1 abnormalities in dysembryoplastic
neuroepithelial tumors. Acta Neuropathol. 131:847–863. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Trisolini E, Wardighi DE, Giry M, Bernardi
P, Boldorini RL, Mokhtari K and Sanson M: Actionable FGFR1 and BRAF
mutations in adult circumscribed gliomas. J Neurooncol.
145:241–245. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jones DT, Kocialkowski S, Liu L, Pearson
DM, Ichimura K and Collins VP: Oncogenic RAF1 rearrangement and a
novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in
activating the MAPK pathway in pilocytic astrocytoma. Oncogene.
28:2119–2123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yde CW, Sehested A, Mateu-Regué À, Østrup
O, Scheie D, Nysom K, Nielsen FC and Rossing M: A new NFIA:RAF1
fusion activating the MAPK pathway in pilocytic astrocytoma. Cancer
Genet. 209:440–444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Janzarik WG, Kratz CP, Loges NT, Olbrich
H, Klein C, Schäfer T, Scheurlen W, Roggendorf W, Weiller C,
Niemeyer C, et al: Further Evidence for a Somatic KRAS Mutation in
a Pilocytic Astrocytoma. Neuropediatrics. 38:61–63. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Guerreiro Stucklin AS, Ryall S, Fukuoka K,
Zapotocky M, Lassaletta A, Li C, Bridge T, Kim B, Arnoldo A,
Kowalski PE, et al: Alterations in ALK/ROS1/NTRK/MET drive a group
of infantile hemispheric gliomas. Nat Commun. 10:43432019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Orillac C, Thomas C, Dastagirzada Y,
Hidalgo ET, Golfinos JG, Zagzag D, Wisoff JH, Karajannis MA and
Snuderl M: Pilocytic astrocytoma and glioneuronal tumor with
histone H3 K27M mutation. Acta Neuropathol Commun. 4:842016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Morita S, Nitta M, Muragaki Y, Komori T,
Masui K, Maruyama T, Ichimura K, Nakano Y, Sawada T, Koriyama S, et
al: Brainstem pilocytic astrocytoma with H3 K27M mutation: Case
report. J Neurosurg. 129:593–597. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
AlShail E, Alahmari AN, Dababo AAM,
Alsagob M, Al-Hindi H, Khalil H, Al Masseri Z, AlSalamah R,
Almohseny E, Alduhaish A, et al: A molecular study of pediatric
pilomyxoid and pilocytic astrocytomas: Genome-wide copy number
screening, retrospective analysis of clinicopathological features
and long-term clinical outcome. Front Oncol. 13:10342922023.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Reinhardt A, Stichel D, Schrimpf D, Sahm
F, Korshunov A, Reuss DE, Koelsche C, Huang K, Wefers AK, Hovestadt
V, et al: Anaplastic astrocytoma with piloid features, a novel
molecular class of IDH wildtype glioma with recurrent MAPK pathway,
CDKN2A/B and ATRX alterations. Acta Neuropathol. 136:273–291. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Konar SK, Shukla D, Nandeesh BN, Prabhuraj
AR and Devi BI: Surgical management and outcome of a bilateral
thalamic pilocytic astrocytoma: Case report and review of the
literature. Pediatr Neurosurg. 54:139–142. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Onorini N, Spennato P, Orlando V, Savoia
F, Calì C, Russo C, De Martino L, de Santi MS, Mirone G, Ruggiero
C, et al: The clinical and prognostic impact of the choice of
surgical approach to fourth ventricular tumors in a single-center,
single-surgeon cohort of 92 consecutive pediatric patients. Front
Oncol. 12:8217382022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sager O, Dincoglan F, Demiral S, Uysal B,
Gamsiz H, Gumustepe E, Ozcan F, Colak O, Gursoy AT, Dursun CU, et
al: Concise review of radiosurgery for contemporary management of
pilocytic astrocytomas in children and adults. World J Exp Med.
12:36–43. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Becker AP, Scapulatempo-Neto C, Carloni
AC, Paulino A, Sheren J, Aisner DL, Musselwhite E, Clara C, Machado
HR, Oliveira RS, et al: KIAA1549: BRAF Gene Fusion and FGFR1
Hotspot Mutations Are Prognostic Factors in Pilocytic Astrocytomas.
J Neuropathol Exp Neurol. 74:743–754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hawkins C, Walker E, Mohamed N, Zhang C,
Jacob K, Shirinian M, Alon N, Kahn D, Fried I, Scheinemann K, et
al: BRAF-KIAA1549 fusion predicts better clinical outcome in
pediatric low-grade astrocytoma. Clin Cancer Res. 17:4790–4798.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu H, Chen Y, Qin X, Jin Z, Jiang Y and
Wang Y: Epidemiology and survival of patients with optic pathway
gliomas: A population-based analysis. Front Oncol. 12:7898562022.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Manoharan N, Liu KX, Mueller S, Haas-Kogan
DA and Bandopadhayay P: Pediatric low-grade glioma: Targeted
therapeutics and clinical trials in the molecular era. Neoplasia.
36:1008572023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lassaletta A, Scheinemann K, Zelcer SM,
Hukin J, Wilson BA, Jabado N, Carret AS, Lafay-Cousin L, Larouche
V, Hawkins CE, et al: Phase II weekly vinblastine for
chemotherapy-naïve children with progressive low-grade glioma: A
canadian pediatric brain tumor consortium study. J Clin Oncol.
34:3537–3543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fangusaro J, Onar-Thomas A, Poussaint TY,
Wu S, Ligon AH, Lindeman N, Campagne O, Banerjee A, Gururangan S,
Kilburn LB, et al: A phase II trial of selumetinib in children with
recurrent optic pathway and hypothalamic low-grade glioma without
NF1: A Pediatric Brain Tumor Consortium study. Neuro Oncol.
23:1777–1788. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fangusaro J, Onar-Thomas A, Young
Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ,
Kilburn LB, Goldman S, et al: Selumetinib in paediatric patients
with BRAF-aberrant or neurofibromatosis type 1-associated
recurrent, refractory, or progressive low-grade glioma: A
multicentre, phase 2 trial. Lancet Oncol. 20:1011–1022. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wen PY, Stein A, van den Bent M, De Greve
J, Wick A, de Vos FYFL, von Bubnoff N, van Linde ME, Lai A, Prager
GW, et al: Dabrafenib plus trametinib in patients with
BRAFV600E-mutant low-grade and high-grade glioma (ROAR): A
multicentre, open-label, single-arm, phase 2, basket trial. Lancet
Oncol. 23:53–64. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bouffet E, Hansford J, Garré ML, Hara J,
Plant-Fox A, Aerts I, Locatelli F, Van der Lugt J, Papusha L, Sahm
F, et al: Primary analysis of a phase II trial of dabrafenib plus
trametinib (dab + tram) in BRAF V600-mutant pediatric low-grade
glioma (pLGG). J Clin Oncol. 40:LBA20022022. View Article : Google Scholar
|
|
60
|
Wright KD, Yao X, London WB, Kao PC, Gore
L, Hunger S, Geyer R, Cohen KJ, Allen JC, Katzenstein HM, et al: A
POETIC Phase II study of continuous oral everolimus in recurrent,
radiographically progressive pediatric low-grade glioma. Pediatr
Blood Cancer. 68:e287872021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Theeler BJ, Ellezam B, Yust-Katz S, Slopis
JM, Loghin ME and de Groot JF: Prolonged survival in adult
neurofibromatosis type I patients with recurrent high-grade gliomas
treated with bevacizumab. J Neurol. 261:1559–1564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wasilewski A and Mohile N: Durable
response to bevacizumab in adults with recurrent pilocytic
astrocytoma. CNS Oncol. 7:CNS262018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kaur A, Doberstein T, Amberker RR, Garje
R, Field EH and Singh N: Immune-related adverse events in cancer
patients treated with immune checkpoint inhibitors. Medicine
(Baltimore). 98:e173482019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Guo X, Pan Y, Xiong M, Sanapala S,
Anastasaki C, Cobb O, Dahiya S and Gutmann DH: Midkine activation
of CD8+ T cells establishes a neuron-immune-cancer axis responsible
for low-grade glioma growth. Nat Commun. 11:21772020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen J, Sinha N, Cobb O, Liu C, Ersen A,
Phillips JJ, Tihan T, Gutmann DH and Dahiya S: Immune cell analysis
of pilocytic astrocytomas reveals sexually dimorphic brain
region-specific differences in T-cell content. Neurooncol Adv.
3:vdab0682021.PubMed/NCBI
|