Introduction
Globally, colorectal cancer (CRC) is among the most
commonly diagnosed malignancies. Despite improvements in operative
techniques and anticancer drugs, CRC ranks as the second highest
cause of all cancer-associated mortality worldwide (1). Liver metastasis (LM) is responsible
for approximately two-thirds of CRC related deaths (2). In total, ~25–50% of patients with CRC
develop LM during the whole course of the disease (3). The 5-year survival rate of patients
with CRC liver metastases (CRLM) is markedly shorter in comparison
with the survival rate of patients without LM (16.9 vs. 70.4%,
respectively) (4). However, the
exact mechanisms underlying CRLM remain to be fully elucidated.
Due to advances in DNA sequencing technology, the
gut microbiota has become an attractive area of research. It has
been estimated that the human gastrointestinal tract may host ~100
trillion microorganisms (5).
Functioning as a hidden organ, the gut microbiota influences organ
structure and homeostasis. Previous studies have proposed a
potential association between the gut microbiome and the occurrence
of cancerous growths (6,7). For instance, individuals with CRC
present variations in the microbial composition of the gut
microenvironment as compared with healthy individuals (8–10).
Notably, several specific microorganisms are involved in the course
of tumor growth and treatment outcomes. Chen et al (11) demonstrated that Fusobacterium
nucleatum (F. nucleatum) facilitated the spread of cancer by
triggering autophagy signaling through the upregulation of card-3
level in mice models. However, Sheng et al (10) demonstrated that the intestinal
microbiota could be used to distinguish different stages of CRC.
For instance, there was suppression of Alistipes bacteria in
stage IV in comparison with stage III CRC. Yu et al
(9) reported that the abundance of
Solobacterium moorei (S. moorei) was increased in
advanced-compared with early-stage CRC. These results highlight
critical biological evolutionary shifts of the gut microbiota
during tumor progression and metastasis. Nevertheless, limited
knowledge is available about the multi-directional alteration of
the gut microbiota in patients with CRC with LM or with no liver
metastasis (NLM).
In the present study, it was hypothesized that there
may be differences in the composition and characteristics of the
microbiome between patients with CRC with or without LM. In
particular, 16S rRNA gene sequencing was utilized to compare the
microbiota communities between LM and NLM. Fresh fecal samples, as
well as tumor tissue samples from different populations were
simultaneously tested and compared. Dissecting the differences, if
any, could highlight potential and novel microbiological markers
that could predict LM. More importantly, a validation cohort was
also used to further reconfirm the findings of the discovery
cohort.
Materials and methods
Study participants and sample
collection
Patients of Chinese descent were enrolled from the
Wuhan Union Hospital of Tongji Medical College, Huazhong University
of Science and Technology, Wuhan, China. The present study was
approved by the Ethics Committee of Tongji Medical College of
Huazhong University of Science and Technology (Approval no.
2014-041). All subjects provided written informed consent prior to
their participation in the study. Samples and data of the
participants were collected from hospital electronic medical
records.
In the present study, 126 patients aged between 20
and 80 years (median age, 60.5 years) diagnosed histologically with
primary colorectal cancer and possessing available clinical data,
were enrolled. A total of 77 cases (61.11%) were female and 49
cases (38.89%) were male. The sample collection period spanned from
June 2016 to December 2017. The patients with LM were assessed
using liver magnetic resonance imaging and with no previous system
treatment history, including chemoradiotherapy or targeted
immunotherapy. Patients with a previous history of familial
adenomatous polyposis, hereditary non-polyposis CRC, inflammatory
bowel disease, metabolic diseases, infectious diseases, or
immunodeficiency diseases, and also patients that had been
administered antibiotics, corticosteroids or probiotics within 3
months prior to specimen collection were also excluded from the
study.
A discovery cohort (cohort 2) and a validation
cohort (cohort 3) were established. Colorectal cancer tissue
samples were also concurrently tested and compared (cohort 1). The
discovery cohort consisted of 18 LM and 36 NLM cases, and the
validation cohort comprised 13 LM and 41 NLM cases. Samples from
both cohorts were fresh feces.
To collect stool samples, ~40 g of feces were
collected using a stool collection kit comprising a specimen
receptacle, disposable sterile spoon, sterile conical tubes and
disposable exam gloves. Participants promptly stored their stool
samples at 4°C for a ≤3 h. Trained study staff aliquoted the stool
samples and preserved them at −80°C for subsequent gut microbiome
analyses. The fecal specimens were obtained during the initial
hospitalization prior to anti-tumor treatment, including surgical
intervention. Additionally, for the collection of tissue samples, a
total of 18 primary carcinoma tissue samples (8 LM and 10 NLM;
cohort 1) were collected. The tissue samples were fixed in formalin
and embedded in paraffin (FFPE). Subsequently, five serial cuts (5
µm) per sample were placed in sterile microtubes and stored at room
temperature until use in subsequent 16S rRNA MiSeq sequencing
(Illumina, San Diego, U.S.A.). The samples were then frozen at
−80°C for subsequent analyses.
DNA extraction and PCR
amplification
DNA was extracted from the collected samples using
the Omega Mag-Bind Soil DNA kit (Omega Bio-Tek, Inc.), following
the manufacturer's instructions. The extracted DNA was quantified
using a Nanodrop (Thermo Fisher Scientific, Inc.). Illumina
sequencing was used to amplify the V3-V4 variable region of 16S
rRNA of bacterial genome using reverse transcription-quantitative
PCR (RT-qPCR). The sense and anti-sense primer sequences were
5′-ACTCCTACGGGAGGCAGCA-3′ and 5′-GGACTACHVGGGTWCTAAT-3′
respectively. The RT-qPCR amplification details have been indicated
in a previous study by the authors (12).
Sequencing data processing
The processing of raw sequencing data involved
filtering, quality assessment, and the removal of query sequences.
Initial filtering of Illumina MiSeq platform-generated data in
FASTQ format utilized a sliding window method, employing a 10 bp
window size and 1 bp step size. This process ensured an average
sequencing accuracy of ≥99, a truncated sequence length of ≥150 bp,
and the exclusion of ambiguous bases (N). Paired-end sequences from
each library were overlapped using FLASH (version 1.2.7;
ccb.jhu.edu/software/FLASH/), with criteria stipulating an
overlapping base length of ≥10 bp and a mismatch base number less
than 10% of the overlapping base length. Identification of mistaken
sequences was conducted using QIIME software (version 1.8.0,
http://qiime.org/), incorporating specifications such
as a sequence length ≥160 bp, the absence of ambiguous bases (N),
and constraints on 5′ primer mismatch base numbers and consecutive
identical base numbers. Detection and removal of chimeric sequences
were carried out through the combined use of USEARCH (version
5.2.236; drive5.com/usearch/) and QIIME software (version 1.9.1,
http://qiime.org/).
Random forest classification
model
Random forest algorithm was used to identify
bacterial species that could distinguish the LM status. The top
distinguishing phyla were included for further analysis. The
ranking according to mean decrease in accuracy were obtained using
default parameters in R (version 3.4.0, R core team, 2017)
environment ‘Random Forest’. To further validate the predictive
flora for LM, receiver operator characteristic (ROC) and area under
curve (AUC) analysis were performed using MedCalc 16.1 (MedCalc
Software bvba).
Bioinformatics analysis
Quantitative Insights into Microbial Ecology 2.0
(QIIME2, version 1.9.1, http://qiime.org/) was used to analyze the obtained
sequences and construct the taxa (13). Microbial alpha-diversity was
analyzed using sampling-based operational taxonomic units OTUs and
presented by Chao1, Faith's_pd and Observed_species indices.
According to the total number of amplicon sequence variants
(ASVs)/OTUs, R software was employed to generate rarefaction curve.
Beta-diversity was assessed according to the microbial community
structures using non-metric multidimensional scaling analysis 2 and
principal coordinate analysis (PCoA) analysis. Linear discriminant
analysis (LDA) effect size (LEfSe) was applied to analyze
significant microbial characteristics. Additionally, the
progression-free survival (PFS) and overall survival (OS) of the
patients with CRC with high and low levels of Bacteroidetes and
Proteobacteria in the primary cancerous tissues were compared.
Statistical analysis
Categorical data were analyzed using the Chi-squared
test or Fisher's exact test, whereas the two independent samples
t-test was used for continuous variables. Analysis was conducted
using SPSS software (version 19.0, IBM SPSS). Kaplan-Meier survival
analysis was employed, and the log-rank test was utilized to assess
and compare the PFS and OS. P<0.05 was considered to indicate a
statistically significant difference.
Results
Study design and participant
information
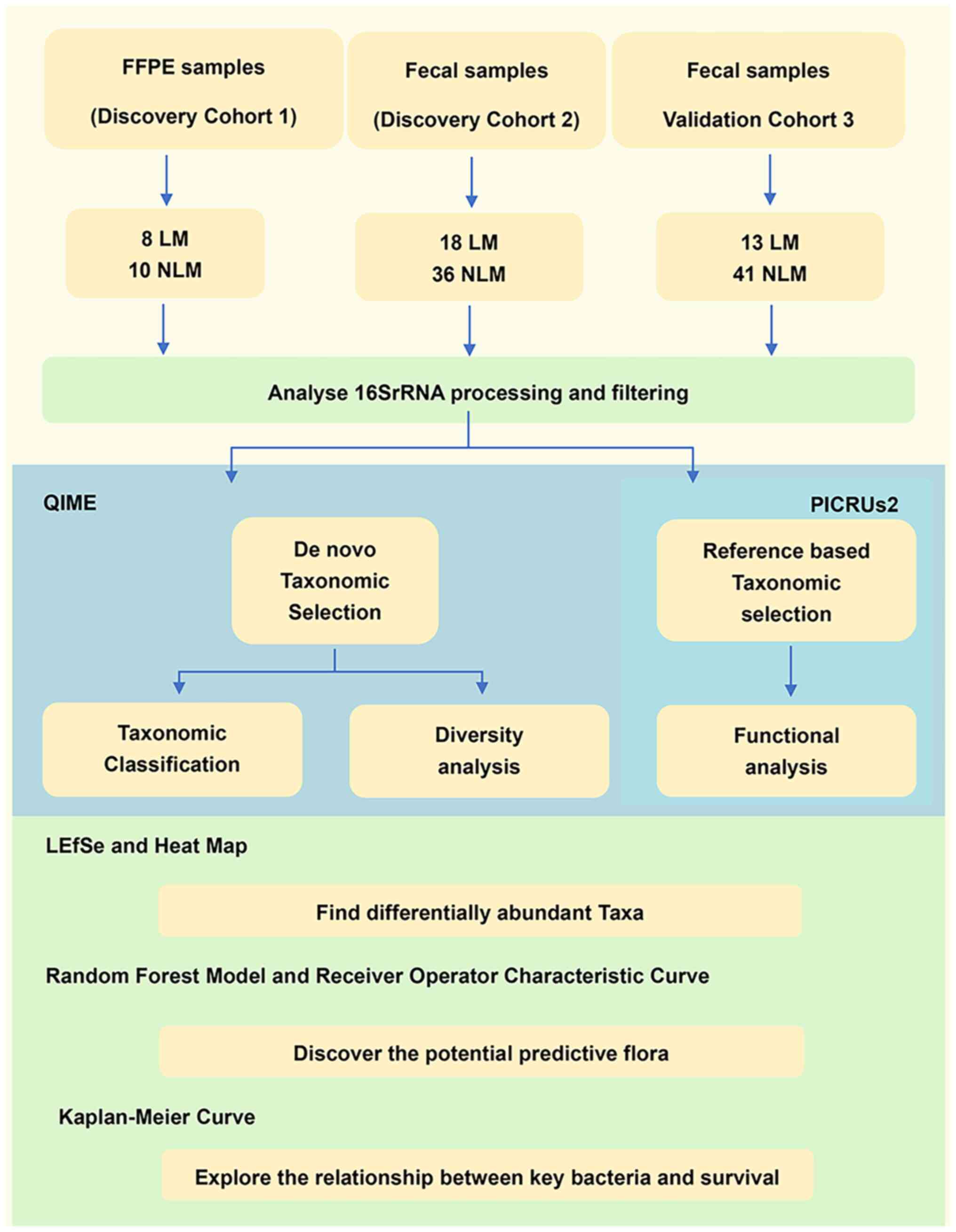
A flow chart of the experimental design is presented
in Fig. 1. A total of 126 samples
were analyzed in the present study. In particular, 54 fecal samples
from the discovery cohort (cohort 2; LM=18 and NLM=36), 54 fecal
samples from the validation cohort (cohort 3; LM=13 and NLM=41) and
18 primary tumor tissues from FFPE cohort as a supplementary
discovery cohort (cohort 1; LM=8 and NLM=10) were included. The
clinicopathological characteristics of the patients with LM and NLM
from the three cohorts were analyzed (Table I). In the discovery cohort 1, there
was no statistically significant differences between LM and NLM
regarding sex, tumor location, and differentiation. Within
discovery cohort 2, there were slight differences in sex and
differentiation between LM and NLM. In validation cohort 3, there
were slight differences in sex between LM and NLM. Our analysis of
gut microbiota features influencing liver metastasis revealed that,
given LM experienced liver metastasis while NLM did not, the
clinical stages of most LM at the time of initial diagnosis
consistently lagged behind (stage IV) in comparison with NLM (stage
III) across all three cohorts.
 | Table I.Demographic sample characteristics of
the patients with CRC. |
Table I.
Demographic sample characteristics of
the patients with CRC.
|
| Cohort 1 (FFPE
samples) |
| Cohort 2 (fecal
samples) |
| Cohort 3 (fecal
samples) |
|
|---|
|
|
|
|
|
|
|
|
|---|
| Characteristic | LM (n=8) | NLM (n=10) | P-value | LM (n=18) | NLM (n=36) | P-value | LM (n=13) | NLM (n=41) | P-value |
|---|
| Age, years
(range) | 58 (32–77) | 59 (26–80) |
| 62 (49–75) | 61 (39–78) |
| 61 (39–78) | 62 (49–75) |
|
| Sex, n (%) |
|
| 0.321 |
|
| 0.018 |
|
| 0.033 |
|
Female | 4 (50.00%) | 2 (20.00%) |
| 3 (16.67%) | 18 (50.00%) |
| 2 (15.38%) | 20 (48.78%) |
|
|
Male | 4 (50.00%) | 8 (80.00%) |
| 15 (83.33%) | 18 (50.00%) |
| 11 (84.62%) | 21 (51.22%) |
|
| Tumor location |
|
| 0.145 |
|
| 0.519 |
|
| 0.937 |
| Left
CRC | 5 (62.50%) | 2 (20.00%) |
| 12 (66.67%) | 27 (75.00%) |
| 10 (76.92%) | 29 (70.73%) |
|
| Right
CRC | 3 (37.50%) | 8 (80.00%) |
| 6 (33.33%) | 9 (25.00%) |
| 3 (23.08%) | 12 (29.27%) |
|
|
Differentiation |
|
| 0.241 |
|
| 0.029 |
|
| 0.818 |
|
Well/moderate | 2 (25.00%) | 3 (30.00%) |
| 5 (27.78%) | 7 (19.44%) |
| 3 (23.08%) | 7 (17.07%) |
|
|
Poor | 6 (75.00%) | 4 (40.00%) |
| 7 (38.89%) | 26 (72.22%) |
| 7 (53.85%) | 26 (63.41%) |
|
|
Unknown | 0 (0.00%) | 3 (30.00%) |
| 6 (33.33%) | 3 (8.33%) |
| 3 (23.08%) | 8 (19.51%) |
|
| AJCC stage, n
(%) |
|
| <0.001 |
|
| <0.001 |
|
| <0.001 |
|
Unknown | 0 (0.00%) | 0 (0.00%) |
| 0 (0.00%) | 0 (0.00%) |
| 0 (0.00%) | 0 (0.00%) |
|
| I | 0 (0.00%) | 0 (0.00%) |
| 0 (0.00%) | 0 (0.00%) |
| 0 (0.00%) | 0 (0.00%) |
|
| II | 0 (0.00%) | 0 (0.00%) |
| 0 (0.00%) | 0 (0.00%) |
| 0 (0.00%) | 0 (0.00%) |
|
|
III | 0 (0.00%) | 10 |
| 1 (5.56%) | 36 |
| 0 (0.00%) | 41 |
|
|
|
| (100.00%) |
|
| (100.00%) |
|
| (100.00%) |
|
| IV | 8 | 0 (0.00%) |
| 17 | 0 (0.00%) |
| 13 | 0 (0.00%) |
|
|
| (100.00%) |
|
| (94.44%) |
|
| (100.00%) |
|
|
Estimation of sequencing depth
DNA was extracted from the primary tumor tissues and
fecal samples. Taxonomic profiling via 16S rRNA gene sequencing was
performed. According to the correspondence between sequence
similarity and bacterial taxonomic status, 97% similarity is
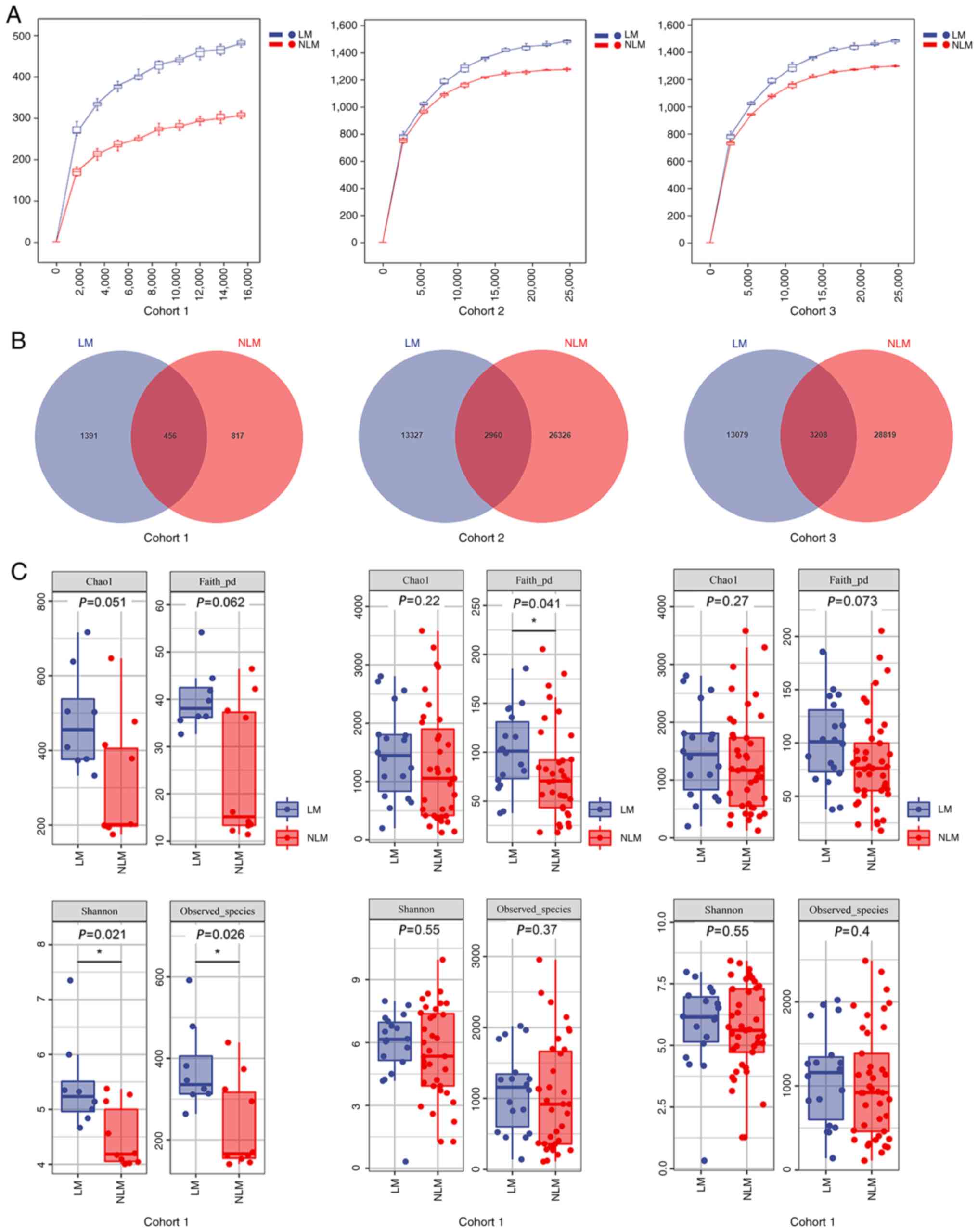
generally considered for species division. The rarefaction curves
displaying the sequencing depth reached a plateau, indicating a
sufficient sequencing depth (Fig.
2A). The Venn diagrams demonstrated the number of shared and
unique OTUs in each group. In total: i) 2,960 shared OTUs were
observed among 42,613 OTUs in the LM and NLM groups of the
discovery cohort; ii) 3,208 shared OUTs in a total number of 45,106
OTUs in the validation cohort and iii) 456 shared OUTs in a total
number of 2,664 OTUs in the LM group and NLM group of the FFPE
sample cohort (Fig. 2B). According
to the Venn diagram, reduced numbers of total OTUs were detected in
primary tumor tissues in comparison with the fecal samples.
Microbiota alpha-diversity and
beta-diversity
Microbiota richness and diversity in the gut and
primary tumors were subsequently compared between the LM and NLM
groups. In the primary cancerous tissues of cohort 1, a discrepancy
of alpha-diversity was observed between two groups, but only Chao1
index and Faith's_pd were not significant (P>0.05; Fig. 2C). In the fecal discovery (cohort 2)
and the validation cohorts (cohort 3), the trend was consistent
with four alpha-diversity index values (Chao1 index, Faith's_pd,
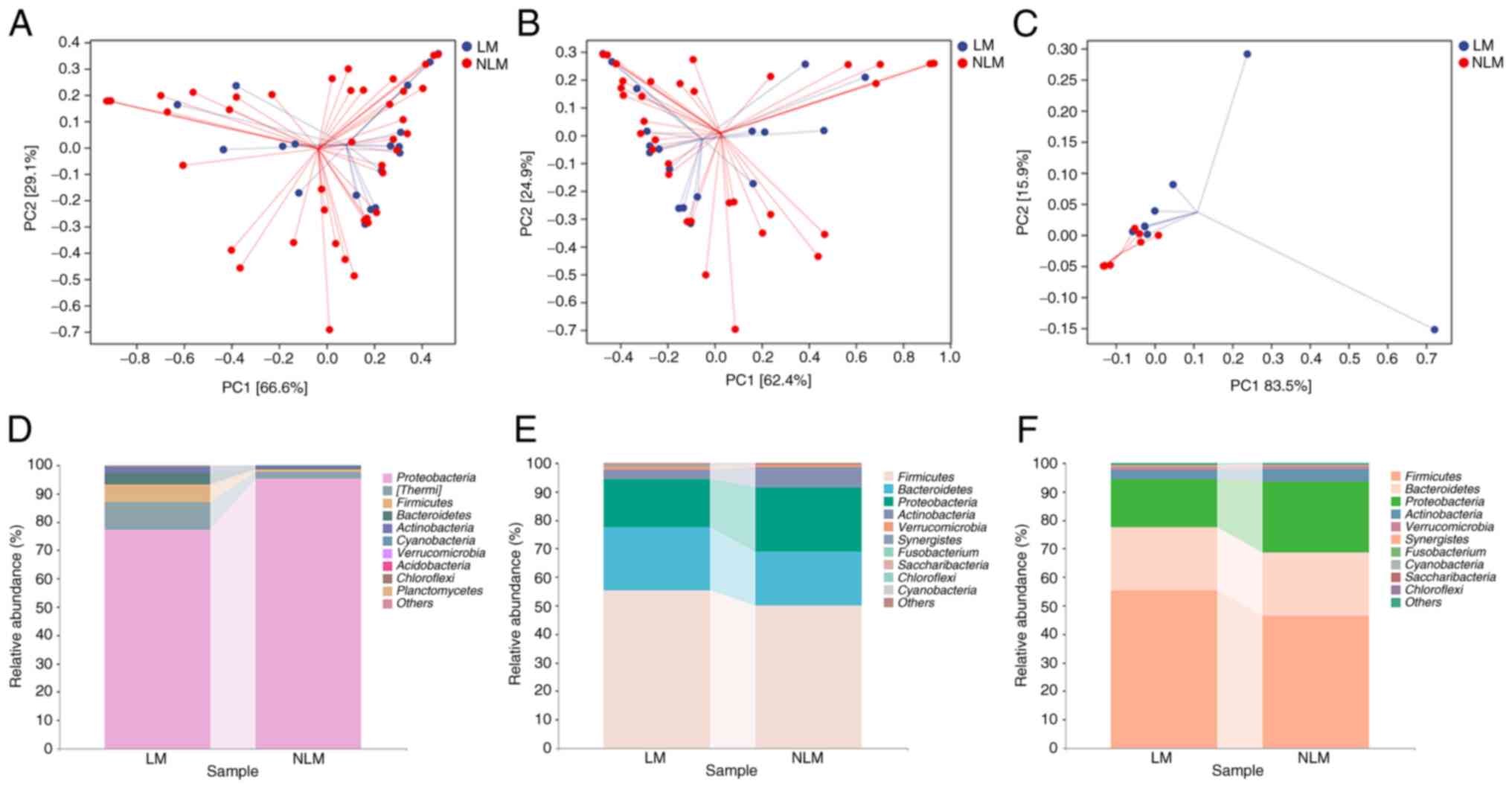
Shannon and Observed_species between LM and NLM; Fig. 2C). Following beta-diversity analysis
using PCoA, a clear separation of the community between the LM and
NLM group in the discovery cohort (cohort 2) was observed (Fig. 3B). In the validation cohort (cohort
3), beta-diversity analysis also exhibited consistent results
(Fig. 3C). These results suggested
a higher microbiota richness and diversity in LM than in NLM
group.
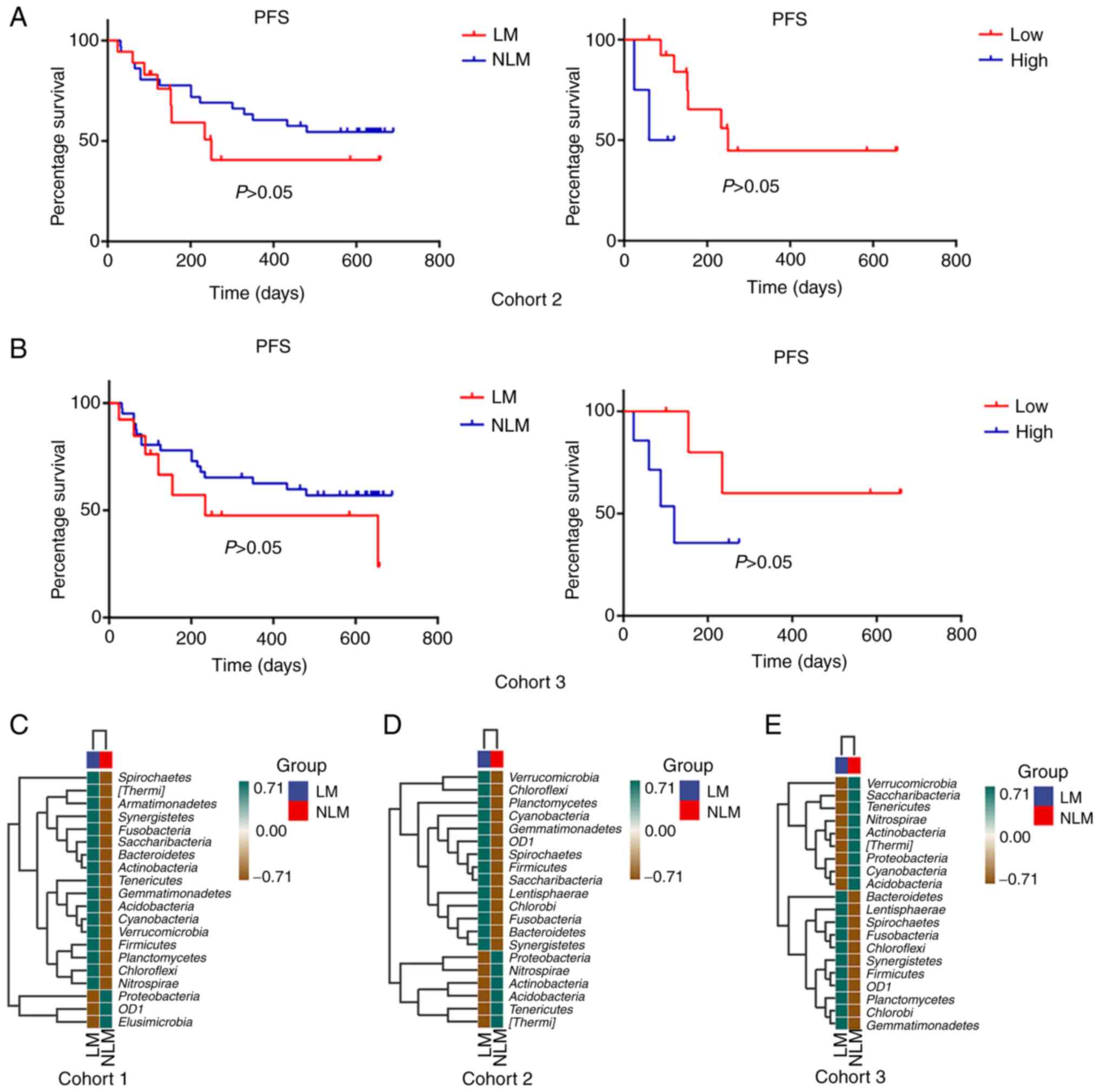
Microbiota composition
Subsequently, it was investigated whether there were
differences in microbiota composition between the LM and NLM group.
Therefore, the general landscape of microbiome was first assessed.
For the top 10 microbiota at phylum level, nine species exhibited
consistent preponderant enrichment in both the discovery and
validation cohorts with fecal samples, including Firmicutes,
Bacteroidetes, Proteobacteria, Actinobacteria, Verrucomicrobia,
Synergistetes, Fusobacterium, Cyanobacteria, Saccharibacteria
and Chloroflexi (Fig. 3E and
F). Firmicutes, Bacteroidetes, Proteobacteria,
Actinobacteria, Verrucomicrobia, Cyanobacteria and
Chloroflexi were also largely enriched in microbial
populations in primary tumor tissues (Fig. 3D). In the validation cohort, the
main microbiota composition analysis at phylum level were
consistent with those of the discovery cohort (Fig. 3D-F).
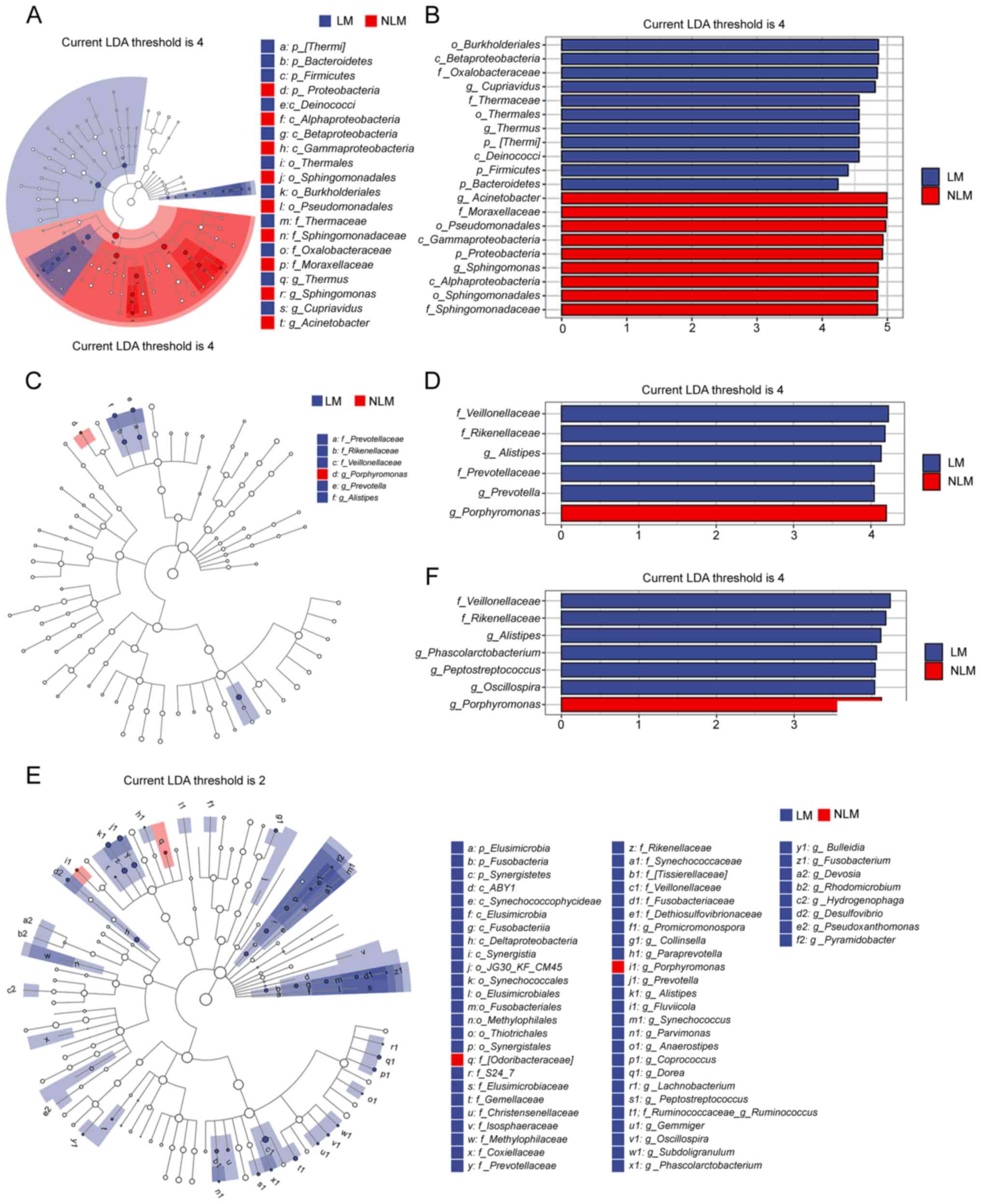
To further investigate these findings, LEfSe
analysis was conducted, in order to detect markedly changed species
with LDA score=4.0. The results indicated that the LM group with
fecal samples was significantly enriched in Veillonellaceae
at the phylum level, whereas the NLM group was significantly
enriched with Porphyromonas at the phylum level (Fig. 4D and F). Additionally, the LM group
of the tumor tissues were rich in Burkholderiales and
Betaproteobacteria at the phylum level (Fig. 4B).
A species composition heatmap was then used to
discover the changed phyla between LM group and NLM. The results
indicated that Bacteroidetes, Planctomycetes, Fusobacterium,
Firmicutes and Bacteroidetes presented consistent
ascending enrichment in LM group compared with the NLM in the two
specimen methods (Fig. 5C-E).
Proteobacteria presented consistent descending enrichment in
LM than NLM group in both tissues and fecal specimen cohorts.
Additionally, OD1 presented reversed trend in LM group in two
specimens (Fig. 5C-E).
Association of Fusobacteria with
survival outcomes
Prognostic analysis was performed in cohort 2 and 3
between the LM and NLM groups and the results did not reveal
statistically significant differences (Fig. 5). For the detection of microbiota
for prognosis, the patients with NLM we stratified into the high
vs. low categories based on the median relative abundance of the
phylum. Fusobacteriumhigh was negatively
associated with a shorter PFS in cohort 2 [hazard ratio (HR, 4.000;
95% confidence interval (CI), 1.1210-203.4000] and cohort 3 (HR,
2.933, 95% CI, 0.6008-15.6400; Fig.
6). The findings further indicated that the tumor microbiota
could serve as a predictor of survival outcomes, suggesting the
potential relevance of the microbiome in mediating CRC
progression.
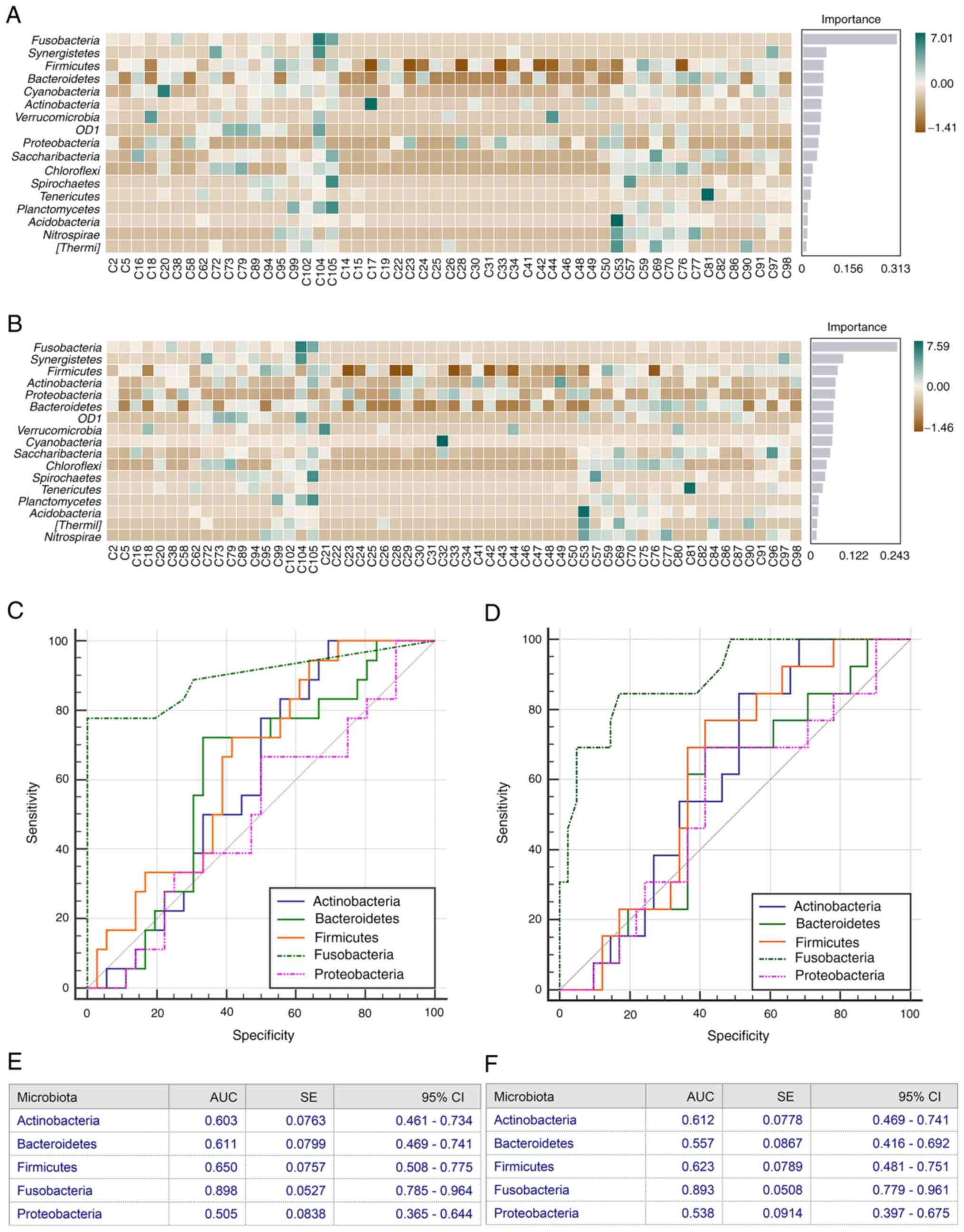
Random forest classification
model
Subsequently, it was investigated whether microbiome
communities could be used as a predictive biomarker for LM based on
OTU abundance at the phylum level. Using the stochastic forest RF
classification model, it was demonstrated that the
Actinobacteria, Bacteroidetes, Firmicutes, Fusobacterium and
Proteobacteria were highly discriminative phyla with the
highest mean decreased score in cohort 2 and cohort 3 (Fig. 6). Thereafter, to define reliable
biomarkers of the gut microbial response to LM, the top altered
bacterial cluster was used to perform AUC-ROC analysis. In the
discovery cohort of fecal samples, a relative high accuracy of
Actinobacteria (AUC=0.603), Bacteroidetes
(AUC=0.611), Firmicutes (AUC=0.650), Fusobacterium
(AUC=0.898) and Proteobacteria (AUC=0.505) in predicting LM
was demonstrated (Fig. 6C and E).
Similarly, in the validation cohort of fecal samples, a relative
high accuracy of Actinobacteria (AUC=0.612),
Bacteroidetes (AUC=0.557), Firmicutes (AUC=0.623),
Fusobacterium (AUC=0.893) and Proteobacteria
(AUC=0.538) in predicting LM was demonstrated (Fig. 6D and F). Notably, Fusobacteria
phylum in both fecal sample cohorts displayed a relatively high
predictive value.
Discussion
In the present study, the differences in diversity
and composition of the gut microbiota between patients with CRC
with or without LM were evaluated using 16S rRNA community
profiling. The primary tumor tissues and fecal samples from two CRC
cohorts were used. In order to enhance the validity of the findings
of the present study in the discovery cohort, the validation cohort
was further used to validate the results obtained. The ‘discovery
cohort’ operates similarly to an experimental group, representing
the set on which the data model is trained. Conversely, the
‘validation cohort’ is designed to validate the model (conclusions)
derived from the discovery cohort. When the sample size of the
discovery cohort is relatively small, the validation cohort becomes
crucial for demonstrating the reproducibility of conclusions across
diverse datasets, thereby enhancing the persuasiveness of research
findings (14). Furthermore, the
majority of the currently reported studies on the gut microbiota
have primarily utilized a single type of specimen (15–18).
Only a limited number of studies have simultaneously compared
analysis data from fecal or tissue specimens, particularly as
regards CRC (19). Through the
comparative analysis of tissue and fecal specimens in the present
study, a reduced number of total OTUs was found in the FFPE samples
of primary tumors compared with fecal samples. Dominating species
in primary tumor FFPE and fecal specimen were nearly consistent at
the phylum level. Additionally, alpha-diversity, beta-diversity,
community composition and metabolic pathways differed to some
extent between the LM group and NLM group. In the two different
specimen analysis, nine phyla including Fusobacterium,
Bacteroidetes, TM7 and Firmicutes presented consistent
and higher enrichment in LM group than NLM group, while eight phyla
including Proteobacteria, Cyanobacteria and Thermi
presented consistent and reduced enrichment in LM than NLM group.
Similar results were obtained from the analysis of the validation
cohort. Of note, the random forest classification model indicated
that Fusobacterium has potential predictive value for LM.
Furthermore, the Fusobacteria phylum was negatively associated with
survival. These data, evaluated for the first time, to the best of
our knowledge, the difference of the tumor-associated microbiome in
CRC with and without LM.
Gut microbes aid towards the maintenance of
intestinal homeostasis, prevent pathogen colonization and release
key nutrients and energy from the diet. Apart from the benefits, it
has been demonstrated in previous studies that gut microbes may
play a role similar to tumor suppressor or oncogenes (20,21).
The microbiome communicates with host via direct and indirect
factors, such as metabolites, proteins and toxins. These
carcinogenic species or substances enter systemic circulation and
affect distant organs (21–23). Similarly, signals released from
tumors could also modulate the microbiome, which possibly induce or
contribute to microbiota dysbiosis or dysfunction (23).
A previous study demonstrated that Fusobacterium
may be a pro-tumorigenic factor in colorectal carcinogenesis
(24–26). Particularly, Fusobacterium
potentiated the biological behavior (proliferation, adhesion and
invasion) of CRC cells by activating relevant cancer-signaling
pathways (27–29). Notably, Bullman et al
(23) discovered that
Fusobacterium, Bacteroides, Selenomonas and
Prevotella were maintained in liver metastases as compared
with primary tumors, thus demonstrating microbiome stability
between paired primary tumor and metastatic tumors.
In the present study, the microbiota distinction was
examined between patients with LM and NLM. A panel of
microorganisms differentially occurring in CRLM was detected,
including Fusobacteria, Firmicutes, Bacteroidetes, Proteobacteria,
Actinobacteria and Verrucomicrobia. Only 6.7% of the FFPE samples
were Fusobacteria-positive, which was consistent with the findings
of previous reports showing that Fusobacterium was detected
in 4.4–13% of the FFPE specimens of patients with primary CRC
(30–32). A higher abundance of
Fusobacterium was also observed in patients with LM than NLM
in both cohorts. The use of LefSe analysis with 4-fold LDA
threshold revealed that the change of Fusobacterium in LM
was not prominent, probably due to a limited sample size and lower
expression rate.
Accumulating evidence indicates that
Fusobacterium abundance is associated with CRC metastasis
(27,33,34).
However, the association between Fusobacterium infection and
the host immune system in CRC metastasis has not yet been
determined. In line with the findings of the present study, Yin
et al (35) revealed that
Fusobacterium administration promoted CRLM and was
associated with the activation of the hepatic immune
microenvironment, revealing the potential role of
Fusobacterium in CRLM. Recently, Sakamoto et al
(30) revealed that
Fusobacterium was associated with a lower density of
CD8+ T-cells and a higher density of myeloid-derived
suppressor cells in LM. Consistent with these results, Yin et
al (35) demonstrated that
Fusobacterium reshaped the immune microenvironment in
metastatic livers by using in vivo murine models of CRC. Xu
et al (36) revealed that
tumor-derived C-C motif chemokine ligand 20 (CCL20) activated by
Fusobacterium not only increased CRC metastasis, but also
participated in the reprograming of the tumor microenvironment.
They also reported that Fusobacterium promoted macrophage
infiltration through CCL20 activation and simultaneously induced M2
macrophage polarization, enhancing the metastasis of CRC (36). A recent study (37) revealed that Fusobacterium
promoted CRC cell metastasis to the liver. Lu et al
(37) revealed that
Fusobacterium upregulated the expression of the lncRNA, long
intergenic non-protein coding RNA 1610, which increased the
metastatic ability of CRC cells in vivo and in vitro.
More importantly, a previous study by the authors demonstrated that
Fusobacterium enrichment was associated with the poor
prognosis of patients with proximal colon cancer (12).
In the microbiological research of CRC, feces and
tissues are the most frequently used samples. However, the
selection between these two types of models remains debatable
(38). Whereas several researchers
believe that bacterial populations in feces and mucous membranes
are completely different with different compositions and diversity
(39), others claim similar
variations between the two samples (19). Zeller et al (19) demonstrated that there was similar
abundance of bacterial species between feces and tissue samples
from patients with CRC, regardless of different patient
nationality, sample source, assay techniques and analysis methods.
FFPE specimens as a special preservation of biopsy or surgical
sample are valuable for cancer research (24). Quality of DNA and RNA isolated from
FFPE biological specimens is reduced, in comparison with fresh
tissue specimens, resulting in certain differences in studies
involving microorganisms (40,41).
Compared with fecal samples, a reduced number of OTUs was detected
in the microbiota in the FFPE tissues of patients with primary CRC.
However, it was revealed that the primary tumor FFPE samples and
fecal samples displayed comparable dominant microbes and similar
diversity in LM populations at the phylum level, albeit with
significant inconsistency at lower taxonomic levels, including the
genus level. The findings of the present study were in accordance
with those of the study by Riquelme et al (42), which revealed similar taxonomic
composition between FFPE and frozen samples of pancreatic
adenocarcinoma using 16S rRNA gene sequencing analysis. Regardless
of FFPE storage, insufficient DNA extraction and potential DNA
contamination, the data of the present study in combination with
those of other previously published studies (42–44)
underline the feasibility of using FFPE CRC tissues in
characterizing NGS-based phylum microbiota.
The present study meticulously selected a population
excluding the microbiota associated with chronic gastrointestinal
diseases and interventions prior to specimen acquisition that may
affect gut microbiota. The cross-sectional comparison of primary
tumor and gut fecal samples from two cohorts was performed to
detect microbiological differences between LM and NLM. However,
future studies are required to also consider possible confounders,
including comorbid symptoms, body mass index and dietary habits.
Secondly, since the two specimen methods exhibited relative
consistency at the phylum level, the majority the analyses in the
present study were based on phylum. The distinctions between the
FFPE samples of primary tumors and fecal microbiota at lower
taxonomic levels, such as the genus level, imply the potential
necessity for employing metagenomic approaches in future studies to
conduct more nuanced analyses. Thirdly, functional analysis was
performed based on bacterial abundance and sequencing with a
limited sample size.
In conclusion, the analysis presented in the present
study uncovered gut microbiota alterations in patients with CRC
with or without LM and identified the potential keystone taxa. The
differences in the microbiota of patients with LM contributes to
the already known tumor-host heterogeneity. There is a necessity
for future studies, in order to design an appropriate diet for the
modulation of the gut microbiota in at-risk subjects for the
prevention and treatment of CRC liver metastasis.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Scientific research project
of Hubei Provincial Health Commission (grant no. WJ2023M92), the
Clinical Research Special Fund of Wu Jieping Medical Foundation
(grant no. 320.6750.2023-19-7) and the National Natural Science
Foundation of China (grant nos. 81472707 and 81602255). The present
study was also supported by the USA National Institutes of Health
(NIH) grant (grant no. R35 CA197735) and Nodal Award (grant no.
2016-02) from the Dana-Farber Harvard Cancer Center.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. All raw data acquired using the MiSeq RNA sequencing
described in this article were uploaded to the SRA database
(Accession no. PRJNA909044).
Authors' contributions
HL conceived and designed the present study. HL, TZ
and SO provided administrative support. HL, MJ, QF and FS provided
the study materials or patient follow-up information. FS, SO and TZ
conceived the study and acquisition of data. MJ and QF performed
the collection and assembly of the data and samples, data analysis
and data interpretation. All authors participated in writing and
correcting the manuscript. All authors have read and approved the
final manuscript. MJ and QF confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Tongji Medical College of Huazhong University of
Science and Technology (approval no. 2014-041). All subjects
provided written informed consent prior to their participation in
the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abdalla EK, Adam R, Bilchik AJ, Jaeck D,
Vauthey JN and Mahvi D: Improving resectability of hepatic
colorectal metastases: Expert consensus statement. Ann Surg Oncol.
13:1271–1280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martin J, Petrillo A, Smyth EC, Shaida N,
Khwaja S, Cheow HK, Duckworth A, Heister P, Praseedom R, Jah A, et
al: Colorectal liver metastases: Current management and future
perspectives. World J Clin Oncol. 11:761–808. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Engstrand J, Nilsson H, Stromberg C, Jonas
E and Freedman J: Colorectal cancer liver metastases-a
population-based study on incidence, management and survival. BMC
Cancer. 18:782018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ursell LK, Metcalf JL, Parfrey LW and
Knight R: Defining the human microbiome. Nutr Rev. 70 (Suppl
1):S38–S44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan X, Jin Y, Chen G, Ma X and Zhang L:
Gut microbiota dysbiosis drives the development of colorectal
cancer. Digestion. 102:508–515. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jobin C: Colorectal cancer: Looking for
answers in the microbiota. Cancer Discov. 3:384–387. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uchino Y, Goto Y, Konishi Y, Tanabe K,
Toda H, Wada M, Kita Y, Beppu M, Mori S, Hijioka H, et al:
Colorectal cancer patients have four specific bacterial species in
oral and gut microbiota in common-a metagenomic comparison with
healthy subjects. Cancers (Basel). 13:33322021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu J, Feng Q, Wong SH, Zhang D, Liang QY,
Qin Y, Tang L, Zhao H, Stenvang J, Li Y, et al: Metagenomic
analysis of faecal microbiome as a tool towards targeted
non-invasive biomarkers for colorectal cancer. Gut. 66:70–78. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheng Q, Du H, Cheng X, Cheng X, Tang Y,
Pan L, Wang Q and Lin J: Characteristics of fecal gut microbiota in
patients with colorectal cancer at different stages and different
sites. Oncol Lett. 18:4834–4844. 2019.PubMed/NCBI
|
|
11
|
Chen Y, Chen Y, Zhang J, Cao P, Su W, Deng
Y, Zhan N, Fu X, Huang Y and Dong W: Fusobacterium nucleatum
promotes metastasis in colorectal cancer by activating autophagy
signaling via the upregulation of CARD3 expression. Theranostics.
10:323–339. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin M, Shang F, Wu J, Fan Q, Chen C, Fan
J, Liu L, Nie X, Zhang T, Cai K, et al: Tumor-associated microbiota
in proximal and distal colorectal cancer and their relationships
with clinical outcomes. Front Microbiol. 12:7279372021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Torres Moral T, Sanchez-Niubo A,
Monistrol-Mula A, Gerardi C, Banzi R, Garcia P, Demotes-Mainard J
and Haro JM; The Permit Group, : Methods for stratification and
validation cohorts: A scoping review. J Pers Med. 12:6882022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Himbert C, Stephens WZ, Gigic B, Hardikar
S, Holowatyj AN, Lin T, Ose J, Swanson E, Ashworth A, Warby CA, et
al: Differences in the gut microbiome by physical activity and BMI
among colorectal cancer patients. Am J Cancer Res. 12:4789–4801.
2022.PubMed/NCBI
|
|
16
|
Han S, Zhuang J, Pan Y, Wu W and Ding K:
Different characteristics in gut microbiome between advanced
adenoma patients and colorectal cancer patients by metagenomic
analysis. Microbiol Spectr. 10:e01593222022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marchesi JR, Dutilh BE, Hall N, Peters WH,
Roelofs R, Boleij A and Tjalsma H: Towards the human colorectal
cancer microbiome. PLoS One. 6:e204472011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen W, Liu F, Ling Z, Tong X and Xiang C:
Human intestinal lumen and mucosa-associated microbiota in patients
with colorectal cancer. PLoS One. 7:e397432012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zeller G, Tap J, Voigt AY, Sunagawa S,
Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, et
al: Potential of fecal microbiota for early-stage detection of
colorectal cancer. Mol Syst Biol. 10:7662014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kalasabail S, Engelman J, Zhang LY,
El-Omar E and Yim HCH: A perspective on the role of microbiome for
colorectal cancer treatment. Cancers (Basel). 13:46232021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rossi T, Vergara D, Fanini F, Maffia M,
Bravaccini S and Pirini F: Microbiota-derived metabolites in tumor
progression and metastasis. Int J Mol Sci. 21:57862020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshimoto S, Loo TM, Atarashi K, Kanda H,
Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et
al: Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bullman S, Pedamallu CS, Sicinska E,
Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T,
et al: Analysis of Fusobacterium persistence and antibiotic
response in colorectal cancer. Science. 358:1443–1448. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Debesa-Tur G, Pérez-Brocal V, Ruiz-Ruiz S,
Castillejo A, Latorre A, Soto JL and Moya A: Metagenomic analysis
of formalin-fixed paraffin-embedded tumor and normal mucosa reveals
differences in the microbiome of colorectal cancer patients. Sci
Rep. 11:3912021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kostic AD, Gevers D, Pedamallu CS, Michaud
M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et
al: Genomic analysis identifies association of Fusobacterium
with colorectal carcinoma. Genome Res. 22:292–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Castellarin M, Warren RL, Freeman JD,
Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P,
Allen-Vercoe E, Moore RA and Holt RA: Fusobacterium
nucleatum infection is prevalent in human colorectal carcinoma.
Genome Res. 22:299–306. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Y, Weng W, Peng J, Hong L, Yang L,
Toiyama Y, Gao R, Liu M, Yin M, Pan C, et al: Fusobacterium
nucleatum increases proliferation of colorectal cancer cells
and tumor development in mice by activating Toll-like receptor 4
signaling to nuclear factor-κB, and up-regulating expression of
MicroRNA-21. Gastroenterology. 152:851–866.e24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G
and Han YW: Fusobacterium nucleatum promotes colorectal
carcinogenesis by modulating E-cadherin/β-catenin signaling via its
FadA adhesin. Cell Host Microbe. 14:195–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Abed J, Emgård JE, Zamir G, Faroja M,
Almogy G, Grenov A, Sol A, Naor R, Pikarsky E, Atlan KA, et al:
Fap2 Mediates Fusobacterium nucleatum colorectal
adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc.
Cell Host Microbe. 20:215–225. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sakamoto Y, Mima K, Ishimoto T, Ogata Y,
Imai K, Miyamoto Y, Akiyama T, Daitoku N, Hiyoshi Y, Iwatsuki M, et
al: Relationship between Fusobacterium nucleatum and
antitumor immunity in colorectal cancer liver metastasis. Cancer
Sci. 112:4470–4477. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nosho K, Sukawa Y, Adachi Y, Ito M,
Mitsuhashi K, Kurihara H, Kanno S, Yamamoto I, Ishigami K, Igarashi
H, et al: Association of Fusobacterium nucleatum with
immunity and molecular alterations in colorectal cancer. World J
Gastroenterol. 22:557–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mima K, Sukawa Y, Nishihara R, Qian ZR,
Yamauchi M, Inamura K, Kim SA, Masuda A, Nowak JA, Nosho K, et al:
Fusobacterium nucleatum and T cells in colorectal carcinoma.
JAMA Oncol. 1:653–661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen S, Su T, Zhang Y, Lee A, He J, Ge Q
and Wang L, Si J, Zhuo W and Wang L: Fusobacterium nucleatum
promotes colorectal cancer metastasis by modulating KRT7-AS/KRT7.
Gut Microbes. 11:511–525. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mima K, Nishihara R, Qian ZR, Cao Y,
Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song M, et al:
Fusobacterium nucleatum in colorectal carcinoma tis sue and
patient prognosis. Gut. 65:1973–1980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yin H, Miao Z, Wang L, Su B, Liu C, Jin Y,
Wu B, Han H and Yuan X: Fusobacterium nucleatum promotes
liver metastasis in colorectal cancer by regulating the hepatic
immune niche and altering gut microbiota. Aging (Albany NY).
14:1941–1958. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu C, Fan L, Lin Y, Shen W, Qi Y, Zhang Y,
Chen Z, Wang L, Long Y, Hou T, et al: Fusobacterium
nucleatum promotes colorectal cancer metastasis through
miR-1322/CCL20 axis and M2 polarization. Gut Microbes.
13:19803472021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu X, Xu Q, Tong Y, Zhang Z, Dun G, Feng
Y, Tang J, Han D, Mao Y, Deng L, et al: Long non-coding RNA EVADR
induced by Fusobacterium nucleatum infection promotes
colorectal cancer metastasis. Cell Rep. 40:1111272022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rezasoltani S, Dabiri H, Asadzadeh-Aghdaei
H, Sepahi AA, Modarressi MH and Nazemalhosseini-Mojarad E: The gut
microflora assay in patients with colorectal cancer: In feces or
tissue samples? Iran J Microbiol. 11:1–6. 2019.PubMed/NCBI
|
|
39
|
Ringel Y, Maharshak N, Ringel-Kulka T,
Wolber EA, Sartor RB and Carroll IM: High throughput sequencing
reveals distinct microbial populations within the mucosal and
luminal niches in healthy individuals. Gut Microbes. 6:173–181.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Imrit K, Goldfischer M, Wang J, Green J,
Levine J, Lombardo J and Hong T: Identification of bacteria in
formalin-fixed, paraffin-embedded heart valve tissue via 16S rRNA
gene nucleotide sequencing. J Clin Microbiol. 44:2609–2611. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim S, Park C, Ji Y, Kim DG, Bae H, van
Vrancken M, Kim DH and Kim KM: Deamination effects in
formalin-fixed, paraffin-embedded tissue samples in the era of
precision medicine. J Mol Diagn. 19:137–146. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Riquelme E, Zhang Y, Zhang L, Montiel M,
Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et
al: Tumor microbiome diversity and composition influence pancreatic
cancer outcomes. Cell. 178:795–806.e12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Carrick DM, Mehaffey MG, Sachs MC,
Altekruse S, Camalier C, Chuaqui R, Cozen W, Das B, Hernandez BY,
Lih CJ, et al: Robustness of next generation sequencing on older
formalin-fixed paraffin-embedded tissue. PLoS One. 10:e01273532015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oh E, Choi YL, Kwon MJ, Kim RN, Kim YJ,
Song JY, Jung KS and Shin YK: Comparison of accuracy of whole-exome
sequencing with formalin-fixed paraffin-embedded and fresh frozen
tissue samples. PLoS One. 10:e01441622015. View Article : Google Scholar : PubMed/NCBI
|