Introduction
Lymphoplasmacytic lymphoma (LPL) is a rare low-grade
non-Hodgkin lymphoma which affects the B-lymphocytes and causes
their abnormal growth and dysregulation (1). The abnormal B-cells show features of
both lymphocytes and plasma cells, hence originates the name of the
disease. Most patients have the clinical syndrome of Waldenström
macroglobulinemia (WM), which is defined as LPL with an associated
immunoglobulin M (IgM) serum monoclonal protein. Roughly 5% of LPL
patients secrete non-IgM paraproteins (e.g., IgG, IgA, kappa,
lambda) or are non-secretory (2).
Due to its extreme rarity (only 5% of all LPL
cases), non-IgM LPL is challenging to diagnose and, also, to treat
and cure. Moreover, those rare cases usually present with a
heterogeneous clinical phenotype and there is generally a lack of
reported cases. To the best of our knowledge, this is the second
non-IgM LPL case reported in scientific literature with kappa chain
production (3). Before that, Cautha
et al (2) reported a case of
a non-IgM LPL with lambda light chain monoclonal paraprotein
expression. In addition, it was presented how the management
strategy was modified from the time of diagnosis until the patient
finally benefitted from a treatment using a Bruton TK inhibitor
(Ibrutinib). This case will be useful in hematological practice to
shorten the time of diagnosis of such complicated and rare cases
and to suggest the most beneficial treatment protocol.
Case report
А 62-year-old female patient was accepted initially
in a regional hospital (September 2022; Hospital ‘St. George’,
Plovdiv, Bulgaria) suspected with a plasma cell neoplasia (PCN). At
that time, flow cytometry of the bone marrow revealed that 75% of
the lymphocytes had phenotype as
CD19+/CD20+/CD79b/CD5−/CD200−/CD23-/CD10−/CD43−.
In total, 16% of the lymphocytes belonged to T-subtype
(CD3+), 11% were Th cells (CD3+CD46) and
4.55% were Tc (CD3+CD8+). The ratio Th/Tc was
2.2; 2.31% were NK cells
(CD3−CD56+CD16+) and 2% of the
cells in the aspirate were plasma cells with a phenotype
CD38+/CD138+/CD19+/−/CD45+/CD56
kappa+. As those results were suspected for B-cell
lymphoma kappa (+), a molecular genetic analysis was recommended.
PCR analysis followed by 2% agarose gel electrophoresis with
ethidium bromide revealed a carriership of a somatic mutation L265P
in MYD88 gene. The flow cytometric and molecular-genetic
analyses were performed before the admission of the patient to our
clinic, and thus, the presented results were retrieved from the
official medical records, which is one of the limitations of the
present study.
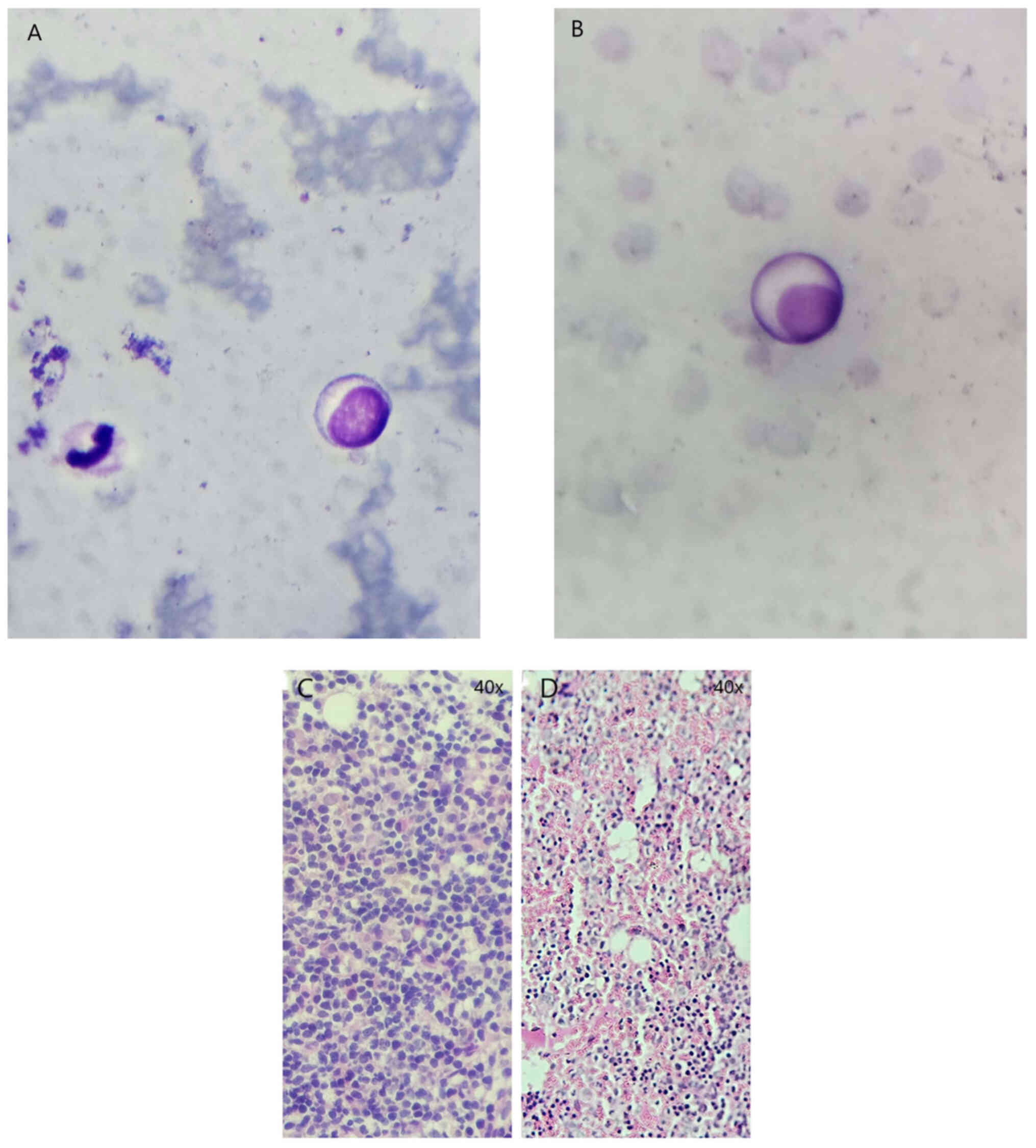
Trepanobiopsy showed cells typical for LPL, bearing
marks both of lymphocytes and plasma cells (Fig. 1A and B). Staining of the bone marrow
sample revealed a massive infiltration by lymphoplasmacytic cell
populations (Fig. 1C). The
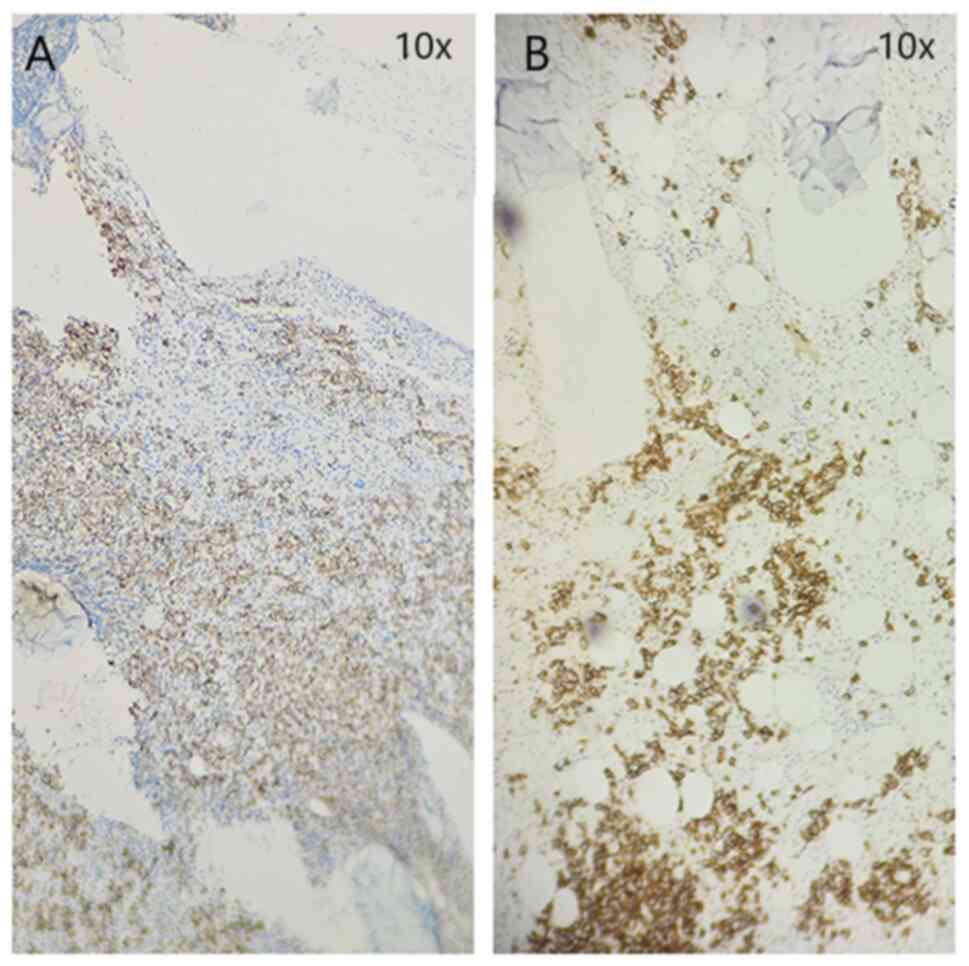
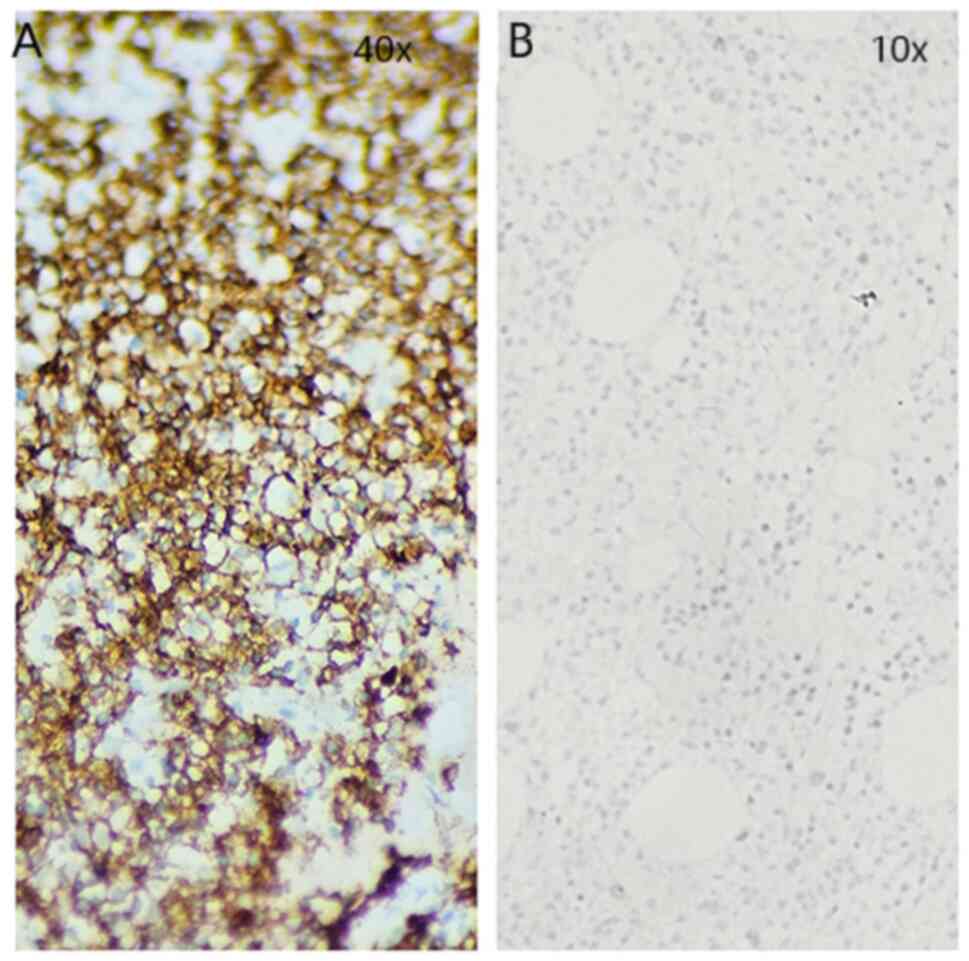
subsequent staining for markers CD138 (Fig. 2A) and CD20 (Fig. 3A) was positive. A substantial part
of the analyzed cells were Pax5-positive (a nuclear marker of
mature B cells), CD5 staining was positive only in a small amount
of dispersed lymphoids, and CD56 staining was positive in
osteoclasts and osteoblasts. MUM1 (a marker of a transition of
mature B cells towards plasma cells) was also expressed in a part
of the cell content; moderate fibrosis was detected (stage MF-2).
The subsequent agarose gel electrophoresis demonstrated a presence
of M gradient. The staining data were taken directly from the
medical records of the patient. An initial therapy was initiated by
one course of Bendamustine/Rituximab. The treatment provoked a
reaction of general kidney failure and anuria and the patient was
accepted at the Nephrology department for treatment.
Later on, upon a reason of her kidney problems, the
patient decided to seek a second opinion and referred to the
Hematology ward of the University Hospital ‘St. Ivan Rilski’ where
she had been admitted. She was assigned additional testing,
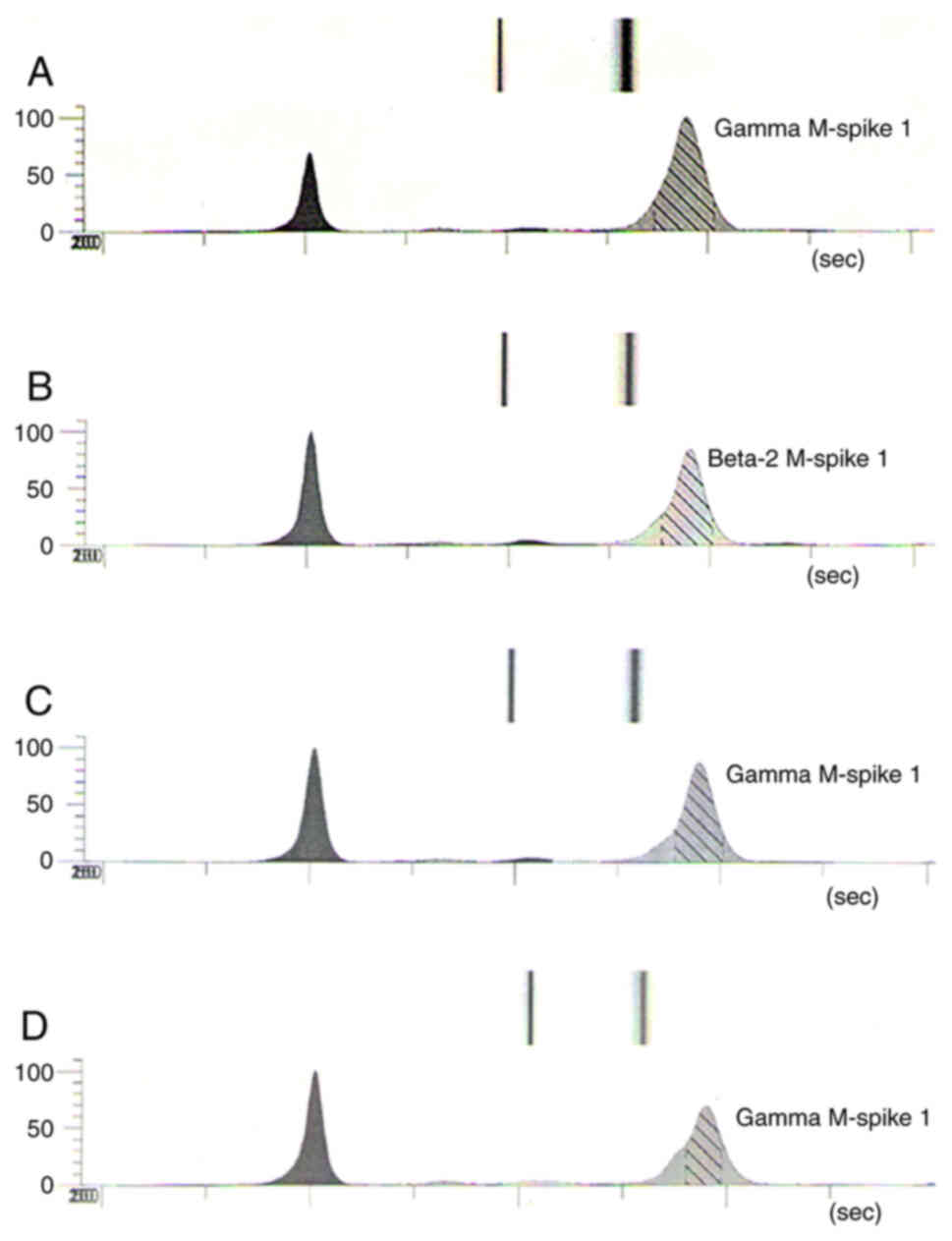
including serum isoelectric focusing (IEF), which demonstrated a
distinct amount of paraprotein in the serum type IgA/kappa (Gamma
M-spike 63.97 g/l, KF 119.94 mg/l) (Fig. 4A) and myelogram that reported
cytological data for LPL with moderate and expressed hypoplasia of
the bone marrow (54% lymphoplasmocytes). Computed tomography (CT)
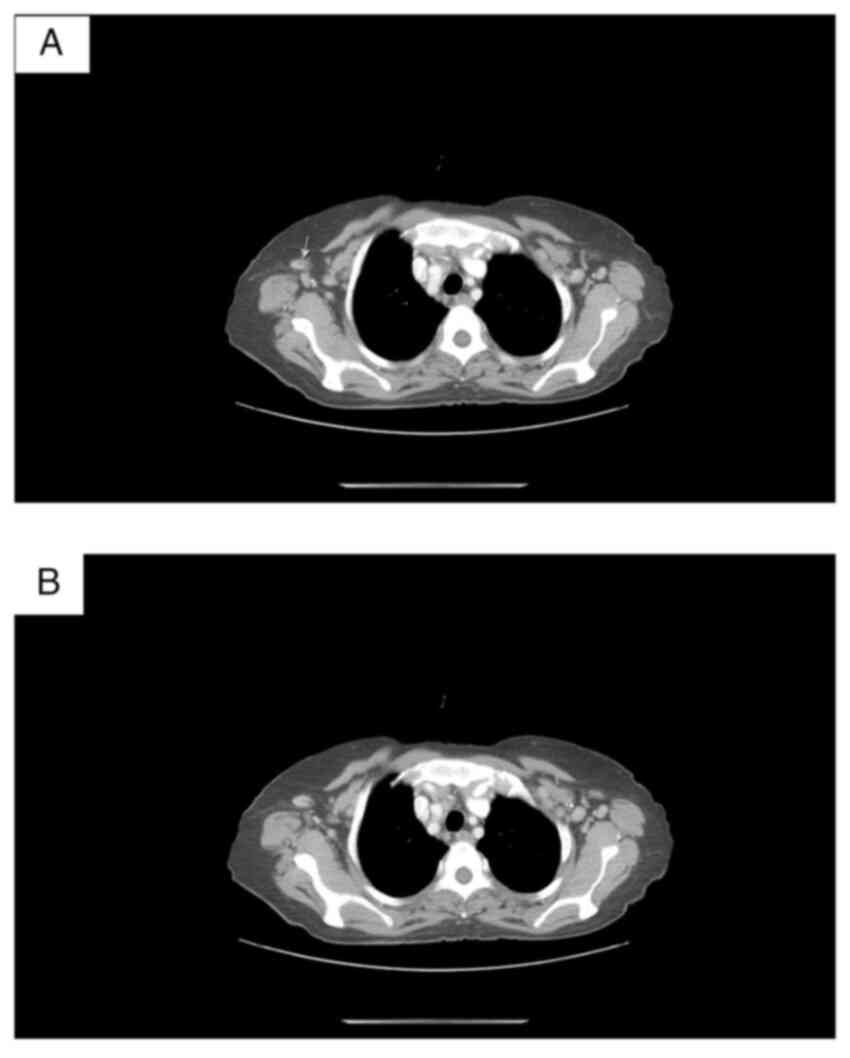
demonstrated rounded cervical lymph nodes with maximum axial
dimensions 8.8/8.5 mm; axial lymph nodes bilaterally [dimensions:
13.5/11 mm from the left side and 15/11 mm from the right (Fig. 5)]. No pleural effusion was
visualized. Тhe trachea had lumen of normal width and wall
thickness. The bronchi bilaterally were freely passable up to the
segmental level. No infiltrative changes or nodular lesions were
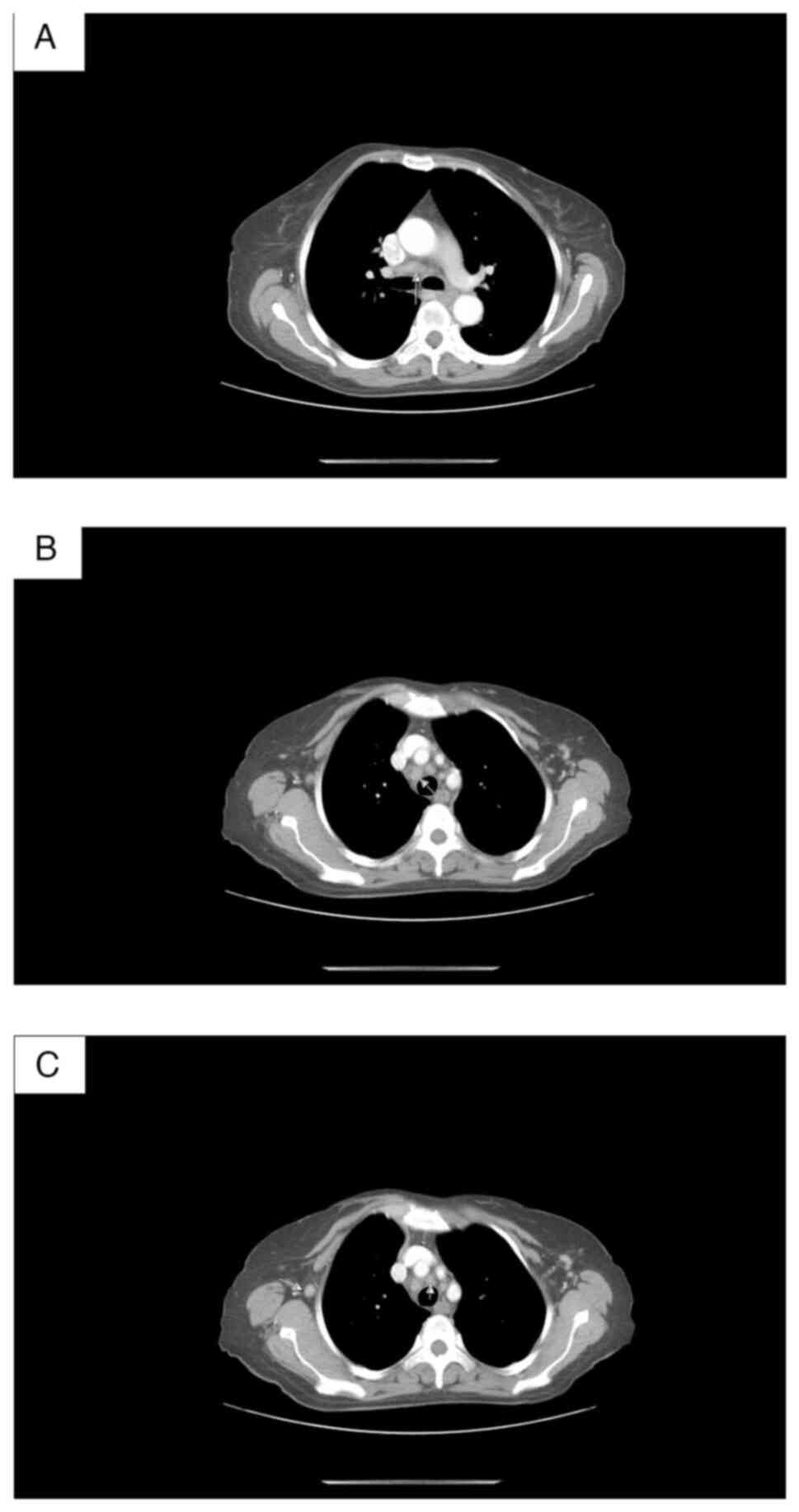
visualized in the lung parenchyma. CT data of enlarged mediastinal
lymph nodes with axial size 27/13 mm (Fig. 6). Тhere were no focal lesions in the
structure of the liver and the spleen; adrenal glands and both
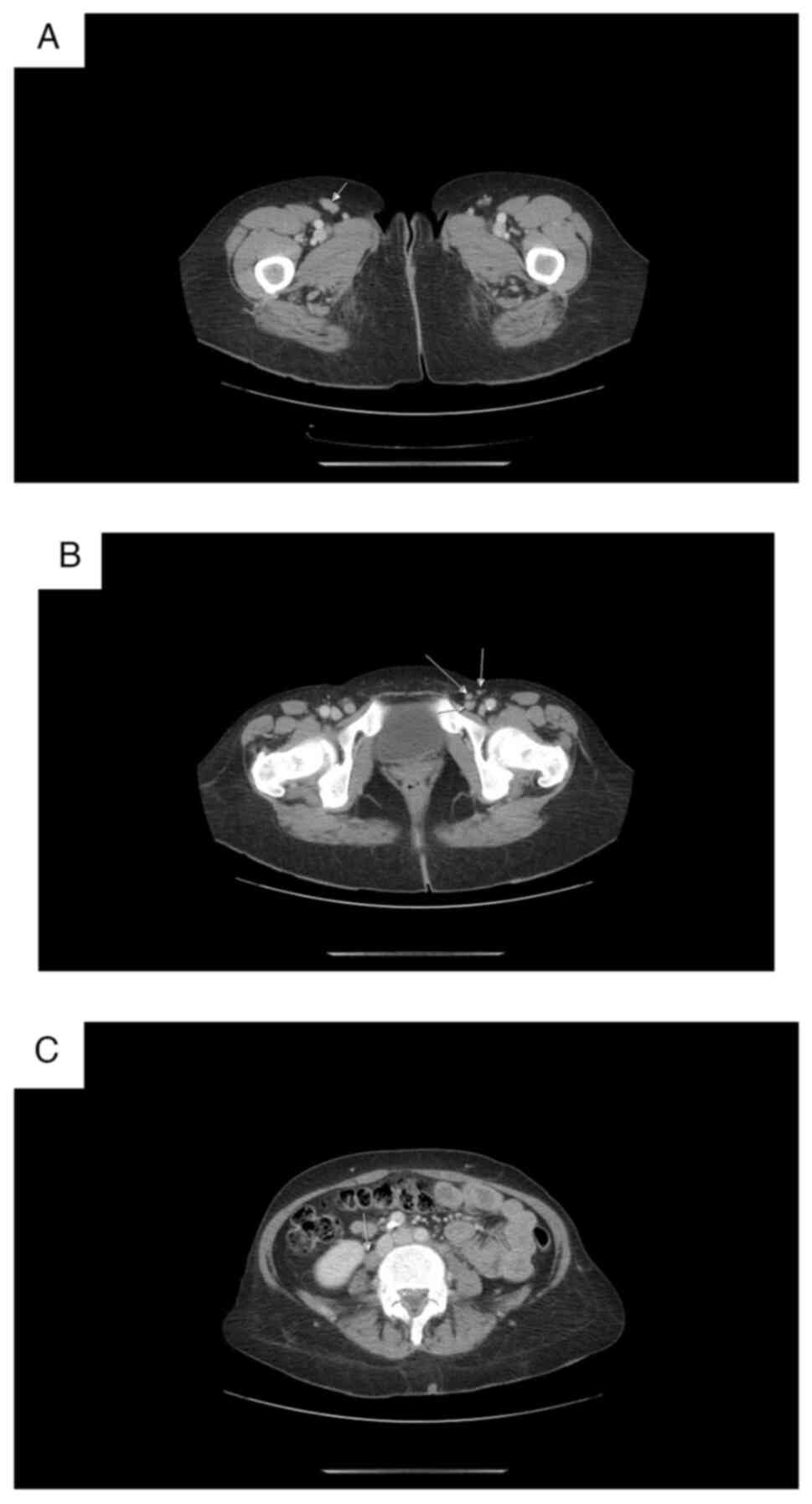
kidneys were intact. CT data for axillar, mediastinal, abdominal
(Fig. 7C), pelvic and inguinal
lymphadenopathy (Fig. 7A and B).
The diagnosis of LPL IV B clinical stage, stage D LPL was
confirmed. The treatment was based on NCCN clinical practice
guidelines in oncology (4) and the
patient was assigned three more courses of Bendamustine/Rituximab
(from November of 2022 to January of 2023). At that stage, the
patient decided to continue her treatment and follow up at the
University Hospital.
In February 2023, the patient was accepted again for
control examinations and re-assessment of the treatment.
Trepanobiopsy provided evidence of partial response (PR) to the
treatment. IEF confirmed the decrease in the paraprotein type
IgA/K-Gamma M-spike in the serum 44.57 g/l (Fig. 4B). Even if there was a partial
reduction, the plasma cell populations were again stained positive
(Fig. 1D). The lymphoplasmocytes
still accounted for >50% of the cell contents (Fig. 2B), indicating that the level of
plasma cell populations in the bone marrow was still high. There
was a complete lack of cells expressing CD20 (Fig. 3B). The results from the WBC are
included in Table I.
 | Table I.Whole blood count of the patient upon
her acceptance in the Hematology ward of ‘St. Ivan Rilski’
University hospital. |
Table I.
Whole blood count of the patient upon
her acceptance in the Hematology ward of ‘St. Ivan Rilski’
University hospital.
| Cell populations | Result | Units | Reference
ranges |
|---|
| WBC | 1.25 |
x109/l | 3.5–10.8 |
| RBC | 3.52 |
x1012/l | 3.76–5.34 |
| HGB | 113 | g/l | 115-150 |
| HTC | 0.305 | l/l | 0.350–0.490 |
| MCV | 86.8 | fL | 85.2–98.5 |
| MCH | 32.0 | pg | 28.4–34.5 |
| MCHC | 368 | g/l | 325-356 |
| PLT | 137 |
x109/l | 112-330 |
| LYM% | 41.9 | % | 15.2–41.9 |
| MONO% | 21.4 | % | 4.9–12.6 |
| EO% | 1.7 | % | Up to 6.2 |
| BASO% | 0.5 | % | Up to 1.3 |
| NEUT% | 34.5 | % | 37.6–78.7 |
| LYM | 0.52 |
x109/l | 1.0–4.5 |
| MONO | 0.27 |
x109/l | 0.4–1.1 |
| EO | 0.02 |
x109/l | 0.04–0.5 |
| BASO | 0.01 |
x109/l | Up to 0.1 |
| NEUT | 0.43 |
x109/l | 2.0–7.0 |
| RDW | 14.9 |
| 11.5–14.0 |
| MPV | 7.5 | fL | 6.2–10.6 |
| Sedimentation
rate | 91 | mm/h | Up to 30 |
| Glucose | 5.54 | mmol/l | 3.5–6.1 |
| Creatinine | 57.0 | µmol/l | Up to 96 |
| Total protein | 87.67 | g/l | 64.0–83.0 |
| β2
microglobulin-serum | 2.8 | mg/l | 0.97–2.64 |
Following the PR to the treatment, it was decided to
continue on the same regimen for another 2 courses of
immuno-chemotherapy. The patient's treatment course included
Rituximab 600 mg at day 1, Bendamustine 100 mg at days 2 and 3,
dexamethasone 20 mg/day for 4 days, Fraxiparine 0.4 mg/day and
additional drugs for her high blood pressure and kidney
failure.
After 6 courses in total with Bendamustine/Riuximab,
the patient was evaluated again with serum IEF exhibiting a
persistence of paraprotein type IgA/kappa in the serum (Gamma
M-spike 44.57 g/l, KF 39.74 mg/l) (Fig.
4C). From April 2023 she started therapy with Bruton kinase
inhibitor [Ibrutinib (brand name, Imbruvica)]-3 tablets/day (total
420 mg/day).
The last control examinations of the patient dated
from August 2023. After four months of treatment with Imbruvica,
she had substantially improved her health condition (Table II) and had lowered the level of IgA
with ~30 g (from October 2022 until July 2023). The serum
electrophoresis reported Gamma M-spike concentration of 29.27 mg/l
(Fig. 4D). Currently the patient is
feeling well; her ECOG PS performance status is 1, as of December
2023. The patient's blood examinations revealed mild anemia (HGB,
109 g/l), neutropenia (Neu, 1.01 g/l) and the biochemical results
revealed slightly elevated levels of total protein 87.8 g/l (normal
range 64–83 g/l). Her condition is continued to be monitored by
regular IEF of blood and urine samples.
| Table II.Whole blood count (WBC) of the
patient after her discharge from the Hematology ward of ‘St. Ivan
Rilski’ University hospital (at her last control examination). |
Table II.
Whole blood count (WBC) of the
patient after her discharge from the Hematology ward of ‘St. Ivan
Rilski’ University hospital (at her last control examination).
| Cell
populations | Result | Units | Reference
ranges |
|---|
| WBC | 1.68 |
x109/l | 3.5–10.8 |
| RBC | 4.11 |
x1012/l | 3.76–5.34 |
| HGB | 124 | g/l | 115-150 |
| HTC | 0.370 | l/l | 0.350–0.490 |
| MCV | 89.9 | fL | 85.2–98.5 |
| MCH | 30.2 | pg | 28.4–34.5 |
| MCHC | 336 | g/l | 325-356 |
| PLT | 243 |
x109/l | 112-330 |
| LYM% | 26.1 | % | 15.2–41.9 |
| MONO% | 17.2 | % | 4.9–12.6 |
| EO% | 5.2 | % | Up to 6.2 |
| BASO% | 1.3 | % | Up to 1.3 |
| NEUT% | 50.3 | % | 37.6–78.7 |
| LYM | 0.44 |
x109/l | 1.0–4.5 |
| MONO | 0.29 |
x109/l | 0.4–1.1 |
| EO | 0.09 |
x109/l | 0.04–0.5 |
| BASO | 0.02 |
x109/l | Up to 0.1 |
| NEUT | 0.85 |
x109/l | 2.0–7.0 |
| RDW | 15 |
| 11.5–14.0 |
| MPV | 9.5 | fL | 6.2–10.6 |
Discussion
WM is a term indicating LPL with an associated
immunoglobulin M (IgM) serum monoclonal protein (5). LPL, associated with IgA paraprotein is
even more rare and those cases mimic a plasma cell neoplasm (PCN)
(6). In a study of 27 cases with
either IgA or IgG secretion, Qiu et al (6) reported that all patients had a median
bone marrow involvement of 10%, light chain was identified in 96%
of the cases measured by flow cytometry, while MYD88
mutation was detected in 17 of 24 cases (71%). The neoplastic
plasma cells were positive for CD45 (100%), CD19 (96%), CD81 (89%),
CD27 (83%), CD56 (16%), whereas CD117 was consistently negative
(6). As differential diagnostic
features of LPL over PCN, the authors point the characteristic
immunophenotypic profile and the presence of MYD88 and/or
CXCR4 mutations. In a study comparing non-IgM LPL cases and
WM controls matched by age and sex, the presence of extramedullary
disease was higher in cases, while neuropathy and hyperviscosity
was higher in controls (7). A
recent study using next-generation sequencing approach reported
MYD88 to be the most common mutation in non-IgM LPL patients
(70%), followed by CXCR4 (20%), KMT2D (10%) and
ARID1A (10%). Mutant allele frequency in MYD88 L265P
did not differ significantly between WM and non-IgM-type LPL. Most
mutations detected by NGS were subclonal following MYD88
L265P. Those results proved similar genetic characteristics in the
two subsets of LPL patients (8).
Nearly 90% of IgM secreting LPL develop a somatic
mutation in the gene MYD88, and even rarer, in CXCR4
(1). MYD88 is a driver gene
in hematologic B-cell malignancies and the missense mutation
(L265P) is a single causative mutation transforming IgM-producing B
cells into malignant B cells (9).
It constitutively activates NF-κB and its associated signaling
pathways, thereby promoting B-cell proliferation and survival
(10). It is a missense
gain-of-function mutation in myeloid differentiation changing the
amino-acid leucine to proline at position 265 of MYD88 (11). MYD88 is playing the role of an
adaptor molecule in the canonical NF-κB pathway coordinating the
assembly of a multisubunit complex of IRAK1 and IRAK4 (IL-1
receptor-associated kinases), which through TRAF6, activate TAK1
(TGFβ-activated kinase 1). The activated NF-κB increases signaling
of IL-6 and IL-10, which further promotes B-cell proliferation and
survival via JAK/STAT signaling cascade (10).
MYD88 L265P is a somatic mutation (9,12)
identified in ~90% of WM, but the demonstration of this mutation is
also a necessary tool for the right WHO diagnosis of LPL (12,13).
It is usually found in blood and/or the bone marrow. However, there
are unusual places of its expression, as the skin in cutaneous form
of WM (14). MYD88 L265P is
usually used to discriminate WM and non-IgM LPL from other B-cell
disorders (15). However, there are
several studies discussing the potential of the mutation as a
prognostic biomarker. It was previously revealed that MYD88
expression was an independent prognostic factor affecting overall
survival in diffuse large B cell lymphoma (16,17).
It was positively correlated with high Ki-67 expression and
promotes tumor proliferation (17).
Pham-Ledard et al (18)
reported that MYD88 L265P was associated with a poorer
outcome of patients with large B-cell lymphoma. Previous studies
could not engage with a statement about the prognostic impact of
the mutation (19,20). In LPL/WM, there is no consensus
about the role of MYD88 L265P as prognostic biomarker.
LPL/WM patients harboring the MYD88 L265P mutation show
clues of longer survival compared with wild-type cases (21). In IgM-secreted monoclonal
gammopathies, the mutation behaves like an adverse risk factor and
exhibits a 5-fold increased risk of progression to LPL/WM (22). According to other authors, the
identification of the MYD88 L265P gene mutation
represented a major advance in the diagnosis of LPL (3,13).
Rossi (23) stated that the
presence of this mutation is just an indicative of a more accurate
diagnosis of LPL and maybe just accompanies the tumor burden on
predicting the disease progression. Cautha et al (2) reported a non-IgM rare case of LPL with
lambda light chain paraprotein expression positive for MYD88 L265P
mutation. It was stated that non-IgM LPL has a poorer outcome yet
does not infer the prognosis to the mutational presence (2). In the present study, the patient shows
a favorable recovery yet, according to the authors, this is rather
due to the treatment strategy than to the prognostic indication of
MYD88 L265P mutation.
Major clinical complications of non-IgM-secreting
LPL have been described as anemia associated symptom (53.8%),
mucocutaneous hemorrhage and superficial lymphadenopathy (15.4%).
At the same time, clinical and biological differences between
non-IgM LPL and WM have not been identified (24). One of the serious complications
associated with high levels of paraproteins in patients with
hematological malignancies is the hyperviscosity syndrome (HVS). It
is demonstrated by symptoms, including nosebleeds, blurring vision,
dizziness, headaches and shortness of breath (1). The treatment of WM (IgM LPL) mainly
aims to monitor the disease and keep it under stable control. In
case of serious symptoms, HVS or deviations from the normal blood
indicators (anemia, neutropenia or thrombocytopenia), the usual
starting treatment consists of a combination of a chemotherapeutic
and an antibody (Rituximab). The common regimens include either DRC
(Dexamethasone, Rituximab and Cyclophosphamide) or Benda-R
(Bendamustine and Rituximab). In the current presented case, the
second treatment option was preferably used. In some cases, Bruton
tyrosine kinase (BTK) inhibitors or proteasome inhibitors give
promising results but in specific circumstances (relapsed lymphoma
or lack of response to previous treatment). When trying to
discriminate non-IgM LPL and WM, there were no discovered
differences in both response and overall survival and they are
similar between the two groups (7).
However, a study from 2018 in Korean patients, reported that
patients with non-IgM LPL demonstrated a higher 5-year mortality
rate and more adverse prognostic factors than those with LPL/WM
(25). Another study of non-WM LPL
cases identified the most common paraprotein being IgG (54%),
followed by IgA (15%) and non-secretory (12%). The authors reported
more adverse prognostic factors such as elevated LDH, anaemia and
lymphocytosis at diagnosis but no difference in overall survival
(26). As the combination of
Bendamustine and Rituximab in the present case did not produce the
desired effect, Ibrutinib (IMBRUVICA®) was included in
the treatment protocol.
Ibrutinib has been approved in combination with
Rituximab for the treatment of adults with WM in 2018. From that
moment on, several studies reported the overall efficacy of
Ibrutinib treatment. In a study of 2017, Helber et al
(27) reported the clinical effect
of Ibrutinib on 23 patients with LPL (most with IgM and one with
IgG secretion). The median maximum IgM decrease was 67%, as for
IgG-the decrease was 37%. It was underlined that the response to
Ibrutinib requires long-term continuation of treatment in most of
the patients (27). According to
the approved therapeutic indications of the drug, it is recommended
for the treatment of elderly patients with CLL (in combination with
Bendamustine and Rituximab) who have received at least one
preceding therapy or for patients with WM (in combination with
Rituximab). In a previously reported case of non-IgM LPL and lambda
light chain expression, the authors reported that, even in case of
an initial response, the treatment of Ibrutinib and Rituximab did
not lead to an improve (the patient succumbed after 8 months of
diagnosis) (2). To the best of the
authors' knowledge, there is only one more case in literature,
reporting the usage of Ibrutinib in a refractory IgA LPL carrying
MYD88 L265P gene mutation (3). In
the cited study, the patient benefited from Ibrutinib treatment for
a period of 4–6 weeks following treatment and the PR was maintained
for ~1 year of therapy. Therefore, the present case is the second
reported kappa positive LPL case positively influenced by Ibrutinib
treatment and the response was detected at the fourth month
following treatment. In the current case, the patient showed a
substantial improvement in her condition and the level of the IgA
paraprotein was decreased ~50% after four months of treatment with
the triple combination (Bendamustine, Rituximab and Ibrutinib).
Therefore, the reported data confirms previously reported single
observations and adds a relevant knowledge to those
challenging-to-diagnose and rare cases of non-IgM IgA kappa
secreting LPLs.
In conclusion, the case presented by our team
describes a very rare hematological malignancy; IgA-secreted LPL
resembling PCN. The processes of the disease diagnosis and
treatment were reported, highlighting the effectiveness of the
triple treatment protocol (Benda-R plus BTK) in the patient of the
present case report. Regardless of the observed improvement, there
is a necessity for a long-term monitoring of the patient condition
to prove the suitability of the selected treatment option.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
DN designed the study, analyzed and interpreted the
data, and drafted the manuscript. AY, LS and AM acquired the data
and participated in the analysis. DN, AR and AM interpreted the
data. AR critically revised the manuscript and made final
suggestions. AY, AM, LS and AR treated and cared for the patient.
AY and AR confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written informed consent upon
admission to the University Hospital ‘St. Ivan Rilski’ for
publication of the data and associated images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
LPL
|
lymphoplasmacytic lymphoma
|
|
WM
|
Waldenström macroglobulinemia
|
|
WBC
|
whole blood count
|
|
PR
|
partial response
|
|
PCN
|
plasma cell neoplasia
|
References
|
1
|
Lymphoma Action: Lymphoplasmacytic
lymphoma and Waldenström's macroglobulinaemia. Lymphoma Action,
Bucks. 2022.https://lymphoma-action.org.uk/types-lymphoma-non-hodgkin-lymphoma/lymphoplasmacytic-lymphoma-and-waldenstroms-macroglobulinaemia#symptoms
|
|
2
|
Cautha S, Gupta S, Hanif A, Moirangthem V
and Jain K: Lymphoplasmacytic lymphoma with only lambda light chain
monoclonal paraprotein expression. Eur J Case Rep Intern Med.
9:0031062022.PubMed/NCBI
|
|
3
|
Quaglia FM, Rigolin GM, Saccenti E,
Negrini M, Volta E, Dabusti M, Ciccone M, Urso A, Laudisi M and
Cuneo A: Response to Ibrutinib of a refractory IgA
lymphoplasmacytic lymphoma carrying the MYD88 L265P gene mutation.
Mediterr J Hematol Infect Dis. 11:e20190572019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar SK, Callander NS, Adekola K,
Anderson LD Jr, Baljevic M, Baz R, Campagnaro E, Castillo JJ,
Costello C, D'Angelo C, et al: Waldenström
macroglobulinemia/lymphoplasmacytic lymphoma, version 2.2024, NCCN
clinical practice guidelines in oncology. J Natl Compr Canc Netw.
22((1D)): e2400012024.PubMed/NCBI
|
|
5
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD and Jaffe ES: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qiu L, Nwogbo OV, Medeiros LJ, Thakral B,
Li S, Xu J, You MJ, Wang W, Quesada AE, Ramos CB, et al:
Lymphoplasmacytic lymphoma with IgG or IgA paraprotein: A study of
29 cases including cases that can mimic plasma cell neoplasms. Hum
Pathol. 130:47–57. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Castillo JJ, Itchaki G, Gustine JN, Meid
K, Flynn CA, Demos MG, Guerrera ML, Jimenez C, Kofides A, Liu X, et
al: A matched case-control study comparing features, treatment and
outcomes between patients with non-IgM lymphoplasmacytic lymphoma
and Waldenström macroglobulinemia. Leuk Lymphoma. 61:1388–1394.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Awata-Shiraiwa M, Yokohama A, Kanai Y,
Gotoh N, Kasamatsu T, Handa H, Saitoh T, Murakami H, Hirato J,
Ikota H and Tsukamoto N: Waldenstrom macroglobulinemia and
non-IgM-type lymphoplasmacytic lymphoma are genetically similar.
Acta Haematol. 146:384–390. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu X, Li W, Deng Q, Li L, Hsi ED, Young
KH, Zhang M and Li Y: MYD88 L265P mutation in lymphoid
malignancies. Cancer Res. 78:2457–2462. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Groen RAL, Schrader AMR, Kersten MJ,
Pals ST and Vermaat JSP: MYD88 in the driver's seat of B-cell
lymphomagenesis: From molecular mechanisms to clinical
implications. Haematologica. 104:2337–2348. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ngo VN, Young RM, Schmitz R, Jhavar S,
Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al:
Oncogenically active MYD88 mutations in human lymphoma. Nature.
470:115–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martinez-Lopez A, Curiel-Olmo S, Mollejo
M, Cereceda L, Martinez N, Montes-Moreno S, Almaraz C, Revert JB
and Piris MA: MYD88 (L265P) somatic mutation in marginal zone
B-cell lymphoma. Am J Surg Pathol. 39:644–651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martino G, Marra A, Ascani S and
Sportoletti P: Uncommon lymphoplasmacytic lymphoma with IgA
paraproteinemia: A challenging clinical diagnosis solved by MYD88
mutation analysis. Ann Hematol. 98:1507–1508. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Minzenmayer AN, Miranda RN, Powell PR and
Parekh PK: An unusual case of cutaneous Waldenström
macroglobulinemia with the MYD88 L265P mutation. J Cutan Pathol.
47:850–853. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao
Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al: MYD88
L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J
Med. 367:826–833. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fernández-Rodríguez C, Bellosillo B,
García-García M, Sánchez-González B, Gimeno E, Vela MC, Serrano S,
Besses C and Salar A: MYD88 (L265P) mutation is an independent
prognostic factor for outcome in patients with diffuse large B-cell
lymphoma. Leukemia. 28:2104–2106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Niu J, Ma Z, Nuerlan A, Li S, Cui W, Gao
H, Abulajiang G, Zhang W and Li X: Prognostic value of MYD88 L265P
mutation in diffuse large B cell lymphoma via droplet digital PCR.
Mol Med Rep. 22:1243–1256. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pham-Ledard A, Beylot-Barry M, Barbe C,
Leduc M, Petrella T, Vergier B, Martinez F, Cappellen D, Merlio JP
and Grange F: High frequency and clinical prognostic value of MYD88
L265P mutation in primary cutaneous diffuse large B-cell lymphoma,
leg-type. JAMA Dermatol. 150:1173–1179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu S, Luo H, Pan M, Palomino LA, Song X,
Wu P, Huang JM and Zhang Z: High frequency and prognostic value of
MYD88 L265P mutation in diffuse large B-cell lymphoma with R-CHOP
treatment. Oncol Lett. 15:1707–1715. 2018.PubMed/NCBI
|
|
20
|
Lee YS, Liu J, Fricano KA, Webb EM,
Toolsie DR, Jones S, Rhoads JA, Vij R, Cashen AF, Abboud CN, et al:
Lack of a prognostic impact of the MyD88 L265P mutation for diffuse
large B cell lymphoma patients undergoing autologous stem cell
transplantation. Biol Blood Marrow Transplant. 23:2199–2204. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Treon SP, Cao Y, Xu L, Yang G, Liu X and
Hunter ZR: Somatic mutations in MYD88 and CXCR4 are determinants of
clinical presentation and overall survival in Waldenstrom
macroglobulinemia. Blood. 123:2791–2796. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Varettoni M, Zibellini S, Arcaini L,
Boveri E, Rattotti S, Pascutto C, Mangiacavalli S, Gotti M,
Pochintesta L, Paulli M and Cazzola M: MYD88 (L265P) mutation is an
independent risk factor for progression in patients with IgM
monoclonal gammopathy of undetermined significance. Blood.
122:2284–2285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rossi D: Role of MYD88 in
lymphoplasmacytic lymphoma diagnosis and pathogenesis. Hematology
Am Soc Hematol Educ Program. 2014:113–118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zou D, Yi S, Liu H, Li Z, Lyu R, Liu W, Ru
K, Zhang P, Chen H, Qi J, et al: Clinical and biological
characteristics of non-IgM lymphoplasmacytic lymphoma. Zhonghua Xue
Ye Xue Za Zhi. 36:493–496. 2015.(In Chinese). PubMed/NCBI
|
|
25
|
Kang J, Hong JY and Suh C: Clinical
features and survival outcomes of patients with lymphoplasmacytic
lymphoma, including non-IgM type, in Korea: A single-center
experience. Blood Res. 53:189–197. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brandefors L, Sander B, Lundqvist K and
Kimby E: Clinical characteristic and outcome of lymphoplasmacytic
lymphoma of non-Waldenstrom macroglobulinemia type: A Swedish
lymphoma registry study. Br J Haematol. 196:1362–1368. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Helber MJ, Moore JE, Williams AM, Meacham
PJ, Rothberg PG and Zent CS: Ibrutinib therapy for
lymphoplasmacytic lymphoma. Am J Hematol. 92:E542–E544. 2017.
View Article : Google Scholar : PubMed/NCBI
|