Introduction
Cervical cancer is the fourth most frequently
diagnosed cancer and the fourth leading cause of cancer-related
mortality in women worldwide (1).
Based on the Global Cancer Statistics resource, there were ~604,000
new cases of cervical cancer and 342,000 deaths resulting from
cervical cancer worldwide in 2020, and the mortality rate was
considerably higher in developing countries compared with developed
countries (1). The main pathogenic
factor of cervical cancer is high-risk human papillomavirus
(HPV)-persistent infection, which has reportedly led to >90% of
cervical cancer cases (2). With the
application of early screening and diagnostic methods, a larger
number of patients can receive a favorable prognosis; however, for
advanced, metastatic and recurrent cervical cancer, the prognosis
is not optimistic. Based on clinical findings, the 12-month
survival rate of patients with advanced cervical cancer has never
exceeded 30%, and the response rate is <15% (3). In recent years, immunotherapy and
tumor-targeting therapies have yielded promising results for the
treatment of solid tumors. However, unlike other solid tumors,
there are few targeted drugs available at present for the treatment
of cervical cancer, and the treatment options for advanced cervical
cancer remains very limited (4,5).
Therefore, the identification of novel therapeutic targets for the
treatment of cervical cancer is urgently needed.
As a central component of the tumor microenvironment
(TME) in solid tumors, the pro-tumorigenic functions of
cancer-associated fibroblasts (CAFs) have been assessed in breast
cancer, colorectal cancer and numerous other cancer types (6). CAFs are typically associated with a
worse prognosis and are currently being studied as a potential
therapeutic target (7). However,
CAFs also exhibit a certain degree of versatility, which may
present certain difficulties in terms of translating research
findings into clinical practice. The current classic classification
includes myofibroblastic CAFs, which are associated with smooth
muscle contraction and collagen precipitation, immune regulatory
CAFs, which are associated with immune cell recruitment and
suppression, and antigen presenting CAFs, which could recruit T
cells, and the composition of CAFs is different in different tumors
(8). In cervical cancer,
fibroblasts have been reported to be more sensitive to HPV-infected
keratinocytes and to secrete fibroblast growth factors (FGFs) such
as FGF-2 and FGF-7, through the paracrine system, which in turn
promotes angiogenesis and tumor proliferation (9). Therefore, an improved understanding of
the biology of CAFs may lead to prognostic and therapeutic
benefits.
Based on the potential of single-cell
(sc)RNA-sequencing (seq) to decipher cellular heterogeneity, the
present study aimed to assess the intra-tumor heterogeneity of CAFs
in cervical cancer using this method to further examine CAF
function. Through the validation of the findings using multiple
databases and clinical data, the preliminary results in the present
study may be useful as a reference point for further research.
Materials and methods
Participants and sample
collection
Clinical data and tissue specimens were collected
from patients with cervical cancer treated at the Women's Hospital,
School of Medicine, Zhejiang University (Hangzhou, China) between
June 2020 and December 2021. The inclusion criteria were as
follows: i) Adult female patients with a sexual history; ii)
diagnosis of squamous cervical cancer; iii) no neoadjuvant therapy
before surgery; and iv) Federation of Gynecology and Obstetrics
(FIGO) 2018 stage ≥IB1 (10). The
exclusion criteria were as follows: i) Patients not diagnosed with
primary squamous cervical cancer; ii) no lesions visible to the
naked eye; and iii) any treatments, such as radiotherapy and
chemotherapy, received before surgery. Patients were considered to
have cervical cancer according to preoperative histological and/or
imaging examinations, such as ultrasound, magnetic resonance
imaging or computed tomography.
Tumor samples of ~0.5×0.5×0.5 cm were collected from
within the visible tumor area during surgery. Para-tumorous
tissues, which were defined as normal tissues according to the
naked eye and located 2 cm away from the cancer margin, were also
collected if available. The size of the sample collected was the
same as that for the tumor tissues. After the sampling was
completed, blood and debris on the surface of the tissue were
cleaned with normal saline that had been pre-cooled at 4°C. Liquid
on the surface of the tissue was then absorbed by dust-free paper
and the tissue samples were transferred to a frozen tube filled
with the pre-cooled tissue preservation solution (MACS®
Tissue Storage Solution; cat. no. 130-100-008; Miltenyi Biotec,
Inc.). The tube cover was tightened, sealed with sealing film, and
then stored and transported at 2–8°C. The fresh tissues collected
were processed within 24 h to prepare for the following steps.
Before the final diagnosis was made according to the postoperative
pathology and if the patient met the inclusion and exclusion
criteria, the sequencing data would be used for subsequent
analysis.
In total, five tumor and three para-tumorous tissues
were obtained (median patient age, 63 years; range, 48–67 years).
The cancer clinical stage was defined according to the
aforementioned FIGO 2018 staging system, and stages I, II and III
were all included in the present study. As the samples were
collected at different times, batch effects were not introduced
when analyzing the scRNA-seq data.
The protocol of the present study was approved by
the Institutional Research Ethics Committee of the Women's
Hospital, School of Medicine, Zhejiang University (approval no.
IRB-20200006-R). All patients provided written informed consent.
The clinical characteristics of the patients and samples, including
age, histological type and presence of metastasis, are detailed in
Table SI.
Cell suspensions from tumor tissues
and cell quality control
Fresh cervical cancer tissue samples were placed in
a sterile RNase-free culture dish containing 3–5 ml calcium-free
and magnesium-free 1X PBS on ice. The tissue samples were
transferred to new culture dishes and cut into 0.5 mm2
pieces. The tissues were subsequently washed with 1X PBS, and as
much irrelevant tissue as possible, including blood stains and
fatty layers, was removed. The tissues were then dissociated into
single cells using dissociation solution (0.35% collagenase I–V5, 2
mg/ml papain, 120 U/ml DNase I) in a water bath at 37°C with
shaking for 20 min (100 rev/min). The digestion was terminated by
the addition of 1X PBS containing 10% fetal bovine serum (v/v), and
the solution was then resuspended 5–10 times using a Pasteur
pipette. The resulting cell suspension was filtered with a 70–30 µm
stacked cell strainer and then centrifuged at 300 × g for 5 min at
4°C (11). The cell pellet was
subsequently resuspended in 100 µl 1X PBS containing 0.04% BSA, to
which 1 ml 1X red blood cell lysis buffer [Red Blood Cell Lysis
Solution (10X); cat. no. 130-094-183; Miltenyi Biotec, Inc.] was
added. The cell suspension was then incubated either at room
temperature or on ice for 2–10 min to lyse the remaining red blood
cells. Following this incubation, the cell suspension was
centrifuged at 300 × g for 5 min at room temperature. The cells
were subsequently resuspended in 100 µl Dead Cell Removal
MicroBeads, and dead cells were removed using the Dead Cell Removal
Kit (cat. no. 130-090-101; Miltenyi Biotec, Inc.). The cells were
then resuspended in 1X PBS containing 0.04% BSA, centrifuged at 300
× g for 3 min at 4°C (this process was repeated twice), before 50
µl 1X PBS containing 0.04% BSA was again added to resuspend the
cell pellet. The overall cell viability was determined by trypan
blue exclusion staining, and cell viability >85% was required
for subsequent experiments. The single-cell suspensions were
counted using a Countess® II Automated Cell Counter
(Invitrogen™; Themo Fisher Scientific, Inc.), and the
cell concentration was adjusted to 700–1,200 cells/µl.
10X Genomics library preparation and
scRNA-seq
Gel beads containing the barcode information
(16-base sequence) were bound to a mixture of the cells to sequence
and enzymes (including reverse transcriptase), and then
encapsulated in droplets of oil surfactant located in a
microfluidic system to form the gel beads in emulsion (GEMs). The
GEMs were subsequently collected in to a reservoir in the equipment
called the chromium controller, and then reverse transcription was
performed in a PCR instrument (Bio-Rad Laboratories, Inc.) to
generate cDNA. In this process, the gel beads were lysed during
heating and released the barcode sequences that could label the
sample cells. At the same time, there were also a large amount of
primer sequences in gel beads. The protocol for this step was 53°C
for 45 min, 85°C for 5 min and a 4°C hold. The GEMs were then
collected, and the cDNA was used as a template for PCR
amplification. Subsequently, the GEMs were disrupted and broken up
into oil droplets. The cDNA was then enzymatically digested into
fragments of 200–300 bp, and the DNA library was finally amplified
by PCR following the library construction process that comprised
traditional second-generation sequencing, including sequencing
connectors and primers. The reagents used in this process were as
follows: i) Chromium Next GEM Single Cell 3′ GEM, Library & Gel
Bead Kit v3.1, 16 rxns PN-1000121 (10X Genomics); and ii) Chromium
Next GEM Chip G Single Cell Kit, 48 rxns PN-1000120 (10X Genomics).
Subsequently, an Agilent 2100 bioanalyzer (Agilent Technologies,
Inc.) was used for cDNA library quality control. When the peak
shape was normally distributed, between 500 and 8,000 bp, and the
main peak was >1,300 bp, follow-up experiments could be
conducted. The cDNA library was quantitatively determined by qPCR
using VAHTS Library Quantification Kit for Illumina (Vazyme NQ101,
NQ105) to the concentration of 150 pM. Finally, libraries were
sequenced using a NovaSeq 6000 Sequencing System at a minimum depth
of 20,000 reads per cell (12)
(paired-end multiplexing run; 150 bop; Illumina, Inc.) by using
NovaSeq 6000 S4 Reagent Kit v1.5 (300 cycles) (cat. no. 20028312).
These experiments which including cDNA library construction and
single-cell sequencing were supported by LC-Bio Technologies
Hangzhou Co., Ltd., and single-cell sequencing related reagents and
instruments were from 10X Genomics.
Sequencing data quality control and
quantification
Cell Ranger (4.0.0) is the official analysis
software of 10X Genomics (https://support.10×genomics.com/single-cell-gene
expression/software/overview/welcome) (13), which is capable of reading raw
downstream sequencing data directly and can output the statistical
results of the sequencing data for each sample. As the present
study involved multiple samples, integration and homogenization of
the multi sample data was required prior to further analysis, and
to obtain uniform UMI abundance information for all the genes in
all cells. After running the whole process, Cell Ranger used
algorithms to calculate the number of cells obtained during the
process based on the barcode and UMI information, and the
expression level of genes was thereby detected in each cell,
providing data that were used for further downstream analyses.
Single cell data filtering and cluster
analysis
The Cell Ranger output was loaded into the Seurat
(version 3.1.1) (http://seurat.r-forge.r-project.org) program for
dimensional reduction, clustering and analysis of the scRNA-seq
data. The quality control threshold parameters were set as follows:
i) Genes that were expressed in <300 cells were removed; ii) the
low cut-off was >500 genes expressed per cell, whereas <5,000
genes expressed per cell was the high cut-off; iii) the number of
UMI counts was ≥500; iv) the proportion of mitochondrial
DNA-derived genes expressed was <25%; and v) the potential
doublets that occurred in the encapsulation step and/or as
occasional pairs of cells were removed using the Doublet Finder
package (version 2.0.2) (https://rdrr.io/github/chris-mcginnis-ucsf/DoubletFinder/src/R/doubletFinder.R).
After the cell filtering process was completed, the
Seurat software was used to normalize the data and to find highly
variable genes using the built-in parameters ‘Normalize Data’ and
‘Find Variable Features’. Seurat was then used to perform a
downstream principal component (PC) analysis (PCA) on these highly
variable genes calculated by ‘Find Variable Features’ to derive the
corresponding PCA values, and subsequently the data were analyzed
according to these values for dimensionality reduction, clustering
and subgrouping. The first 20 PCs of the PCA were then used for the
subsequent clustering and subgrouping analysis.
To visualize the data, the dimensionality of all
33,090 cells from five tumor samples was further reduced using
Seurat, and the non-linear dimensional reduction technique,
t-distributed Stochastic Neighbor Embedding, was used to project
the cells into two-dimensional (2D) space. The principle was to
drop the expression matrix data of the cells onto a 2D plane by
means of dimensionality reduction, and to differentiate the data
according to similarity to obtain the statistical results of the
cell clustering.
Finding marker genes based on the
clustering of subgroups
To find clusters, the weighted Shared Nearest
Neighbor graph-based clustering method was selected. Marker genes
for each cluster were identified using the ‘Find All Markers’
function in Seurat (14) and the
Wilcoxon rank sum test was performed (default parameter: ‘bimod’
Likelihood-ratio test). This function allowed the pre-processed
data to be limited to a certain range, thus eliminating undesirable
effects caused by exceptionally large or small datasets. The
following were parameters selected for the genes: i) Genes
expressed in >10% of the cells in a cluster; ii) adjusted
P<0.01; and iii) an average log2 fold change (FC) of
>0.26, which implied a fold change of >1.3. Due to the large
amount of single-cell data, this difference was already very
notable. Typically, logFC >0.25 is acceptable in single-cell
analysis; however, the criteria were made more stringent in the
present study to increase the reliability (15–17).
Subsequently, cell clusters in the resulting 2D representation were
annotated to known biological cell types using canonical marker
genes that have been reported in previously published studies
(18–20). Finally, marker genes of the clusters
were revealed by heatmap analysis, and heatmaps were generated
using the pheatmap function (1.0.12) from the R package (R Core
Team). Furthermore, when comparing differential genes of two groups
of cells, the basic principles of statistics was similar with
finding marker genes of several clusters and the function in Seurat
was called ‘Find Marker’.
Pathway and functional annotation
analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathway functional enrichment analyses were
performed using the Cluster Profiler R package (version 4.0.5),
through which enriched biological processes, molecular functions,
cellular components and pathways of identified hub genes in the
cluster of interest were identified (9). P<0.05 was considered to indicate a
statistically significant enrichment (21). The results of the enrichment
analysis are shown as scatterplots, and the ggplot2:Rich factor was
used to indicate the number of differential genes/total number of
genes, with the greater the Rich factor, the higher the degree of
enrichment.
The Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb)
(genesets c2.cp.kegg.v7.0.symbols and h.all.v7.0.symbols) was used
in conjunction with the fgsea R package (version 1.18.0) for gene
set enrichment analysis (GSEA) (22,23) to
assess the different KEGG pathways and hallmark gene sets between
the high- and low-risk CAF groups. Pathways with adjusted P<0.05
were regarded as being statistically significantly enriched
(18). The results of the GSEA were
visualized using the enrichplot R package (version 1.12.3).
Cell-cell communication analysis
Cell communication analysis was performed after
1,000 permutation tests based on gene expression levels of the
major cell types, and the cell-cell interactions among them were
visualized by heatmaps using CellPhoneDB (version 2.1.2; www.cellphonedb.org), a publicly available repository
of curated receptors, ligands and their interactions. Cell-cell
interactions within identical cellular lineages were excluded, and
receptor-ligand pairs between the relevant cell types were
identified. A combined P<0.05 was required, and the
ligand/receptor needed to have been expressed in >10% of cells
(24).
Database bulk transcriptome RNA-seq
data collection and processing
The bulk transcriptome RNA-seq data and
corresponding clinical data were obtained from The Cancer Genome
Atlas (TCGA) (https://portal.gdc.cancer.gov) cervical cancer (CESC;
309 samples), head and neck squamous cancer (HNSC; 546 samples),
bladder cancer (BLCA; 430 samples), colon adenocarcinoma (COAD; 512
samples) and prostate cancer (PRAD; 551 samples) databases using
the UCSC Xena browser (GDC hub; http://gdc.xenahubs.net) (25). Additionally, the transcriptomic data
of 300 cervical cancer samples in GSE44001 were obtained as the
external validation cohorts via the Gene Expression Omnibus
database (https://www.ncbi.nlm.nih.gov/geo/) (26).
Molecular prognostic signature
construction
Focusing on the hub genes of extracellular marked
CAFs and differentially expressed genes (DEGs) between CAFs and
fibroblasts in para-tumors, a prognostic signature was constructed
for patients with cervical cancer. Overall survival (OS) rate was
identified as the primary outcome. First, univariate Cox regression
analysis was performed to identify prognostic ecCAF-associated
genes in TCGA-CESC dataset. P<0.05 was considered to indicate a
statistically significant result. To minimize the overfitting risk,
the least absolute shrinkage and selection operator (LASSO) Cox
regression model was then applied using the glmnet 4.1 4 R package
(27), and the CAF signature was
calculated according to the following formula: CAF risk score=Σ(βi
× Expi), where βi represents the LASSO coefficient of the gene and
Expi is the expression value of the candidate gene (28). Patients in TCGA-CESC database were
classified into high- and low-risk groups according to the median
CAF risk score, and their association with OS was evaluated using
Kaplan-Meier (KM) plot analysis (29). Heatmap analysis was used to
visualize the association between CAF risk scores and candidate
genes. Similarly, the CAF signature was tested in other external
validation cohorts, including the aforementioned cervical cancer
cohort GSE44001 and other HPV-associated cancer datasets, including
TCGA-HNSC, TCGA-BLCA, TCGA-COAD and TCGA-PRAD datasets (21).
TME infiltration estimation
The Estimation of Stromal and Immune cells in
Malignant Tumors using Expression data (ESTIMATE) algorithm in R
package (version 1.0.13) was used to calculate the immune scores
and tumor purity of the patients. In addition, the Wilcoxon
rank-sum test was used to compare immune scores and tumor purity
between the CAF high- and low-risk score groups (30).
The infiltration levels of different immune cells
between the CAF high- and low-risk score groups were calculated
using the following two methods: i) The combination of Cell type
Identification By Estimating Relative Subsets Of RNA Transcripts
(CIBERSORT) via ‘CIBERSORT.R’ and LM22 (leukocyte signature matrix)
was used to evaluate the proportions of the 22 human leukocyte cell
subsets; and ii) single-sample (ss)GSEA analysis was applied via
the GSVA R package (version 1.42.0) (31) to assess the proportions of 28 types
of infiltrating immune cells of the tumor samples (32). Subsequently, the Wilcoxon rank-sum
test was used to compare the differences between the two groups,
and the ggplot R package was used for visualization of the
results.
Immunotherapeutic responses and
chemotherapeutic sensitivity predictions
The immunotherapeutic responses of patients in
TCGA-CESC database and other cancer databases were estimated using
the online algorithm, Tumor Immune Dysfunction and Exclusion (TIDE;
http://tide.dfci.harvard.edu/) (33). Furthermore, patient chemotherapeutic
sensitivities were predicted using the oncoPredict R package
(version 0.2) by building a ridge regression model (34). Several common anticancer drugs for
cervical cancer (cisplatin, docetaxel, 5-fluorouracil, paclitaxel
and cyclophosphamide) were tested, and the training genetic
profiles were obtained from the largest publicly accessible
pharmacogenomics database, Genomics of Drug Sensitivity in Cancer
(GDSC; http://www.cancerrxgene.org/)
(21,35). GDSC contains two datasets, the newer
dataset, GDSC2, was used in the present study.
Nomogram construction and
validation
To identify which clinical characteristics were
associated with the prognosis of patients with cervical cancer,
univariate and multivariate Cox proportional hazards regression
analyses were performed using SPSS 26.0 (IBM Corp.). After passing
the Schoenfeld residual plot of proportional hazards test, the
significant variables were tested using the proportional hazards
assumption (36). Nomograms were
then constructed based on the independent prognostic factors in the
Cox regression to predict the 1-, 3- and 5-year OS rates of
patients with cervical cancer (37). Every variable in the nomogram was
expressed as a line, and the length of the lines reflected the
weight of the variables in the model; the value of the variable was
equal to a point on the line. After adding the points of all
variables, the survival rates at different time nodes were obtained
(21).
In addition, the concordance index (C-index) was
calculated to evaluate the level of discrimination of the model,
and calibration curves were plotted to assess the consistency
between the predicted 1-, 3- and 5-year OS rate probabilities and
the observed rates, the accuracies of which were derived from the
nomogram (bootstrap-based 1,000 iterations, with resampling of
validations) (38).
Human Protein Atlas (HPA)
database
Immunohistochemical results were obtained from the
HPA online database (https://www.proteinatlas.org/) (39), to compare the expression levels of
the signature genes at the protein level in normal cervix and
cervical cancer tissues.
Statistical analysis
All statistical analyses and visualizations were
performed using the R v4.1.1 (https://www.r-project.org/) and SPSS 26.0 software
packages. KM survival curves and the log-rank test were used to
compare the OS rates between high- and low-risk groups, and these
analyses were performed using survival and survminer R packages
(40). Furthermore, the survival
results of only one gene were obtained from the Gene Expression
Profiling Interactive Analysis database (http://gepia.cancer-pku.cn). The LASSO-Cox regression
model was used to construct the extracellular (ec)CAF signature.
Time-dependent receiver operating characteristic (ROC) curves were
used to evaluate the predictive performance of the model. Wilcoxon
and unpaired Student's t-tests were used to compare differences
between two groups, depending on whether the data were normally
distributed. χ2 tests were used to compare constituent
ratios between two groups. Finally, Pearson's correlation
coefficient was used to assess correlations between two variates.
P<0.05 was considered to indicate a statistically significant
difference.
Results
scRNA-seq analysis and CAF subgroup
identification
A flow diagram depicting the overall steps of the
present study is presented in Fig.
1. A total of five fresh tumor tissues and three available
para-tumorous tissues were collected from patients with cervical
cancer, and scRNA-seq was subsequently performed with all samples.
Following quality control assessment and the removal of batch
effects, a total of 33,090 single cells from tumor samples were
sorted into 20 major clusters (Figs.
2A and S1A). Heatmap analysis
was then conducted to reveal the top 10 hub genes of the 20
clusters (Fig. 2B), and a bar-plot
was constructed to demonstrate the sample source of the clusters
(Fig. 2C). Subsequently, the cell
types were annotated by finding canonical markers in
cluster-specific genes (Fig. 2D):
Epithelial cellular adhesion molecule was used for identifying
epithelial cells (Fig. 2E); protein
tyrosine phosphatase, receptor type C was used for identifying
immune cells (Fig. 2F); claudin-5
was used for endothelial cells (Fig.
2G); and collagen, type I, α1 was used for CAFs. Two clusters,
namely cluster 10 and cluster 16 (C10 and C16, respectively), were
recognized as CAFs (Fig. 2H).
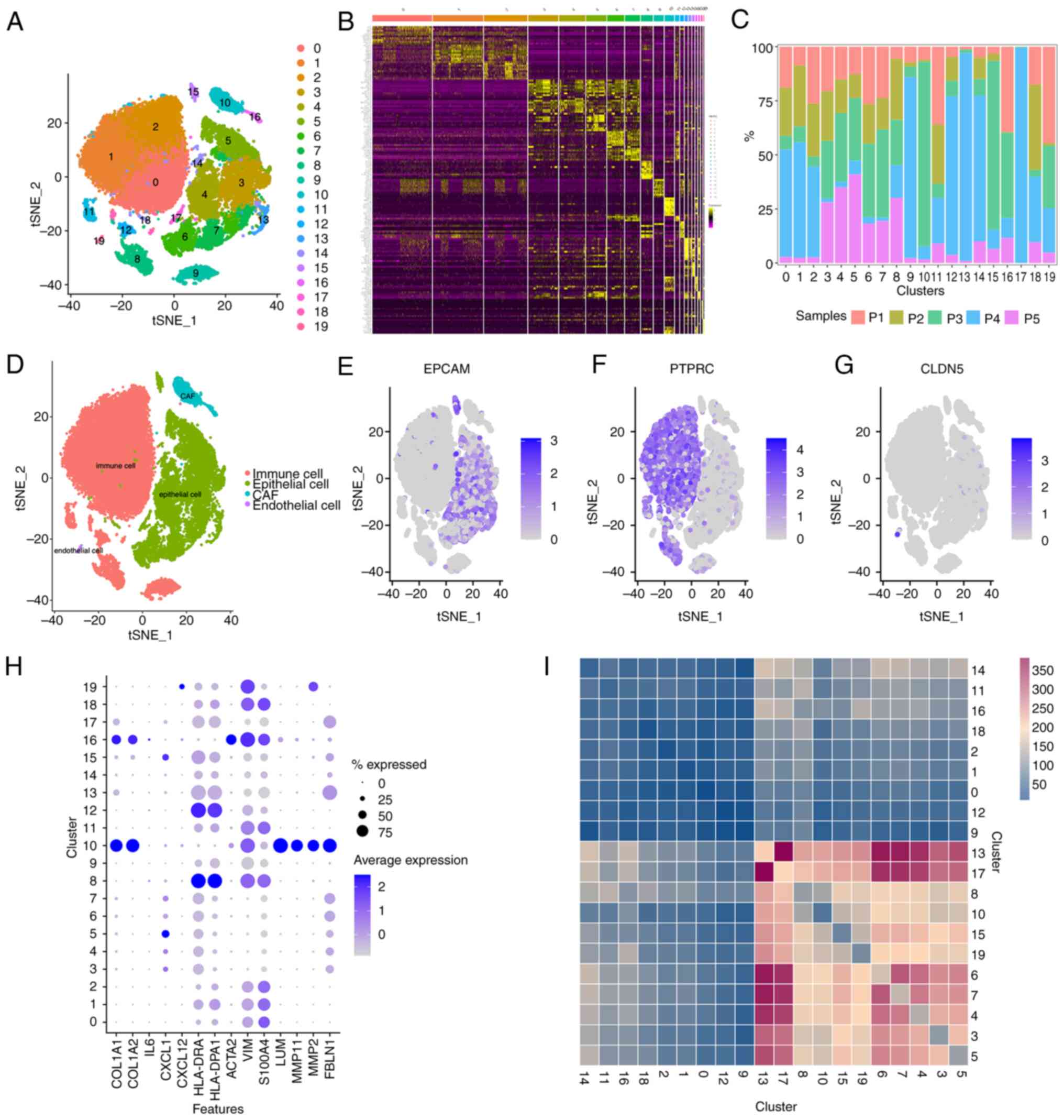 | Figure 2.Single-cell RNA sequencing from
33,090 cells from 5 cervical cancer tissues and cell type
identification. (A) A total of 20 separate clusters according to
tSNE. (B) Heatmap of the top 10 marker genes in each cluster. (C)
Sample source of the 20 clusters. (D) Canonical markers identified
cell types in the TME: (E) Epithelial cells (EPCAM), (F) immune
cells (PTPRC) and (G) endothelial cells (CLDN5). (H) The expression
of CAF markers in all 20 clusters. (I) Cell-cell communication
analysis among all clusters in cervical cancer microenvironment.
tSNE, t-distributed Stochastic Neighbor Embedding; TME, tumor
microenvironment; EPCAM, epithelial cellular adhesion molecule;
PTPRC, protein tyrosine phosphatase, receptor type C; CLDN5,
claudin-5; CAF, cancer-associated fibroblast; COL1A1, collagen,
type I, α1; COL1A1, collagen, type I, α2; IL6, interleukin-6; CXCL,
chemokine (C-X-C motif) ligand; HLA-DRA, major histocompatibility
complex, class II, DRα; HLA-DPA1, major histocompatibility complex,
class II, DPα1; VIM, vimentin; S100A4, S100 calcium binding protein
A4; LUM, lumican; MMP11, matrix metalloproteinase 11; MMP2, matrix
metallopeptidase 2; FBLN1, fibulin 1. |
Subsequently, the heterogeneity of CAFs in a
cervical cancer environment was evaluated by assessing the gene
expression profile of the two clusters. One difference identified
was in the expression of α-smooth muscle actin (α-SMA; also known
as ACTA2), a classical marker of myofibroblastic (my)CAFs (41). ACTA2 was expressed at notable levels
in C16, whereas the expression level in C10 was comparatively low
(Fig. 2E). With the exception of
ACTA2, C16 also exhibited notably higher expression levels of other
intracellular markers, including vimentin and ferroptosis
suppressor protein 1 (also known as S100A4) compared with C10;
whereas C10 expressed extracellular markers, including lumican
(LUM), matrix metalloproteinase (MMP)11, MMP2 and fibulin 1 (FBLN1)
(Fig. 2H). Previous studies have
suggested that MMPs remodel the extracellular matrix (ECM) via the
proteolysis of collagens, fibrin, fibronectin, laminins and
vitronectin, and through promotion of the invasion of tumor cells
in an HPV-infected microenvironment (42,43).
For these reasons, the C10 CAF subgroup was classified as ecCAFs in
the present study. In addition, certain genes that are
characteristic of CAF subpopulations that have been reported in
other types of tumor were also assessed, including immune
regulatory CAFs and antigen-presenting CAFs (8). However, these types of CAFs did not
appear to exist in the cervical cancer microenvironment, as both of
the clusters did not express classical marker,s such as IL6, CXCL1,
CXCL12, HLA-DRA and HLA-DPA1 (Fig.
2H).
Subsequently, cell-cell communication analysis was
performed to assess the interactions among different cell types in
the cervical cancer microenvironment (Fig. 2I). The results demonstrated that
ecCAFs had a notably higher number of interactions with other cell
types compared with myCAFs (C16), as a greater number of
ligand-receptor pairings were detected. The majority of clusters
that made connections with ecCAFs were cancer cells (clusters 3, 4,
5, 6, 7, 13, 14, 15 and 17; cancer cells originated from epithelial
cells that highly express EPCAM), followed by monocytes (cluster 8,
which was associated with a high expression level of CD14). This
suggested that, when compared with myCAFs, ecCAFs may more notably
contribute towards promoting cancer progression.
ecCAFs demonstrate more robust
pro-tumorigenic effects compared with myCAFs
As myCAFs are the major subtype of activated
fibroblasts in the TME, they are thought to have a pro-tumorigenic
role (8). myCAFs were therefore
used as the reference cells in the present study, to assess the
function of ecCAFs in the cervical cancer microenvironment. First,
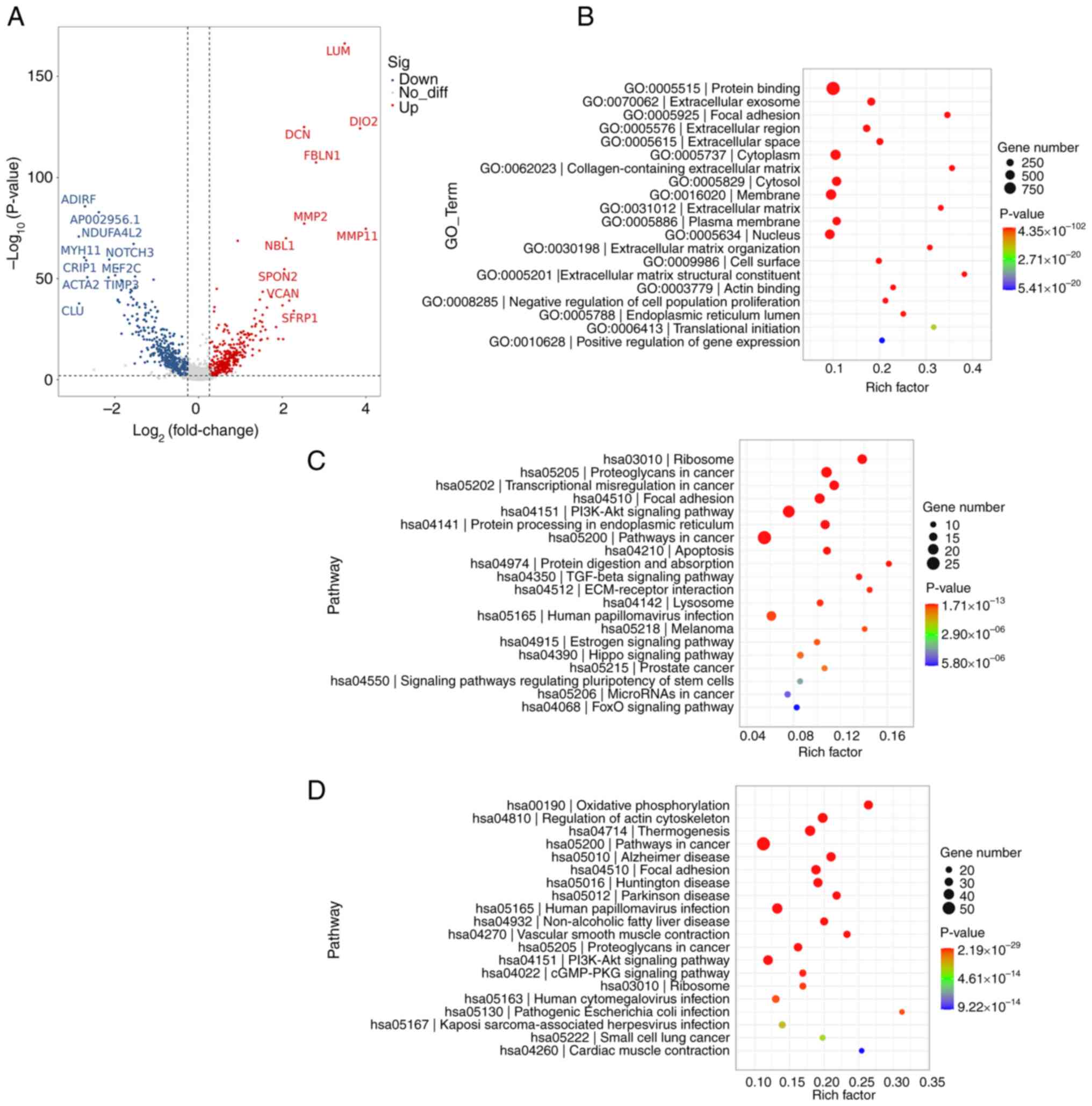
the DEGs were compared between the two subgroups. Volcano plots
were constructed to demonstrate the most significantly up and
downregulated genes in ecCAFs compared with myCAFs (P<0.01;
Fig. 3A). In addition to LUM and
MMP11, numerous other genes that fulfill a crucial role in the ECM
were found to be upregulated, including decorin (44), FBLN1 (45), MMP2, neuroblastoma 1 and spondin 2
(46), whereas genes that serve
regulatory roles in certain intercellular signaling pathways and
mitochondrial functions, such as NADH dehydrogenase 1 α subcomplex,
4 like 2 (47) and neurogenic locus
notch homolog protein 3 (48), were
downregulated. Moreover, genes that control myogenesis such as
myosin heavy chain 11 (49) and
myocyte-specific enhancer factor 2C (50), were also demonstrated to be
upregulated in myCAFs and downregulated in ecCAFs.
GO and KEGG enrichment analyses were then performed
for the DEGs identified between ecCAFs and myCAFs. In the GO
enrichment analysis, the DEGs were found to be closely associated
with ECM organization (Fig. 3B).
KEGG enrichment analysis revealed that, even though the two types
of CAFs were both associated with cancer, ecCAFs were more abundant
in cancer-linked signaling pathways and cancer-associated
components (P<0.05; Fig. 3C),
such as ‘PI3K-Akt signaling pathway’, ‘TGF-beta signaling pathway’,
‘Pathways in cancer’, ‘Proteoglycans in cancer’, ‘Transcriptional
misregulation in cancer’ and ‘MicroRNAs in cancer’. These findings
corroborated that the novel extracellular marker CAFs, namely
ecCAFs, had a more significant role in cervical cancer progression
compared with the classic intracellular marker CAFs (or myCAFs).
Moreover, the ECM may be an important location for ecCAFs in terms
of their support of tumor progression, as focal adhesion and
ECM-receptor interactions were also found to be significantly
enriched (Fig. 3C and D). Notably,
the two subgroups of CAFs were also demonstrated to be
significantly associated with HPV infection.
Construction of a prognostic signature
based on the hub genes of ecCAFs
Given the cancer-promoting functions of ecCAFs,
ecCAFs were subsequently used to predict the prognosis of patients
with cervical cancer. TCGA cervical cancer cohort, which contained
296 samples, was used to construct the model, and the OS rate was
chosen as the primary outcome. Both the hub genes of ecCAFs and the
differential gene profiles of CAFs in cervical cancer tissues and
fibroblasts in para-tumorous tissues on the single-cell level were
taken into consideration. Finally, 51 intersecting genes, which
were hub genes only in ecCAFs when compared with myCAFs, and
fibroblasts in paratumor were obtained. Univariate Cox proportional
risk models were then used to analyze the intersecting genes, and
16 genes were identified (P<0.05; Fig. 4A). The LASSO Cox regression
algorithm was then performed on these genes, and the
λmin values (namely, the value of λ that gave the
minimum mean cross-validated error) was determined as the optimal λ
value by tenfold cross validations (Fig. 4B). A total of 12 prognostic genes
with non-zero coefficients were identified. A 12-gene ecCAF
signature was subsequently constructed based on the expression
level of each gene, and its coefficient was as follows: Risk
score=[0.228614961999191 × heparan sulfate proteoglycan 2 (HSPG2)]
+ [0.280769639745156 × leptin receptor (LEPR) overlapping
transcript (OT)] + (0.164309473932873 × LEPR)-[(0.101889575739853 ×
LIM domain only 4 (LMO4)]-[(0.0924809661366454 × vascular cell
adhesion molecule 1 (VCAM1)]-[(0.120237842213106 × regulator of G
protein signaling 5 (RGS5)] + [(0.0176198675612701 × prostaglandin
endoperoxide synthase 2 (PTGS2)] + [(0.398174149944109 ×
transmembrane protein 9 (TMEM9)] + [(0.0569947847625145 × collagen,
type IV, α1 (COL4A1)]-[(0.11168153683065 × NMYC
downstream-regulated gene 2 (NDRG2)] + [(0.0959648255524675 × Egl 9
family hypoxia inducible factor 3 (EGLN3)] + [(0.0896858331705779 ×
SEC23 homolog A, COPII coat complex component (SEC23A)]. Among the
prognostic genes, eight genes, HSPG2, LEPROT, LEPR, PTGS2, TMEM9,
COL4A1, EGLN3 and SEC23A, were regarded as risk-associated genes
(HR>1), whereas the other four genes were considered as
protective genes (HR <1). Fig.
4C shows the change in survival status (dead/alive) of patients
in the TCGA-CESC cohort with the increase of risk score, and the
patients are divided into high risk and low risk groups by median.
Based on the aforementioned risk formula, the ecCAF risk score of
each patient was calculated and visualized by heatmap analysis
(Fig. 4D). Patients in TCGA-CESC
cohort were subsequently divided into low and high ecCAF risk
groups according to their median risk scores. The Wilcoxon rank-sum
test demonstrated that the ecCAF risk score in the group of dead
patients was significantly higher compared with the group of live
patients (P=3×10−8; Fig.
4E). The pairwise comparison of OS in the different risk groups
was then assessed using the log-rank test. KM curves revealed that
the high ecCAF risk group had a significantly more unfavorable
survival outcome compared with the low ecCAF risk group
(P<0.001; Fig. 4F). ROC curves
were subsequently used to evaluate the sensitivity and specificity
of the OS model. The overall area under the ROC curve (AUC) value
was found to be 0.724 (Fig. 4G),
and for the 1-, 3- and 5-year survival prognoses, the constructed
risk model predicted AUC values of 79.92, 79.26 and 81.68,
respectively (Fig. 4H).
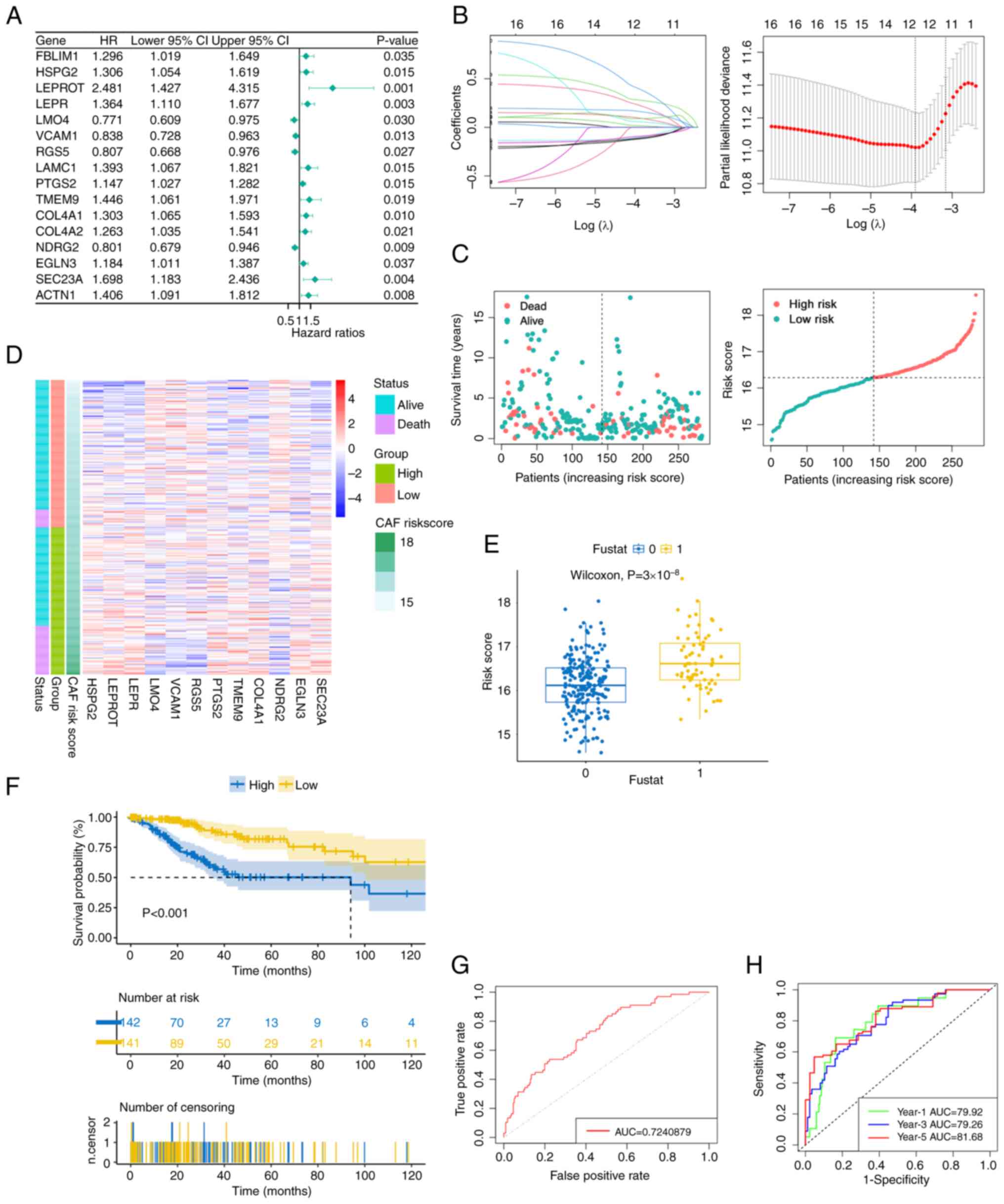 | Figure 4.A 12-gene signature correlated with
overall survival in The Cancer Genome Atlas-CESC based on ecCAFs,
constructed using LASSO Cox regression analysis. (A) Forest plot of
the results from the univariate Cox regression. (B) LASSO Cox
regression model constructed, with λmin as the optimal
λ. (C) CAF risk score calculated in CESC according to the LASSO Cox
regression model, with samples grouped by median CAF risk score.
(D) Heatmap visualizing the expression levels of model genes with
the CAF risk scores. (E) CAF risk scores in dead patient group
(1) and alive patient group (0).
(F) Overall survival in high- and low-risk group by Kaplan-Meier
curves. (G) Receiver operating characteristic curve for the overall
AUC value. (H) Receiver operating characteristic curve for the 1-,
3- and 5-year AUC values. LASSO, least absolute shrinkage and
selection operator; CESC, cervical cancer; CAF, cancer-associated
fibroblast; fustat, fundamental state; ecCAF, extracellular CAF;
AUC, area under the curve; HR, hazard ratio; CI, confidence
interval. |
Subsequently, the reliability of the ecCAF signature
was further assessed. When building this signature, the OS rate was
defined as the primary outcome; however, when changing the survival
outcome to other parameters, including to disease-specific
survival, disease free interval and progression free interval, the
model also demonstrated robustness in terms of its forecast
performance when dividing the patients according to the median
ecCAF risk score (P<0.01; Fig.
5A-C). Cervical cancer information is limited in the public
datasets selected, and therefore the GSE44001 dataset, which
contained information regarding cancer recurrence, was selected as
the validation dataset. The signature was also able to predict the
prognosis of patients when the cut-off point was set at the upper
quartile of the ecCAF risk score (P=0.012; Fig. 5D). Furthermore, the survival results
of every model gene were obtained from the Gene Expression
Profiling Interactive Analysis database. The survival results
revealed that 7/12 genes (COL4A1, EGLN3, LEPR, LEPROT, SEC23A,
TMEM9 and VCAM1) had prognostic functions that were consistent with
the risk model results of the present study when the cut-off point
was set at the median value (Fig.
S2). Patients with higher VCAM1 expression levels had an
improved prognosis, whereas a higher expression level of other
genes indicated a worse prognosis for survival. Taken together,
these results supported the prediction for the OS rate of patients
with CESC according to the constructed model.
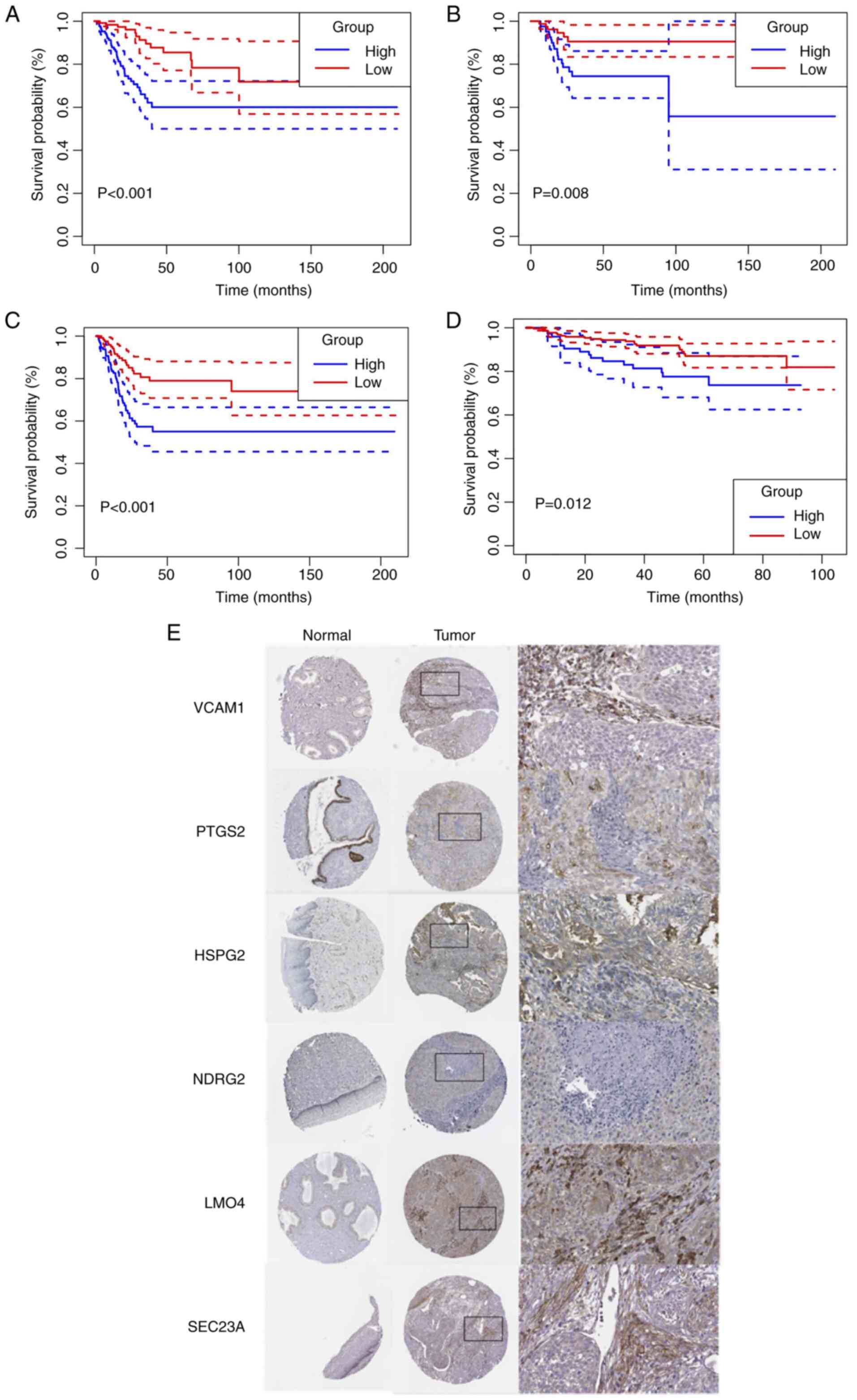 | Figure 5.Verification the prognostic effect of
the singnature based on TCGA-CESC and GSE4401. (A) Disease-specific
survival, (B) DFI and (C) progression-free interval in the high-
and low-risk groups by Kaplan-Meier curves, with data from The
Cancer Genome Atlas-cervical cancer cohort. (D) DFI in the high-
and low-risk groups by Kaplan-Meier curves, with data from
GSE44001. (E) Immunohistochemical results demonstrating the
expression of six model genes in normal tissues and cervical
cancer. Images are from the Human Protein Atlas database. DFI,
disease-free interval. VCAM1, vascular cell adhesion molecule 1;
PTGS2, prostaglandin-endoperoxide synthase 2; HSPG2, heparan
sulfate proteoglycan 2; NDRG2, NDRG family member 2; LMO4, LIM
domain only 4; SEC23A, SEC23 homolog A, COPII coat complex
component. |
Furthermore, to evaluate the protein expression
levels of the model genes in normal and tumor fibroblasts,
immunohistochemical results were obtained from the HPA database. In
total, 6/12 genes (SEC23A, NDRG2, PTGS2, VCAM1, LMO4 and HSPG2)
were found to have immunohistochemical results, and SEC23A, VCAM1,
LMO4 and HSPG2 were found to be expressed at a notably higher level
in cervical cancer stroma compared with normal cervical stroma
(Fig. 5E). These findings were
consistent with the results of single cell sequencing showing that
the expression of these genes was upregulated in ecCAFs compared
with that in fibroblasts in para-tumor tissues. By contrast, there
was no notable difference in the protein expression levels of NDRG2
and PTGS2 between the tumor and normal groups in HPA.
Higher ecCAF risk indicates an immune
exclusive environment and does not respond well to immunotherapy or
chemotherapy
The influence of the ecCAF risk score on the TME was
evaluated by ESTIMATE (51). In
TCGA-CESC cohort, patients with a higher ecCAF risk score exhibited
significantly higher tumor purity (Wilcoxon rank-sum test, P=0.003;
Fig. 6B) and lower immune cell
infiltration (Wilcoxon rank-sum test, P<0.001; Fig. 6A), compared with patients with a
lower risk score. Subsequently, CIBERSORT and the ssGSEA algorithms
were used to assess the relationship between the ecCAF risk score
and TME constituents at the bulk RNA-seq level. The results
demonstrated that a high ecCAF risk score was significantly
associated with a higher level of macrophages 0, which represents
the initial state of the macrophage, compared with a low-risk
score, whereas there was a deficit of a majority of immune
contents, including several subtypes of B cells, CD4 T cells, CD8 T
cells and dendritic cells, all of which usually have antitumor
functions in the tumor microenviroment (52–54)
(Fig. 6C-E). In conclusion, a
higher ecCAF risk score may indicate a microenvironment that is
more exclusive to immune cells.
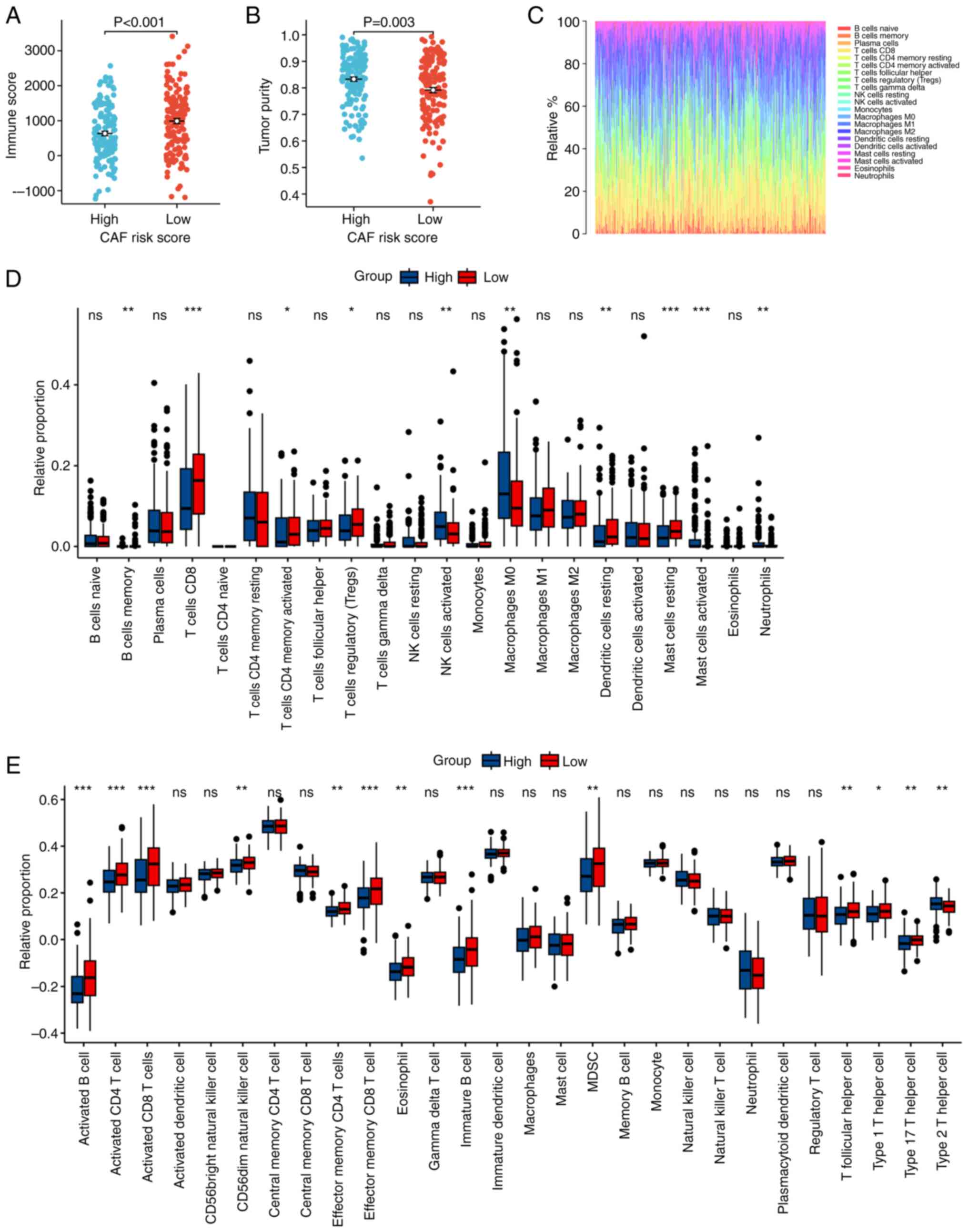 | Figure 6.Influence of CAF risk score on the
TME. A higher CAF risk score was significantly associated with a
(A) lower immune score and (B) higher tumor purity, as determined
by Estimation of Stromal and Immune cells in Malignant Tumors using
Expression. (C) Visualization of the CIBERSORT results of
TCGA-CESE. The influence of CAF risk score on TME, determined by
(D) CIBERSORT and (E) ssGSEA. *P<0.05, **P<0.01,
***P<0.001. CAF, cancer-associated fibroblast; TME, tumor
microenvironment; CIBERSORT, Cell type Identification By Estimating
Relative Subsets Of RNA Transcripts; ssGSEA, single-sample gene set
enrichment analysis; NK, natural killer; MDSC, myeloid-derived
suppressor cell; ns, not significant. |
Since it was determined that the ecCAF risk score
may influence the TME outlook, particularly in terms of immune
cells, the practicability of the model in predicting the response
to immunotherapy in cervical cancer was evaluated. According to the
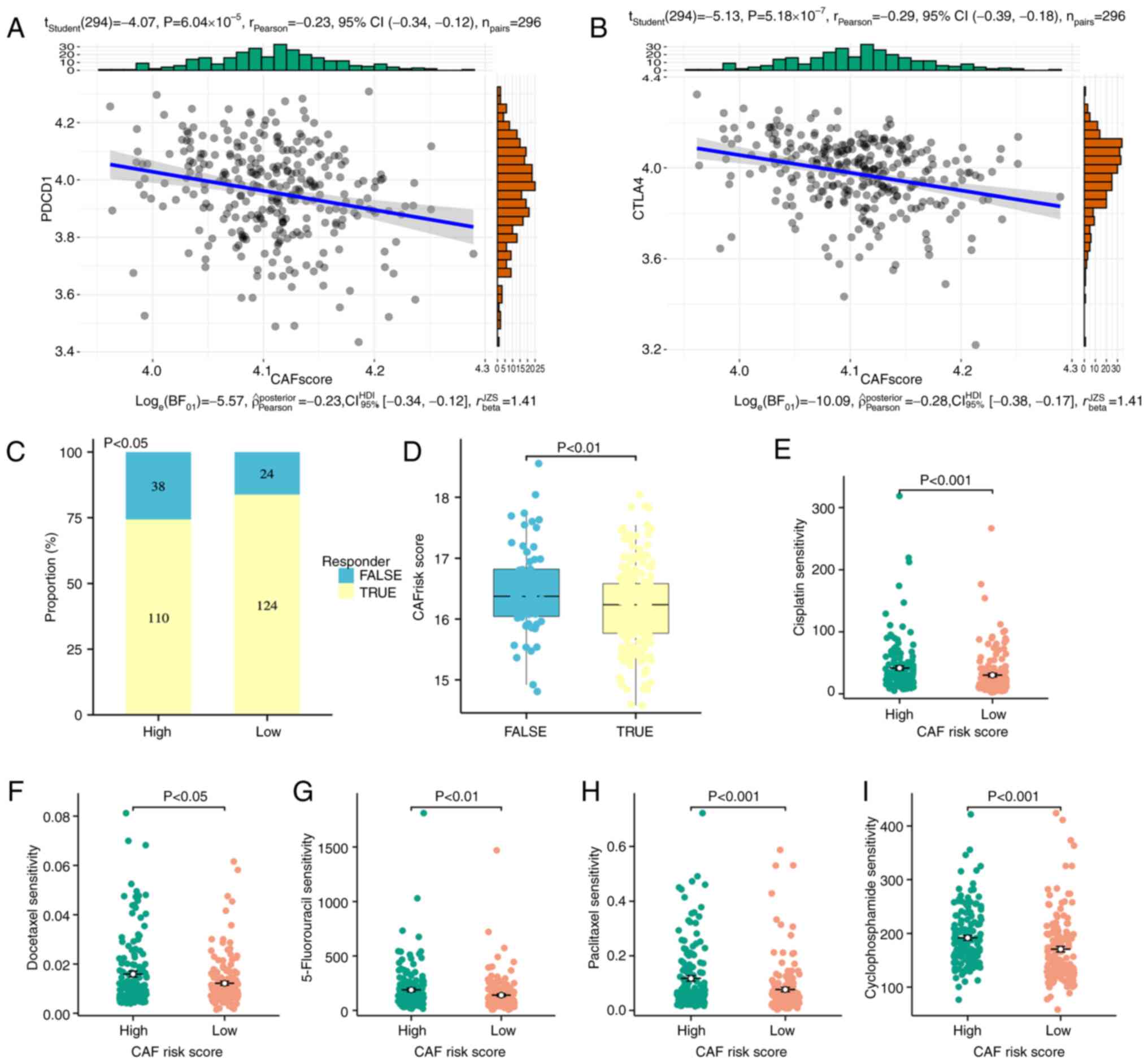
Pearson's correlation coefficient analysis, the ecCAF signature
score was significantly but weakly negatively correlated with the
expression levels of programmed cell death protein 1 (P<0.001;
r=−0.23; Fig. 7A) and cytotoxic
T-lymphocyte associated protein 4 (P<0.001; r=−0.29; Fig. 7B). The probability of samples in the
CESC cohorts yielding a response to immune-checkpoint inhibitors
was predicted using the TIDE online algorithm. Patients in the low
ecCAF risk group (148/296; non-responders, n=24) were significantly
more reactive to immunotherapy compared with patients in the
high-ecCAF risk group (148/296; non-responders, n=38)
(χ2 test, P<0.05; Fig.
7C). In addition, the non-responders had a significantly higher
ecCAF risk score compared with responders (Student's t-test,
P<0.01; Fig. 7D). These results
demonstrated that the constructed model could be used to provide an
indication as to whether a patient may be an immunotherapy
responder.
Subsequently, the ability of the constructed
signature to predict response to chemotherapy was evaluated. The
oncoPredict algorithm (36) was
used to estimate the sensitivity of five frequently used drugs in
cervical cancer therapy (cisplatin, docetaxel, 5-fluorouracil,
paclitaxel and cyclophosphamide), and a training cohort was
obtained from the GDSC2 dataset (a resource for therapeutic
biomarker discovery in cancer cells). Of all five drugs assessed,
docetaxel and paclitaxel were significantly associated with
improved therapeutic effects compared with the other drugs, as
their sensitivity scores were relatively small. In addition, the
samples with a higher ecCAF risk score had significantly lower
sensitivity than those with a lower risk score (Wilcoxon rank-sum
test, all P<0.05; Fig.
7E-I).
Clinical applications of the ecCAF
signature
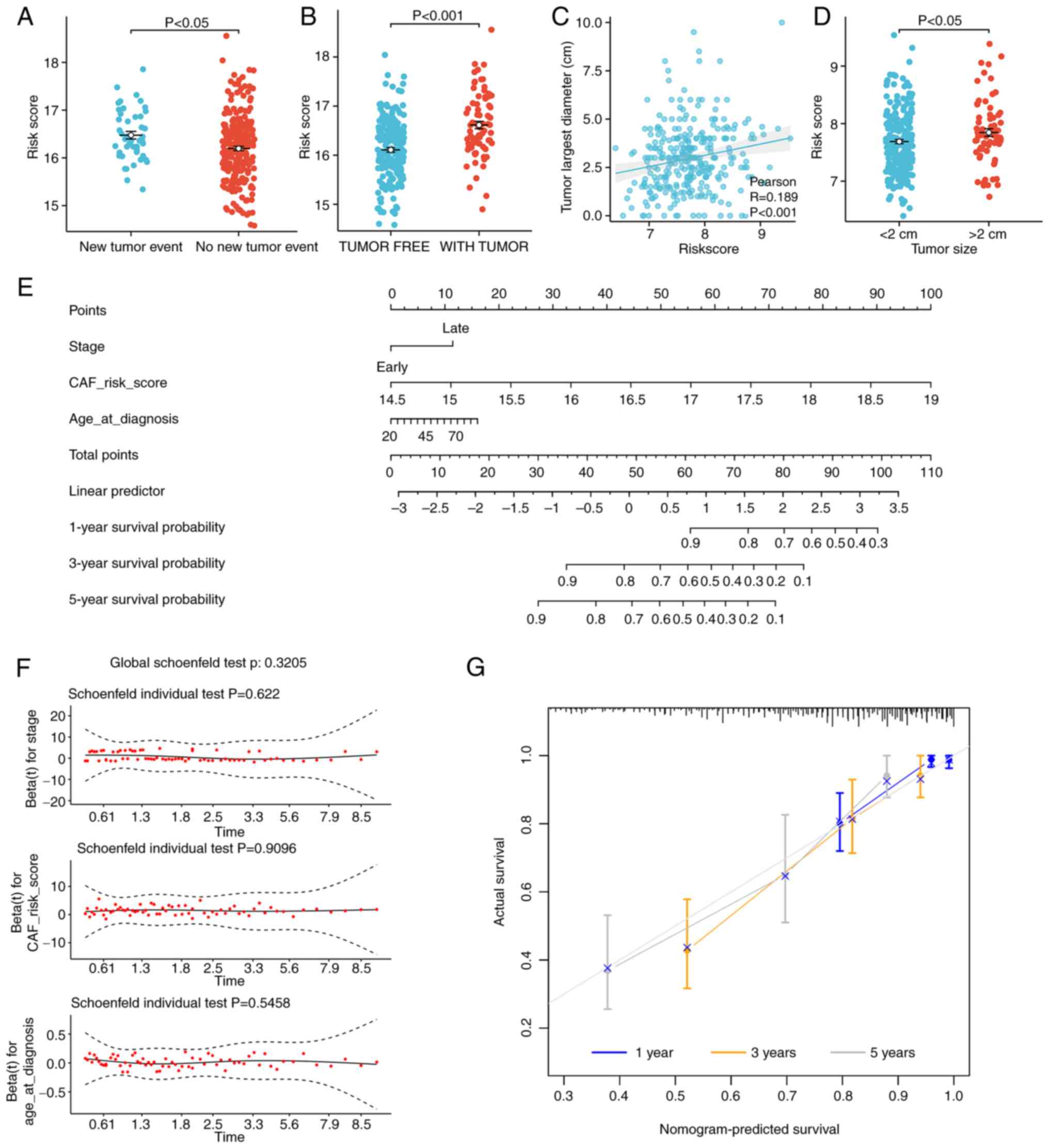
The association between the risk score and the
clinicopathological characteristics of patients was assessed to
expand its applicability value. First, the risk scores of the 5
patients whose tissue samples were collected for single cell
sequencing were calculated using the constructed signature, and it
was demonstrated that 2 patients with stage III cancer had higher
risk scores than the other 3 patients (Table SI). Based on TCGA-CESC dataset,
higher risk scores were significantly associated with both a
greater number of new tumor events (distant metastasis,
locoregional recurrence and new primary tumors; Student's t-test,
P<0.05; Fig. 8A) and the tumors
were more likely to be incompletely removed by surgery (Student's
t-test, P<0.001; Fig. 8B).
According to the GSE44001 dataset, patients with higher risk scores
tended to have significantly larger tumors (Pearson's correlation
coefficient, P<0.001; r=0.189; Fig.
8C; and Wilcoxon rank-sum test, P<0.05; Fig. 8D).
Subsequently, based on TCGA data,
clinicopathological characteristics were considered in the
construction of a nomogram. A univariate Cox regression model was
established to screen possible prognostic factors. A multivariate
Cox model was then established to identify independent risk factors
affecting the OS rate of patients with cervical cancer. The
univariate and multivariate Cox regression results are presented in
Table I. It was demonstrated that
age (<65 vs. ≥65 years), FIGO stage [early (I–II) vs. late
(III–IV)] and risk score (upper vs. lower 50%) were independent
risk factors. Based on the Schoenfeld residual plot of proportional
hazards test, the P-values of the three variables were all >0.05
(Fig. 8F), which implied that they
met the prerequisites of the proportional hazard assumption. This
indicated that these variables could be used to establish a
nomogram, and the resultant model is presented in Fig. 8E. The nomogram detailed the impact
of each factor on CESC and demonstrated that the ecCAF risk score
was the greatest influencing factor with respect to the age and
stage of cancer of the patient. The C-index was found to be 0.795
(95% confidence interval, 0.743–0.846), which implied that the
nomogram had a good discrimination level. The calibration curves of
the model demonstrated that the predictive values of the nomogram
were highly consistent with the observed values (Fig. 8G), which indicated that the nomogram
had a good level of accuracy.
 | Table I.Univariate and multivariate Cox
proportional hazards regression analysis of the association of
clinicopathologic features and overall survival. |
Table I.
Univariate and multivariate Cox
proportional hazards regression analysis of the association of
clinicopathologic features and overall survival.
|
| Cox proportional
hazards regression analysis |
|---|
|
|
|
|---|
|
| Univariate | Multivariate |
|---|
|
|
|
|
|---|
| Characteristic | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Age (≥65 vs. <65
years) | 2.049
(1.153–3.641) | 0.014 | 1.920
(1.081–3.413) | 0.026 |
| Stage (late vs.
early) | 2.364
(1.426–3.919) | 0.001 | 2.287
(1.376–3.801) | 0.001 |
| CAF risk score
(upper vs. lower 50%) | 3.732
(2.157–6.428) | 0.000 | 3.676
(2.131–6.342) | <0.001 |
| Ethnicity |
|
|
|
|
|
White | Reference | - | - | 0.826 |
|
Black | 1.127
(0.532–2.387) | 0.755 | - | 0.547 |
|
Asian | 0.683
(0.165–2.827) | 0.599 | - | 0.488 |
|
Other | 1.222
(0.615–2.429) | 0.567 | - | 0.739 |
| Histological
type |
|
|
|
|
|
Squamous carcinoma | Reference | - | - | 0.686 |
|
Adenocarcinoma | 0.949
(0.483–1.865) | 0.880 | - | 0.635 |
|
Adenosquamous carcinoma | 1.988
(0.271–14.565) | 0.499 | - | 0.481 |
ecCAF signature is closely associated
with tumor-linked pathways
Since the designed ecCAF signature was correlated
with adverse prognosis and refractory therapeutic responses, the
functional pathways in this model were subsequently assessed using
GSEA. Patients in the TCGA-CESC cohort were separated into high-
and low-risk groups based on their median ecCAF risk scores. DEGs
between the high and low ecCAF score groups were mainly enriched in
cancer- and metabolism-associated pathways, including xenobiotic
metabolism by cytochrome, oxidative phosphorylation, the TGF-β
signaling pathway, and extracellular matrix- and immune-related
pathways (P.adjusted <0.05; Fig.
9A; Table SII). Notably, the
majority of the signaling pathways were enriched in the high ecCAF
score group, whereas the immune-associated pathways were more
enriched in the low ecCAF score group. The results obtained using
hallmark gene sets were found to be similar; cancer-associated gene
sets, including epithelial-mesenchymal transition, hypoxia and TNFA
signaling via NFKB, were also significantly enriched in the high
ecCAF risk group (P.adjusted<0.05; Fig. 9B).
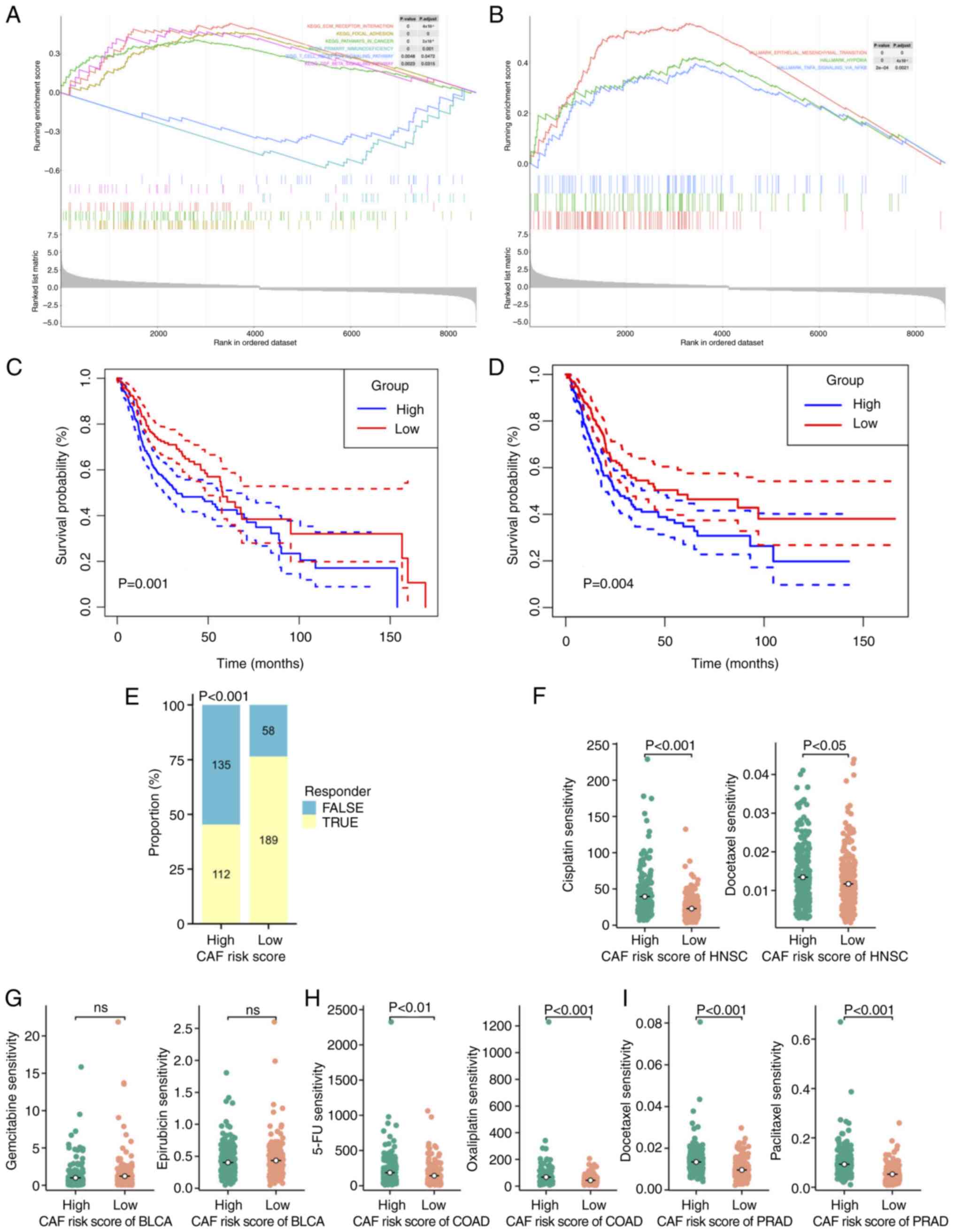
Role of the ecCAF-based signature in
other HPV-associated tumors
In aforementioned results (Fig. 3C and D), KEGG enrichment analysis
demonstrated that the HPV infection pathway was enriched in ecCAFs.
In addition to cervical cancer, HPV infection has been proposed to
contribute to the emergence of a series of different cancer types
(55), including HNSC (56), bladder urothelial carcinoma
(57), COAD (58) and prostate adenocarcinoma (59). Therefore, a pan-cancer analysis was
performed to assess whether the ecCAF-based signature was effective
in predicting the prognosis of other HPV-associated cancers. As
previously, the OS rate was chosen as the primary outcome. The
survival analysis demonstrated that the high ecCAF risk group had a
significantly worse prognosis in HNSC (P=0.001) (Fig. 9C) and BLCA (P=0.004) (Fig. 9D), when the observation period was
170 months, whereas no significant differences were identified in
terms of COAD or PRAD (Fig.
S1B).
The predictive effect of the model on immunotherapy
was also evaluated in terms of the four HPV-associated cancers
(namely HNSC, COAD, BLCA and PRAD). As demonstrated in Fig. 9E, there were significantly more
non-responders in the HNSC high ecCAF risk group compared with the
low-risk group, which implied unfavorable effects resulting from
therapy (P<0.05); however, for BLCA, COAD and PRAD, there were
no differences between the two different risk groups (Fig. S1C).
In terms of chemotherapy, several different
chemotherapeutic drugs that are frequently used to treat the four
types of HPV-associated cancers were selected for analysis
(56,60–62),
and significant differences were identified between the low and
high ecCAF risk score groups in the HNSC, COAD and PRAD cohorts
(Wilcoxon rank-sum test, all P<0.05; Fig. 9F-I). Specifically, patients in the
high-risk group had a worse chemotherapeutic response compared with
the low-risk group, which indicated that the model had potential in
terms of its wider applicability. However, the difference in the
BLCA cohort was not found to be significant.
Discussion
Cervical cancer is the most common gynecological
cancer worldwide and it remains a major health problem among women
(63). As reported by the World
Health Organization (WHO), 341,831 women died from cervical cancer
in 2020, accounting for 7.7% of the total number of cancer-related
deaths in women worldwide (1).
Currently, first-line treatments after initial diagnosis of the
disease are typically surgery or a combination of chemotherapy and
radiation, depending on the stage of cancer and other
clinicopathological risk factors of the patient (64). However, as one of the vital
components in cervical cancer therapy, chemotherapy rarely proves
to be curative; moreover, it is associated with a number of adverse
side effects and a narrow therapeutic window (65). For example, first-line
cisplatin-based chemotherapy yields only a 13% response rate when
cisplatin alone is administered, whereas the response rate is 36%
for cisplatin double therapy (66,67).
The development of effective treatment therapies for cervical
cancer, especially advanced cervical cancer, is therefore urgently
required.
As an important component of the solid TME, CAFs
have long been considered as a potential therapeutic target for
tumors. However, numerous therapeutic strategies for CAFs have
failed in the clinic, and research in this field of cervical cancer
is comparatively lacking. One important reason for this is that
CAFs are quite heterogeneous, and the composition and function of
CAFs in different types of tumors remain poorly understood
(68). However, analysis based on
single-cell sequencing may effectively solve these problems.
According to the scRNA-seq results of the present study, the
composition of CAFs in the cervical cancer microenvironment was
relatively simple, and the tumor-promoting effects of ecCAFs were
clearly observed. Furthermore, the present study demonstrated the
pro-tumorigenic function of ecCAFs using data from additional
databases, and this effect was demonstrated to be correlated with
several clinical indicators that may be useful in terms of
predicting patient prognosis and treatment response.
According to the WHO and the Centers for Disease
Control and Prevention, HPV is the second most common carcinogenic
infectious agent, ranked only after Helicobacter pylori. HPV
induces 31.1% of all cases of infection-caused cancers (69) and ~5% of all cases of cancer
(9). As ecCAFs are associated with
HPV infection and appear to co-occur in several HPV-associated
tumors, it is possible to discuss the role of HPV infection in
shaping the tumor stroma. According to a previous study (70), bidirectional crosstalk occurs
between HPV-infected epithelial cells and the constituents of the
microenvironment. HPV+ cells influence the production of
a laminin-rich matrix and are also associated with reduced levels
of fibronectin and collagen in fibroblasts (71), increasing the expression levels of
pro-angiogenic genes in cells within the adjacent stroma (72) and the direct expression of
pro-inflammatory genes in fibroblasts or other stromal cell types
(73). Conversely, stromal cells
affect the proliferation and differentiation of HPV-infected
epithelial cells, and they were reported to serve a key role in an
HPV-associated cancer xenograft model (74). The mechanism responsible for
crosstalk may be reciprocal secretion and uptake of extracellular
vesicles through exosomes. Either the HPV E6 and E7 transcripts or
proteins were shown to be transferred to cells via microvesicles
(75). These findings not only
provide a potential theoretical basis for epithelial-stromal
crosstalk, but also represent a major paradigm shift in terms of
the understanding of HPV-associated diseases. From this
perspective, stromal cells may have certain commonality among
different types of HPV-associated cancer, which may prove to be
helpful in terms of understanding and controlling HPV-associated
cancer.
Moreover, it has been suggested that the ECM may
participate in preventing patients from benefiting from the
available immunotherapeutic treatments via the exclusion of immune
cells. When the tumor stroma is rich in collagen, fibronectin and
several proteoglycans (for example, hyaluronic acid and versican),
it will have a ‘trapping’ effect on T cells, resulting in
inhibition of T cell motility (76). The protease-independent nature of
T-cell migration typically results in their movement along the path
of least resistance of collagen fibers (77). This is consistent with the findings
of the present study where patients with higher ecCAF risk scores
tended to have a lower level of immune cell infiltration and were
consequently less likely to benefit from immunotherapy. Therefore,
targeting ecCAFs in cervical cancer and other HPV-associated tumors
may not only have direct antitumor effects, but may also enhance
the response rate of patients to immunotherapy. However, this
merits further study.
Subsequently, the function of model genes in cancer
was assessed in the present study to provide indicators for further
research. Although the level of research on cervical cancer remains
comparatively deficient, the genes in question have been reported
to fulfill roles in other types of cancer. Regarding the eight
genes in the signature that were positively correlated with a poor
prognosis in the present study, HSPG2 is an extracellular
proteoglycan that orchestrates prostate cancer angiogenesis,
proliferation, differentiation and invasion (78). Increased levels of HSPG2 expression
may therefore be used to independently predict poor OS in
neurological tumors and leukemia (79,80).
Both LEPROT and LEPR are upregulated in breast cancer tumor
tissues, and LEPR has previously been reported to be associated
with poor prognosis in breast cancer (81). LEPROT may communicate with the TME,
thereby regulating inflammatory or immune signals, and has been
reported both to affect cancer development and to serve as a
potential prognostic marker or a therapeutic target in pan-cancer
(82). In numerous types of cancer,
such as lung and prostate cancer, PTGS2 is secreted by CAFs,
macrophage type 2 cells and tumor cells, and is therefore reported
to have pleiotropic and multifaceted roles in terms of both
carcinogenesis and cancer cell resistance to chemotherapy and
radiotherapy (83). In liver and
colorectal cancer, TMEM9 was reported to hyperactivate Wnt
signaling in tumorigenesis through the lysosomal degradation of APC
(84). As a component of basement
membranes, COL4A1 has been reported to function as a therapeutic
target for cancers (85) due to its
interaction with other ECM components, thereby participating in
epithelial-to-mesenchymal transition, and serving widely
pro-tumorigenic roles in different types of cancer. EGLN3 regulates
tumor cell apoptosis and proliferation in glioma (86). SEC23A has been reported to function
either as an oncogene or as a tumor suppressor gene in different
types of cancer and is a potential biomarker for the therapeutic
efficacy of docetaxel and vandetanib (87,88).
As for the four protective genes (LMO4, VCAM1, RGS5 and NDRG2) in
the model constructed in the present study, NDRG2 is widely
recognized as a tumor suppressor in several types of cancer
(89), whereas the functions of the
other three genes remain controversial. RGS5, which co-localizes
with platelet/endothelial cell adhesion molecule-1/CD31, platelet
derived growth factor receptor-β or α-SMA, typically supports a
pro-angiogenic microenvironment (90). Although research in this area is
largely lacking, VCAM1 secreted from CAFs was reported in a study
to enhance the growth and invasion of lung cancer cells through AKT
and MAPK signaling (91). Finally,
LMO4 has been reported to participate in tumor proliferation and
invasion in numerous types of cancer, and it was also reported to
mediate trastuzumab resistance in breast cancer (92).
It should be noted that there were both innovative
aspects and limitations associated with the present study. The
novelty of the present study is as follows: First, the
heterogeneity of CAFs in the cervical cancer microenvironment were
assessed and a new subgroup was defined, which was termed
‘extracellular CAF’ on the basis of the scRNA-seq data. As an
important component of the cervical cancer microenvironment, ecCAFs
have a notable tumor promoting effect. Second, based on the results
of the present study, ecCAFs may be prevalent stroma cells in
several HPV-associated tumors and directly related to patient
prognosis. This suggests the potential value of ecCAFs as a
therapeutic target for HPV-associated tumors, which warrants
further study. However, it should be acknowledged that the present
study has certain limitations: First, the sample size for scRNA-seq
analysis was relatively small as it was restricted by research
conditions such as lack of funding. Second, the present study
preliminarily assessed the function of ecCAFs based on sequencing
and public database data, which requires further verification
through in vitro and in vivo experiments.
In conclusion, based on the single-cell sequencing
of clinical samples, the present study demonstrated the
heterogeneity of fibroblasts in the cervical cancer
microenvironment, and, to the best of our knowledge, is the first
study to identify a new subgroup of CAFs that may be closely
associated with tumor progression. A prognostic signature was
developed according to the hub genes of ecCAFs, and its clinical
significance was analyzed. ecCAFs may be a potential therapeutic
target for cervical cancer and other HPV-associated cancers, and
these findings may assist in guiding clinical practice and
providing the direction of subsequent research.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Mr. Zhuangchou Han
(from the laboratory of LC-Bio Technologies Hangzhou Co., Ltd.) for
their support with single-cell sequencing and analyzing data.
Funding
The present work was supported by the Key Research and
Development Plan in Zhejiang Province (grant no. N0.2020C03025),
Natural Science Foundation of Zhejiang Province (grant no.
LGF21H180011) and Zhejiang Provincial Educational Project (grant
no. Y202045540).
Availability of data and materials
The raw sequence data from the 5 patients generated
in the present study may be found in the Genome Sequence Archive
(Genomics, Proteomics & Bioinformatics 2021) in National
Genomics Data Center (Nucleic Acids Res 2022), China National
Center for Bioinformation/Beijing Institute of Genomics, Chinese
Academy of Sciences under accession number, HRA004457, or at the
following link https://ngdc.cncb.ac.cn/gsa-human/browse/HRA004457
(93,94). All other data generated in the
present study may be requested from the corresponding author.
Authors' contributions
MX collected the clinical samples. YW and YF
supervised the single-cell sequencing process and performed
preliminary analysis of the raw data. YW and YF confirm the
authenticity of all the raw data. YW used the software to complete
the main analysis process and wrote the original draft. YY and MX
helped with data analysis and interpretation. The fundings was
provided by YF, YY and XW. SZ and YL were responsible for
validating the results, and supervising the research, and took part
in designing the structure of the article. XW and FY provided the
conception of the article and reviewed the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was performed according to the
guidelines of the Declaration of Helsinki and approved by the
Institutional Review Board (Ethics Committee) of Women's Hospital,
School of Medicine, Zhejiang University (Hangzhou, China; approval
no. IRB 20200006 R; 2020-01-19). Written informed consent was
obtained from the patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TCGA
|
The Cancer Genome Atlas
|
|
CAF
|
cancer-associated fibroblast
|
|
HPA
|
Human Protein Atlas
|
|
TME
|
tumor microenvironment
|
|
FGF
|
fibroblast growth factor
|
|
OS
|
overall survival
|
|
LASSO
|
least absolute shrinkage and
selection operator
|
|
TIDE
|
Tumor Immune Dysfunction and
Exclusion
|
|
GDSC
|
Genomics of Drug Sensitivity in
Cancer
|
|
ROC
|
receiver operating characteristic
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cancer Genome Atlas Research Network:
Albert Einstein College of Medicine; Analytical Biological
Services; Barretos Cancer Hospital; Baylor College of Medicine;
Beckman Research Institute of City of Hope; Buck Institute for
Research on Aging; Canada's Michael Smith Genome Sciences Centre;
Harvard Medical School, . Helen F, et al: Integrated genomic and
molecular characterization of cervical cancer. Nature. 543:378–384.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tewari KS and Monk BJ: Gynecologic
oncology group trials of chemotherapy for metastatic and recurrent
cervical cancer. Curr Oncol Rep. 7:419–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liontos M, Kyriazoglou A, Dimitriadis I,
Dimopoulos MA and Bamias A: Systemic therapy in cervical cancer: 30
Years in review. Crit Rev Oncol Hematol. 137:9–17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mutlu L, Tymon-Rosario J, Harold J and
Menderes G: Targeted treatment options for the management of
metastatic/persistent and recurrent cervical cancer. Expert Rev
Anticancer Ther. 22:633–645. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li C, Teixeira AF, Zhu HJ and ten Dijke P:
Cancer associated-fibroblast-derived exosomes in cancer
progression. Mol Cancer. 20:1542021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu L, Liu L, Yao HH, Zhu ZQ, Ning ZL and
Huang Q: Stromal myofibroblasts are associated with poor prognosis
in solid cancers: A meta-analysis of published studies. PLoS One.
11:e01599472016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lavie D, Ben Shmuel A, Erez N and Scherz
Shouval R: Cancer-associated fibroblasts in the single-cell era.
Nat Cancer. 3:793–807. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan Y, Cai X, Shen F and Ma F: HPV
post-infection microenvironment and cervical cancer. Cancer Lett.
497:243–254. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wright JD, Matsuo K, Huang Y, Tergas AI,
Hou JY, Khoury-Collado F, St Clair CM, Ananth CV, Neugut AI and
Hershman DL: Prognostic performance of the 2018 international
federation of gynecology and obstetrics cervical cancer staging
guidelines. Obstet Gynecol. 134:49–57. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garré JM, Silva HM, Lafaille JJ and Yang
G: CX3CR1+ monocytes modulate learning and
learning-dependent dendritic spine remodeling via TNF-α. Nat Med.
23:714–722. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Y, Chen S, Li K, Zhang Y, Huang X, Li
T, Wu S, Wang Y, Carey LB and Qian W: Overdosage of balanced
protein complexes reduces proliferation rate in aneuploid cells.
Cell Syst. 9:129–142.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diskin B, Adam S, Cassini MF, Sanchez G,
Liria M, Aykut B, Buttar C, Li E, Sundberg B, Salas RD, et al:
PD-L1 engagement on T cells promotes self-tolerance and suppression
of neighboring macrophages and effector T cells in cancer. Nat
Immunol. 21:442–454. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cano Gamez E, Soskic B, Roumeliotis TI, So
E, Smyth DJ, Baldrighi M, Willé D, Nakic N, Esparza Gordillo J,
Larminie CGC, et al: Single-cell transcriptomics identifies an
effectorness gradient shaping the response of CD4+ T
cells to cytokines. Nat Commun. 11:18012020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xin Z, Lin M, Hao Z, Chen D, Chen Y, Chen
X, Xu X, Li J, Wu D, Chai Y and Wu P: The immune landscape of human
thymic epithelial tumors. Nat Commun. 13:54632022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang C, Siebert JR, Burns R, Gerbec ZJ,
Bonacci B, Rymaszewski A, Rau M, Riese MJ, Rao S, Carlson KS, et
al: Heterogeneity of human bone marrow and blood natural killer
cells defined by single-cell transcriptome. Nat Commun.
10:39312019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng Y, Zheng Y, Li D, Hong Q, Zhang M, Li
Q, Fu B, Wu L, Wang X, Shen W, et al: Expression characteristics of
interferon-stimulated genes and possible regulatory mechanisms in
lupus patients using transcriptomics analyses. EBioMedicine.
70:1034772021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Z, Zhou L, Liu L, Hou Y, Xiong M,
Yang Y, Hu J and Chen K: Single-cell RNA sequencing highlights the
role of inflammatory cancer-associated fibroblasts in bladder
urothelial carcinoma. Nat Commun. 11:50772020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He D, Wang D, Lu P, Yang N, Xue Z, Zhu X,
Zhang P and Fan G: Single-cell RNA sequencing reveals heterogeneous
tumor and immune cell populations in early-stage lung
adenocarcinomas harboring EGFR mutations. Oncogene. 40:355–368.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wagner J, Rapsomaniki MA, Chevrier S,
Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst
S, Soysal SD, et al: A single-cell atlas of the tumor and immune
ecosystem of human breast cancer. Cell. 177:1330–1345.e18. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng H, Liu H, Ge Y and Wang X:
Integrated single-cell and bulk RNA sequencing analysis identifies
a cancer associated fibroblast-related signature for predicting
prognosis and therapeutic responses in colorectal cancer. Cancer
Cell Int. 21:5522021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liberzon A, Birger C, Thorvaldsdóttir H,
Ghandi M, Mesirov JP and Tamayo P: The molecular signatures
database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sergushichev AA: An algorithm for fast
preranked gene set enrichment analysis using cumulative statistic
calculation. bioRxiv. 0600122016.
|
|
24
|
Wang Z, Li Z, Zhou K, Wang C, Jiang L,
Zhang L, Yang Y, Luo W, Qiao W, Wang G, et al: Deciphering cell
lineage specification of human lung adenocarcinoma with single-cell
RNA sequencing. Nat Commun. 12:65002021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goldman MJ, Craft B, Hastie M, Repečka K,
McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al:
Visualizing and interpreting cancer genomics data via the Xena
platform. Nat Biotechnol. 38:675–678. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee YY, Kim TJ, Kim JY, Choi CH, Do IG,
Song SY, Sohn I, Jung SH, Bae DS, Lee JW and Kim BG: Genetic
profiling to predict recurrence of early cervical cancer. Gynecol
Oncol. 131:650–654. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Simon N, Friedman J, Hastie T and
Tibshirani R: Regularization paths for Cox's proportional hazards
model via coordinate descent. J Stat Softw. 39:1–13. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan Y, Zhang M and Xu S and Xu S:
Identification of an immune gene expression signature for
predicting lung squamous cell carcinoma prognosis. Biomed Res Int.
2020:50249422020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gong Z, Hong F, Wang H, Zhang X and Chen
J: An eight-mRNA signature outperforms the lncRNA-based signature
in predicting prognosis of patients with glioblastoma. BMC Med
Genet. 21:562020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang X, Wu W, Pan Y, Zhou Q, Xu J and Han
S: Immune-related genes in tumor-specific CD4+ and
CD8+ T cells in colon cancer. BMC Cancer. 20:5852020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hänzelmann S, Castelo R and Guinney J:
GSVA: Gene set variation analysis for microarray and RNA-seq data.
BMC Bioinformatics. 14:72013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang D, Zhang Y, Wang X, Zhang L and Xu S:
Construction and validation of an aging-related gene signature
predicting the prognosis of pancreatic cancer. Front Genet.
14:10222652023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X,
Li Z, Traugh N, Bu X, Li B, et al: Signatures of T cell dysfunction
and exclusion predict cancer immunotherapy response. Nat Med.
24:1550–1558. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Arina A, Idel C, Hyjek EM, Alegre ML, Wang
Y, Bindokas VP, Weichselbaum RR and Schreiber H: Tumor-associated
fibroblasts predominantly come from local and not circulating
precursors. Proc Natl Acad Sci USA. 113:7551–7556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang W, Soares J, Greninger P, Edelman EJ,
Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et
al: Genomics of drug sensitivity in cancer (GDSC): A resource for
therapeutic biomarker discovery in cancer cells. Nucleic Acids Res.
41:(Database Issue). D955–D961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maeser D, Gruener RF and Huang RS:
oncoPredict: An R package for predicting in vivo or cancer patient
drug response and biomarkers from cell line screening data. Brief
Bioinform. 22:bbab2602021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li Y, Lu S, Lan M, Peng X, Zhang Z and
Lang J: A prognostic nomogram integrating novel biomarkers
identified by machine learning for cervical squamous cell
carcinoma. J Transl Med. 18:2232020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Long J, Chen P, Lin J, Bai Y, Yang X, Bian
J, Lin Y, Wang D, Yang X, Zheng Y, et al: DNA methylation-driven
genes for constructing diagnostic, prognostic, and recurrence
models for hepatocellular carcinoma. Theranostics. 9:7251–7267.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Uhlén M, Fagerberg L, Hallström BM,
Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C,
Sjöstedt E, Asplund A, et al: Proteomics. Tissue-based map of the
human proteome. Science. 347:12604192015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song K, Hao J, Ge Z and Chen P: Clinical
and survival impact of sex-determining region Y-Box 2 in colorectal
cancer: An integrated analysis of the immunohistochemical study and
bioinformatics analysis. J Oncol. 2020:37615352020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoshida GJ: Regulation of heterogeneous
cancer-associated fibroblasts: The molecular pathology of activated
signaling pathways. J Exp Clin Cancer Res. 39:1122020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Au Yeung CL, Tsang TY, Yau PL and Kwok TT:
Human papillomavirus type 16 E6 induces cervical cancer cell
migration through the p53/microRNA-23b/urokinase-type plasminogen
activator pathway. Oncogene. 30:2401–2410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sato T, Sakai T, Noguchi Y, Takita M,
Hirakawa S and Ito A: Tumor-stromal cell contact promotes invasion
of human uterine cervical carcinoma cells by augmenting the
expression and activation of stromal matrix metalloproteinases.
Gynecol Oncol. 92:47–56. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang W, Ge Y, Cheng Q, Zhang Q, Fang L
and Zheng J: Decorin is a pivotal effector in the extracellular
matrix and tumour microenvironment. Oncotarget. 9:5480–5491. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schulz WA, Ingenwerth M, Djuidje CE, Hader
C, Rahnenführer J and Engers R: Changes in cortical cytoskeletal
and extracellular matrix gene expression in prostate cancer are
related to oncogenic ERG deregulation. BMC Cancer. 10:5052010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lucarelli G, Rutigliano M, Bettocchi C,
Palazzo S, Vavallo A, Galleggiante V, Trabucco S, Di Clemente D,
Selvaggi FP, Battaglia M and Ditonno P: Spondin-2, a secreted
extracellular matrix protein, is a novel diagnostic biomarker for
prostate cancer. J Urol. 190:2271–2277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kubala JM, Laursen KB, Schreiner R,
Williams RM, van der Mijn JC, Crowley MJ, Mongan NP, Nanus DM,
Heller DA and Gudas LJ: NDUFA4L2 reduces mitochondrial respiration
resulting in defective lysosomal trafficking in clear cell renal
cell carcinoma. Cancer Biol Ther. 24:21706692023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen PH, Shen WL, Shih CM, Ho KH, Cheng
CH, Lin CW, Lee CC, Liu AJ and Chen KC: The CHAC1-inhibited Notch3
pathway is involved in temozolomide-induced glioma cytotoxicity.
Neuropharmacology. 116:300–314. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Deaton RA, Bulut G, Serbulea V, Salamon A,
Shankman LS, Nguyen AT and Owens GK: A new autosomal
Myh11-CreERT2 smooth muscle cell lineage tracing and
gene knockout mouse model-brief report. Arterioscler Thromb Vasc
Biol. 43:203–211. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Loumaye A, Lause P, Zhong X, Zimmers TA,
Bindels LB and Thissen JP: Activin a causes muscle atrophy through
MEF2C-dependent impaired myogenesis. Cells. 11:11192022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yoshihara K, Shahmoradgoli M, Martínez E,
Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW,
Levine DA, et al: Inferring tumour purity and stromal and immune
cell admixture from expression data. Nat Commun. 4:26122013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chiossone L, Dumas PY, Vienne M and Vivier
E: Natural killer cells and other innate lymphoid cells in cancer.
Nat Rev Immunol. 18:671–688. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Giles JR, Globig AM, Kaech SM and Wherry
EJ: CD8+ T cells in the cancer-immunity cycle. Immunity.
56:2231–2253. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Speiser DE, Chijioke O, Schaeuble K and
Münz C: CD4+ T cells in cancer. Nat Cancer. 4:317–329.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wei E, Reisinger A, Li J, French LE,
Clanner-Engelshofen B and Reinholz M: Integration of scRNA-Seq and
TCGA RNA-Seq to analyze the heterogeneity of HPV+ and HPV-cervical
cancer immune cells and establish molecular risk models. Front
Oncol. 12:8609002022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primers. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shaker OG, Hammam OA and Wishahi MM: Is
there a correlation between HPV and urinary bladder carcinoma?
Biomed Pharmacother. 67:183–191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pelizzer T, Dias CP, Poeta J, Torriani T
and Roncada C: Colorectal cancer prevalence linked to human
papillomavirus: A systematic review with meta-analysis. Rev Bras
Epidemiol. 19:791–802. 2016.(In Portuguese, English). View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen AC, Waterboer T, Keleher A, Morrison
B, Jindal S, McMillan D, Nicol D, Gardiner RA, McMillan NA and
Antonsson A: Human papillomavirus in benign prostatic hyperplasia
and prostatic adenocarcinoma patients. Pathol Oncol Res.
17:613–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kamat AM, Hahn NM, Efstathiou JA, Lerner
SP, Malmström PU, Choi W, Guo CC, Lotan Y and Kassouf W: Bladder
cancer. Lancet. 388:2796–2810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gelibter AJ, Caponnetto S, Urbano F,
Emiliani A, Scagnoli S, Sirgiovanni G, Napoli VM and Cortesi E:
Adjuvant chemotherapy in resected colon cancer: When, how and how
long? Surg Oncol. 30:100–107. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Schatten H: Brief overview of prostate
cancer statistics, grading, diagnosis and treatment strategies. Adv
Exp Med Biol. 1095:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shanmugaraj B, Malla A, Bulaon CJI,
Phoolcharoen W and Phoolcharoen N: Harnessing the potential of
plant expression system towards the production of vaccines for the
prevention of human papillomavirus and cervical cancer. Vaccines
(Basel). 10:20642022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
National Comprehensive Cancer Network and
Clinical practice guidelines in oncology cervical cancer, . version
4. 2019. https://www.nccn.org/professionals/physician_gls/pdf/cervical.pdfMarch
29–2019.
|
|
65
|
Mauricio D, Zeybek B, Tymon-Rosario J,
Harold J and Santin AD: Immunotherapy in cervical cancer. Curr
Oncol Rep. 23:612021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Long HJ III, Bundy BN, Grendys EC Jr,
Benda JA, McMeekin DS, Sorosky J, Miller DS, Eaton LA and Fiorica
JV; Gynecologic Oncology Group Study, : Randomized phase III trial
of cisplatin with or without topotecan in carcinoma of the uterine
cervix: A gynecologic oncology group study. J Clin Oncol.
23:4626–4633. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Monk BJ, Sill MW, McMeekin DS, Cohn DE,
Ramondetta LM, Boardman CH, Benda J and Cella D: Phase III trial of
four cisplatin-containing doublet combinations in stage IVB,
recurrent, or persistent cervical carcinoma: A gynecologic oncology
group study. J Clin Oncol. 27:4649–4655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen Y, McAndrews KM and Kalluri R:
Clinical and therapeutic relevance of cancer-associated
fibroblasts. Nat Rev Clin Oncol. 18:792–804. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Szymonowicz KA and Chen J: Biological and
clinical aspects of HPV-related cancers. Cancer Biol Med.
17:864–878. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Spurgeon ME and Lambert PF: Human
papillomavirus and the stroma: Bidirectional crosstalk during the
virus life cycle and carcinogenesis. Viruses. 9:2192017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Fullár A, Dudás J, Oláh L, Hollósi P, Papp
Z, Sobel G, Karászi K, Paku S, Baghy K and Kovalszky I: Remodeling
of extracellular matrix by normal and tumor-associated fibroblasts
promotes cervical cancer progression. BMC Cancer. 15:2562015.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Pilch H, Schlenger K, Steiner E,
Brockerhoff P, Knapstein P and Vaupel P: Hypoxia-stimulated
expression of angiogenic growth factors in cervical cancer cells
and cervical cancer-derived fibroblasts. Int J Gynecol Cancer.
11:137–142. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Woodby B, Scott M and Bodily J: The
interaction between human papillomaviruses and the stromal
microenvironment. Prog Mol Biol Transl Sci. 144:169–238. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Murata T, Mizushima H, Chinen I, Moribe H,
Yagi S, Hoffman RM, Kimura T, Yoshino K, Ueda Y, Enomoto T and
Mekada E: HB-EGF and PDGF mediate reciprocal interactions of
carcinoma cells with cancer-associated fibroblasts to support
progression of uterine cervical cancers. Cancer Res. 71:6633–6642.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chiantore MV, Mangino G, Iuliano M,
Zangrillo MS, De Lillis I, Vaccari G, Accardi R, Tommasino M,
Columba Cabezas S, Federico M, et al: Human papillomavirus E6 and
E7 oncoproteins affect the expression of cancer-related microRNAs:
Additional evidence in HPV induced tumorigenesis. J Cancer Res Clin
Oncol. 142:1751–1763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Evanko SP, Potter-Perigo S, Bollyky PL,
Nepom GT and Wight TN: Hyaluronan and versican in the control of
human T-lymphocyte adhesion and migration. Matrix Biol. 31:90–100.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bougherara H, Mansuet-Lupo A, Alifano M,
Ngô C, Damotte D, Le Frère-Belda MA, Donnadieu E and Peranzoni E:
Real-time imaging of resident T cells in human lung and ovarian
carcinomas reveals how different tumor microenvironments control T
lymphocyte migration. Front Immunol. 6:5002015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Grindel BJ, Martinez JR, Tellman TV,
Harrington DA, Zafar H, Nakhleh L, Chung LW and Farach Carson MC:
Matrilysin/MMP-7 cleavage of perlecan/HSPG2 complexed with
semaphorin 3A supports FAK-mediated stromal invasion by prostate
cancer cells. Sci Rep. 8:72622018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kazanskaya GM, Tsidulko AY, Volkov AM,
Kiselev RS, Suhovskih AV, Kobozev VV, Gaytan AS, Aidagulova SV,
Krivoshapkin AL and Grigorieva EV: Heparan sulfate accumulation and
perlecan/HSPG2 up-regulation in tumour tissue predict low
relapse-free survival for patients with glioblastoma. Histochem
Cell Biol. 149:235–244. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhou X, Liang S, Zhan Q, Yang L, Chi J and
Wang L: HSPG2 overexpression independently predicts poor survival
in patients with acute myeloid leukemia. Cell Death Dis.
11:4922020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li L, Meng X, Liu L, Xiang Y, Wang F, Yu
L, Zhou F, Zheng C, Zhou W, Cui S, et al: Single-nucleotide
polymorphisms in LEP and LEPR associated with breast cancer risk:
Results from a multicenter case-control study in Chinese females.
Front Oncol. 12:8095702022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li B, He Y, Li P and Chen X: Leptin
receptor overlapping transcript (LEPROT) is associated with the
tumor microenvironment and a prognostic predictor in pan-cancer.
Front Genet. 12:7494352021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hashemi Goradel N, Najafi M, Salehi E,
Farhood B and Mortezaee K: Cyclooxygenase-2 in cancer: A review. J
Cell Physiol. 234:5683–5699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Jung YS, Stratton SA, Lee SH, Kim MJ, Jun
S, Zhang J, Zheng B, Cervantes CL, Cha JH, Barton MC and Park JI:
TMEM9-v-ATPase activates Wnt/β-catenin signaling via APC lysosomal
degradation for liver regeneration and tumorigenesis. Hepatology.
73:776–794. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhang H, Wang Y and Ding H: COL4A1,
negatively regulated by XPD and miR-29a-3p, promotes cell
proliferation, migration, invasion and epithelial-mesenchymal
transition in liver cancer cells. Clin Transl Oncol. 23:2078–2089.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mao K, You C, Lei D and Zhang H: Potential
regulation of glioma through the induction of apoptosis signaling
via Egl-9 family hypoxia-inducible factor 3. Oncol Lett.
13:893–897. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wang Y, Lieberman R, Pan J, Zhang Q, Du M,
Zhang P, Nevalainen M, Kohli M, Shenoy NK, Meng H, et al: miR-375
induces docetaxel resistance in prostate cancer by targeting SEC23A
and YAP1. Mol Cancer. 15:702016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lassalle S, Zangari J, Popa A, Ilie M,
Hofman V, Long E, Patey M, Tissier F, Belléannée G, Trouette H, et
al: MicroRNA-375/SEC23A as biomarkers of the in vitro efficacy of
vandetanib. Oncotarget. 7:30461304782016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chen XL, Lei L, Hong LL and Ling ZQ:
Potential role of NDRG2 in reprogramming cancer metabolism and
epithelial-to-mesenchymal transition. Histol Histopathol.
33:655–663. 2018.PubMed/NCBI
|
|
90
|
Silini A, Ghilardi C, Figini S, Sangalli
F, Fruscio R, Dahse R, Pedley RB, Giavazzi R and Bani M: Regulator
of G-protein signaling 5 (RGS5) protein: A novel marker of cancer
vasculature elicited and sustained by the tumor's proangiogenic
microenvironment. Cell Mol Life Sci. 69:1167–1178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhou Z, Zhou Q, Wu X, Xu S, Hu X, Tao X,
Li B, Peng J, Li D, Shen L, et al: VCAM-1 secreted from
cancer-associated fibroblasts enhances the growth and invasion of
lung cancer cells through AKT and MAPK signaling. Cancer Lett.
473:62–73. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ding K, Wu Z, Li X, Sheng Y, Wang X and
Tan S: LMO4 mediates trastuzumab resistance in HER2 positive breast
cancer cells. Am J Cancer Res. 8:594–609. 2018.PubMed/NCBI
|
|
93
|
Chen T, Chen X, Zhang S, Zhu J, Tang B,
Wang A, Dong L, Zhang Z, Yu C, Sun Y, et al: The genome sequence
archive family: toward explosive data growth and diverse data
types. Genomics Proteomics Bioinformatics. 19:578–583. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
CNCB-NGDC Members, . Partners Database
resources of the national genomics data center, China national
center for bioinformation in 2022. Nucleic Acids Res. 50(D1):
D27–D38. 2022. View Article : Google Scholar : PubMed/NCBI
|