Introduction
Succinate dehydrogenase (SDH)-deficient renal cell
carcinoma (RCC) is a rare form of RCC that has a hereditary
component (1). Renal cancer, a
common tumor of the urinary system, accounts for 2–3% of adult
malignancies globally (2). However,
SDH-deficient RCC accounts for 0.05–0.20% of all cases of renal
cancer (3). This form of RCC is
rare compared to more common subtypes such as clear cell RCC, which
accounts for about 70–80% of all RCC cases. In 2004, Vanharanta
et al (4) first reported
SDH-deficient RCC. SDH-deficient RCC was subsequently included as a
new subtype of RCC in the 2016 edition of the World Health
Organization classification of renal tumors (5).
SDH-deficient RCC is an autosomal dominant disease
caused by an SDH gene mutation. SDH, also known as mitochondrial
complex II, is formed from four protein subunits [SDH complex
flavoprotein subunit A (SDHA), SDHB, SDHC and SDHD], which is
involved in the tricarboxylic acid cycle and the respiratory
electron transport chain (6,7). The
function of SDH is to catalyze the energy-dependent conversion of
succinate to fumarate (6). The four
subunits are encoded by four genes respectively, and mutations in
any one of the genes could cause the decrease, and even loss, of
SDH activity (8), which results in
the intracytoplasmic accumulation of succinate and potentially
predisposes patients to neoplastic transformation (9,10).
SDHB mutations are the most common, followed by SDHC and SDHA
mutations (1). SDHD mutations are
rare compared with other mutations, and ~30% are multifocal or
bilateral renal tumors (3,5,11).
Studies have reported that succinate, the catalytic substrate of
SDH, is involved in signal transduction pathways in renal cell
cancer (7–9,12), but
the exact mechanism of the SDH gene mutation that leads to
tumorigenesis is currently unclear. Mutations in the SDH gene are
also associated with certain types of hereditary tumors, such as
pheochromocytomas, gastrointestinal stromal tumors, familial
paragangliomas and partial pituitary adenomas (13,14).
SDH-deficient RCC is more commonly diagnosed in
young adults (age, 22 to 72 years) and slightly more prevalent in
men (3,11). The diagnosis of SDH-deficient RCC
relies upon pathology, immunohistochemistry (IHC) and genetic
testing (3). Macroscopically, the
tumors are well-circumscribed, with mass or cystic growth that is
grayish-red or grayish-brown in appearance and can be accompanied
by hemorrhage. Microscopically, the tumor cells typically appear
cuboidal or oval and are arranged in nests or tubules, often
exhibiting cystic changes (15).
The tumor cells typically have round or oval nuclei, scattered
flocculent chromatin and inconspicuous nucleoli (13,15).
The most prominent histological feature of SDH-deficient RCC is the
presence of cytoplasmic vacuoles or inclusion bodies (13).
The present study reported the case of a 49-year-old
patient with a highly malignant tumor who developed distant bone
metastasis upon initial examination. The postoperative pathological
diagnosis was at first unclear and, in combination with the
patient's clinical manifestations and imaging data, hereditary
leiomyomatosis and RCC syndrome (HLRCC) was considered. Finally,
genetic screening identified a germline mutation in the SDHA gene,
and IHC demonstrated loss of SDHB expression. The imaging and
pathological data of the patient were similar to that of HLRCC and
differential diagnosis was challenging. To the best of our
knowledge, misdiagnosed with HLRCC due to microscopic resemblance,
but genetic testing later revealed an SDHA mutation, confirming
SDHA-deficient RCC, which has not been reported before and
emphasizes the necessity for further genetic screening.
Case report
The present report was conducted in accordance with
the Surgical CAse REport guidelines (16). A 49-year-old male patient presented
to Linyi People's Hospital, Linyi, China) in November 2022 with a
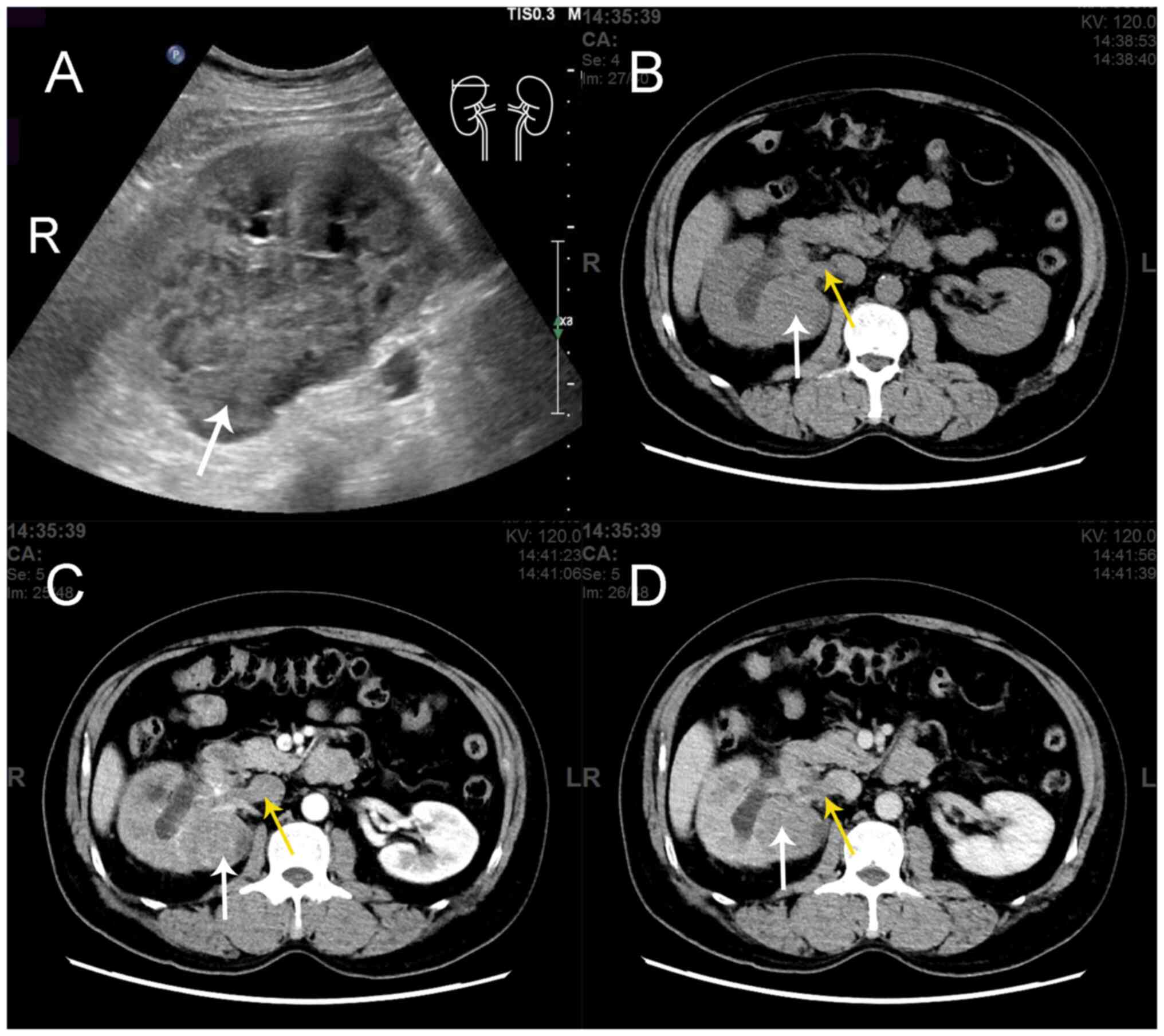
soft-tissue mass on his right kidney, detected by B-mode ultrasound
imaging during a health check-up (Fig.
1). The patient had a history of fractures, with a left humerus
fracture occurring 1 month prior to admission due to minor external
force, with no risk factors for malignancy and no family history of
malignant disease. Upon physical examination, the patient
demonstrated no abnormalities in both kidneys. Contrast-enhanced
computed tomography (CT) scan indicated a 55×68 mm mass in the
right kidney along with a filling defect in the right renal vein
and inferior vena cava (Fig. 1),
which was suspected to indicate an intravascular tumor thrombus. A
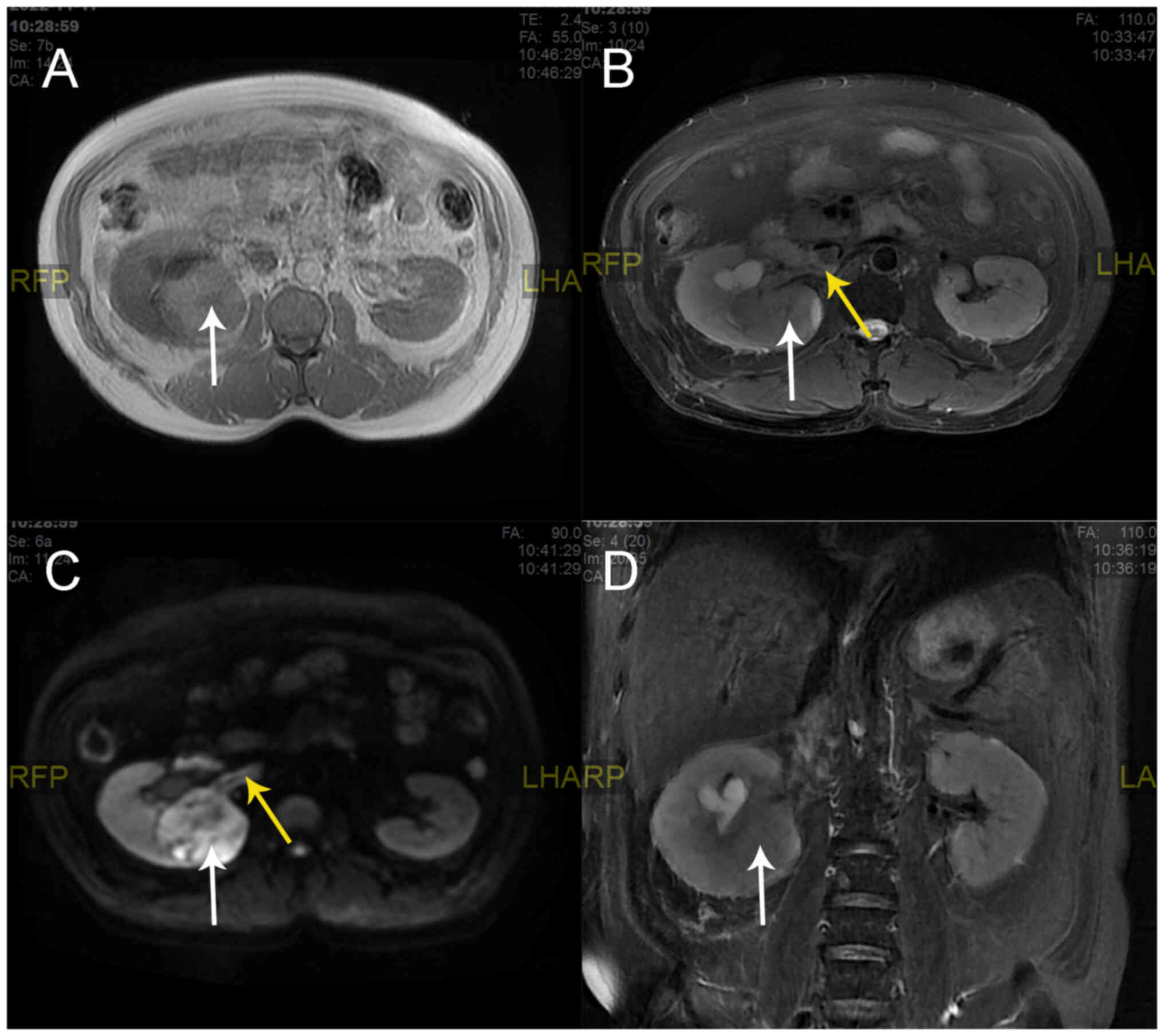
renal magnetic resonance imaging (MRI) scan showed enlargement of
the right kidney with abnormal round signals covering an area of
55×68 mm, which included a mixed, slightly high T1 weighted image
(T1WI) signal, a mixed T2 weighted image (T2WI) signal and an
inhomogeneous high diffusion-weighted imaging (DWI) signal
(Fig. 2). The perirenal contour was
well circumscribed. The right renal pyelectasis was accompanied by
a patchy homogeneous T1 signal, a high T2 signal and DWI exhibited
a high signal (Fig. 2). Digital
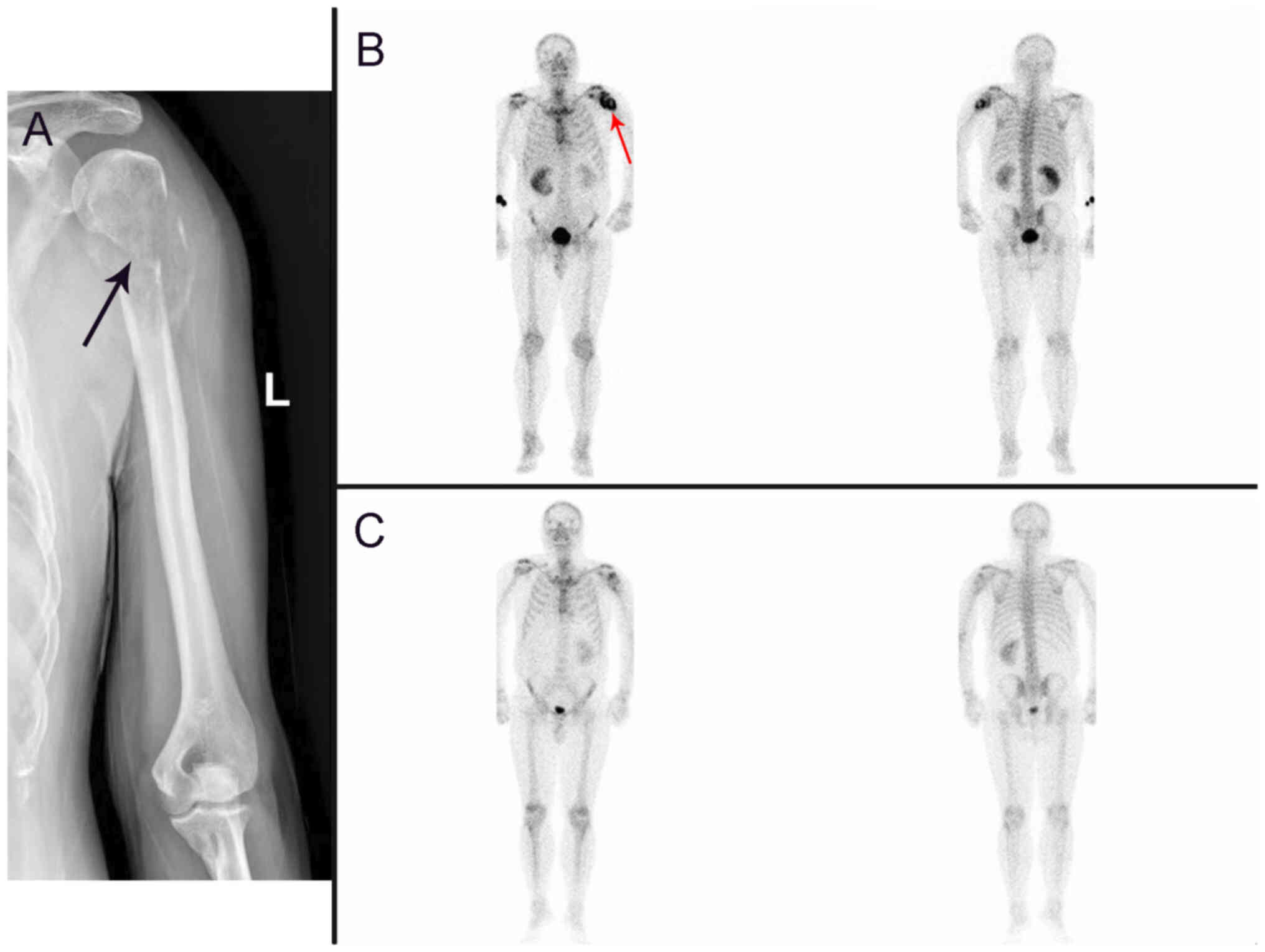
radiography demonstrated discontinuity in the cortical bone at the
proximal end of the left humerus (Fig.
3A). A whole-body bone scan demonstrated radionuclide
accumulation at the upper end of the left humerus (Fig. 3B), which indicated possible
pathological bone destruction.
The preoperative diagnosis was malignant tumor of
the right kidney, accompanied by tumor cell emboli in the right
renal vein and inferior vena cava. Subsequently, surgical resection
of the tumor was performed. Pathological assessment was performed
on the formalin-fixed, paraffin-embedded (FFPE) tissue block of
surgical specimen stained with hematoxylin and eosin. Postoperative
pathology demonstrated a high-grade RCC with lymphatic and vascular
involvement, as well as invasion of the renal sinus but not
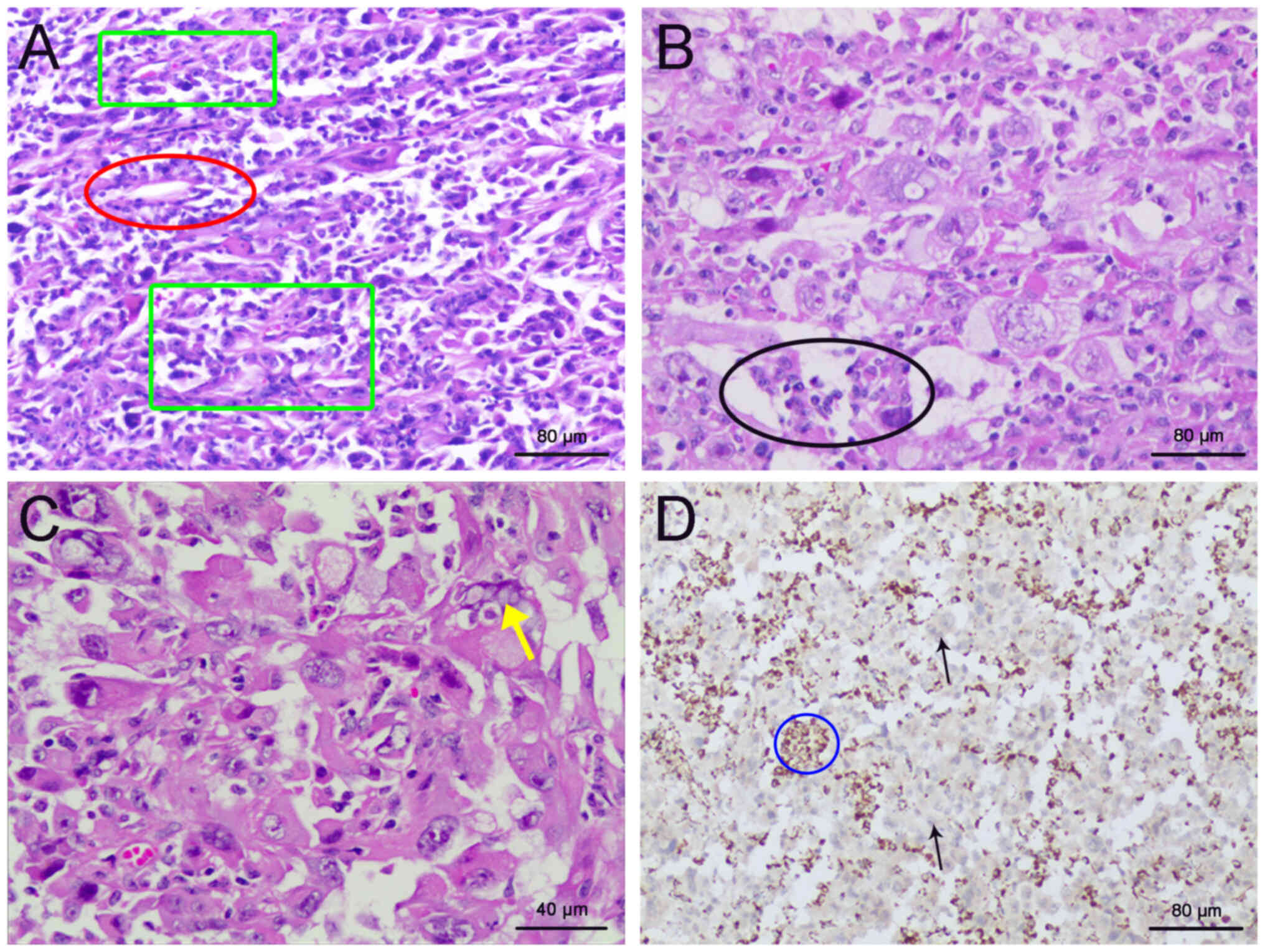
perirenal fat (Fig. 4A and B).
Microscopically, the tumor cells exhibited a mixed growth pattern
arranged in nests, papillae and tubular cysts with prominent
nucleoli, increased mitotic figures and marked nuclear atypia.
Immunohistochemical analysis was conducted on formalin-fixed
paraffin-embedded (FFPE) tissue blocks. Specimens were fixed in 10%
neutral buffered formalin at room temperature for 16–24 h. Tumor
sections, cut to a thickness of 4 µm, were deparaffinized and
rehydrated through a series of solutions: xylene I for 15 min,
xylene II for 15 min, 100% ethanol for 5 min, 95% ethanol for 5
min, 80% ethanol for 2 min, and 70% ethanol for 2 min. The sections
were washed in phosphate-buffered saline (PBS) three times, each
wash lasting 10 min. Antigen retrieval was performed in a pressure
cooker using 0.01 M citrate buffer (pH 6.0) at 95°C for 10 min.
Following this, the slides were washed three times with 0.01 M PBS
(pH 7.4), each wash for 5 min. Endogenous peroxidase activity was
blocked by incubating the slides with 3% hydrogen peroxide at 37°C
for 30 min, followed by three PBS washes, each lasting 10 min.
Non-specific binding was blocked by incubating the
sections with 10% goat serum (Fuzhou Maixin Biotechnology
Development Co., Ltd.) at room temperature for 30 min, followed by
three PBS washes, each for 10 min. Primary antibodies were applied
at room temperature for 30 min and then at 4°C for 12 h. The
primary antibodies used included Cytokeratin 7 (ZM-0071),
α-methylacyl-coenzyme A racemase (ZA-0228), E-cadherin (ZM-0092),
CD10 (ZM-0092), carbonic anhydrase IX (TA500623), Pax-2 (ZA-0467),
CD117 (ZA-0523), Anaplastic Lymphoma kinase (ALK)-1A4/1H7
(ZM-0248), cytokeratin 20 (ZA-0574), integrase interactor 1
(ZA-0696), tumor protein 63 (ZM-0406), GATA binding protein 3
(ZA-0661), S100 calcium-binding protein P (ZM-0494),
Vimentin(ZM-0260), and octamer-binding transcription factor 4
(ZM-0233). All primary antibodies were provided as ready-to-use
formulations, requiring no further concentration or dilution, and
were supplied by Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd. Following incubation, the slides were brought to room
temperature for 30 min and then washed three times with PBS, each
wash for 10 min. A horseradish peroxidase-conjugated goat
anti-rabbit IgG secondary antibody (ZB-5301, 1:5,000, provided by
Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.) was added,
and the slides were incubated at room temperature for 1 h.
Diaminobenzidine (DAB) was used as the chromogen for visualization.
The sections were counterstained with hematoxylin for 2 min at room
temperature and then blued in ammonia water for 1 min, sealed with
Permount Mounting Medium, and examined under a LEICA DM2000 light
microscope. IHC demonstrated the following: Cytokeratin 7 (−),
α-methylacyl-coenzyme A racemase (+), E-cadherin (+), CD10 (+),
carbonic anhydrase IX (+), Pax-2 (+), CD117 (−), ALK-1A4/1H7 (−),
cytokeratin 20 (−), integrase interactor 1 (+), tumor protein 63
(−), GATA binding protein 3 (−), S100 calcium-binding protein P
(−), Vimentin (+), octamer-binding transcription factor 4 (−) and
CK (+) (Fig. 5). The tumor stage
was pT3bN0M1. Due to the microscopic appearance of the cancer cells
closely resembling hereditary leiomyomatosis and renal cell
carcinoma (HLRCC), and the characteristic markers of SDH-deficient
RCC-cytoplasmic vacuolization or cytoplasmic inclusions-were
difficult to identify in this case. Pathological examination could
not confirm the diagnosis and HLRCC was considered. The diagnosis
of HLRCC requires genetic sequencing for confirmation.
Consequently, genetic testing was conducted on the patient.
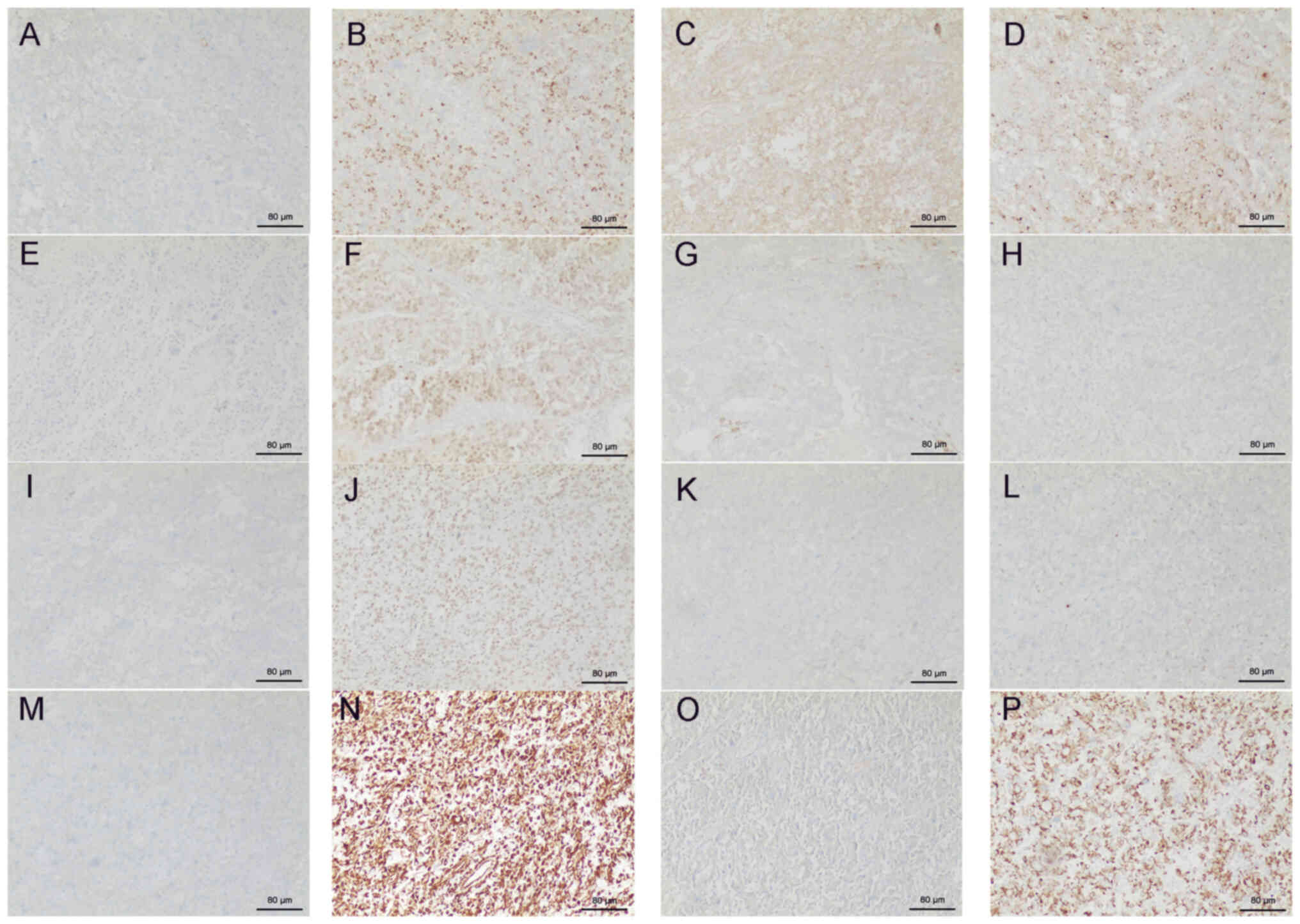 | Figure 5.Immunohistochemistry of renal cancer
tissues excised from the patient. (A) Cytokeratin 7 (−), (B)
α-methylacyl-coenzyme A racemase (+), (C) E-cadherin (+), (D) CD10
(+), (E) carbonic anhydrase IX (+), (F) Pax-2 (+), (G) CD117 (−),
(H) ALK-1A4/1H7 (−), (I) cytokeratin 20 (−), (J) integrase
interactor 1 (+), (K) tumor protein 63 (−), (L) GATA binding
protein 3 (−), (M) S100 calcium-binding protein P (−), (N) Vimentin
(+), (O) octamer-binding transcription factor 4 (−) and (P) CK (+).
Scale bar, 40 mm. +, positive; -, negative |
In this case, high-throughput sequencing was
performed. The sequencing process was contracted out to a certified
company, ensuring high-quality and reliable data. The procedure
employed high-throughput sequencing combined with targeted region
capture technology. The DNA/RNA samples for sequencing were
prepared using the MagPure Buffy Coat DNA Midi KF Kit, which is
catalogued under D3537-02#; Magen Biotech. Agarose gel
electrophoresis was employed to verify the quality/integrity of
processed samples. The sequencing was executed using the
MGISEQ-2000RS High-Throughput Sequencing Kit (catalog number
1000012554# (two-part), supplied by MGI. The kit supports
paired-end sequencing (PE100+100+10). The sequencing strategy
employed was paired-end sequencing, with a sequence length of 100
base pairs. Finally, the loading concentration of the final library
was measured at 26.42 fmol/ul using the Qubit ssDNA Assay.
Postoperative high-throughput sequencing was performed and
indicated a mutation in the SDHA gene [c.1A>G (p. Met1?)]. On
this basis, the disease was diagnosed as SDHA-deficient RCC. Human
genetic counseling indicated that this specific mutation leads to
the alteration of the protein translation start codon, resulting in
a change to the initiation site of protein synthesis and ultimately
impacting the function of the encoded protein.
In order to identify histopathological features and
confirm the lack of SDHB expression in cancer cells, the
pathological tissue excised from the patient was reprocessed into
histological sections and subjected to hematoxylin & eosin
(H&E) staining as well as IHC staining. The specimen was fixed
using a 10% neutral buffered formalin solution. The fixation
process was carried out at room temperature for a duration ranging
from 16 to 24 h. The tissue sections were cut to a thickness of 4
µm. Following sectioning, the slides were stained using Harris
hematoxylin solution. The staining was performed at room
temperature and the sections were left in the stain for 5 min.
After rinsing, the sections were counterstained with eosin for 1
min. Similar to hematoxylin application, the eosin staining process
was also conducted at room temperature. The stained slides were
examined under a light microscope. Due to the extremely scarce
presence of vacuoles in paraffin-embedded specimens, they were very
difficult to detect. Upon examination, the rare characteristic
cytoplasmic vacuoles were identified (Fig. 4C). IHC for SDHB revealed its loss of
expression (Fig. 4D).
The patient underwent radiotherapy and targeted
therapy 1 month after the surgery. The patient is prescribed
Sunitinib Malate capsules at a dosage of 50 mg daily, administered
orally. The regimen consists of taking the medication continuously
for 4 weeks followed by a 2-week break. Immediate discontinuation
of the drug is advised in the event of severe adverse reactions.
Targeted radiotherapy was administered to a metastatic lesion
located at the upper end of the left humerus. The radiation field
measured 12×9 cm with a depth of 8 cm. The treatment utilized a
Source-to-Axis Distance (SAD) technique with anterior and posterior
opposed fields, employing 6 MV X-rays. A total dose of 50 Gy was
delivered over 25 fractions. The patient underwent three
radiotherapy sessions. Chest and abdominal CT scans performed 3
months after the surgery showed no signs of metastasis while a
whole-body bone scan demonstrated reduced radionuclide
concentration at the upper end of the left humerus (Fig. 3C). At the time of writing (July
2023), the patient was under observation for 6 months and their
condition remained stable.
Discussion
In the present case, the patient was diagnosed with
a right renal mass during a health check-up without any particular
clinical symptoms, and no tumor was identified in other parts of
the body during preoperative examination, therefore secondary
SDH-deficient RCC was excluded. The patient was postoperatively
diagnosed with SDH-deficient RCC with tumor emboli and bone
metastasis following identification of an SDHA germline mutation by
genetic screening. To the best of our knowledge, this is the first
report of such a case according to a search using MEDLINE via
PubMed using the terms ‘SDHA-deficient RCC’, ‘tumor emboli’ and
‘bone metastasis’. A previous study reported two cases of
SDHA-deficient RCC with bone metastases (17). However, based on the currently
available literature, there have been no reports of an
SDHA-deficient RCC case concurrently presenting with both
intravascular tumor emboli and bone metastasis. Therefore, the
present case is rare.
Preoperatively differentiating SDH-deficient RCC
from other types of renal cancer based solely on clinical
manifestations and imaging data is challenging. Diagnosis of
SDH-deficient RCC in the present case used genetic screening and
IHC to confirm the loss of SDHB expression. Histologically, these
tumors tend to exhibit solid, nested or tubular structures, often
accompanied by varying degrees of cysts (3). Typically, nucleoli are not prominent
and there is low nuclear atypia, whilst the cytoplasm appears
eosinophilic and contains characteristic flocculent inclusions or
vacuoles (15). However, in the
present case, the observed tumor cells were of a high nuclear
grade, and characteristic cytoplasmic vacuoles or inclusions were
rare, resembling HLRCC morphologically, which distinguished the
tumor cells from typical SDH-deficient RCC presentation.
The occurrence of HLRCC is related to the fumarate
hydratase gene mutation (18).
Histologically, HLRCC tumor cells typically present a mixed pattern
of growth, including papillary, tubular, tubulopapillary, solid and
cystic elements (18). The
morphologic hallmark of HLRCC tumors is proposed to be the
characteristic feature of a large nucleus with a very prominent
inclusion-like, eosinophilic nucleolus which is surrounded by a
perinucleolar halo (19). The tumor
cell morphology in the present case resembled the aforementioned
HLRCC microscopic features. HLRCC tumor cells typically show
fumarate hydrated enzyme deletion, 2-butylcysteine overexpression
and positive SDHB staining, IHC features which could be used to
distinguish HLRCC diagnosis from SDH-deficient RCC (20).
At present, there is no consensus on the optimal
treatment of SDH-deficient RCC (13,21).
Surgical intervention is the primary treatment option.
Nephron-sparing surgery can be considered in the early stage of
treatment when the tumor is smaller. In cases of advanced stage
tumors, it is recommended to conduct a comprehensive examination to
determine the tumor status, followed by consideration of surgical
intervention or molecular targeted therapy based on the patient's
overall condition (15).
It is unclear whether the prognosis of patients with
SDH-deficient RCC depends upon the specific SDH subtype affected.
According to the currently available literature, postoperative
histopathology suggests that SDH-deficient RCC tumors exhibiting
low-grade nuclear morphology are indolent following complete
resection (12). On the contrary,
tumors with a high nuclear grade accompanied by cellular necrosis
or displaying features of dedifferentiation often signify
aggressive invasion, high malignancy grade and poor prognosis
(13,15). The predominant nuclear grade of
tumor cells in SDHB-deficient RCC is low (Furhman grade 1-2), which
suggests a comparatively lower degree of malignancy, and ~11% of
patients may present with metastasis (1). There is no unified conclusion on the
degree of malignancy of SDHA-deficient RCC. Yakirevich et al
(22) reported a case of a
54-year-old patient with a 10 cm tumor in the upper pole of the
right kidney. The postoperative pathology suggested unclassified
stage III RCC (pT3aNxMx), and lung metastasis occurred 22 months
postoperatively. Kamai et al (17) reported three cases of SDHA-deficient
RCC with systemic metastasis within 5 years from surgery, and
systemic treatment was ineffective on the metastatic lesions.
McEvoy et al (21) reported
a case of SDHA-deficient RCC with no obvious metastasis indicated
by preoperative CT and a 11 cm tumor. Postoperative pathology
confirmed SDHA-deficient RCC, and further genetic testing showed
two SDHA gene mutations: 91C>T (p. Arg31*) and 1765C>T (p.
Arg589Trp). A positron emission-CT scan was performed at 10 months
following surgery which demonstrated the presence of metastasis in
the peritoneum, retroperitoneum and lung. Reports of RCC caused by
SDHC and SDHD gene mutations are limited (17,23).
Thus far, only the mutational profile of SDHD gene mutations has
been described and the prognosis of patients with RCC carrying an
SDHD gene mutation remains unclear (23). Therefore, based on the available
data and the present case, a potential preliminary conclusion could
be that SDHA-deficient RCC is aggressive and associated with poor
prognosis due to the high nuclear grade and dedifferentiation of
the tumor cells. A larger patient cohort is still needed to confirm
this.
In conclusion, SDHA-deficient RCC is a rare entity
that can present challenges for differential diagnosis,
particularly when the histological features closely resemble those
of HLRCC. For this reason, it could be recommended that patients
with suspected SDH-deficient RCC or HLRCC undergo postoperative
genetic screening to determine the presence and subtype of gene
mutations, as well as highlight the necessity for postoperative
complementary therapy (targeted therapy, radiotherapy,
chemotherapy, or immunotherapy). Due to the rarity of this
hereditary condition and the uncertainty of long-term prognosis, it
is recommended to conduct prolonged follow-up on affected patients.
Family members of affected patients should potentially be advised
to seek consultation with a genetic specialist and establish a
cancer screening profile. Meanwhile, further exploration of the
underlying mechanisms of SDH deficiency-induced tumorigenesis is
warranted.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The present manuscript
contains original data generated using next-generation sequencing,
which was uploaded to the BioProject database under the accession
no. PRJNA1100966 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1100966).
Authors' contributions
ZD, XW and YZ acquired the patient's data. YQ and JL
interpreted the data. ZD, XW, YZ and YQ drafted and wrote the
manuscript. JL revised the manuscript and acted as corresponding
author. ZD and XW managed patient relations. YZ and YQ collected
and organized the literature. All authors read and approved the
final version of the manuscript. ZD, XW and YZ confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The authors certify that all necessary consents were
taken from the patient. A formal ethical approval from the
institutional review board was not required as this is an
individual case report.
Patient consent for publication
The patient provided written informed consent to
publish their personal and medical information for the present case
report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gill AJ: Succinate dehydrogenase
(SDH)-deficient neoplasia. Histopathology. 72:106–116. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gill AJ, Hes O, Papathomas T, Šedivcová M,
Tan PH, Agaimy A, Andresen PA, Kedziora A, Clarkson A, Toon CW, et
al: Succinate dehydrogenase (SDH)-deficient renal carcinoma: A
morphologically distinct entity: A clinicopathologic series of 36
tumors from 27 patients. Am J Surg Pathol. 38:1588–1602. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vanharanta S, Buchta M, McWhinney SR,
Virta SK, Peçzkowska M, Morrison CD, Lehtonen R, Januszewicz A,
Järvinen H, Juhola M, et al: Early-onset renal cell carcinoma as a
novel extraparaganglial component of SDHB-associated heritable
paraganglioma. Am J Hum Genet. 74:153–159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moch H, Cubilla AL, Humphrey PA, Reuter VE
and Ulbright TM: The 2016 WHO classification of tumours of the
urinary system and male genital organs-Part A: Renal, Penile, and
testicular tumours. Eur Urol. 70:93–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gill AJ: Succinate dehydrogenase (SDH) and
mitochondrial driven neoplasia. Pathology. 44:285–292. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barletta JA and Hornick JL: Succinate
dehydrogenase-deficient tumors: Diagnostic advances and clinical
implications. Adv Anat Pathol. 19:193–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rizza S, Montagna C, Cardaci S, Maiani E,
Di Giacomo G, Sanchez-Quiles V, Blagoev B, Rasola A, De Zio D,
Stamler JS, et al: S-nitrosylation of the mitochondrial chaperone
TRAP1 sensitizes hepatocellular carcinoma cells to inhibitors of
succinate dehydrogenase. Cancer Res. 76:4170–4182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eng C, Kiuru M, Fernandez MJ and Aaltonen
LA: A role for mitochondrial enzymes in inherited neoplasia and
beyond. Nat Rev Cancer. 3:193–202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gottlieb E and Tomlinson IP: Mitochondrial
tumour suppressors: A genetic and biochemical update. Nat Rev
Cancer. 5:857–866. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Williamson SR, Eble JN, Amin MB, Gupta NS,
Smith SC, Sholl LM, Montironi R, Hirsch MS and Hornick JL:
Succinate dehydrogenase-deficient renal cell carcinoma: Detailed
characterization of 11 tumors defining a unique subtype of renal
cell carcinoma. Mod Pathol. 28:80–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuroda N, Yorita K, Nagasaki M, Harada Y,
Ohe C, Jeruc J, Raspollini MR, Michal M, Hes O and Amin MB: Review
of succinate dehydrogenase-deficient renal cell carcinoma with
focus on clinical and pathobiological aspects. Pol J Pathol.
67:3–7. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsai TH and Lee WY: Succinate
dehydrogenase-deficient renal cell carcinoma. Arch Pathol Lab Med.
143:643–647. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eijkelenkamp K, Osinga TE, Links TP and
van der Horst-Schrivers ANA: Clinical implications of the
oncometabolite succinate in SDHx-mutation carriers. Clin Genet.
97:39–53. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang G and Rao P: Succinate
dehydrogenase-deficient renal cell carcinoma: A short review. Arch
Pathol Lab Med. 142:1284–1288. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Agha RA, Franchi T, Sohrabi C, Mathew G
and Kerwan A; SCARE Group, : The SCARE 2020 guideline: Updating
consensus surgical CAse REport (SCARE) guidelines. Int J Surg.
84:226–230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kamai T, Higashi S, Murakami S, Arai K,
Namatame T, Kijima T, Abe H, Jamiyan T, Ishida K, Shirataki H and
Yoshida KI: Single nucleotide variants of succinate dehydrogenase A
gene in renal cell carcinoma. Cancer Sci. 112:3375–3387. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ooi A: Advances in hereditary
leiomyomatosis and renal cell carcinoma (HLRCC) research. Semin
Cancer Biol. 61:158–166. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Merino MJ, Torres-Cabala C, Pinto P and
Linehan WM: The morphologic spectrum of kidney tumors in hereditary
leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg
Pathol. 31:1578–1585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen YB, Brannon AR, Toubaji A, Dudas ME,
Won HH, Al-Ahmadie HA, Fine SW, Gopalan A, Frizzell N, Voss MH, et
al: Hereditary leiomyomatosis and renal cell carcinoma
syndrome-associated renal cancer: Recognition of the syndrome by
pathologic features and the utility of detecting aberrant
succination by immunohistochemistry. Am J Surg Pathol. 38:627–637.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McEvoy CR, Koe L, Choong DY, Leong HS, Xu
H, Karikios D, Plew JD, Prall OW, Fellowes AP and Fox SB:
SDH-deficient renal cell carcinoma associated with biallelic
mutation in succinate dehydrogenase A: Comprehensive genetic
profiling and its relation to therapy response. NPJ Precis Oncol.
2:92018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yakirevich E, Ali SM, Mega A, McMahon C,
Brodsky AS, Ross JS, Allen J, Elvin JA, Safran H and Resnick MB: A
novel SDHA-deficient renal cell carcinoma revealed by comprehensive
genomic profiling. Am J Surg Pathol. 39:858–863. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Andrews KA, Ascher DB, Pires DEV, Barnes
DR, Vialard L, Casey RT, Bradshaw N, Adlard J, Aylwin S, Brennan P,
et al: Tumour risks and genotype-phenotype correlations associated
with germline variants in succinate dehydrogenase subunit genes
SDHB, SDHC and SDHD. J Med Genet. 55:384–394. 2018. View Article : Google Scholar : PubMed/NCBI
|