Introduction
Rhabdomyosarcoma (RM), a prevalent soft-tissue
malignancy in children, consists of rhabdomyoblasts at various
stages of differentiation. RM predominantly affects the head and
neck, genitourinary tract, retroperitoneum and extremities
(1). The occurrence of RM,
particularly spindle cell RM (SCRM), in the adult thorax is
exceptionally rare. To the best of our knowledge, only one case of
SCRM in the thoracic cavity has been reported to date (2). The present study details a case of
primary thoracic SCRM in an adult patient and includes a literature
review to enhance the understanding of this rare tumor type.
RM is a common soft-tissue sarcoma in children and
adolescents, and accounts for 3% of all pediatric tumors (3,4). The
World Health Organization (2013) classifies soft-tissue tumors into
four subtypes based on their morphology: Acinus-shape, embryonal,
pleomorphic and sclerotic/SC RM (5). SCRM, first described by Cavazzana
et al (6) in 1992, is a
specific and rare subtype of RM that primarily occurs in the
paradidymal region, followed by the head and neck, in children. The
first adult case of SCRM was reported by Rubin et al
(7) in 1998. Unlike in children,
adult SCRM predominantly occurs in the head and neck region, with
cases also reported in the prostate, uterus and bones (8–10).
However, primary thoracic SCRM is extremely rare in clinical
settings, with only one case involving a 5-year-old female patient
reported to date (2), to the best
of our knowledge.
The present study aims to enhance the understanding
and awareness of this rare tumor by providing a detailed report of
a case of a giant SCRM in the thorax of an adult. By describing the
clinical characteristics, pathological findings and treatment
outcomes of this case, the study offers valuable insights for the
diagnosis and management of similar cases. Additionally, it
contributes to the early clinical identification of this tumor and
supports the development of individualized therapeutic
strategies.
Case report
A 24-year-old female patient was admitted to the
Affiliated Hospital of Zunyi Medical University (Zunyi, China) in
November 2012 due to right chest pain for 10 months and aggravation
for 4 months. The patient initially experienced dull pain in the
right side of the chest without any apparent cause. The patient had
no symptoms, such as a cough, phlegm, cold, fever, abdominal pain
or distension. At 10 months prior to admission, the patient was
treated at Affiliated Hospital of Guiyang Medical College (Guiyang,
China) for dull right-sided chest pain. A chest and abdominal
computed tomography (CT) scan revealed a large space in the right
upper diaphragm and right pleural effusion. Despite 6 months of
treatment for right-sided tuberculous pleurisy, the symptoms
persisted, and a mass puncture performed 4 months prior to the
current admission identified a spindle cell tumor. In the last 3
months, the patient's chest pain on the right side continued to
worsen, prompting a transfer to the Affiliated Hospital of Zunyi
Medical University for further treatment. The patient was admitted
with a diagnosis of a right thoracic tumor. Upon physical
examination, the following findings were noted: Decreased
respiratory motion of the right lung, pain induced by light
pressure on the right chest wall, a solid sound on percussion of
the right lung and a leftward shift of the relative border of
cardiac dullness. Laboratory tests for tumor markers and
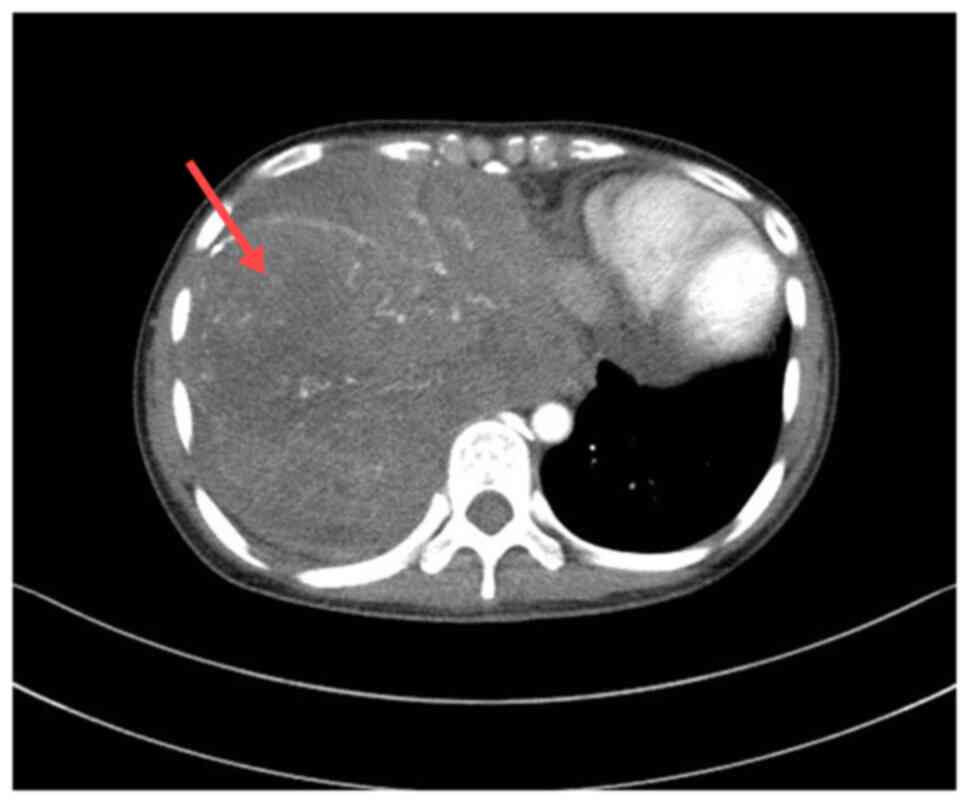
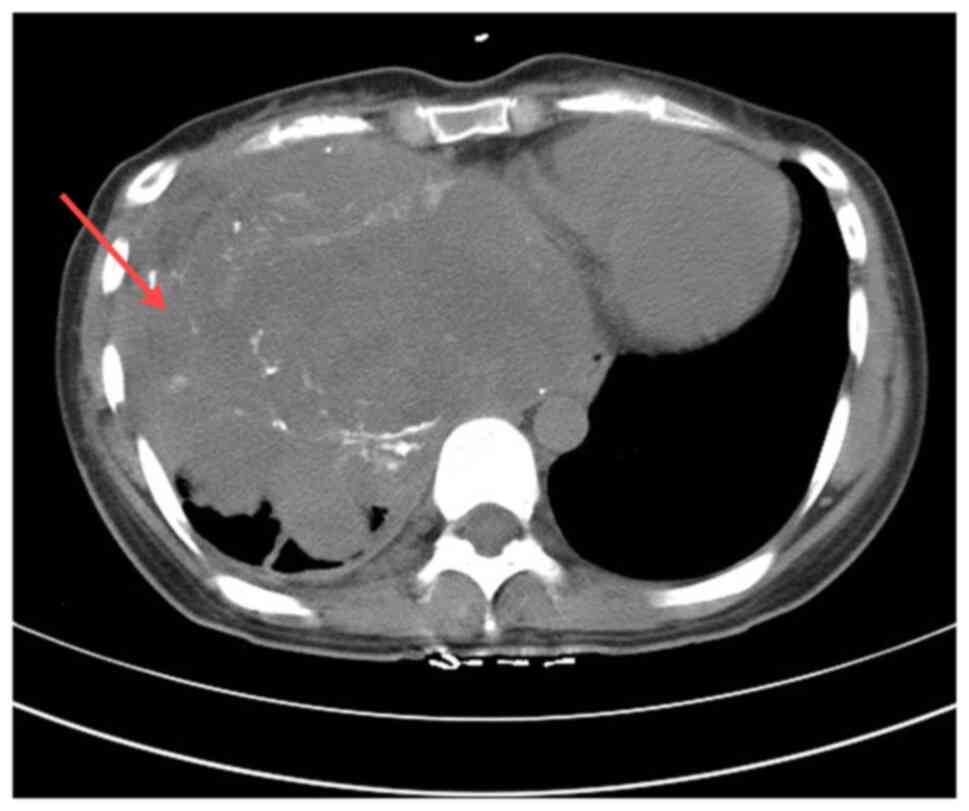
biochemical indicators were normal. A chest CT scan showed
irregular masses in the right lower thorax, the right middle
mediastinum and the diaphragm area, with unclear borders and uneven
density. CT values ranged from 25–65 Hounsfield units, with a
maximum cross-sectional area of ~173×140 mm. Multiple nodular and
small dot-like high-density shadows were observed. Enhancement
scans indicated heterogeneous enhancement, significant compression
and deformation of the right inferior pulmonary vein and right
atrium, a poor display of the right atrium, and a leftward shift of
the mediastinum and heart. No adjacent bone destruction was
observed (Fig. 1). The patient had
no specific past medical or family history. The patient underwent a
thoracotomy for a suspected primary thoracic tumor.
Intraoperatively, yellowish effusion was noted in the right thorax,
and the tumor occupied approximately three-quarters of the right
thorax, displaying a large lobulated morphology. The tumor
exhibited aggressive growth, invading the diaphragm, lower lung,
mediastinum and part of the chest wall, with an incomplete capsule.
The tumor protruded downward into the abdominal cavity, but did not
invade the liver and heart, with a clear demarcation between the
tumor and the pericardium.
The pathological findings were of a mass of
gray-white and gray-red fragmented tissue measuring 25.0×20.0×8.0
cm, with some well-defined areas. The cut surface had a fish
meat-like appearance, gray-white and gray-red in color, with a
solid and soft texture. The specimens were fixed in 4% neutral
formalin at room temperature for 12 h, followed by routine
dehydration, paraffin embedding and sectioning at a thickness of 5
µm. Hematoxylin and eosin staining was then performed at room
temperature for 5 min each. Examination under a light microscopic
examination revealed an incomplete tumor capsule with infiltrative
growth, and tumor cells were observed to invade the surrounding
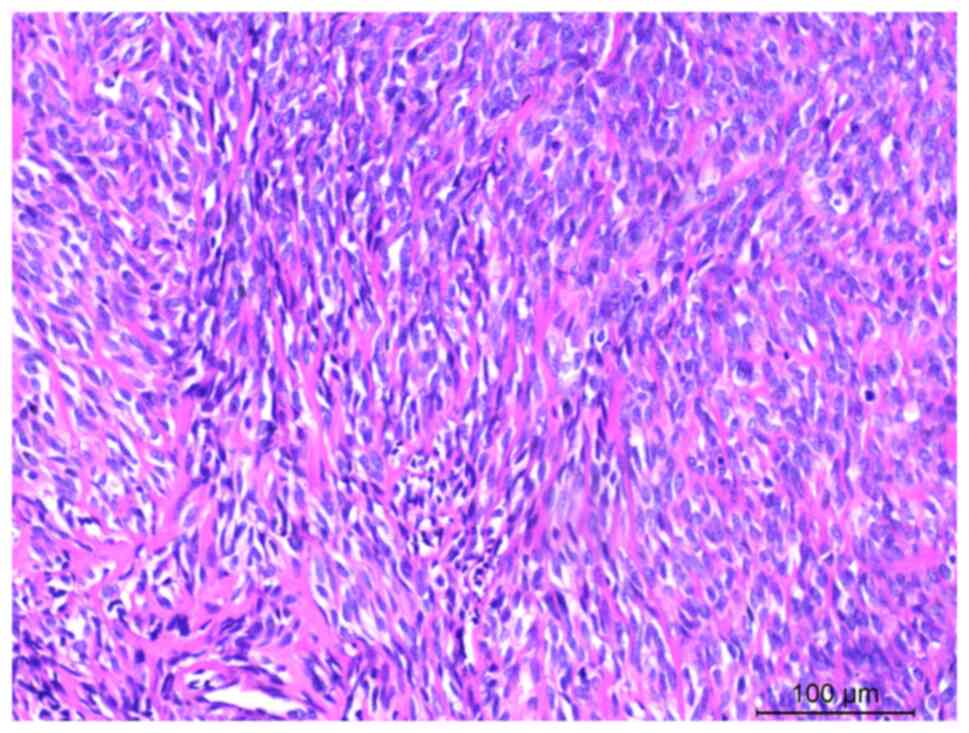
muscle and adipose tissue. The tumor predominantly consisted of
long spindle cells arranged in bundles, featuring darkly stained
nuclei, inconspicuous nucleoli, mitosis and eosinophilic cytoplasm
(Fig. 2). In a few regions, the
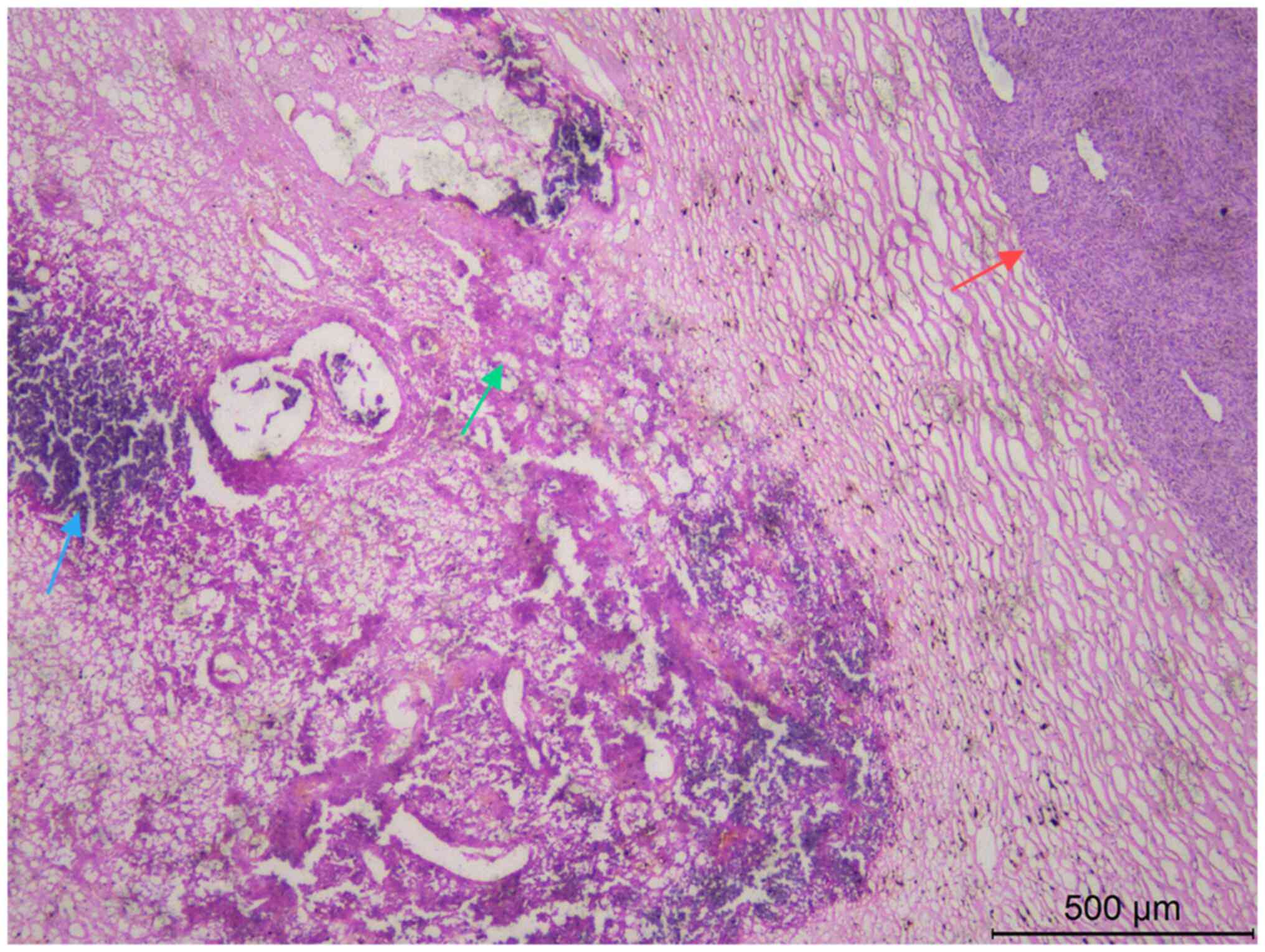
tumor cells were naive, stellate or irregularly shaped, with
interstitial mucinous edema-like changes. Some tumor cells showed
lamellar necrosis and calcification (Fig. 3).
The specimens were fixed in 10% neutral formalin,
followed by routine dehydration, paraffin embedding and sectioning
at a thickness of 3 µm. Immunohistochemistry using the Envision
two-step method was employed to assess the expression of relevant
proteins in the tumor tissue. The staining procedures were
performed strictly according to the manufacturer's instructions
(all primary antibodies used were rabbit and mouse anti-human
monoclonal antibodies, purchased from Fuzhou Maixin Biotechnology
Development Co. Ltd., and were used at a working concentration of
1:100). The primary antibodies were added to the sections and
incubated overnight (12 h) at 4°C. Immunohistochemical staining
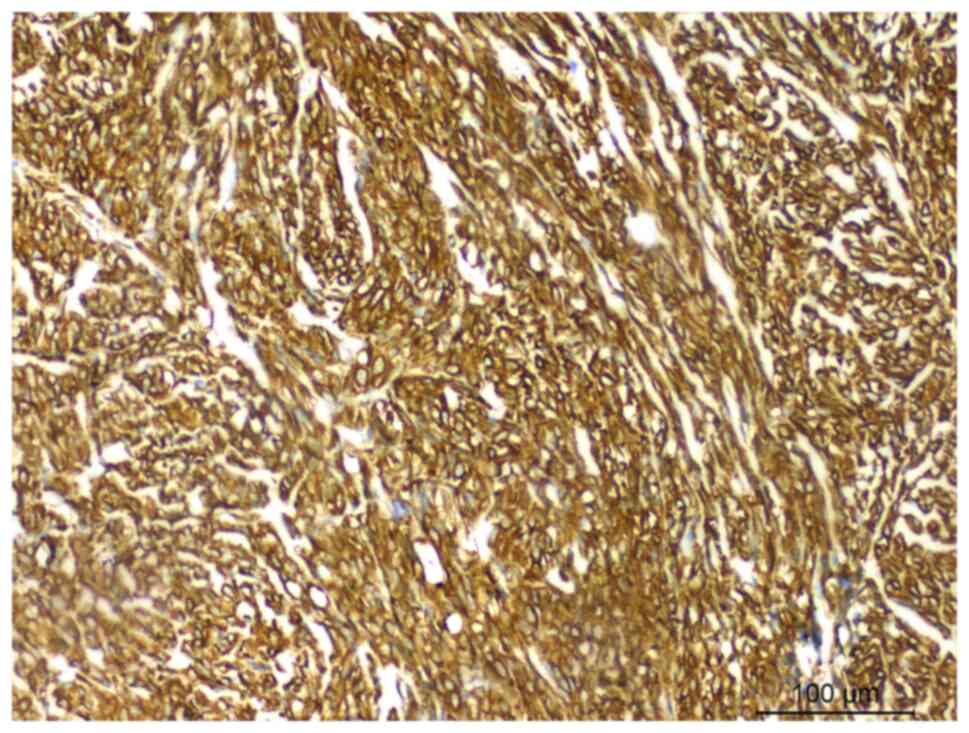
revealed, under a light microscope, that the tumor cells expressed
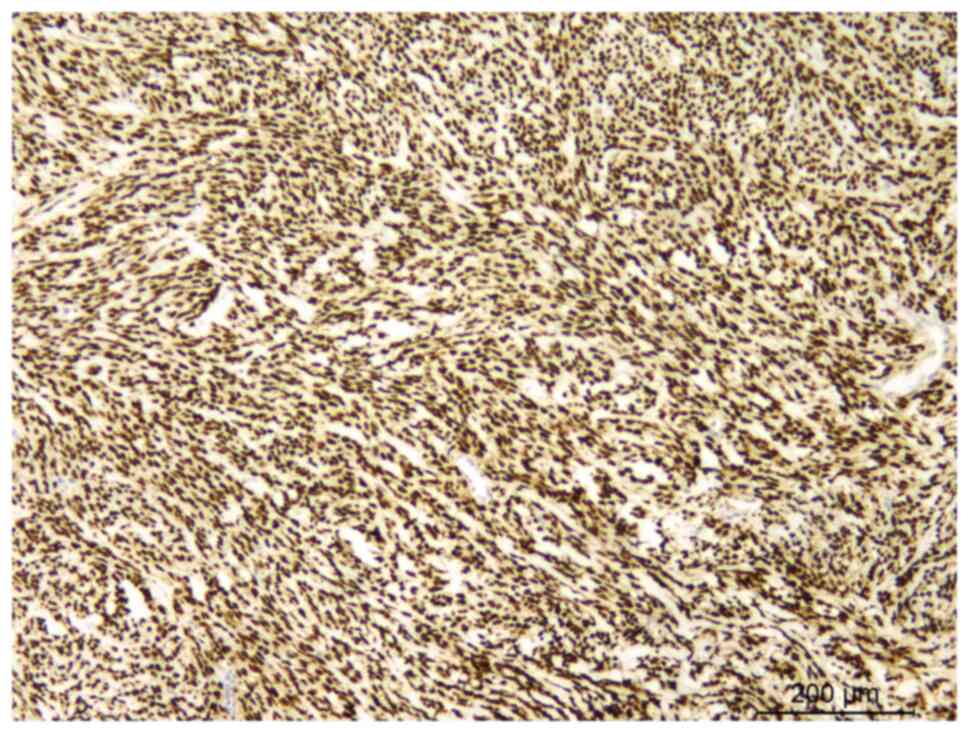
vimentin (catalog no. RMA-0547) (Fig.
4), myoblast determination protein 1 (MyoD1) (catalog no.
MAB-0822) (Fig. 5) and desmin
(catalog no. MAB-0766) (Fig. 6),
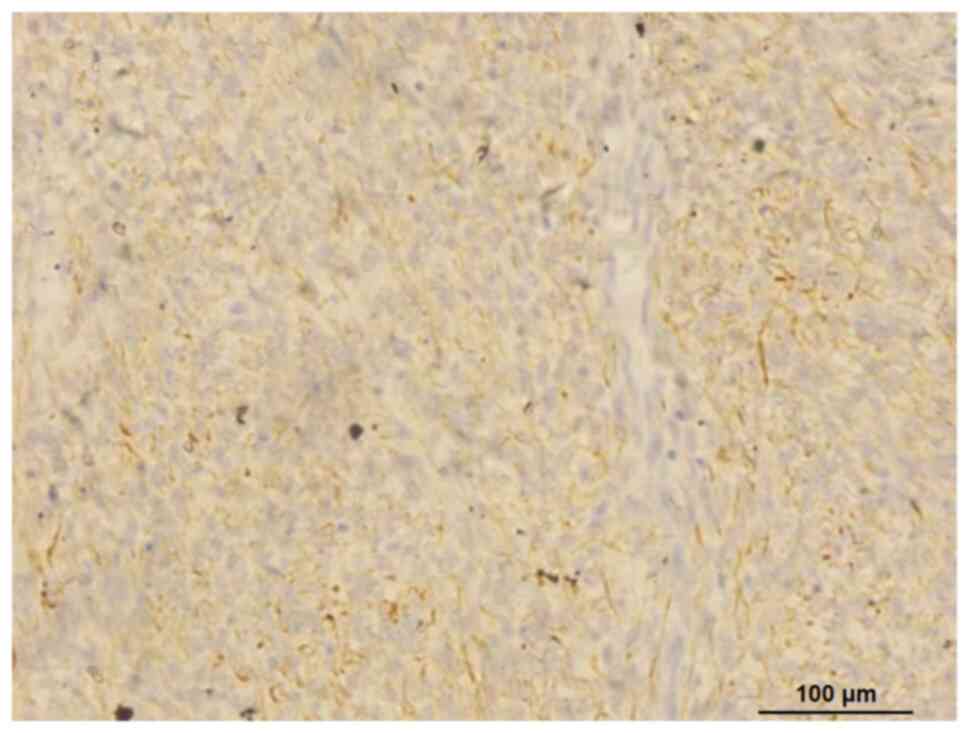
but did not express CD117 (catalog no. Kit-0029), CD34 (catalog no.
MAB-1076), CD68 (catalog number: Kit-0026), epithelial membrane
antigen (EMA) (catalog no. Kit-0011), smooth muscle actin (catalog
no. ZM-0003), pan cytokeratin (AE1/AE3) (catalog no. Kit-0009),
cytokeratin (CK)7 (catalog no. MAB-0828), CK19 (catalog no.
MAB-0829), CD99 (catalog no. MAB-1012), transcription factor SOX-10
(SOX-10) (catalog no. RMA-1058), synaptophysin (catalog no.
MAB-0742), neuron-specific enolase (catalog no. MAB-0791), S100
(catalog no. RAB-0150) and anaplastic lymphoma kinase (catalog no.
MAB-0848) (Fig. S1). The Ki-67
index was ~30%. The patient was pathologically diagnosed with a
right thoracic SCRM.
The patient was in good condition after surgery, and
telephone follow-ups were performed at 1, 3 and 5 years after
surgery. However, due to personal economic conditions and other
factors, the patient declined postoperative radiotherapy and
chemotherapy, and regular physical examinations. After 5 years, the
patient exhibited symptoms of chest pain and dyspnea. A chest CT
scan at the 5-year follow-up visit suggested a recurrence of the
thoracic tumor (Fig. 7), and the
patient continued to refuse treatment. The patient has been lost to
follow-up.
Discussion
The clinical presentation of primary thoracic SCRM
lacks specificity. The severity of symptoms depends on primary site
and size of the tumor, the degree of compression and infiltration,
and the extent of tissue destruction caused by the tumor cells. A
preoperative diagnosis of SCRM is challenging due to the
non-specific nature of imaging findings (2,11,12).
In the present case report, the patient primarily presented with
chest pain, without additional symptoms such as hemoptysis or a
cough. Microscopically, the tumor predominantly consisted of
spindle cells arranged in interlacing bundles, resembling
fibrosarcoma and leiomyosarcoma. The spindle cells exhibited
abundant red-stained cytoplasm, oval or elongated nuclei with deep
staining, and inconspicuous or small nucleoli. Additionally, a
small number of spindle or polygonal rhabdomyoblasts were
interspersed among the spindle cells. The presence of
rhabdomyoblasts suggested a diagnosis of SCRM, with mitotic figures
ranging from 1 to 30 per 10 high-power fields. Immunohistochemical
staining demonstrated varying degrees of expression of myogenic
markers, including desmin and MyoD1, in the SCRM (13), with strong positivity for MyoD1.
However, epithelial markers (such as CK and EMA) and neurogenic
markers (such as S-100 and SOX-10) were not expressed.
Molecular genetic studies have identified genetic
differences between young children with SCRM and older children or
adults with the same condition. Young children often present with
vestigial-like family member 2, serum response factor, TEA domain
transcription factor 1 or nuclear receptor coactivator 2-associated
gene fusions, which are associated with a better prognosis
(3). By contrast, older children
and adults frequently have mutations in the MYOD1 gene, leading to
a poorer prognosis. Tsai et al (14) reported the cases of a group of
patients aged 8–64 years with SCRM, finding that the mutation rate
in MYOD1 was 30–67%. MYOD1 was diffusely expressed, and myogenin
showed patchy expression in all MYOD1-mutated patients.
Additionally, Dashti et al (10) reported a case of bone SCRM with
fused in sarcoma-transcription factor cellular promoter 2 gene
fusion. Further research on SCRM is expected to uncover more
molecular genetic alterations, providing a basis for improved
treatment strategies. However, no genetic analysis was performed in
the present case for economic reasons.
Primary thoracic SCRM must be distinguished from the
following tumors: i) Fibromatosis: Occurring primarily in adults
with aggressive growth, fibromatosis features long, spindle-shaped
tumor cells with minimal cellular atypia, low mitotic activity and
abundant interstitial collagen fibers. Immunohistochemically, SMA
and catenin are expressed, while MyoD1 and myogenin are not
(15–18). ii) Adult-type fibrosarcoma:
Comprised of fibroblasts, these tumors present with long,
spindle-shaped cells with pointed nuclei arranged in bundles or a
herringbone pattern, and abundant interstitial collagen.
Hemangiopericytoma-like structures are common in congenital
fibrosarcoma. Immunohistochemical markers are positive for vimentin
but negative for desmin, MyoD1 and myogenin (19,20).
iii) Leiomyosarcoma: Primarily occurring in the retroperitoneum,
extremities, trunk, head and neck of adults, leiomyosarcoma
consists of fasciculated spindle cells with abundant eosinophilic
cytoplasm arranged longitudinally and transversely. Tumor cells
feature rod-shaped nuclei with blunt ends. Immunohistochemical
assays typically show SMA positivity and MyoD1 negativity (21,22).
iv) Synovial sarcoma: Often found around large joints in patients
aged 15–40 years, synovial sarcoma consists of epithelial and
spindle cell components. Spindle cells are uniform with scant
cytoplasm, ovoid nuclei, and inconspicuous nucleoli; localized
hemangiopericytoma-like structures are common. Poorly
differentiated synovial sarcoma cells can resemble RM.
Immunohistochemically, CD99 and BCL-2 are positive, while myogenic
markers are negative (23,24). Fluorescence in situ
hybridization assays frequently reveal synaptotagmin gene
translocation (25,26). v) Mixed malignant tumors of
neuroepithelial origin: Affecting the extremities, head and neck,
retroperitoneum, abdominal wall, perineum, scrotum and brain, these
tumors exhibit multiple differentiations, including ganglion cells,
neuroblastoma cells and RM cells. RM is characterized by the
absence of a neuroepithelial component (27).
RM is primarily treated with surgery combined with
chemoradiotherapy. The study by Yasui et al (5) emphasized that complete resection of
the tumor, along with adjuvant chemotherapy and radiotherapy, could
prevent local recurrence. Prognostic factors for RM include the
location of the tumor, the completeness of its resection, its size
and its histological subtype (28,29).
The highly aggressive nature of SCRM in adults contributes to a
poor prognosis (30). A previous
study has shown that adult RM generally has a worse prognosis
compared with that of pediatric RM, with 24.6% of patients dying
from the cancer or treatment-associated complications, The overall
5-year survival and metastasis-free survival rates were recorded as
52.9 and 62.9%, respectively, The sole predictor of metastasis was
the National Federation of Cancer Centers tumor grade (31). Although most RM cases present as
large tumors, lymph node or distant metastases are rare at the time
of diagnosis. In one study, RM initially showed a good response to
vincristine, actinomycin and cyclophosphamide chemotherapy, but
>50% of tumors recurred or progressed. These data suggest that
SCRM has a worse prognosis compared with the infantile fetal
variation (5). In the present adult
patient, despite complete resection of the tumor, no standardized
radiotherapy and chemotherapy regimen was available, and disease
progression was observed over a 5-year follow-up period. Therefore,
standardized postoperative radiotherapy and chemotherapy are
crucial components of the treatment plan.
In conclusion, RM is a rare soft-tissue malignancy.
Adult SCRM is particularly aggressive and associated with a poor
prognosis. Due to its rarity, the clinicopathological features,
molecular genetic characteristics and biological behavior of SCRM
are not well understood. Consequently, large-sample analyses are
essential to enhance the understanding of this tumor and facilitate
the development of more effective precision medicine
strategies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
YQL and JJW analyzed the data and were the primary
author of the manuscript. SL acquired the CT scan images. YL
performed the immunohistochemical staining. XM analyzed patient
data. JJW and YQL confirm the authenticity of the data. JJW and XH
conducted the histopathological evaluation and assisted in writing
the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
This case report was approved by the Ethics
Committee of the Affiliated Hospital of Zunyi Medical University
(Zunyi, China; approval no. KLLY-2020-064).
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of this case report and any
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Skapek SX, Ferrari A, Gupta AA, Lupo PJ,
Butler E, Shipley J, Barr FG and Hawkins DS: Rhabdomyosarcoma. Nat
Rev Dis Primers. 5:12019. View Article : Google Scholar
|
|
2
|
Su F, Li S, Shou J, Hong Q and Zhang ZX:
Giant spindle cell rhabdomyosarcoma of children in the thoracic
cavity: A case report. Zhonghua Zhong Liu Za Zhi. 42:779–780.
2020.(In Chinese).
|
|
3
|
Smith MH, Atherton D, Reith JD, Islam NM,
Bhattacharyya I and Cohen DM: Rhabdomyosarcoma, spindle
cell/sclerosing variant: A clinical and histopathological
examination of this rare variant with three new cases from the oral
cavity. Head Neck Pathol. 11:494–500. 2017. View Article : Google Scholar
|
|
4
|
Amer KM, Thomson JE, Congiusta D, Dobitsch
A, Chaudhry A, Li M, Chaudhry A, Bozzo A, Siracuse B, Aytekin MN,
et al: Epidemiology, incidence, and survival of rhabdomyosarcoma
subtypes: SEER and ICES database analysis. J Orthop Res.
37:2226–2230. 2019. View Article : Google Scholar
|
|
5
|
Yasui N, Yoshida A, Kawamoto H, Yonemori
K, Hosono A and Kawai A: Clinicopathologic analysis of spindle
cell/sclerosing rhabdomyosarcoma. Pediatr Blood Cancer.
62:1011–1016. 2015. View Article : Google Scholar
|
|
6
|
Cavazzana AO, Schmidt D, Ninfo V, Harms D,
Tollot M, Carli M, Treuner J, Betto R and Salviati G: Spindle cell
rhabdomyosarcoma. A prognostically favorable variant of
rhabdomyosarcoma. Am J Surg Pathol. 16:229–235. 1992. View Article : Google Scholar
|
|
7
|
Rubin BP, Hasserjian RP, Singer S, Janecka
I, Fletcher JA and Fletcher CD: Spindle cell rhabdomyosarcoma
(so-called) in adults: Report of two cases with emphasis on
differential diagnosis. Am J Surg Pathol. 22:459–464. 1998.
View Article : Google Scholar
|
|
8
|
Schildhaus HU, Lokka S, Fenner W, Küster
J, Kühnle I and Heinmöller E: Spindle cell embryonal
rhabdomyosarcoma of the prostate in an adult patient-case report
and review of clinicopathological features. Diagn Pathol.
11:562016. View Article : Google Scholar
|
|
9
|
McCluggage WG, Lioe TF, McClelland HR and
Lamki H: Rhabdomyosarcoma of the uterus: Report of two cases,
including one of the spindle cell variant. Int J Gynecol Cancer.
12:128–132. 2002. View Article : Google Scholar
|
|
10
|
Dashti NK, Wehrs RN, Thomas BC, Nair A,
Davila J, Buckner JC, Martinez AP, Sukov WR, Halling KC, Howe BM
and Folpe AL: Spindle cell rhabdomyosarcoma of bone with FUS-TFCP2
fusion: Confirmation of a very recently described rhabdomyosarcoma
subtype. Histopathology. 73:514–520. 2018. View Article : Google Scholar
|
|
11
|
Przygodzki RM, Moran CA, Suster S and Koss
MN: Primary pulmonary rhabdomyosarcomas: A clinicopathologic and
immunohistochemical study of three cases. Mod Pathol. 8:658–661.
1995.
|
|
12
|
Travis WD: Sarcomatoid neoplasms of the
lung and pleura. Arch Pathol Lab Med. 134:1645–1658. 2010.
View Article : Google Scholar
|
|
13
|
Agaram NP: Evolving classification of
rhabdomyosarcoma. Histopathology. 80:98–108. 2022. View Article : Google Scholar
|
|
14
|
Tsai JW, ChangChien YC, Lee JC, Kao YC, Li
WS, Liang CW, Liao IC, Chang YM, Wang JC, Tsao CF, et al: The
expanding morphological and genetic spectrum of MYOD1-mutant
spindle cell/sclerosing rhabdomyosarcomas: A clinicopathological
and molecular comparison of mutated and non-mutated cases.
Histopathology. 74:933–943. 2019. View Article : Google Scholar
|
|
15
|
Goldstein JA and Cates JM: Differential
diagnostic considerations of desmoid-type fibromatosis. Adv Anat
Pathol. 22:260–266. 2015. View Article : Google Scholar
|
|
16
|
Skubitz KM: Biology and treatment of
aggressive fibromatosis or desmoid tumor. Mayo Clin Proc.
92:947–964. 2017. View Article : Google Scholar
|
|
17
|
Riedel RF and Agulnik M: Evolving
strategies for management of desmoid tumor. Cancer. 128:3027–3040.
2022. View Article : Google Scholar
|
|
18
|
Garcia-Ortega DY, Martín-Tellez KS,
Cuellar-Hubbe M, Martínez-Said H, Álvarez-Cano A, Brener-Chaoul M,
Alegría-Baños JA and Martínez-Tlahuel JL: Desmoid-type
fibromatosis. Cancers (Basel). 12:18512020. View Article : Google Scholar
|
|
19
|
Bahrami A and Folpe AL: Adult-type
fibrosarcoma: A reevaluation of 163 putative cases diagnosed at a
single institution over a 48-year period. Am J Surg Pathol.
34:1504–1513. 2010. View Article : Google Scholar
|
|
20
|
Folpe AL: Fibrosarcoma: A review and
update. Histopathology. 64:12–25. 2014. View Article : Google Scholar
|
|
21
|
Serrano C and George S: Leiomyosarcoma.
Hematol Oncol Clin North Am. 27:957–974. 2013. View Article : Google Scholar
|
|
22
|
Bayçelebi D, Kefeli M, Yıldız L and
Karagöz F: Comprehensive immunohistochemical analysis based on the
origin of leiomyosarcoma. Pol J Pathol. 73:233–243. 2022.
View Article : Google Scholar
|
|
23
|
Fisher C: Synovial sarcoma. Ann Diagn
Pathol. 2:401–421. 1998. View Article : Google Scholar
|
|
24
|
Fiore M, Sambri A, Spinnato P, Zucchini R,
Giannini C, Caldari E, Pirini MG and De Paolis M: The biology of
synovial sarcoma: state-of-the-art and future perspectives. Curr
Treat Options Oncol. 22:1092021. View Article : Google Scholar
|
|
25
|
Sun Y, Sun BC, Liu YX, Zhang SW, Zhao XL,
Wang J and Hao XS: Diagnostic value of SYT-SSX fusion gene
detection by fluorescence in-situ hybridization for synovial
sarcoma. Zhonghua Bing Li Xue Za Zhi. 37:660–664. 2008.(In
Chinese).
|
|
26
|
Shahi F, Alishahi R, Pashaiefar H,
Jahanzad I, Kamalian N, Ghavamzadeh A and Yaghmaie M:
Differentiating and categorizing of liposarcoma and synovial
sarcoma neoplasms by fluorescence in situ hybridization. Iran J
Pathol. 12:209–217. 2017. View Article : Google Scholar
|
|
27
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar
|
|
28
|
Yechieli RL, Mandeville HC, Hiniker SM,
Bernier-Chastagner V, McGovern S, Scarzello G, Wolden S, Cameron A,
Breneman J, Fajardo RD and Donaldson SS: Rhabdomyosarcoma. Pediatr
Blood Cancer. 68 (Suppl 2):e282542021. View Article : Google Scholar
|
|
29
|
Dasgupta R, Fuchs J and Rodeberg D:
Rhabdomyosarcoma. Semin Pediatr Surg. 25:276–283. 2016. View Article : Google Scholar
|
|
30
|
Carroll SJ and Nodit L: Spindle cell
rhabdomyosarcoma: A brief diagnostic review and differential
diagnosis. Arch Pathol Lab Med. 137:1155–1158. 2013. View Article : Google Scholar
|
|
31
|
Stock N, Chibon F, Binh MB, Terrier P,
Michels JJ, Valo I, Robin YM, Guillou L, Ranchère-Vince D,
Decouvelaere AV, et al: Adult-type rhabdomyosarcoma: analysis of 57
cases with clinicopathologic description, identification of 3
morphologic patterns and prognosis. Am J Surg Pathol. 33:1850–1859.
2009. View Article : Google Scholar
|