Introduction
Glioblastoma (GBM) is the most common type of
malignant tumor in the central nervous system among adults and is
associated with a poor survival rate (1). Despite significant advancements in
surgical techniques, radiotherapy and chemotherapy, the prognosis
for GBM remains dismal (2). The
malignant behavior of cancer cells is regulated by oncogenes and
tumor suppressor genes through various signaling pathways, which
significantly affect the efficacy of clinical treatments and
patient outcomes (3,4). The development of GBM requires
specific molecular aberrations, including mutations in the P53 and
retinoblastoma signaling pathways, as well as alterations in the
receptor tyrosine kinase/Ras/phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT) signaling pathways and the epithelial
growth factor receptor (5,6). Despite efforts to target these
abnormal changes, GBM treatment has not yielded favorable results.
Hence, there is an urgent need to identify novel biomarkers and
therapeutic targets in GBM.
A previous study revealed that NIMA related kinase 2
(NEK2) overexpression was significantly correlated with the grade,
proliferation and prognosis of GBM (7). In addition, it enhances malignancy
through the NIK/NF-κB pathway (8).
The NEK family is a group of protein kinases that share
similarities with NIMA kinase, found in higher eukaryotes (9). This family comprises 11 members,
designated as NEK 1 to 11, with NEK2 exhibiting the highest
similarity to NIMA (9). NEK2 is
primarily located in cell centrosomes and is involved in various
cellular processes, including centrosomal circulation during cell
mitosis, formation of bipolar mitotic spindles (10) and stabilization of microtubules
(11). Abnormal overexpression of
NEK2 has been observed in several types of human cancers, including
non-small cell lung cancer (12),
myeloma (13), pancreatic (14) and breast cancer (15). This overexpression has been
associated with various aspects of malignant transformation, such
as tumorigenesis, therapy resistance and tumor progression
(16).
Cyclin A2 (CCNA2) is a gene related to the cell
cycle that has been suggested as a possible molecular marker in
low-grade gliomas (17). This gene
is in the Q27 region of human chromosome 4 and plays a crucial role
in promoting the transition through the G1/S and G2/M phases of the
cell cycle by binding and interacting with cyclin-dependent kinases
CDK1 and CDK2 (18). Furthermore,
the complete absence of CCNA2 leads to embryonic lethality in mice
(19). CCNA2 is differentially
expressed in several types of tumors in the digestive, urinary and
central nervous systems (20). It
has been found to play a regulatory role in the development of
kidney cancer (20), breast cancer
(21), lung cancer (22), colorectal cancer (23) and other types of tumors. Moreover,
CCNA2 has been demonstrated to regulate epithelial-mesenchymal
transition in distinct types of cancer, including oral squamous
cell carcinoma (24), colorectal
cancer (25) and bladder cancer
(26). CCNA2 and NEK2 have been
also identified as crucial genes in cancer transformation induced
by hepatitis B (27) and the
progression and prognosis of pancreatic cancer (28). However, the specific roles and
correlation of CCNA2 and NEK2 with GBM have not been yet
elucidated.
In the present study, it was observed that high
expression of CCNA2 and NEK2 in glioma indicates poor clinical
outcomes. Furthermore, CCNA2 and NEK2 were significantly
co-expressed in neural progenitor cells (NPCs) at the single-cell
level. Moreover, CCNA2 and NEK2, along with NPCs, were closely
linked to the cell cycle in GBM and controlled the malignant
advancement of tumor cells. Based on these findings, a
comprehensive nomogram that supports a clinical prognosis analysis
has been developed, which may prompt the development of new
treatment strategies.
Materials and methods
Data collection
A total of 693 expression profiles of bulk
sequencing and relevant clinical data for glioma were extracted
from the Chinese Glioma Genome Atlas (CGGA; http://www.cgga.org.cn/) (29) and The Cancer Genome Atlas Program
(TCGA; http://www.cancer.gov/ccg/research/genome-sequencing/tcga)
(30). The single cell RNA
sequencing (scRNA-seq) expression profile and relevant clinical
data of nine isocitrate dehydrogenase (IDH) wildtype GBM samples
were collected from the Gene Expression Omnibus (GEO) database,
with accession number GSE131928 (31). Data were obtained using 10X
scRNA-seq.
Cellular clustering, gene analysis and
cell type annotation of scRNA-seq
The gene expression matrix and corresponding
clinical information for the nine IDH wildtype GBM samples were
imported into RStudio (v.4.3.0; r-project.org) and analyzed using
the Seurat package (v.4.0; R; r-project.org) (32). Downstream analysis was performed on
the primary expression data, removing low-quality single cells.
Cells with <300 expressed genes, >10% mitochondrial
transcripts, >0.5% red blood cell transcripts, or genes that
were expressed in fewer than three individual cells were also
excluded. After excluding 2,841 low-quality cells, the expression
data from the remaining 13,360 single cells were normalized using
the ‘LogNormalize’ method. The top 5,000 highly variable features
were then selected using the ‘FindVariableFeatures’ method, with
variance stabilizing transformation. Next, the gene expression data
were transformed using the z-score method and scaled by the
‘ScaleData’. The linear method was used to scale and center the top
5,000 highly variable features in this dataset, after which the
principal component analysis was performed to reduce the
dimensionality of the data. The top 5,000 most variable genes in
the dataset were used for this analysis. The first 20 principal
components (PCs) were analyzed using the ‘JackStrawPlot’ and
‘ElbowPlot’. These 20 PCs were then used for downstream
calculations based on ‘FindNeighbors’ with default parameters. The
resolution parameter applied to identify clusters by ‘FindClusters’
was 0.5, which was determined based on the range of 0.1 to 1 for
the single-cell datasets. The t-distributed stochastic neighbor
embedding (t-SNE) was employed to perform non-linear dimensionality
reduction and visualize different single-cell clusters. Marker
genes for each cluster were identified using ‘FindAllMarkers’. Only
the top five significantly upregulated genes were selected for
presentation in the heatmap, based on the following criteria:
Adjusted P-value <0.05, minimal percentage >0.25 and
log2-fold change (log2FC) >0.25. Subsequently,
SingleR (Bioconductor-SingleR) and Cellmarker 2.0 (http://bio-bigdata.hrbmu.edu.cn/CellMarker/index.html)
were used together to annotate cell types for the different
single-cell clusters (33,34).
Evaluation of stemness in single-cell
clusters using scRNA-seq
Cellular trajectory reconstruction analysis using
gene counts and expression (CytoTRACE) is an emerging computational
method for evaluating the transcriptional diversity of each
single-cell cluster in terms of differential or stemness status
based on scRNA-seq. This method has been validated in large-scale
datasets and has exceeded pre-existing computational strategies for
evaluating stemness (35). The
CytoTRACE package (v.0.3.3; CytoTRACE) was used to calculate the
CytoTRACE score for each single-cell cluster. This score ranges
from 0 to 1, with a higher score indicating greater stemness or
fewer differentiation characteristics.
Pseudo-time trajectory analysis of
scRNA-seq
The Monocle 2 package (v.2.28.0; Monocle;
cole-trapnell-lab.github.io) was utilized to perform trajectory
analysis, assuming that the one-dimensional variable ‘time’
captures the high-dimensional expression characteristics and
reveals the transformation of cell status based on scRNA-seq data
(36). The cell types identified as
astrocytes and NPC clusters were further analyzed for trajectory
features, using Monocle 2. The ‘newCellDataSet’ function was
applied to establish an analysis purpose with the parameter
‘expressionFamily=negbinomial.size’. Subsequently, the highly
variable genes generated from the ‘VariableFeatures’ were utilized
to reduce dimensions and sort cells in pseudo-time, using the
‘reduceDimension’ function with the ‘DDRTree’ algorithm. To
identify candidate genes that separate cells into branches, a
filtering criterion of ‘mean expression ≥0.5’ and ‘dispersion
empirical ≥1× dispersion_fit’ was used. The branch expression
analysis modeling (BEAM) was employed to analyze expression data
and identify significant genes with a Q-value <0.0001. These
genes were then grouped into different subgroups based on their
expression patterns.
Estimating the cell cycle phase of
scRNA-seq data
The tricycle package (v.1.8.0; bioconductor.org) was
used to evaluate cell cycle phases by leveraging critical
characteristics of cell cycle biology. This method has been
positively compared with gold-standard experimental assays and has
demonstrated significant predictive potential with multiple cell
types or tissues in single-cell datasets (37). The preprocessed scRNA-seq dataset
was evaluated for cell cycle phase, using the default parameters of
the tricycle package. This package calculated a cell cycle
position, represented by polar coordinates ranging from 0 to 2π,
indicating distinct phases.
Estimating the proportion of positive
NPCs (P-NPCs) in bulk sequencing samples
P-NPCs were defined as NPCs with CCNA2 and NEK2
expression levels >0. The marker genes were identified using
‘FindMarkers’, with a threshold of log2FC >1, an
adjusted P-value <0.05 and a minimum percentage >0.70. The
proportion of P-NPCs in bulk sequencing samples was evaluated
through single-sample gene set enrichment analysis (ssGSEA), using
marker genes.
Analysis of differentially expressed
genes (DEGs) in bulk sequencing
Preprocessing of the expression profile from bulk
sequencing included background correction, gene symbol
transformation and normalization using RStudio programming.
Significant DEGs in these datasets were found using the limma
package (version 3.48.3; Bioconductor). Genes were considered as
upregulation with adjusted P-value <0.05 and
log2-fold change (log2FC) >1.5 and the
downregulated genes had an adjusted P-value of <0.05 and a
log2-fold change (log2FC) <-1.5.
Gene ontology (GO), Kyoto Encyclopedia
of Genes and Genomes (KEGG), gene set enrichment analysis (GSEA)
and gene set variation analysis (GSVA)
Candidate genes resulting from bulk sequencing
analysis or scRNA-seq analysis were used to conduct GO and KEGG
analyses, using the clusterprofiler package (v.4.8.1;
Bioconductor). Gene sets were evaluated according to the hallmark
gene sets in the MSigDB database
(gsea-msigdb.org/gsea/msigdb/index.jsp) (38). The results met the requirements,
with an adjusted P-value of <0.05 (39). Furthermore, the GSEA was utilized to
identify enriched pathways from the previous analyses of bulk
sequencing or scRNA-seq. The significantly enriched results were
subsequently validated using GSVA.
Establishing and assessing the
predictive nomogram
To construct a nomogram that predicts the prognosis
of patients with GBM, a multivariate Cox regression analysis to
identify significant independent risk and protective factors was
conducted. Based on these factors, a comprehensive nomogram was
developed. The predictive potential of the nomogram was also
demonstrated by the calibration curve. The suitability of the
current predictors used in the nomogram was tested by evaluating
the Schoenfeld residuals and deviance residuals to assess the
proportional hazards assumption and identify outliers. Decision
curve analysis (DCA) was also performed to assess the clinical
applicability of the nomogram and the net benefit of diverse
prediction models at different threshold probabilities. This was
achieved by adding the benefits and minimizing the harms. The
time-dependent receiver operating characteristic (ROC) curve was
used to detect the discrimination of the nomogram and the
Kaplan-Meier curve was used to estimate the prognostic value of the
nomogram score.
Acquisition of glioma tissue
A total of 18 samples included World Health
Organisation (WHO) II, III and IV grade gliomas were obtained from
patient samples with a pathological diagnosis, who underwent
craniotomy at the First Affiliated Hospital of Xi'an Jiao Tong
University from April 2022 to December 2022. Of 18 patients, 8 were
female and 10 were male. The mean age was 56 years (range, 42–69
years). The grades of the 18 glioma samples were confirmed by two
independent pathologists. Inclusion criteria: i) Glioma patients
meeting the 5th edition of the World Health Organization
classification of tumors of the central nervous system in 2021; ii)
All patients received conventional imaging examination within 1
week before surgery and were diagnosed with glioma; iii) the first
diagnosed case without any invasive or non-invasive treatment; iv)
Age above 18 years old, regardless of gender; v) The tumor was
supratentorial. Exclusion criteria: i) Patients with other types of
brain tumors; ii) patients who could not receive surgical treatment
due to physical, financial or other reasons. The research involving
the utilization of these biological specimens received ethical
approval from the Ethics Committee of the hospital located in
Xi'an, Shaanxi, China (approval no. 2020-G13). Informed consents
for the utilization of clinical samples were approved and signed by
patients.
Immunohistochemistry staining
For the immunohistochemical study, tissue blocks
were fixed in 4% paraformaldehyde (PFA) at 4°C for 48 h, embedded
in paraffin and sectioned at a thickness of 4 µm. The processed
sections were blocked with 5% bovine serum albumin (BSA,
EZ2811C238; BioFroxx) for 1 h at room temperature. Then the tissue
sections were incubated with primary antibodies (1:100) to NEK2 and
CCNA2 overnight at 4°C, after which they were incubated with
biotinylated secondary antibodies (1:2,000) at room temperature for
1 h. Next, sections were incubated with horseradish
peroxidase-conjugated avidin (PK4002; NEOBIOSCIENCE) at room
temperature for 1 h, washed with PBS and stained with
3,3-diaminobenzidine (30 mg dissolved in 100 ml of Tris buffer
containing 0.03% H2O2) at room temperature
for 5 min. The sections were then rinsed in water and
counterstained with hematoxylin at room temperature for 3 min. For
the evaluation of NEK2 and CCNA2 expression, 10 visual fields per
section were randomly selected and examined by light microscopy.
The present study utilized the following antibodies: Mouse
anti-NEK2 primary antibodies (1:100, sc-55601; Santa Cruz
Biotechnology, Inc.) and rabbit anti-CCNA2 primary antibodies
(1:100, ab181591; Abcam). Goat anti-rabbit IgG (1:2,000, ab97051;
Abcam) and Goat anti-mouse IgG (1:2,000, PK4002; NEOBIOSCIENCE) was
used as the secondary antibody. The results were analyzed using the
ImageJ (v.1.8.0) software (National Institutes of Health).
Western blot analysis
The samples were prepared in RIPA buffer; Servicebio
Biological) containing a protease inhibitor cocktail
(MilliporeSigma) were used for western blot analysis. And the
protein concentration was measured using a BCA protein assay kit
(Beyotime Biotechnology). Equal amounts of protein lysates were
loaded onto the wells (20 µg/lane) of a 10% precast SDS-PAGE gel
and transferred to a PVDF membrane (Thermo Fisher Scientific, Inc.;
Invitrogen). The membrane was then incubated with 5% skimmed milk
for 1 h at 25°C, followed by overnight treatment with the target
antibodies (anti-NEK2, 1:500; anti-CCNA2 primary antibody, 1:1,000)
at 4°C. Following three washes with TBS-Tween 20 (TBST; 0.1% Tween)
for 10 min each, the membrane was incubated with horseradish
peroxide-conjugated secondary antibodies (1:5,000) for 1 h at room
temperature. The Amersham ECL Western Blot System (Cytiva) was
applied to visualize the protein expression levels of each sample,
using GAPDH (1:5,000) as the loading control. The following
antibodies were used: Mouse anti-NEK2 primary antibody, rabbit
anti-CCNA2 primary antibody, mouse GAPDH antibody (1:5,000,
sc-47724; Santa Cruz Biotechnology, Inc.), Goat Anti-Mouse
secondary antibody (1:5,000, DY60203; DIYIBio) and mouse
anti-rabbit secondary antibody (1:5,000, sc-2357; Santa Cruz
Biotechnology). Western blot analysis results were analyzed using
the ImageJ (v.1.8.0) software.
Statistical analysis
Statistical analyses were performed using RStudio.
Data were presented as the mean ± standard deviation of triplicate
determinations. The normality of the data distribution was
evaluated by the Shapiro-Wilk test. Unpaired two-tailed Student's
t-tests were performed to evaluate statistical differences in two
groups and one-way ANOVA analyses following Tukey's multiple
comparisons test was applied for comparisons between multiple
groups and other statistical analysis methods were consistent with
R packages listed in the manuscript. The log-rank test was utilized
to conduct the Kaplan-Meier survival analysis. Multivariate Cox
stepwise regression was selected for further analysis of survival.
The P-value <0.05 indicated statistically significant
differences.
Results
Highly expression of CCNA2 and NEK2 in
glioma
The TCGA and CGGA databases were utilized to
investigate the functions of CCNA2 and NEK2 in glioma. Results
indicated a significant increase in the expression levels of CCNA2
and NEK2 with the progression of WHO grade in GBM (Fig. 1A, B, D and E). A significant
positive correlation between the expression of CCNA2 and NEK2 in
GBM was also observed (Fig. 1C and
F). The Kaplan-Meier survival analysis revealed that patients
with high expression of CCNA2 and NEK2 had a shorter overall
survival time in GBM and all grades (Fig. 1G-K). To investigate the consistency
of CCNA2 and NEK2 expression levels in clinical glioma samples, IHC
staining was performed on glioma samples. Results indicated that
CCNA2 and NEK2 gene expression levels significantly increased with
the progression of WHO grade in glioma (Fig. 1L). The western blot analysis of
clinical samples yielded comparable results (Fig. 1M). Original figures of western blot
were presented in Fig. S1.
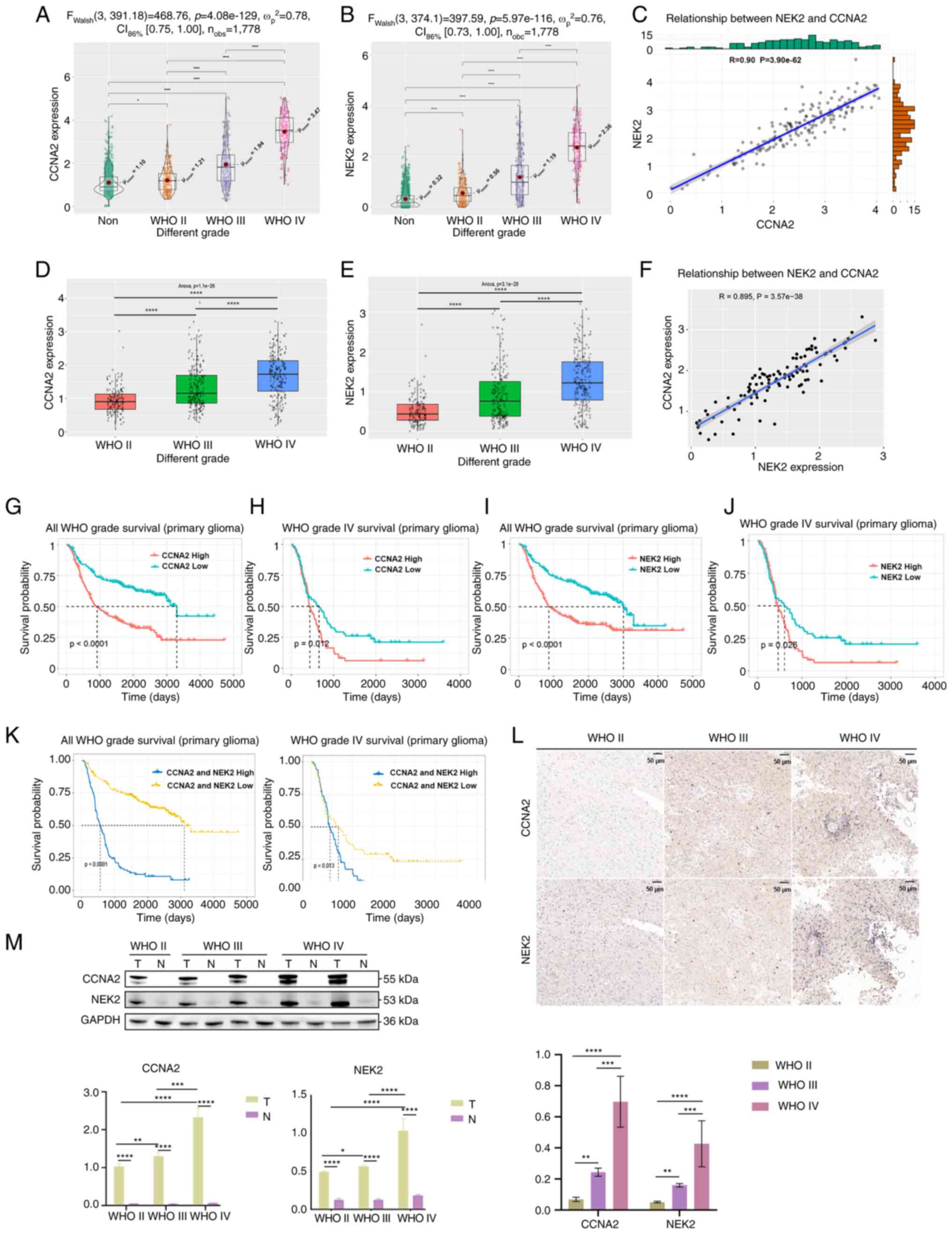 | Figure 1.Highly expressed CCNA2 and NEK2 in
glioma. (A) Analysis of NEK2 expression in different WHO grades of
glioma, using TCGA database (*P<0.05 and ****P<0.0001, with
independent t-test). (B) Analysis of NEK2 expression in different
WHO grades of glioma, using TCGA database. (C) Co-expression
analysis of CCNA2 and NEK2 expression in GBM, using TCGA database
(P<0.0001, with Pearson correlation coefficient). (D) Analysis
of NEK2 expression in different WHO grades of glioma, using the
CGGA database. (E) Analysis of NEK2 expression in different WHO
grades of glioma, using the CGGA database (****P<0.0001, with
one-way ANOVA followed by Tukey's multiple comparison test). (F)
Co-expression analysis of CCNA2 and NEK2 expression in GBM, using
the CGGA database (P<0.0001, with Pearson correlation
coefficient). (G) The Kaplan-Meier analysis of overall survival in
patients with glioma based on CCNA2 expression, using the CGGA
database (P<0.0001, with log-rank test). (H) The Kaplan-Meier
analysis of overall survival in patients with GBM based on CCNA2
expression, using the CGGA database (P=0.012, with log-rank test).
(I) Kaplan-Meier analysis of overall survival in patients with
glioma based on NEK2 expression, using the CGGA database
(P<0.0001, with log-rank test). (J) The Kaplan-Meier analysis of
overall survival in patients with GBM based on NEK2 expression,
using the CGGA database (P=0.026, with log-rank test). (K) The
Kaplan-Meier analysis of overall survival in patients with glioma
and GBM based on CCNA2 and NEK2 expression, using the CGGA database
(P<0.0001, for all glioma and P=0.013, for GBM, respectively,
with log-rank test). (L) Immunohistochemical staining of CCNA2 and
NEK2 in GBM samples, compared with adjacent normal tissue. (M)
Protein expression of CCNA2 and NEK2 in GBM samples, compared with
adjacent normal tissue. T, tumor tissue; N, adjacent normal tissue.
Original blots are presented in Fig.
S1. Western blot and immunohistochemical analyses were
conducted to detect CCNA2 and NEK2 protein levels in GBM samples
(*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, with
independent t-test). Data represent the mean ± SD of triplicate
determinations from three independent experiments. CCNA2, cyclin
A2; NEK2, NIMA related kinase 2; WHO, World Health Organization;
TCGA, The Cancer Genome Atlas; GBM, glioblastoma; CGGA, Chinese
Glioma Genome Atlas. |
CCNA2 and NEK2 are co-expressed in NPC
subtypes
Although a consistent expression of CCNA2 and NEK2
was demonstrated, the heterogeneity of GBM contributed to ambiguity
regarding the reciprocal regulation within the same cells (40). Therefore, the expression pattern of
CCNA2 and NEK2 was explored using scRNA-seq information collected
from the GEO database (dataset no. GSE131928) to identify whether
they were expressed in the same cell types. After conducting
quality control, low-quality cells were eliminated and 13,360 cells
were retained for further analysis (Fig. S2). Cells from nine patients were
clustered into 16 distinct groups, using t-SNE dimensionality
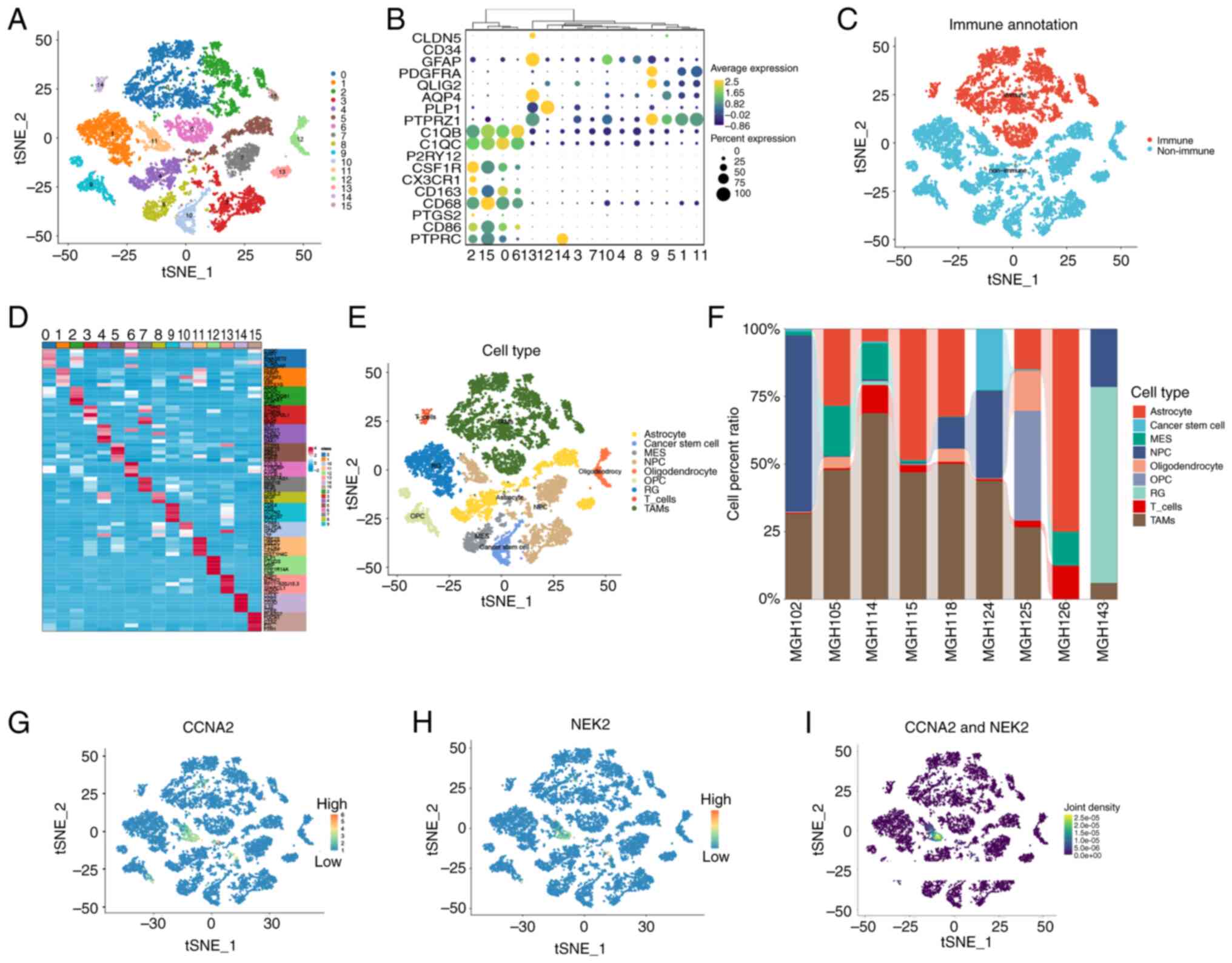
reduction (Fig. 2A). To
differentiate between cell types in the GBM microenvironment,
immune and non-immune clusters were classified based on marker
genes identified in previous studies (41–58).
Genes such as C1QB (41), CD68
(42), CSF1R (43), PTPRC (44), C1QC (45), P2RY12 (46), CX3CR1 (47), CD163 (48), PTGS2 (49) and CD86 (50) were used to identify immune clusters;
whereas genes AQP4 (51), PTPRZ1
(52), CLDN5 (53), CD34 (54), GFAP (55), FDGFRA (56), OLIG2 (57) and PLP1 (58) were used to identify non-immune
clusters. Clusters 0, 2, 6, 14 and 15 were identified as immune
cells, whereas clusters 1, 3, 4, 5, 7, 8, 9, 10, 11, 12 and 13 were
identified as non-immune cells based on the average expression of
PTPRC, as demonstrated in Fig. 2B and
C. The top five significantly DEGs (marker genes) among these
16 clusters are presented in Fig.
2D. Based on the information provided by the singleR and
CellMarker databases, nine distinct cell types out of the 16
clusters that were identified through the use of ‘FindAllMarkers’
were accurately annotated (Fig.
2E). According to Neftel et al (31), the following cell types were
considered malignant: Astrocytes, cancer stem cells, mesenchymal,
NPCs, oligodendrocytic precursor cells and radial glia. Notably,
the results showed significant heterogeneity in the proportions of
various cell types present among different patients (Fig. 2F). Consequently, gene expression
levels were analyzed in various cell types, revealing that CCNA2
and NEK2 were expressed in NPCs, belonging to cluster 11, which
were derived from patient ‘MGH143’ (Fig. 2G-I). Although clusters 3, 11 and 13
were defined as NPCs, only cluster 11 expressed CCNA2 and NEK2,
indicating that different subtypes of NPCs may possess distinct
functions. CCNA2 is a ubiquitously expressed member of the cyclin
family (59,60), it is commonly associated with cell
proliferation and it is expressed at high levels in many cancers
(61). NEK2 is a core component of
the human centrosome (62), where
it regulates a key step in the centrosome cycle, namely centrosome
disjunction (63), which has been
associated with the progression of a variety of cancers (64). Therefore, it was hypothesized that
cluster 11, which co-expressed CCNA2 and NEK2, has the potential to
stimulate cell proliferation and differentiation.
NPCs exhibit significant stemness in
GBM
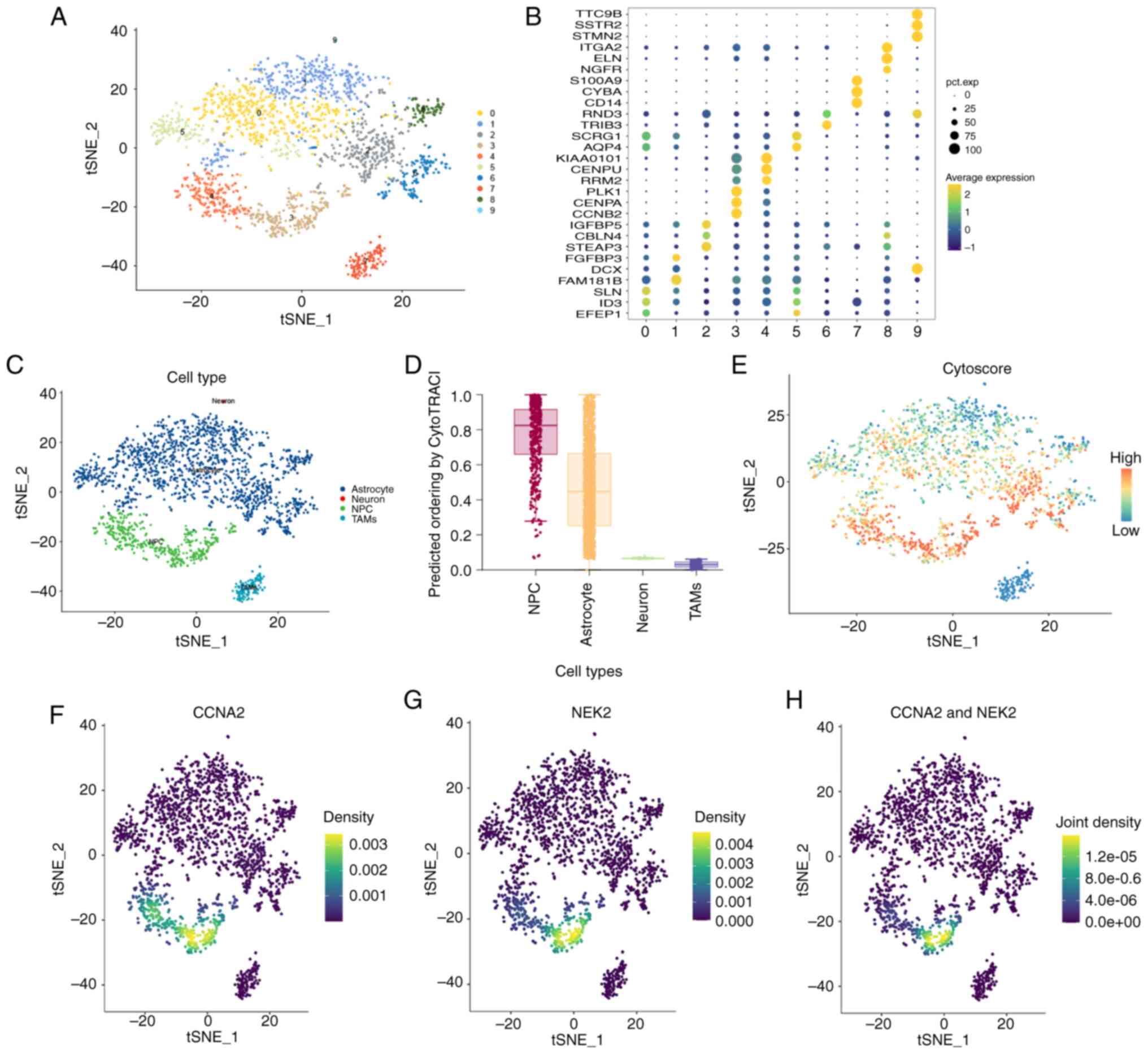
As aforementioned, it was revealed that cluster 11,
which exhibited high expression of CCNA2 and NEK2, was upregulated
in the ‘MHG143’ patient. GBM cells obtained from this patient were
then divided into 10 clusters, using t-SNE dimensionality reduction
(Fig. 3A). Marker genes were
identified as the top three differential genes in each cluster
(Fig. 3B). According to the SingleR
and CellMarker databases, four cell types including NPC,
astrocytes, neurons and tumor-associated macrophages (TAMs) were
annotated. Clusters 3 and 4 were identified as NPCs (Fig. 3C. Since NPCs may function as the
initiating cells of GBM (65,66),
these four cell types were evaluated using the CytoTRACE package.
The results demonstrated that NPCs had a higher potential to
proliferate and differentiate (Fig. 3D
and E). Moreover, when projected into these four cell types,
CCNA2 and NEK2 were co-expressed in cluster 3 (Fig. 3F-H).
The trajectory of GBM cell states
reveals branched progression
Based on the aforementioned results, it was found
that NPCs showed a higher level of stemness, indicating that these
cell types may contribute to the initiation and progression of GBM.
In addition, CCNA2 and NEK2 expression levels significantly
increased in NPCs. To comprehend the function of CCNA2 and NEK2 in
GBM, the pseudo-time analysis was utilized to delineate the
differentiation pathways of astrocytes and NPCs from the ‘MHG143’
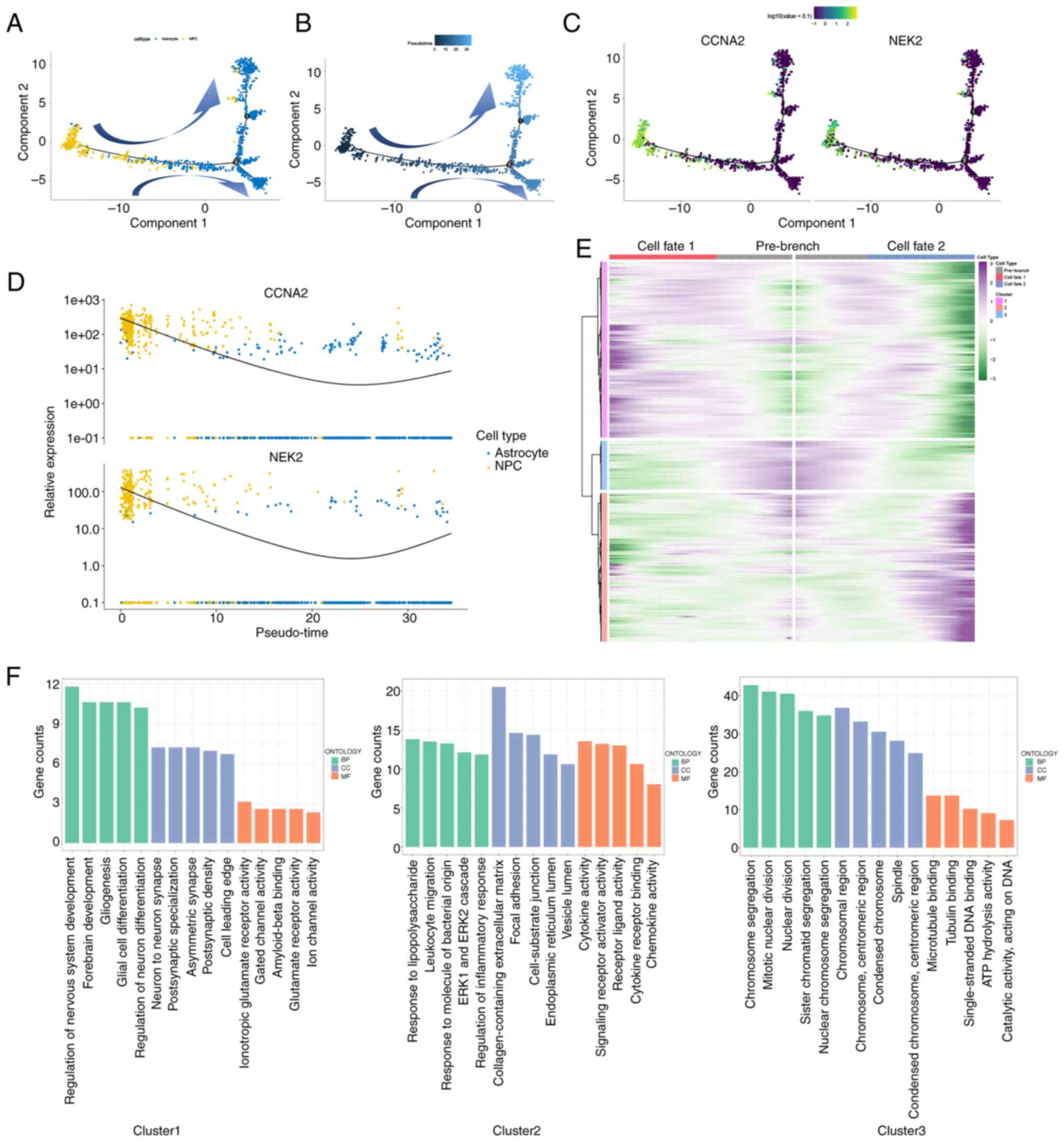
patient, employing the Monoce 2 package. As depicted in Fig. 4A and B, it was observed that
astrocytes and NPCs followed a trajectory of differentiation with
two branched progressions originating from NPCs. It was observed
that NPCs were mainly in the first half of pseudo-time trajectory,
whereas astrocytes were mainly in the second half. Furthermore,
NPCs highly expressed CCNA2 and NEK2, which were related to cell
cycle. It was hypothesized that NPCs may differentiate into
astrocytes, but the specific differentiation mechanism remains
unclear. Furthermore, when examining the different clusters, it was
observed that clusters 3 and 4 (NPCs) were primarily located in the
starting position, whereas the remaining clusters were dispersed
throughout various time periods (Fig.
S3). This supported the hypothesis that NPC contributes to the
onset of GBM. Moreover, as demonstrated in Fig. 4C, it can be observed that CCNA2 and
NEK2 expression levels were higher at the beginning and end stages
of GBM and exhibited a ‘U’-shaped pattern of distribution (Fig. 4D). It was hypothesized that CCNA2
and NEK2-highly expressing NPCs may have an advantage to grow in
the middle and later development of GBM, which has polyclonal
sources (67), but it requires
further research to investigate it.
As aforementioned, astrocytes and NPCs follow two
different paths at a distinct bifurcation point. According to
earlier studies, cells on different trajectories exhibit distinct
cellular functions or procedural changes. Therefore, analyzing
bifurcation points in cells will help understand the underlying
genes responsible for these changes (68,69).
The BEAM analysis was performed on this bifurcation point,
resulting in the clear division of 1,542 genes into three clusters
with distinct expression patterns. Cluster 3 comprises 190 genes,
including CCNA2 and NEK2, which were found to be overexpressed in
the primary stage, based on a branched expression pattern in
pseudo-time dimension. This suggested that these genes may play a
role in the progression of GBM (Fig.
4E). Furthermore, the GSEA analysis revealed that these
clusters exhibited distinct functions. Specifically, cluster 3 was
associated with chromosome segregation, mitotic nuclear division,
sister chromatid segregation, nuclear chromosome segregation and
nuclear division (Fig. 4F), which
are related to the cell cycle.
GBM prognosis may be associated with
P-NPCs
As aforementioned (Figs.
2 and 4), CCNA2 and NEK2 were
co-expressed in a group of early active cells known as NPCs. To
investigate the clinical significance of these cells in patients
with GBM, marker genes that were differentially expressed between
P-NPCs and negative cells in the patient ‘MGH143’ were identified
(Fig. 5A). The ‘AddModileScore’ was
utilized to assess all single-cell datasets and validate the
suitability of marker genes (Fig.
S4). The results were consistent with those shown in Fig. 2I, thus indicating that this set of
genes accurately represents P-NPCs. Subsequently, ssGSEA was
employed to investigate the expression of marker genes in gliomas
of varying grades in the CGGA database. Results indicated a
significant difference in expression levels with increasing WHO
grades, as demonstrated in Fig. 5B.
Comparable results were observed in TCGA database (Fig. 5C). As revealed in Fig. 5D, the goodness-of-fit was
investigated, and the results indicated that the proportion of
P-NPCs was more appropriate in GBM, suggesting that P-NPCs may be
more representative in GBM. The Kaplan-Meier analysis demonstrated
that patients with high expression of marker genes exhibited a
shorter overall survival in GBM and gliomas of all grades (Fig. 5E). These results indicated that
marker genes, which may represent P-NPCs, may be utilized to
differentiate the WHO grade of GBM patients and assess their
prognosis.
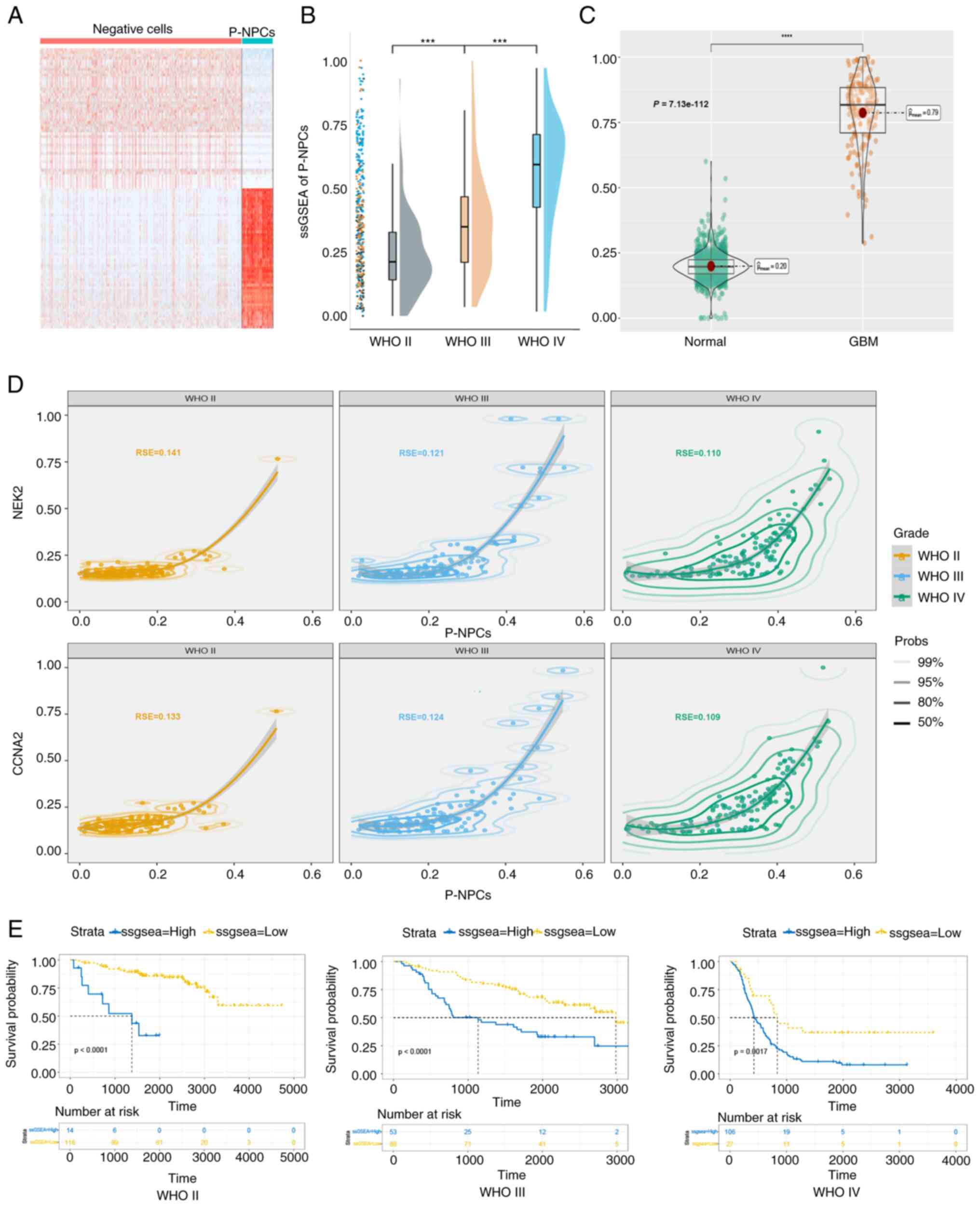 | Figure 5.GBM prognosis may be associated with
P-NPCs. (A) Heatmap of gene expression differences between
negative-cells and P-NPCs. (B) ssGSEA score of P-NPCs in the
Chinese Glioma Genome Atlas database across various grades of
glioma (***P<0.001, with independent t-test). (C) The ssGSEA
score of P-NPCs derived from both normal and GBM patient samples in
The Cancer Genome Atlas database (****P<0.0001, with independent
t-test). (D) Suitability of P-NPCs and CCNA2 and NEK2 expression in
glioma grades. (E) Association of high expression of marker genes
with shorter overall survival in patients with glioma, according to
the Kaplan-Meier analysis (P<0.0001 for WHO II, P<0.0001 for
WHO III and P=0.0017 for WHO IV, respectively, with log-rank test).
GBM, glioblastoma; P-NPCs; positive neural progenitor cells;
ssGSEA, single-sample gene set enrichment analysis; CCNA2, cyclin
A2; NEK2, NIMA related kinase 2; WHO, World Health
Organization. |
P-NPCs are associated with the
co-expression of CCNA2 and NEK2 in the G2M checkpoint pathway
Based on the aforementioned survival analysis for
patients with GBM in the CGGA database (Fig. 5), a total of 27 patients with poor
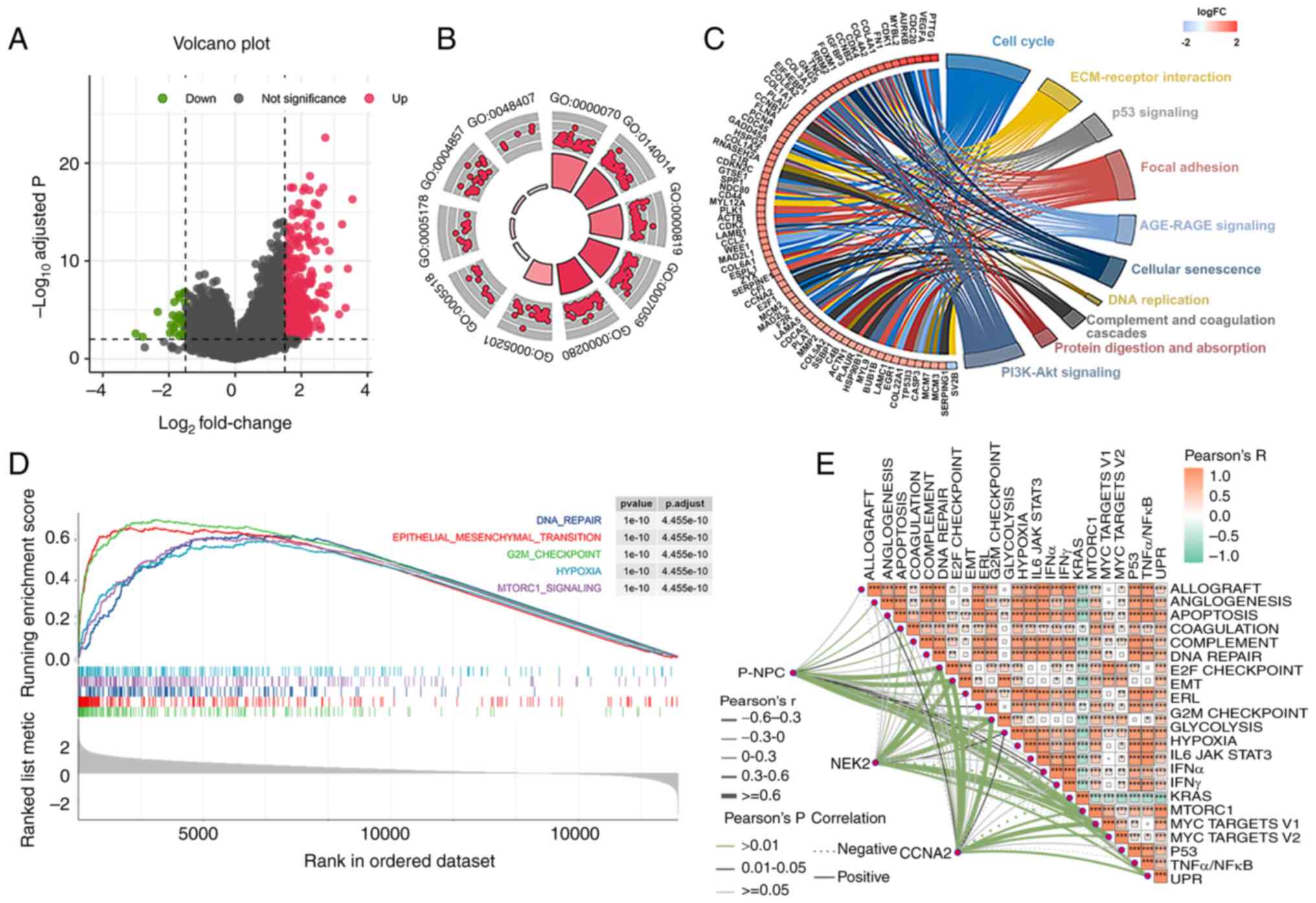
survival and 106 patients with improved survival rates were
observed. An analysis of DEGs in patients with varying prognoses
was then conducted. Results indicated that 455 genes were
significantly upregulated, whereas 29 genes were significantly
downregulated (Fig. 6A). Enrichment
analysis of GEO and KEGG pathways for the DEGs indicated that the
low survival rate of patients with GBM was associated with
increased activity in mitosis and cell cycles (Fig. 6B and C). Subsequently, the GSEA
analysis revealed that five pathways, namely DNA repair,
epithelial-mesenchymal transition, G2M checkpoint, hypoxia and
MTORC1 signaling, were significantly activated in patients with
poor survival (Fig. 6D). To
validate the aforementioned results, ssGSEA analysis was utilized
to reevaluate them. As a result, a total of 23 pathways, which
included the five aforementioned pathways, were found to be
significantly enriched (Fig. S5).
Furthermore, a correlation between the ssGSEA score of P-NPCs and
the expression of CCNA2 and NEK2 in the G2M checkpoint pathway was
observed (Fig. 6E). These results
suggested that the differential prognosis of these patients may be
related to the role of P-NPCs in driving the cell cycle and that
the G2M checkpoint may be a key factor in these differences.
G2M checkpoint pathway is activated in
P-NPCs
NPCs from patient ‘MGH143’ exhibited high activity
levels, but the specific functions of the other cells remain
unclear. To further examine the underlying pathways in scRNA-seq,
TAMs and neurons were removed. Next, the marker genes of the
remaining eight clusters were used to conduct significant
differential pathway analysis through GSEA. As a result, hallmark
pathways were significantly enriched, including mTORC1, hypoxia,
Myc-targets-v1, TNFα-signaling-via-NF-κB and G2M checkpoint
(Fig. 7A). The top four pathways
(mtorc1, cell cycle, oxidative, and NF-κB) were selected to
generate butterfly plots, based on the enrichment score of each
cluster (Fig. 7B). Clusters 3 and 4
belonged to NPCs, as demonstrated in Fig. 3C. These clusters revealed a strong
proliferation of E2F targets and G2M checkpoints, as depicted in
Fig. 7A. Furthermore, they were
assigned to cell cycle quadrants, as illustrated in Fig. 7B. However, they were not equally
distributed among the four quadrants. Cluster 3, as compared with
cluster 4, exhibited significantly enriched pathways, including the
mitotic spindle and the G2M checkpoint (Fig. 7C). This indicated a stronger
association with the cell cycle. The tricycle analysis (37) was used to evaluate the cell cycle
position of single-cell data from patients. The position of cells
in the cell cycle was denoted by an angle θ and cells in the same
stage always appeared at a similar θ. To accurately represent the
position of cells in the cell cycle, a circular color scale that
considers the circular nature of the cell cycle was utilized, with
positions ‘wrap around’ from 0 to 2π (Fig. 7D). For instance, cells at the G2/M
stage are represented by colors centered at 1.75π (Fig. 7D). It was revealed that cluster 11
(NPCs) accounted for a high proportion of cells in the G2/M phase
(Fig. 7D). In general, NPCs or a
subset of them (cluster 3) were strongly enriched at the G2M
checkpoint. These cells may contribute to GBM cell population
proliferation and disease progression.
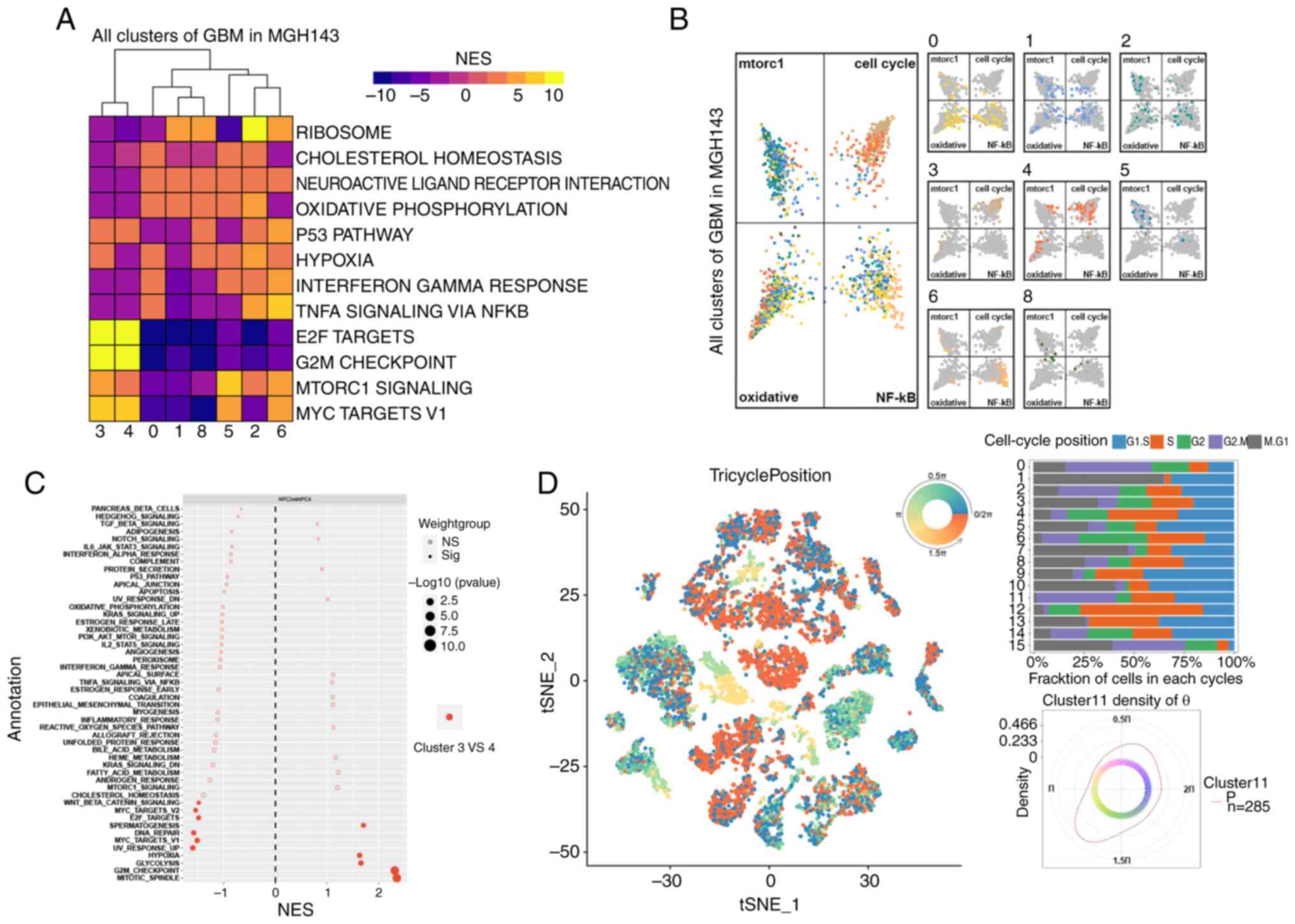 | Figure 7.Activation of the G2M checkpoint
pathway in positive neural progenitor cells. (A) Hallmark pathways,
including mTORC1, hypoxia, Myc-targets-v1, TNFα-signaling-via-NF-κB
and G2M checkpoint, were enriched. (B) Four pathways (mTORC1, cell
cycle, oxidative and NF-κB) were selected for butterfly plots based
on cluster enrichment scores. (C) Cluster 3 had enriched pathways,
including mitotic spindle and G2M checkpoint, as compared with
cluster 4. (D) Tricycle analysis of the cell cycle position of
single-cell data. GBM, glioblastoma; t-SNE, t-distributed
stochastic neighbor embedding; NES, normalized enrichment
score. |
Establishment and evaluation of a
nomogram with the TCGA dataset
The forest plot presented P-NPCs, age and
chemotherapy score as independent risk factors (Fig. S6). As radiotherapy has been widely
used in clinical practice and its effectiveness has been previously
reported in the literature (70,71),
it was included in the present study. The Schoenfeld residual test
indicated that all variables equally satisfied the assumption of
proportional hazards (Fig. S6).
Outliers were not observed, based on the deviance residual test
(Fig. S6). After considering all
the aforementioned significant predictive factors, a comprehensive
nomogram that includes P-NPCs, age, chemotherapy and radiotherapy
score was developed (Fig. 8A). The
calibration curves for one, two and three years revealed a
satisfactory calibration efficiency. A closer alignment with the
dashed line indicated an improved prediction performance (Fig. 8B). The DCA was used to evaluate the
clinical application of the nomogram and the net benefit of various
prediction models at different threshold probabilities. This was
achieved by weighing the benefits against the harms and minimizing
the latter. The comprehensive nomogram demonstrated a more
favorable probability and an improved net benefit compared with
P-NPCs and clinical index, as shown in Fig. 8C-E. Subsequently, the nomogram score
model and clinical index score model were evaluated using the
concordance index. Results demonstrated that the nomogram score had
a higher prediction accuracy than the clinical index score
(Fig. 8F). Furthermore, the
time-dependent ROC curve analysis demonstrated that the predictive
performance of the nomogram gradually improved over time (Fig. 8G). The Kaplan-Meier analysis
indicated that a higher nomogram score was associated with a poorer
prognosis for patients with GBM (Fig.
8H). Therefore, a comprehensive nomogram was established based
on multiple prognostic factors that exceeded the predictive power
of each factor alone. The nomogram may assist clinicians in making
more precise assessments of patient prognosis.
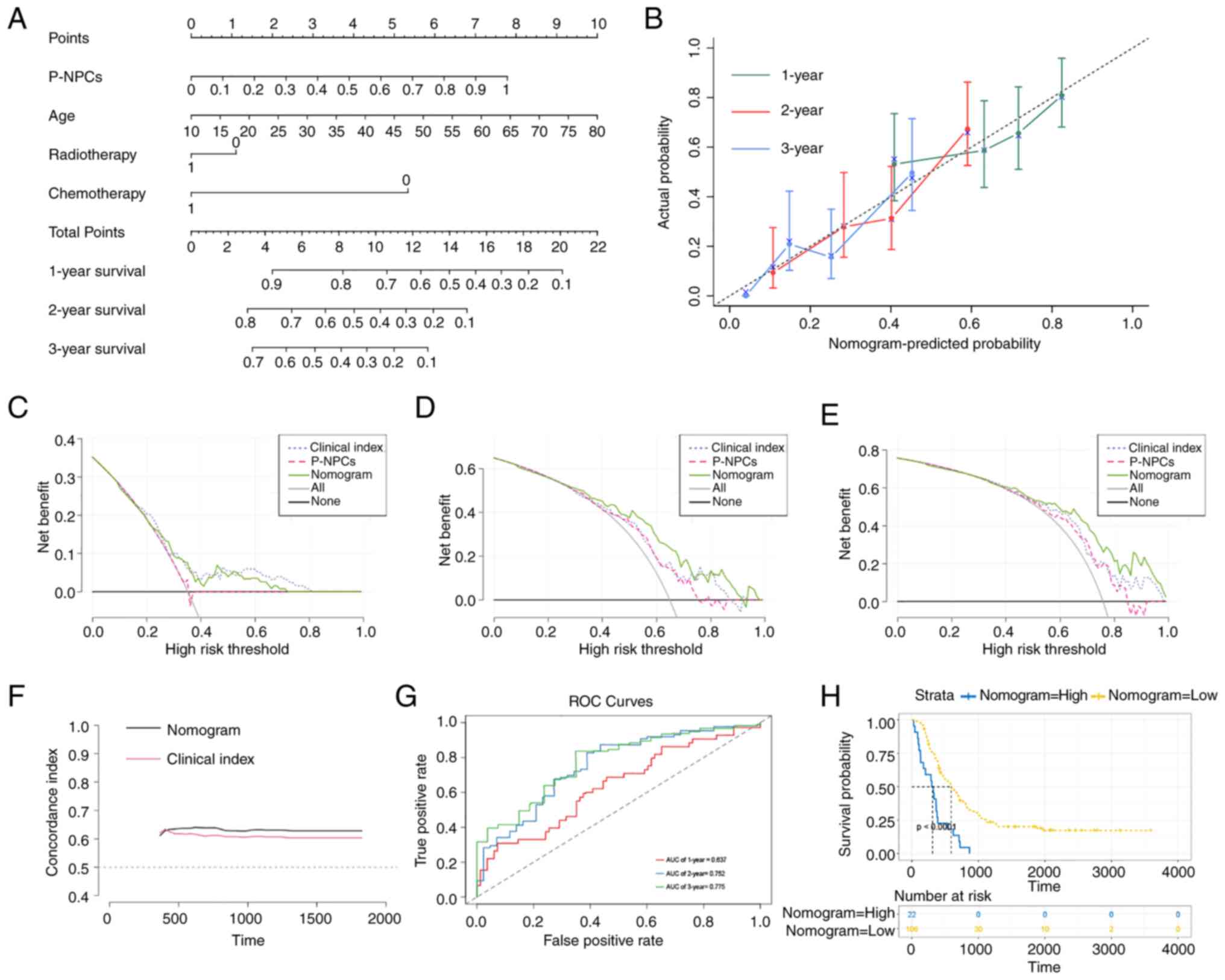 | Figure 8.Establishment and evaluation of a
nomogram with TCGA dataset. (A) A nomogram predicts 1-, 2- and
3-year overall survival probability in GBM by considering P-NPCs,
age, chemotherapy and radiotherapy scores. (B) The calibration
curves for 1, 2 and 3 years showed improved calibration potential
in TCGA cohort. The black dotted lines represent the ideal
predictive model, whereas the solid lines represent the nomogram
models for each respective year. (C-E) The decision curve analysis
was used to evaluate the clinical application of the nomogram and
the net benefit of various prediction models when the threshold
probability is between 0 and 0.80. (F) The concordance index was
used to evaluate the accuracy and discrimination of the nomogram's
predicted values vs. the clinical index model. (G) Time-dependent
ROC curve analysis was conducted for the nomogram at 1, 2 and 3
years in TCGA cohort. (H) The Kaplan-Meier analysis found that
higher nomogram scores were linked to poorer prognosis in patients
with GBM (P<0.0001 with log-rank test). TCGA, The Cancer Genome
Atlas; GBM, glioblastoma; P-NPCs, positive neural progenitor cells;
ROC, receiver operating characteristic. |
Discussion
The cell cycle, a complex process tightly regulated
by various proteins (67), is
closely linked to the development and advancement of cancer
(72,73). NEK2 is a member of the NIMA-related
family of serine/threonine protein kinases, which are considered to
play a role in regulating the cell cycle (74). Upregulation of NEK2 in human cells
leads to premature splitting of the centrosome, whereas
overexpression of a NEK2 kinase-dead mutant results in centrosome
abnormalities and aneuploidy (10,75).
These factors are significant contributors to tumorigenesis
(76). NEK2 has two splice
variants, namely NEK2A and NEK2B. NEK2A is necessary for centrosome
separation during the G2/M transition (77). The abnormal expression of NEK2A may
play a role in regulating genetic stability and tumorigenesis. In
the present study, a positive correlation was observed between the
expression of CCNA2 and NEK2 and the progression of glioma.
Furthermore, CCNA2 and NEK2 were upregulated in a subset of P-NPCs
in GBM, which exhibited significant proliferation and progression
properties. CCNA2 is located on chromosome 4 and is encoded by the
human CCNA2 gene. Belonging to the highly conserved cyclin family,
this protein promotes progression through the S-phase and
transition from G2 to M phase by binding to CDK in the mitotic cell
cycle (78). Jiang et al
(79) proposed that CCNA2 promotes
proliferation, migration, invasion and regulates macrophage
polarization in glioma. It is recognized that changes in cell cycle
proteins may trigger cancer. CCNA2 and NEK2 are classified as cell
cycle regulatory genes, which are crucial in the advancement and
prognosis of pancreatic cancer (28). However, the correlation and
underlying mechanism between CCNA2 and NEK2 has not been fully
explored in GBM. The present study presented a novel finding on the
co-expression of CCNA2 and NEK2 in GBM and their association with
NPCs, thus suggesting their involvement in regulating the cell
cycle.
GBM are the most common malignant tumors in the
nervous system, which are known for their rapid progression and
short survival rates (80,81). Therapeutic approaches for GBM have
slowly progressed, unlike other multimodal therapies for tumors
(82). Ongoing efforts are being
made to understand the highly heterogeneous nature of GBM.
Differences have been observed among various tumor types,
individuals with identical diagnoses, non-tumor cell types and
states and individual tumor cell clones (83). NPCs are the precursor cells of the
central nervous system (CNS) that generate many, if not all, of the
glial and neuronal cell types that populate the CNS (84). It has been proposed that NPCs are
the preferred cell of origin for GBM (85). In addition, NPCs have a strong
potential to migrate (86), renew
(87) and maintain the population
(88,89). In the present study, patients with
GBM were analyzed at the single-cell level and it was discovered
that CCNA2 and NEK2 were co-expressed in NPCs. These findings
suggested that they may have a significant role in the development
and progression of GBM. Furthermore, the abundance of NPCs in
patient ‘MGH143’ was higher during the initial stages of
tumorigenesis and was similar to the peak expression of CCNA2 and
NEK2. Therefore, it was hypothesized that changes in CCNA2 and NEK2
may drive the progression of NPCs and ultimately contribute to the
development of GBM. CCNA2 and NEK2 were found to be differentially
expressed in NPC subtypes, indicating cellular heterogeneity in the
glioma-associated microenvironment. This finding agrees with
previous research on NPCs heterogeneity (90).
In the present study, NPCs exhibiting high
expression levels of CCNA2 and NEK2 as P-NPCs were defined.
Moreover, the marker genes based on P-NPCs significantly
distinguish the prognosis of patients with GBM. In GBM, NPCs highly
expressed stemness-associated cell membrane antigens such as CD133,
CD15/SSEA, CD44, or A2B5 and intracellular markers such as Sox2 and
Nestin (91,92) and have demonstrated the potential
for cellular transitions (31).
Similar to normal neurogenesis processes, NPCs may generate more
differentiated phenotypes with astrocytic features in GBM (91). As aforementioned, CCNA2 and NEK2 are
closely related to the cell cycle, which is responsible for cell
differentiation and fate (93). The
origin of glioma cells is polyclonal (40) and in the development of cancer, the
population with competitive advantages will be selected to develop
(94). Therefore, it was
hypothesized by the authors that CCNA2 and NEK2 are only expressed
in some NPC subtypes, which may have a competitive advantage, but
the specific process requires further discussion. In the analysis
of different pathways among these patients with varying prognoses,
it was found that the G2M checkpoint was highly enriched. The G2/M
checkpoint prevents cells with damaged DNA from entering mitosis,
allowing repair of DNA that was damaged during the late S or G2
phases before mitosis. A weakened G2/M checkpoint under therapeutic
conditions may trigger cell death through mitotic catastrophe in
cells with irreparable DNA damage and faulty mitotic machinery
(95). It was revealed that the G2M
checkpoint is significantly increased in patients with poor
prognosis. This increase may inhibit mitotic catastrophe, which in
turn promotes rapid tumor growth. Currently, there are numerous
drugs that target the G2M checkpoint (96). The present study may offer a novel
approach to the pharmacological mechanism. By recognizing the
significance of P-NPCs, a comprehensive nomogram that incorporates
clinical characteristics to predict patient prognosis more
accurately has been developed.
However, the data on the regulatory relationship
between NEK2 and CCNA2 expression in GBM are limited, which cannot
accurately reflect the heterogeneity of glioma. At the same time,
single-cell sequencing loses the information of spatial location,
thus it is impossible to characterize their co-localization
relationship in spatial location. Mapping research through multiple
samples will be conducted. Moreover, the authors have marginal
information on the regulatory association between CCNA2 and NEK2 in
GBM and the role they play in different stages of the cell cycle is
unclear, thus more experiments are needed to determine their
regulatory role in GBM. To date, it is not feasible to precisely
determine the composition of these cells before surgery. This
information is only inferred through postoperative sequencing. To
address this practical issue, a non-invasive preoperative
assessment method should be developed for these cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82072781 and 81602207) and Medical
Foundation-Clinical Integration Program of Xi'an Jiaotong
University (grant no. YXJLRH2022040).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MDW and PM conceptualized and designed the study.
HYZ, TW and WW developed methodology. HYZ, WW, YYC and YCW acquired
data. HYZ, BCZ and WW analysed and interpreted the data. HYZ, WW
and YYC wrote the manuscript. YYC performed experiments. HYZ and
YCW confirm the authenticity of all the raw data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
The WHO grade II, III and IV gliomas were obtained
from patient samples with a pathological diagnosis who underwent
craniotomy at the First Affiliated Hospital of Xi'an Jiao Tong
University (Shaanxi, China). The research involving the utilization
of these biological specimens received ethical approval from the
Ethics Committee of the hospital located in Xi'an, Shaanxi, China
(approval no. XJTU1AF2022LSK-428). Informed consent was obtained
through the signing of consent forms for the utilization of
clinical samples, all experimental protocols used in the present
study were in accordance with the guidelines of the Declaration of
Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Price M, Neff C, Cioffi G,
Waite KA, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2015–2019. Neuro Oncol. 24 (Suppl
5):v1–v95. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu S, Tang L, Li X, Fan F and Liu Z:
Immunotherapy for glioma: Current management and future
application. Cancer Lett. 476:1–12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu Y, Lang F and Yang C: NRF2 in human
neoplasm: Cancer biology and potential therapeutic target.
Pharmacol Ther. 217:1076642021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Asad AS, Candia AJN, González N, Zuccato
CF, Seilicovich A and Candolfi M: Current non-viral gene therapy
strategies for the treatment of glioblastoma. Curr Med Chem.
28:7729–7748. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shergalis A, Bankhead A III, Luesakul U,
Muangsin N and Neamati N: Current challenges and opportunities in
treating glioblastoma. Pharmacol Rev. 70:412–445. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang PH, Xu AM and White FM: Oncogenic
EGFR signaling networks in glioma. Sci Signal. 2:re62009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu H, Liu B, Hou X, Pang B, Guo P, Jiang
W, Ding Q, Zhang R, Xin T, Guo H, et al: Overexpression of
NIMA-related kinase 2 is associated with poor prognoses in
malignant glioma. J Neurooncol. 132:409–417. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiang J, Alafate W, Wu W, Wang Y, Li X,
Xie W, Bai X, Li R, Wang M and Wang J: NEK2 enhances malignancies
of glioblastoma via NIK/NF-κB pathway. Cell Death Dis. 13:582022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xia J, Franqui Machin R, Gu Z and Zhan F:
Role of NEK2A in human cancer and its therapeutic potentials.
Biomed Res Int. 2015:8624612015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Faragher AJ and Fry AM: Nek2A kinase
stimulates centrosome disjunction and is required for formation of
bipolar mitotic spindles. Mol Biol Cell. 14:2876–2889. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jeong Y, Lee J, Kim K, Yoo JC and Rhee K:
Characterization of NIP2/centrobin, a novel substrate of Nek2, and
its potential role in microtubule stabilization. J Cell Sci.
120:2106–2116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhong X, Guan X, Liu W and Zhang L:
Aberrant expression of NEK2 and its clinical significance in
non-small cell lung cancer. Oncol Lett. 8:1470–1476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu Z, Xia J, Xu H, Frech I, Tricot G and
Zhan F: NEK2 promotes aerobic glycolysis in multiple myeloma
through regulating splicing of pyruvate kinase. J Hematol Oncol.
10:172017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X, Huang X, Xu J, Li E, Lao M, Tang
T, Zhang G, Guo C, Zhang X, Chen W, et al: NEK2 inhibition triggers
anti-pancreatic cancer immunity by targeting PD-L1. Nat Commun.
12:45362021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee J and Gollahon L: Mitotic
perturbations induced by Nek2 overexpression require interaction
with TRF1 in breast cancer cells. Cell Cycle. 12:3599–3614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fang Y and Zhang X: Targeting NEK2 as a
promising therapeutic approach for cancer treatment. Cell Cycle.
15:895–907. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qi C, Lei L, Hu J, Wang G, Liu J and Ou S:
Serine incorporator 2 (SERINC2) expression predicts an unfavorable
prognosis of low-grade glioma (LGG): Evidence from bioinformatics
analysis. J Mol Neurosci. 70:1521–1532. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pagano M, Pepperkok R, Verde F, Ansorge W
and Draetta G: Cyclin A is required at two points in the human cell
cycle. EMBO J. 11:961–971. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Murphy M, Stinnakre MG, Senamaud-Beaufort
C, Winston NJ, Sweeney C, Kubelka M, Carrington M, Bréchot C and
Sobczak-Thépot J: Delayed early embryonic lethality following
disruption of the murine cyclin A2 gene. Nat Genet. 15:83–86. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang A, Zhou Y, Gong W, Pan X, Gan X, Wu
Z, Liu B, Qu L and Wang L: CCNA2 as an immunological biomarker
encompassing tumor microenvironment and therapeutic response in
multiple cancer types. Oxid Med Cell Longev. 2022:59105752022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Zhong Q, Li Z, Lin Z, Chen H and
Wang P: Integrated profiling identifies CCNA2 as a potential
biomarker of immunotherapy in breast cancer. Onco Targets Ther.
14:2433–2448. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Z, Zhang Y, Zhou Y, Wang F, Yin C, Ding
L and Zhang S: Tanshinone IIA suppresses the progression of lung
adenocarcinoma through regulating CCNA2-CDK2 complex and AURKA/PLK1
pathway. Sci Rep. 11:236812021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gan Y, Li Y, Li T, Shu G and Yin G: CCNA2
acts as a novel biomarker in regulating the growth and apoptosis of
colorectal cancer. Cancer Manag Res. 10:5113–5124. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai Y and Yang W: PKMYT1 regulates the
proliferation and epithelial-mesenchymal transition of oral
squamous cell carcinoma cells by targeting CCNA2. Oncol Lett.
23:632022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bendris N, Arsic N, Lemmers B and
Blanchard JM: Cyclin A2, Rho GTPases and EMT. Small GTPases.
3:225–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Ying Y, Xie H, Jin K, Yan H, Wang S,
Xu M, Xu X, Wang X, Yang K, et al: Dual regulatory role of CCNA2 in
modulating CDK6 and MET-mediated cell-cycle pathway and EMT
progression is blocked by miR-381-3p in bladder cancer. FASEB J.
33:1374–1388. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Liu X, Zhou W, Lu S, Wu C, Wu Z,
Liu R, Li X, Wu J, Liu Y, et al: Identification of key genes
associated with the process of hepatitis B inflammation and cancer
transformation by integrated bioinformatics analysis. Front Genet.
12:6545172021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou Z, Cheng Y, Jiang Y, Liu S, Zhang M,
Liu J and Zhao Q: Ten hub genes associated with progression and
prognosis of pancreatic carcinoma identified by co-expression
analysis. Int J Biol Sci. 14:124–136. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao Z, Zhang KN, Wang Q, Li G, Zeng F,
Zhang Y, Wu F, Chai R, Wang Z, Zhang C, et al: Chinese glioma
genome atlas (CGGA): A comprehensive resource with functional
genomic data from Chinese glioma patients. Genomics Proteomics
Bioinformatics. 19:1–12. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Neftel C, Laffy J, Filbin MG, Hara T,
Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM,
et al: An integrative model of cellular states, plasticity, and
genetics for glioblastoma. Cell. 178:835–849.e21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Satija R, Farrell JA, Gennert D, Schier AF
and Regev A: Spatial reconstruction of single-cell gene expression
data. Nat Biotechnol. 33:495–502. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aran D, Looney AP, Liu L, Wu E, Fong V,
Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al:
Reference-based analysis of lung single-cell sequencing reveals a
transitional profibrotic macrophage. Nat Immunol. 20:163–172. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu C, Li T, Xu Y, Zhang X, Li F, Bai J,
Chen J, Jiang W, Yang K, Ou Q, et al: CellMarker 2.0: An updated
database of manually curated cell markers in human/mouse and web
tools based on scRNA-seq data. Nucleic Acids Res. 51(D1):
D870–D876. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Z, Wang ZX, Chen YX, Wu HX, Yin L,
Zhao Q, Luo HY, Zeng ZL, Qiu MZ and Xu RH: Integrated analysis of
single-cell and bulk RNA sequencing data reveals a pan-cancer
stemness signature predicting immunotherapy response. Genome Med.
14:452022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Trapnell C, Cacchiarelli D, Grimsby J,
Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS and
Rinn JL: The dynamics and regulators of cell fate decisions are
revealed by pseudotemporal ordering of single cells. Nat
Biotechnol. 32:381–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng SC, Stein-O'Brien G, Augustin JJ,
Slosberg J, Carosso GA, Winer B, Shin G, Bjornsson HT, Goff LA and
Hansen KD: Universal prediction of cell-cycle position using
transfer learning. Genome Biol. 23:412022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liberzon A, Birger C, Thorvaldsdóttir H,
Ghandi M, Mesirov JP and Tamayo P: The Molecular signatures
database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Querec TD, Akondy RS, Lee EK, Cao W,
Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, et al:
Systems biology approach predicts immunogenicity of the yellow
fever vaccine in humans. Nat Immunol. 10:116–125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kinoshita M, Uchida T, Sato A, Nakashima
M, Nakashima H, Shono S, Habu Y, Miyazaki H, Hiroi S and Seki S:
Characterization of two F4/80-positive Kupffer cell subsets by
their function and phenotype in mice. J Hepatol. 53:903–910. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang L, Li Z, Skrzypczynska KM, Fang Q,
Zhang W, O'Brien SA, He Y, Wang L, Zhang Q, Kim A, et al:
Single-cell analyses inform mechanisms of myeloid-targeted
therapies in colon cancer. Cell. 181:442–459.e29. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haber AL, Biton M, Rogel N, Herbst RH,
Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et
al: A single-cell survey of the small intestinal epithelium.
Nature. 551:333–339. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qiu J, Qu X, Wang Y, Guo C, Lv B, Jiang Q,
Su W, Wang L and Hua K: Single-cell landscape highlights
heterogenous microenvironment, novel immune reaction patterns,
potential biomarkers and unique therapeutic strategies of cervical
squamous carcinoma, human papillomavirus-associated (HPVA) and
non-HPVA adenocarcinoma. Adv Sci (Weinh). 10:e22049512023.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
van der Poel M, Ulas T, Mizee MR, Hsiao
CC, Miedema SSM, Adelia Schuurman KG, Helder B, Tas SW, Schultze
JL, et al: Transcriptional profiling of human microglia reveals
grey-white matter heterogeneity and multiple sclerosis-associated
changes. Nat Commun. 10:11392019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bassez A, Vos H, Van Dyck L, Floris G,
Arijs I, Desmedt C, Boeckx B, Vanden Bempt M, Nevelsteen I, Lambein
K, et al: A single-cell map of intratumoral changes during anti-PD1
treatment of patients with breast cancer. Nat Med. 27:820–832.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wendisch D, Dietrich O, Mari T, von
Stillfried S, Ibarra IL, Mittermaier M, Mache C, Chua RL, Knoll R,
Timm S, et al: SARS-CoV-2 infection triggers profibrotic macrophage
responses and lung fibrosis. Cell. 184:6243–6261.e27. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gong Z, Li Q, Shi J, Li P, Hua L, Shultz
LD and Ren G: Immunosuppressive reprogramming of neutrophils by
lung mesenchymal cells promotes breast cancer metastasis. Sci
Immunol. 8:eadd52042023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kennedy A, Waters E, Rowshanravan B, Hinze
C, Williams C, Janman D, Fox TA, Booth C, Pesenacker AM, Halliday
N, et al: Differences in CD80 and CD86 transendocytosis reveal CD86
as a key target for CTLA-4 immune regulation. Nat Immunol.
23:1365–1378. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Amiry-Moghaddam M: AQP4 and the fate of
gliomas. Cancer Res. 79:2810–2811. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J,
Wang K, Wang Z, Nam Y, Jiang B, et al: Mutational landscape of
secondary glioblastoma guides MET-targeted trial in brain tumor.
Cell. 175:1665–1678.e18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kakogiannos N, Ferrari L, Giampietro C,
Scalise AA, Maderna C, Ravà M, Taddei A, Lampugnani MG, Pisati F,
Malinverno M, et al: JAM-A acts via C/EBP-α to promote claudin-5
expression and enhance endothelial barrier function. Circ Res.
127:1056–1073. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hosmann A, Jaber M, Roetzer-Pejrimovsky T,
Timelthaler G, Borkovec M, Kiesel B, Wadiura LI, Millesi M, Mercea
PA, Phillips J, et al: CD34 microvascularity in low-grade glioma:
Correlation with 5-aminolevulinic acid fluorescence and patient
prognosis in a multicenter study at three specialized centers. J
Neurosurg. 138:1281–1290. 20232PubMed/NCBI
|
|
55
|
Agostini M, Amato F, Vieri ML, Greco G,
Tonazzini I, Baroncelli L, Caleo M, Vannini E, Santi M, Signore G
and Cecchini M: Glial-fibrillary-acidic-protein (GFAP) biomarker
detection in serum-matrix: Functionalization strategies and
detection by an ultra-high-frequency surface-acoustic-wave
(UHF-SAW) lab-on-chip. Biosens Bioelectron. 172:1127742021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gai QJ, Fu Z, He J, Mao M, Yao XX, Qin Y,
Lan X, Zhang L, Miao JY, Wang YX, et al: EPHA2 mediates PDGFA
activity and functions together with PDGFRA as prognostic marker
and therapeutic target in glioblastoma. Signal Transduct Target
Ther. 7:332022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Suvà ML, Rheinbay E, Gillespie SM, Patel
AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et
al: Reconstructing and reprogramming the tumor-propagating
potential of glioblastoma stem-like cells. Cell. 157:580–594. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang Y, Chu L, Zeng Z, Xu S, Yang H, Zhang
X, Jia J, Long N, Hu Y and Liu J: Four specific biomarkers
associated with the progression of glioblastoma multiforme in older
adults identified using weighted gene co-expression network
analysis. Bioengineered. 12:6643–6654. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bendris N, Loukil A, Cheung C, Arsic N,
Rebouissou C, Hipskind R, Peter M, Lemmers B and Blanchard JM:
Cyclin A2: A genuine cell cycle regulator? Biomol Concepts.
3:535–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Loukil A, Cheung CT, Bendris N, Lemmers B,
Peter M and Blanchard JM: Cyclin A2: At the crossroads of cell
cycle and cell invasion. World J Biol Chem. 6:346–350. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liang W, Guan H, He X, Ke W, Xu L, Liu L,
Xiao H and Li Y: Down-regulation of SOSTDC1 promotes thyroid cancer
cell proliferation via regulating cyclin A2 and cyclin E2.
Oncotarget. 6:31780–31791. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Andersen JS, Wilkinson CJ, Mayor T,
Mortensen P, Nigg EA and Mann M: Proteomic characterization of the
human centrosome by protein correlation profiling. Nature.
426:570–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
O'regan L, Blot J and Fry AM: Mitotic
regulation by NIMA-related kinases. Cell Div. 2:252007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Huang X, Zhang G, Tang T, Gao X and Liang
T: One shoot, three birds: Targeting NEK2 orchestrates
chemoradiotherapy, targeted therapy, and immunotherapy in cancer
treatment. Biochim Biophys Acta Rev Cancer. 1877:1886962022.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Alcantara Llaguno S, Chen J, Kwon CH,
Jackson EL, Li Y, Burns DK, Alvarez-Buylla A and Parada LF:
Malignant astrocytomas originate from neural stem/progenitor cells
in a somatic tumor suppressor mouse model. Cancer Cell. 15:45–56.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu C, Sage JC, Miller MR, Verhaak RG,
Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L
and Zong H: Mosaic analysis with double markers reveals tumor cell
of origin in glioma. Cell. 146:209–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hydbring P, Malumbres M and Sicinski P:
Non-canonical functions of cell cycle cyclins and cyclin-dependent
kinases. Nat Rev Mol Cell Biol. 17:280–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Huang J, Li Q, Peng Q, Xie Y, Wang W, Pei
C, Zhao Y, Liu R, Huang L, Li T, et al: Single-cell RNA sequencing
reveals heterogeneity and differential expression of decidual
tissues during the peripartum period. Cell Prolif. 54:e129672021.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhao T, Fu Y, Zhu J, Liu Y, Zhang Q, Yi Z,
Chen S, Jiao Z, Xu X, Xu J, et al: Single-cell RNA-Seq reveals
dynamic early embryonic-like programs during chemical
reprogramming. Cell Stem Cell. 23:31–45.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Balducci M, Chiesa S, Diletto B,
D'Agostino GR, Mangiola A, Manfrida S, Mantini G, Albanese A,
Fiorentino A, Frascino V, et al: Low-dose fractionated radiotherapy
and concomitant chemotherapy in glioblastoma multiforme with poor
prognosis: A feasibility study. Neuro Oncol. 14:79–86. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kong L, Gao J, Hu J, Lu R, Yang J, Qiu X,
Hu W and Lu JJ: Carbon ion radiotherapy boost in the treatment of
glioblastoma: A randomized phase I/III clinical trial. Cancer
Commun (Lond). 39:52019.PubMed/NCBI
|
|
72
|
Kops GJ, Weaver BA and Cleveland DW: On
the road to cancer: Aneuploidy and the mitotic checkpoint. Nat Rev
Cancer. 5:773–785. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Nicholson JM and Cimini D: How mitotic
errors contribute to karyotypic diversity in cancer. Adv Cancer
Res. 112:43–75. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Fry AM, O'Regan L, Sabir SR and Bayliss R:
Cell cycle regulation by the NEK family of protein kinases. J Cell
Sci. 125:4423–4433. 2012.PubMed/NCBI
|
|
75
|
Fry AM, Meraldi P and Nigg EA: A
centrosomal function for the human Nek2 protein kinase, a member of
the NIMA family of cell cycle regulators. EMBO J. 17:470–481. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li JJ and Li SA: Mitotic kinases: the key
to duplication, segregation, and cytokinesis errors, chromosomal
instability, and oncogenesis. Pharmacol Ther. 111:974–984. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Helps NR, Luo X, Barker HM and Cohen PT:
NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase
localized to centrosomes, is complexed to protein phosphatase 1.
Biochem J. 349:509–518. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chotiner JY, Wolgemuth DJ and Wang PJ:
Functions of cyclins and CDKs in mammalian gametogenesis†. Biol
Reprod. 101:591–601. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jiang F, Luo F, Zeng N, Mao Y, Tang X,
Wang J, Hu Y and Wu C: Characterization of fatty acid
metabolism-related genes landscape for predicting prognosis and
aiding immunotherapy in glioma patients. Front Immunol.
13:9021432022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ostrom QT, Bauchet L, Davis FG, Deltour I,
Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh
KM, et al: The epidemiology of glioma in adults: A ‘state of the
science’ review. Neuro Oncol. 16:896–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sturm D, Pfister SM and Jones DTW:
Pediatric gliomas: Current concepts on diagnosis, biology, and
clinical management. J Clin Oncol. 35:2370–2377. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ashby LS and Ryken TC: Management of
malignant glioma: Steady progress with multimodal approaches.
Neurosurg Focus. 20:E32006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Andersen BM, Faust Akl C, Wheeler MA,
Chiocca EA, Reardon DA and Quintana FJ: Glial and myeloid
heterogeneity in the brain tumour microenvironment. Nat Rev Cancer.
21:786–802. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Martínez-Cerdeño V and Noctor SC: Neural
progenitor cell terminology. Front Neuroanat. 12:1042018.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhu Y, Guignard F, Zhao D, Liu L, Burns
DK, Mason RP, Messing A and Parada LF: Early inactivation of p53
tumor suppressor gene cooperating with NF1 loss induces malignant
astrocytoma. Cancer Cell. 8:119–130. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zarco N, Norton E, Quiñones-Hinojosa A and
Guerrero-Cázares H: Overlapping migratory mechanisms between neural
progenitor cells and brain tumor stem cells. Cell Mol Life Sci.
76:3553–3570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zheng H, Ying H, Yan H, Kimmelman AC,
Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al: p53
and Pten control neural and glioma stem/progenitor cell renewal and
differentiation. Nature. 455:1129–1133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Jung E, Alfonso J, Osswald M, Monyer H,
Wick W and Winkler F: Emerging intersections between neuroscience
and glioma biology. Nat Neurosci. 22:1951–1960. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Schonberg DL, Lubelski D, Miller TE and
Rich JN: Brain tumor stem cells: Molecular characteristics and
their impact on therapy. Mol Aspects Med. 39:82–101. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kohwi M and Doe CQ: Temporal fate
specification and neural progenitor competence during development.
Nat Rev Neurosci. 14:823–838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yabo YA, Niclou SP and Golebiewska A:
Cancer cell heterogeneity and plasticity: A paradigm shift in
glioblastoma. Neuro Oncol. 24:669–682. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Liu L, Michowski W, Kolodziejczyk A and
Sicinski P: The cell cycle in stem cell proliferation, pluripotency
and differentiation. Nat Cell Biol. 21:1060–1067. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Meacham CE and Morrison SJ: Tumour
heterogeneity and cancer cell plasticity. Nature. 501:328–337.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Roninson IB, Broude EV and Chang BD: If
not apoptosis, then what? Treatment-induced senescence and mitotic
catastrophe in tumor cells. Drug Resist Updat. 4:303–313. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Castro-Gamero AM, Pezuk JA, Brassesco MS
and Tone LG: G2/M inhibitors as pharmacotherapeutic opportunities
for glioblastoma: The old, the new, and the future. Cancer Biol
Med. 15:354–374. 2018. View Article : Google Scholar : PubMed/NCBI
|