Introduction
Chordoma is a rare primary malignant bone tumor that
originates from the embryonic remnants of the notochord. Chordoma
typically arises in the skull base or sacrum, with an annual
incidence rate of ~0.8 cases per million individuals, accounting
for ~1% of all bone tumors (1).
Chordomas can manifest at any age, but the majority of patients are
diagnosed between the ages of 40 and 50 years. Clinical
manifestations primarily depend on the location and extent of the
lesions, with intracranial chordomas often causing headaches or
facial numbness due to their proximity to cranial nerves, while
sacrococcygeal chordomas may lead to lumbosacral pain or bowel
dysfunction due to compression of the sacral nerve roots (1).
Chordoid meningioma, on the other hand, is a rare
subtype of meningioma, accounting for 0.5–1% of all meningiomas
(2). This tumor is more commonly
found in young female patients and is typically located in the
supratentorial region or at the skull base (3). Chordoid meningiomas can invade
surrounding tissues, leading to symptoms such as headaches,
vomiting and visual disturbances (2,3).
Given the overlapping clinical and pathological
features of chordoma and chordoid meningioma, accurate
differentiation between these two entities is crucial for
appropriate treatment and prognosis. Misdiagnosis can lead to
inappropriate therapeutic strategies, potentially affecting patient
outcomes (4). The present case
report aims to highlight the challenges in differentiating between
chordoma and chordoid meningioma, emphasizing the importance of
immunohistochemical markers and imaging studies in achieving an
accurate diagnosis.
Case report
In March 2022, a 59-year-old male patient presented
to the Qingdao Municipal Hospital (Qingdao, China) with a 2-month
headache, dizziness and numbness in the right hand. The symptoms
intensified when the patient lowered their head and rolled their
neck. Notably, the severity of these symptoms had increased over
the past month. The physical examination revealed normal limb
movements, gait and consciousness, as well as clear speech. The
bilateral pupils were equal in size and shape, and exhibited a
light reaction, and the patient displayed symmetrical forehead
lines and nasolabial grooves on both sides. Tongue extension was
centered. Muscle strength in the limbs was rated at level 5, muscle
tone was normal (5) and blood
pressure readings were within the normal range. The patient had no
abnormal results in routine laboratory tests. Magnetic resonance
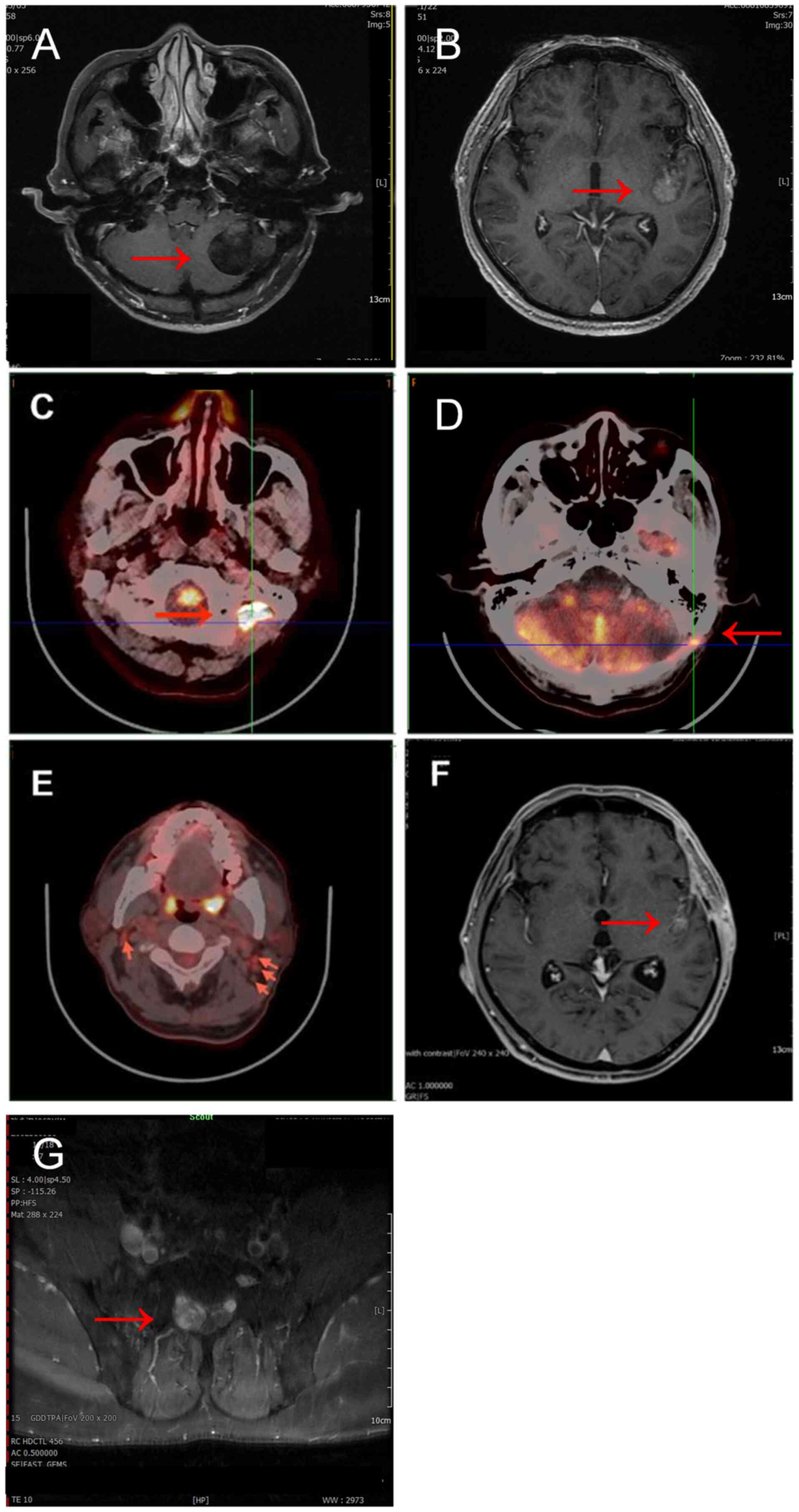
imaging (MRI) revealed a round lesion in the left cerebellar
hemisphere, characterized by long T1 and long T2 signals, with
T2-weighted-fluid-attenuated inversion recovery exhibiting high
signal intensity and unevenness. The lesion measured ~39 mm in
diameter, had clear boundaries and was located adjacent to the
lateral margin of the cerebellum, with diffusion-weighted imaging
(DWI) showing an iso-low signal. MRI enhancement demonstrated
moderate, uneven enhancement of the left cerebellar hemisphere
lesion, featuring a central non-enhanced area and mild enhancement
of the surrounding wall. The MRI findings were indicative of a
‘space-occupying lesion in the left cerebellar hemisphere’
(Fig. 1A). There was no prior
history of tumors. The patient underwent an intracranial tumor
resection in March 2022. Following the procedure, the vital signs
and overall condition of the patient were stable. Subsequently, the
planning target volume (PTV) for radiotherapy following the first
operation was 5,040 cGy/180 cGy/28 fractions, and mannitol
injection (20%) in a volume of 150 ml once was provided to reduce
intracranial pressure.
The gross resected specimen was formed of
grey-white-brown broken tissue, with a total size of 2.5×2×2 cm,
and the local tissue section was soft and spongy. Postoperative
pathology (Data S1) revealed that
the tumor was composed of cords of epithelioid cells with
vacuolated cytoplasm and collagen fibers within a basophilic stroma
(Fig. 2A and B).
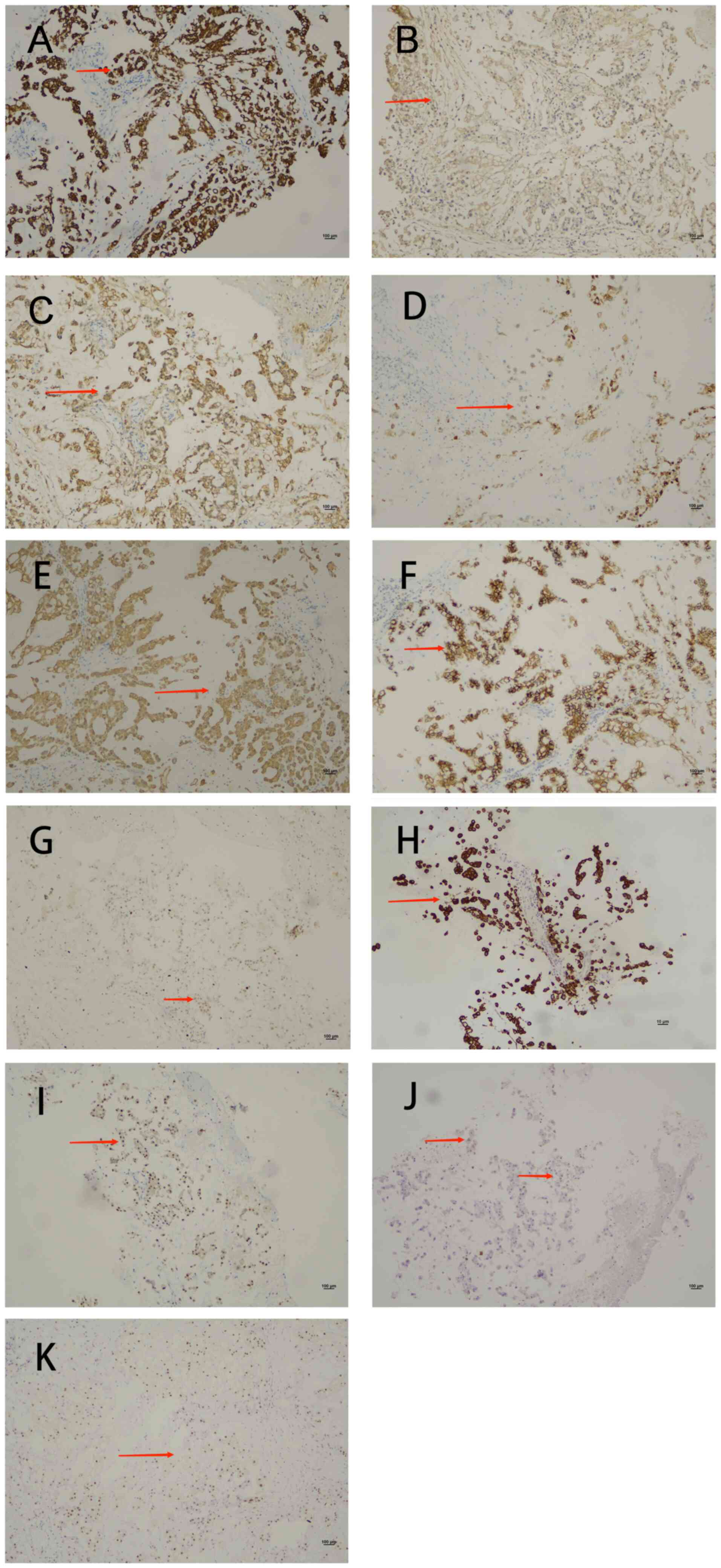
Immunohistochemical analysis (Data
S1) revealed positivity for cytokeratin (CK), cytokeratin 8/18
(CK8/18), vimentin, epithelial membrane antigen (EMA),
synaptophysin and E-cadherin, with a Ki-67 proliferation index of
3% (Fig. 3A-G). The tumor was
negative for S100, glial fibrillary acidic protein (GFAP),
progesterone receptor (PR), D2-40 and SOX-10 (data not shown).
Based on the pathological and immunohistochemical findings, a
diagnosis of chordoma-like meningioma [World Health Organization
(WHO) Grade 2] (6) was made.
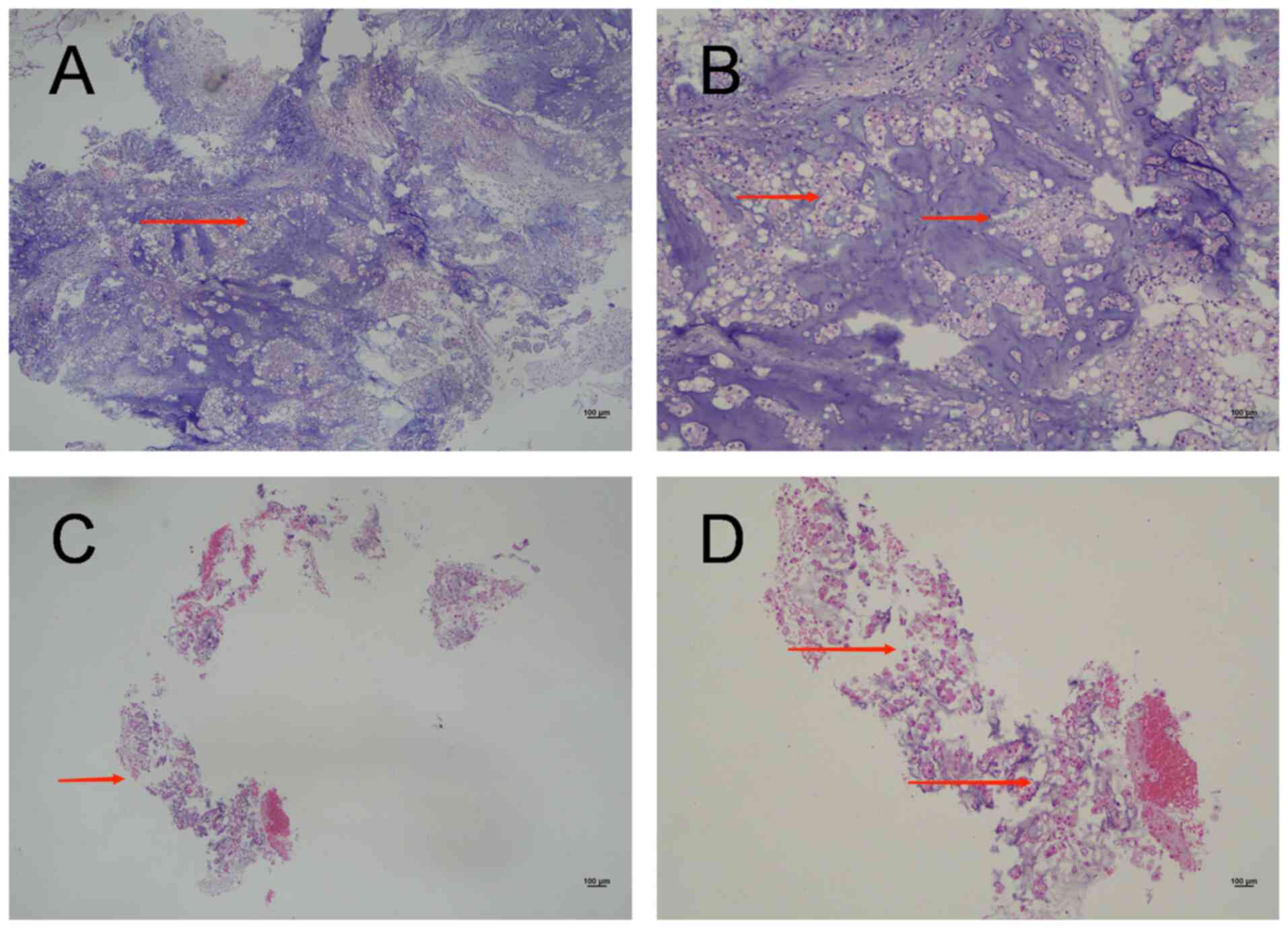 | Figure 2.H&E images of two tumors. (A) An
H&E image from 2022. Magnification, ×4. Under low
magnification, the tissue appears fragmented, with the tumor
exhibiting a pattern of cords and nests within the blue myxoid
matrix (arrow). (B) An H&E image from 2022. Magnification, ×10.
Under medium magnification, the tumor is composed of cords of
epithelioid cells with vacuolated cytoplasm and collagen fibers
within a basophilic stroma (arrows). (C) An H&E image from
2023. Magnification, ×4. Under low magnification, the tissue
appears fragmented and is accompanied by a small amount of
blue-stained mucus (arrow). (D) A 2023 H&E image.
Magnification, ×10. Under medium magnification, the tumor is
composed of cords and isolated epithelioid cells, which exhibit
intracytoplasmic vacuoles within a myxoid matrix (arrows). H&E,
haematoxylin and eosin. |
In November 2023, the patient experienced numbness
on the right side of the face, without accompanying symptoms of
headaches or dizziness. Neurological examination revealed no
abnormalities. An MRI revealed irregular mass enhancement in the
left Sylvian fissure cistern, with a cross-sectional area of ~22×18
mm, suggestive of an intracranial occupying lesion (Fig. 1B). In November 2023, the patient
underwent another intracranial tumor resection. Postoperatively,
the condition of the patient remained stable. The patient received
radiotherapy for intracranial lesions and the lumbosacral area in
December 2023. The PTV for radiotherapy after the second operation
was 4,000 cGy/200 cGy/20 fractions for the intracranial lesion and
7,000 cGy/200 cGy/35 fractions for the lumbosacral lesion. At 1
month after the completion of radiotherapy, the efficacy evaluation
indicated a partial response.
The gross specimen of the second surgery consisted
of grey and broken tissue, with a diameter of 0.3 cm. The pathology
results revealed that the tumor was composed of cords and isolated
epithelioid cells, which exhibited intracytoplasmic vacuoles within
a myxoid matrix (Fig. 2C and D).
Immunohistochemical analysis revealed positive CK (Fig. 3H), Brachyury (Fig. 3I) and weakly-positive S100 (Fig. 3J) expression. These
immunohistochemical findings suggested a diagnosis of chordoma.
A review of the hematoxylin and eosin slices from
the first surgery in 2022 indicated that the epithelioid cells were
arranged in a cord-like distribution within an eosinophilic
mucinous matrix. Some cells exhibited eosinophilic cytoplasm, while
others appeared vacuolated. Classic meningioma structures were
absent, and there was no evidence of lymphocyte or plasma cell
infiltration at the tumor periphery in a layered pattern. Repeat
immunohistochemistry confirmed that the original slices were
Brachyury-positive (Fig. 3K),
further supporting the diagnosis of chordoma.
Postoperative follow-up in March 2024 revealed a
patchy, low-density area in the left cerebellar surgical site,
measuring ~24.7×19.4 mm. Additionally, soft-tissue nodules on the
periphery and below, with a cross-section of ~25.6×15.0 mm,
fluorodeoxyglucose hypermetabolic lesions in the peripheral and
inferior regions of the left cerebellar surgical site and multiple
hypermetabolic lymph nodes in the bilateral neck were noted on
positron emission tomography-computed tomography (PET-CT) (Fig. 1C-E). Given the possibility of tumor
recurrence and metastasis, palliative radiotherapy at 6,000 cGy/200
cGy/30 fractions was administered to the affected area. The
efficacy evaluation indicated progressive disease 1 month after the
completion of radiotherapy. In September 2024, the patient came to
the hospital for re-examination due to dizziness and constipation.
MR enhanced scanning examination showed the formation of softening
lesions in the left cerebellar hemisphere and abnormal enhancement
in the left insular-Sylvian cistern area (Fig. 1F). Follow-ups were organized and the
patient was advised to return if symptoms worsened.
In November 2024, the patient presented to the
hospital with paroxysmal headaches and dizziness. An enhanced MRI
of the lumbosacral spine revealed nodular abnormal signal foci in
the sacral canal at the S1-2 level, indicating a chordoma (Fig. 1G). Due to the progression of the
disease in the brain, cranial radiotherapy was deemed
inappropriate. Although the patient was advised to undergo
comprehensive genetic testing, followed by targeted therapy and
immunotherapy, the patient declined all these recommendations. The
patient received a low dose of imatinib mesylate tablets (0.4 g,
once daily) as targeted therapy, which resulted in slight
alleviation of the dizziness (Table
I). The patient sought treatment in Beijing following this
visit and did not return to the hospital for a follow-up until
February 2025, with the primary complaint still being dizziness.
The clinician prescribed imatinib mesylate tablets (0.4 g, once
daily) as a treatment.
 | Table I.Summary table of patient
presentation. |
Table I.
Summary table of patient
presentation.
| Date | Patient
presentation |
|---|
| March, 2022 | The patient
presented to the hospital with complaints of headaches and
dizziness. MRI revealed a space-occupying lesion in the left
cerebellar hemisphere. A resection of the skull base lesion was
subsequently performed. The pathological diagnosis was chordoid
meningioma. Postoperative radiotherapy was then administered. |
| November, 2023 | The patient
presented to the hospital with facial numbness on the right side.
MRI revealed an intracranial space-occupying lesion. The patient
subsequently underwent resection of the skull base lesion, and the
pathological diagnosis was chordoma. |
| December, 2023 | The patient
underwent local radiotherapy for intracranial lesions and the
sacral vertebra following surgery, with the treatment's efficacy
assessed as a partial response. |
| March, 2024 | Positron emission
tomography-computed tomography scans revealed a space-occupying
lesion in the left cerebellar surgical site. Given the potential
for tumor recurrence, palliative radiotherapy was administered to
the area of recurrence, and the efficacy of this treatment was
evaluated as progressive disease. |
| November, 2024 | The patient
experienced episodes of paroxysmal dizziness, and MRI enhancement
revealed nodular abnormal signal foci within the sacral canal. The
possibility of chordoma recurrence was considered, and targeted
therapy with imatinib was administered. |
Discussion
Chordoma is a rare primary malignant bone tumor that
originates from the embryonic remnants of the notochord. During
embryonic development, the upper end of the notochord is situated
in the sphenoid and occipital bones at the base of the skull, while
the lower end is located in the sacrum. Consequently, chordomas
typically arise in the skull base or sacrum. The annual incidence
rate of chordoma is ~0.8 per million individuals, accounting for
~1% of all bone tumors. Chordoma can manifest at any age; however,
the majority of patients are diagnosed between the ages of 40 and
50 years, with <5% of cases occurring in individuals <20
years (1,7).
Clinical manifestations primarily depend on the
location and extent of the lesions. Intracranial chordomas
typically result in headaches or facial numbness due to the
proximity of the tumor to cranial nerves, whereas sacrococcygeal
chordomas lead to lumbosacral pain or bowel dysfunction as a
consequence of compression of the sacral nerve roots (7).
Meningioma is a type of tumor that arises from the
meninges, the protective membranes surrounding the brain and spinal
cord. Chordoid meningiomas account for 0.5–1% of all meningiomas
and are more commonly found in young female patients (8). These tumors are typically located in
the supratentorial region or at the skull base, where they invade
surrounding tissues, leading to associated symptoms such as
headaches, vomiting and visual disturbances (3).
The clinical symptoms of chordoma and chordoid
meningioma can often present similarly. The current patient
presented at the hospital with complaints of dizziness, headaches
and numbness in the right hand. There were no distinguishing
symptoms to differentiate between the two tumors. While chordoma is
known for its propensity to recur, meningioma typically does not
exhibit this tendency. The recurrence of the tumor within <2
years aligns with the biological behavior of chordoma, prompting
re-evaluation and revision of the initial pathology report.
Chordoma commonly originates from the
sphenoid-occipital symphysis region and primarily grows in the
midline area, typically involving the midline structures in the
slope region. There is marked bone destruction at the skull base,
particularly in areas such as the saddle dorsal and slope, and the
tumor can extend into the adjacent basal cistern. Chordoma
typically presents as a destructive lobulated mass on CT, which
invades adjacent tissues and is associated with lytic bone
destruction and soft-tissue expansion (9). MRI, with its high soft-tissue
resolution, accurately depicts the location of the tumor, tissue
structure and surrounding structures that it invades. T1-weighted
imaging (T1WI) indicates low signal lesions, while T2-weighted
imaging (T2WI) shows high signal lesions accompanied by low signal
septations, resulting in a honeycomb-like pattern of uneven
enhancement (9–11).
Chordoid meningioma in general is located in the
supratentorial region and typically exhibits aggressive behavior on
imaging, often invading surrounding bone while maintaining an
intact capsule. Involvement of the brain parenchyma is rare
(3). A CT scan typically reveals
isodensity, while MRI often shows low signal using T1WI and high
signal using T2WI. Some lesions may exhibit mixed signals with long
T1 and long T2 characteristics, along with marked enhancement
following contrast administration (11). Caudal meningeal signs may be
present, and restricted diffusion on DWI is not limited (12).
The lesion in the present case was located adjacent
to the skull base area of the posterior cranial fossa; however,
there was no evident bone destruction in the adjacent skull, nor
were there any signs of bleeding, necrosis, irregular calcification
or other abnormalities within the lesion itself. The DWI signal of
the lesion was not elevated, indicating that it was not prominently
visible on imaging. There was no clear evidence of restricted
diffusion, which suggests that the imaging findings did not align
with typical clinical presentations of chordomas. The lesion in the
present case presented as a hemicystic, low-density round mass on
the plain CT scan. MRI signal characteristics revealed that T1WI
predominantly exhibited low signal intensity, while T2WI
demonstrated slightly elevated and uneven signal intensity, with
additional patches of slightly low signal observed internally. The
T2-fluid attenuated inversion recovery image displayed
heterogeneous high signal intensity. Notably, most lesions on the
enhanced scan did not exhibit enhancement, although an eccentric
marginal area of pronounced heterogeneous enhancement was evident.
These imaging characteristics are not consistent with typical
clinical presentations of meningiomas. Therefore, this discrepancy
contributed to the misdiagnosis.
The initial diagnosed lesion in the present case was
situated in the left cerebellar hemisphere. The location of its
onset is not a typical site for meningiomas in adults. Furthermore,
the lesion did not exhibit the general characteristics associated
with intracranial and extracerebral tumors on imaging. These
characteristics include, but are not limited to, the white matter
collapse sign, the meningeal tail sign, adjacent subarachnoid space
widening and cerebrospinal fluid clefts (12). Additionally, the remaining lesions
did not show a close relationship with the dura mater or the
skull.
According to the 5th edition of the WHO
classification of bone and soft-tissue tumors, chordoma is
categorized into three types: Classic type, dedifferentiated type
and poorly differentiated type (7).
The pathology of classic chordoma is characterized by a fine
fibrovascular connective tissue stroma, which contains trabecular
and nest-like epithelioid cells within the lobules. The cell sizes
vary, and the cells include multinucleated, stellate and vacuolated
cells with abundant and intensely eosinophilic cytoplasm. The
background features an abundant extracellular myxoid matrix, and
necrosis is often observed, while mitotic figures are uncommon.
Classic chordoma may exhibit both chordoma and chondroid features,
previously referred to as chondroid chordoma; the 5th edition of
the WHO classification of bone and soft tissue tumors includes
chondroid chordoma under classic chordoma (4).
Dedifferentiated chordoma is a biphasic tumor
characterized by the presence of two distinct components: A classic
chordoma component and a high-grade sarcoma component. The
high-grade sarcoma component usually manifests as either high-grade
undifferentiated spindle cell sarcoma or high-grade osteosarcoma.
Tumor cells may display binucleation or multinucleation, and
mitotic figures are often observable (4). The 5th edition of the WHO
classification of bone and soft-tissue tumors categorizes poorly
differentiated chordoma as a new subtype. This subtype has a low
incidence rate, primarily occurring in children and young adults,
and is commonly located at the skull base. Pathological features
include a solid growth pattern and notable cellular atypia, which
is characterized by the loss of integrase interactor 1 (INI1)
expression (4).
The pathology of chordoid meningioma is
characterized by tumor cells arranged in nests or trabecular
patterns, featuring eosinophilic cytoplasm and intracellular
vacuoles. The stroma has a myxoid appearance, and mitotic figures
are rare. Classic areas of meningioma are frequently observed in
the surrounding tissue, displaying a swirling, bundle-like pattern,
while pure chord meningioma is uncommon (13).
When the chordoid areas predominate or when tumors
exhibit chondroid metaplasia, accurate morphological diagnosis can
be problematic as the differential diagnosis broadens to include
chordoid glioma, skeletal/extraskeletal myxoid chondrosarcomas,
chondrosarcomas and enchondromas. The immunohistochemical
evaluation is particularly effective for differentiating these
tumors (13) (Table II).
| Table II.Pathological distinctions of tumors
of the central nervous system with marked histological overlap. |
Table II.
Pathological distinctions of tumors
of the central nervous system with marked histological overlap.
| Tumor | Site | Tumor
structure | Tumor cells | Tumor stroma | Positive
immunohistochemistry results | Genetics | (Refs.) |
|---|
| Chordoma | Skull base or
sacrum | Lobulated
structure | The cell sizes
vary, and the cells include multinucleated, stellate and vacuolated
cells with abundant and intensely eosinophilic cytoplasm | Extracellular
myxoid matrix | CK8, CK18, CK19,
EMA, S100, Brachyury | Mutations in
Brachyury, CDKN2A, PTEN, SMARCB1 | (16,36) |
| Chordoid
meningiomas | Supratentorial
region or at the skull base | Tumor cells
arranged in cords or nests | The tumor cells
arranged in nests or trabecular patterns, featuring eosinophilic
cytoplasm and intracellular vacuoles | Myxoid matrix | Vimentin, EMA,
PR | Deletion of 1p,
2q/NF2 and CDKN2A/B | (2,8) |
| Chordoid
glioma | Third
ventricle | Lobulated
structure, with tumor cells arranged in clusters and cords | The tumor cell
displays characteristics of epithelial cells, including abundant,
deeply stained cytoplasm and oval or round nuclei | Myxoid or
vacuolated matrix | GFAP, TTF-1 | p.D463H mutation in
the PRKCA gene | (37,38) |
|
Skeletal/extraskeletal myxoid
chondrosarcomas | Deep soft tissues
of the proximal limbs, particularly in the lower limbs | Lobulated
structure, with tumor cells arranged in cords, small clusters or
grids | The nuclei of the
tumor cells are small and exhibit deep staining, while the
cytoplasm is eosinophilic and partially vacuolated | Myxoid or
myxochondral matrix | Vimentin, S100,
EMA, CD99, SOX9 | >75% of cases
contain the t(9;22)(q22-31;q11-12) chromosome translocation. The
NR4A3 fusion gene can be detected in >90% of cases | (39,40) |
|
Chondrosarcomas | Pelvis and proximal
femur | Lobulated
structure | Tumor cells are
dense at the edges of the lobules and sparse in the center. Tumor
cells are observed to be round, triangular or star-shaped and are
located within cartilage cavity | Cartilage
matrix | S100, SOX-10 | ~50% of the tumors
presented heterozygous missense mutations in IDH1 R132 and IDH2
R172 | (41,42) |
| Enchondroma | Metaphyseal region
of the long bones | Lobulated hyaline
cartilage nodules | The nuclei of tumor
cells are small, round and darkly stained, while the cytoplasm is
abundant in vacuoles | Cartilage
matrix | Lacks specific
immunohistochemical features | Mutations in the
IDH1/2 genes | (42) |
Chordomas and chordoid meningiomas exhibit marked
histological similarities. In the present study, the pathology
report from the first surgery revealed cord-like epithelioid cells,
myxoid stroma and meningioma-like regions in certain adjacent
areas. However, due to the limited number of gross specimens
available, there were insufficient regions for accurate
identification, leading to a misdiagnosis.
Chordoma expresses CK8, CK18 and CK19, but does not
express CK7 and CK20. Both EMA and S100 protein are positive in
chordomas (14,15). Brachyury, a transcription factor
encoded by the T gene located on chromosome 6q27, serves a crucial
role in embryonic notochord development and is considered a key
driver of chordoma (16). When
utilized for the diagnosis of chordoma, Brachyury demonstrates high
specificity and sensitivity, with a positive rate reaching as high
as 99%. In poorly differentiated chordoma, tumor cells are positive
for broad-spectrum CK and Brachyury, whereas in dedifferentiated
chordoma, expression of all markers may be negative (17,18).
Chordoid meningioma typically exhibits positivity for vimentin, EMA
and PR, while S100, CK and GFAP are generally negative. Sangoi
et al (13) reported a
higher positive rate of D2-40 in chordoid meningioma, highlighting
its diagnostic importance.
Chordoma and chordoid meningioma share numerous
immunohistochemical similarities. Brachyury, an antibody that has
received focus in recent years, is infrequently utilized for
diagnoses other than chordoma. Therefore, numerous pathologists may
not fully recognize its importance. This lack of awareness is also
the reason why Brachyury was not included in the initial pathology
assessment for the present patient.
In addition to Brachyury, mutations in genes such as
cyclin-dependent kinase inhibitor 2A (CDKN2A), PTEN and INI1 have
also been identified in chordoma (19–21).
INI1 is a tumor suppressor protein that serves as a component of
the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex, which
regulates gene expression through the remodeling of chromatin
structure. Deletion or mutation of the INI1 gene results in
decreased protein expression, thereby impairing the normal function
of the SWI/SNF complex. This impairment may subsequently lead to
abnormal cell proliferation and tumor formation (22). A recent study demonstrated that the
immunohistochemical loss of INI1 protein in poorly differentiated
chordoma primarily results from a homozygous deletion of the 22q11
locus (4). The loss of INI1 protein
may provide a rationale for assessing the efficacy of novel
targeted therapies, such as enhancer of zeste homolog 2 (EZH2)
inhibitors. EZH2 serves as the catalytic subunit of the histone
methyltransferase polycomb repressive complex 2, and its
upregulation has been linked to the promotion of carcinogenesis.
EZH2 inhibitors have demonstrated the ability to induce tumor
regression and enhance radiosensitivity in SWI/SNF-related BAF
chromatin remodeling complex subunit B1/INI1-deficient tumor models
(23). INI1 immunohistochemistry
was performed on the two surgical specimens of the patient, both of
which demonstrated weak positivity in a few scattered cells (data
not shown). Horbinski et al (24) reported that deletions of 1p36 and 9p
in chordoma were linked to decreased overall survival (OS).
Furthermore, Wei et al (25)
demonstrated that reduced H3K27me3 epigenetic modification in
chordoma was associated with a poor prognosis. H3K27me3 is a main
epigenetic modification, primarily mediated by EZH2, the catalytic
subunit of the histone methyltransferase PRC2 complex. H3K27me3 is
essential for the regulation of gene expression and is frequently
associated with gene silencing (26).
A previous study identified microRNA (miR)-1, miR-31
and miR-148a as promising prognostic and therapeutic markers for
chordoma (27). Additionally,
Brachyury vaccines and molecular targeted therapies have been
explored as potential treatment options for this condition
(28). The preferred treatment
method for chordoma is surgical intervention; however, the
proximity of vascular and nerve structures to the tumor often
hinders complete resection, resulting in a high risk of recurrence.
Chordomas exhibit a poor response to conventional radiotherapy and
chemotherapy, which renders these modalities ineffective as
standard treatment options (29). A
study involving 357 patients with chordoma performed in the United
States revealed OS rates of 80.5, 68.4 and 39.2% at 3, 5 and 10
years, respectively. Additionally, the disease-specific survival
rates at these intervals were 89.0, 80.9 and 60.1%, respectively.
It was observed that when patients were >60 years old or
received non-surgical treatment due to metastasis, the OS rate
declined (30).
Daoud et al (31) identified that common copy number
variations in chordoid meningioma included a deletion of 1p,
whereas deletions of 22q/NF2, moesin-ezrin-radixin-like tumor
suppressor and CDKN2A/B were molecular alterations associated with
tumor recurrence and progression. In the present study, the patient
refused to undergo molecular testing, and the small size of the
tumor tissue rendered pathological testing unfeasible. It is
essential for physicians to collect additional specimens during
surgery for molecular testing, thereby offering patients a broader
range of treatment options.
The preferred treatment for chordoid meningioma is
surgical intervention, and the grade of meningioma resection, as
categorized by the Simpson grading system, serves as a prognostic
factor. For patients in whom a Simpson grade II resection cannot be
achieved, postoperative radiotherapy is necessary (32). Ren et al (33) indicated that the progression-free
survival (PFS) of patients with chordoid meningioma was improved
compared with that of patients with non-chordoid WHO Grade 2
meningioma, yet worse than that of patients with WHO Grade 1
meningioma. Additionally, the OS of chordoid meningioma was
superior to that of non-chordoid WHO Grade 2 meningioma.
Furthermore, recurrence status and adjuvant radiotherapy were
identified as independent prognostic factors for both PFS and OS
(33).
Although the misdiagnosis did not affect the
surgical method in the present study, it influenced the treatment
approach and prognosis. If the initial diagnosis had been accurate
and radiotherapy for chordoma had been administered promptly, the
prognosis of the patient would likely have been more favorable than
it currently is. The lesion was situated in the cerebellum, and the
primary approach was resection of the skull base lesion. The
planning target volume (PTV) for radiotherapy following the first
operation was 5,040 cGy/180 cGy/28 fractions, while the PTV for
radiotherapy after the second operation was 4,000 cGy/200 cGy/20
fractions for intracranial lesion, 7,000 cGy/200 cGy/35 fractions
for lumbosacral. If chordoma was confirmed during the first
operation, an appropriate radiotherapy could have been utilized to
decrease the recurrence rate.
Pathology was the primary reason for the
misdiagnosis and subsequent mistreatment of the present patient.
Initially, the workflow of the pathology was flawed; the report was
issued by a single pathologist without review by a specialist or
general discussion. Due to a lack of experience in diagnosing
chordoma in the brain, the pathologist mistakenly classified the
chord-like cells as chordoid meningiomas, overlooking the potential
diagnosis of chordoma. Consequently, immunohistochemical staining
for vimentin, EMA, GFAP and S100, along with a negative result for
CD34, supported the erroneous conclusion of chordoid meningioma.
Unaware of the distinctions between chordoma and chordoid
meningioma, the pathologist did not perform the Brachyury test.
Furthermore, no imaging studies were performed to assess the
positional relationship between the tumor and the meninges.
Following recurrence, the original pathologist reviewed the initial
pathology and, upon consultation with a specialist, raised the
possibility of chordoma and recommended the Brachyury test for
confirmation. The positive result validated the diagnosis of
chordoma and highlighted the initial misdiagnosis. Rekhi and
Karmarkar (34) demonstrated that
immunohistochemistry is crucial for differentiating between these
two entities, recommending the use of CK, GFAP, vimentin, EMA,
S100, PR, D2-40 and Brachyury. Therefore, caution is advised when
certain immunohistochemical markers are absent, and it is prudent
to seek consultation from a more experienced pathologist if
necessary.
From a clinical perspective, the current patient
presented with a lack of specific clinical symptoms, and the site
of onset was not typical for a chordoma. Consequently, after
receiving a pathological diagnosis of meningioma, the clinician did
not engage in further communication with the pathologist.
In terms of imaging, the two preoperative MR
diagnoses indicated only intracranial space occupation without
specifying the tumor type or the potential for benign or malignant
characteristics. Following the second recurrence of the tumor and
subsequent pathological diagnosis, PET-CT revealed multiple
metastases throughout the body and suggested a diagnosis of
chordoma. The misdiagnosis prompted the development of a novel
pathology process: All reports must now be diagnosed by two
pathologists. Additionally, in challenging cases, it is essential
to communicate with imaging specialists and clinicians, and to
conduct a multidisciplinary team meeting when necessary. Brachyury
has been incorporated into the meningioma immunohistochemistry
panel to aid in the differentiation between meningiomas and
chordomas.
There are limitations to the present case report.
Molecular/genetic testing was not performed in the current case
report and limited tissue was available for diagnosis. While double
staining for S100 and Brachyury could assist in identifying similar
cases, both antibodies used in Qingdao Municipal Hospital stain the
nucleus. Currently, technology does not allow for the double
staining of antibodies that both target the nucleus. Consequently,
this method was not employed to further verify the diagnosis.
To the best of our knowledge, the current case
represents the first reported instance of such a misdiagnosis.
Previous discussions have predominantly concentrated on the
pathological differentiation between chordoma and chordoid glioma,
with only one reported case of a chordoid meningioma being
misdiagnosed as a chordoma (35).
Therefore, the present case serves to enhance the understanding of
chordoma and chordoid meningioma, ultimately improving diagnostic
accuracy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
DL analyzed and interpreted the patient data, and
contributed to writing the manuscript. CJ and MMZ performed the
pathological examination. DL, MMZ and CJ confirm the authenticity
of all the raw data. TW made substantial contributions to the
acquisition and interpretation of all imaging data (MRI, PET-CT),
critical analysis of radiological-pathological data and revision of
the manuscript for important intellectual content regarding imaging
findings. PZ provided analysis of clinical data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Written informed patient consent was obtained. The
present study was conducted with the approval of the Independent
Ethics Committee of the Qingdao Municipal Hospital.
Patient consent for publication
Written informed consent was obtained from the
patient for publication of the present case report.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MRI
|
magnetic resonance imaging
|
|
PFS
|
progression-free survival
|
|
OS
|
overall survival
|
References
|
1
|
Karpathiou G, Dumollard JM, Dridi M, Dal
Col P, Barral FG, Boutonnat J and Peoc'h M: Chordomas: A review
with emphasis on their pathophysiology, pathology, molecular
biology, and genetics. Pathol Res Pract. 216:1530892020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Couce ME, Aker FV and Scheithauer BW:
Chordoid meningioma: A clinicopathologic study of 42 cases. Am J
Surg Pathol. 24:899–905. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nambiar A, Pillai A, Parmar C and Panikar
D: Intraventricular chordoid meningioma in a child: Fever of
unknown origin, clinical course, and response to treatment. J
Neurosurg Pediatr. 10:478–481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oakley GJ, Fuhrer K and Seethala RR:
Brachyury, SOX-9, and podoplanin, new markers in the skull base
chordoma vs chondrosarcoma differential: A tissue microarray-based
comparative analysis. Mod Pathol. 21:1461–1469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Medical Research Council: Nerve Injuries
Committee, . M.R.C. War Memorandum: Aids to the investigation of
peripheral nerve injuries. Physical Therapy. 23:1401943. View Article : Google Scholar
|
|
6
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi JH and Ro JY: The 2020 WHO
classification of tumors of soft tissue: Selected changes and new
entities. Adv Anat Pathol. 28:44–58. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin JW, Ho JT, Lin YJ and Wu YT: Chordoid
meningioma: A clinicopathologic study of 11 cases at a single
institution. J Neurooncol. 100:465–473. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santegoeds RGC, Temel Y,
Beckervordersandforth JC, Van Overbeeke JJ and Hoeberigs CM:
State-of-the-art imaging in human chordoma of the skull base. Curr
Radiol Rep. 6:162018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Golden L, Pendharkar A and Fischbein NJc:
Chapter 7 - Imaging Cranial Base Chordoma and Chondrosarcoma.
Chordomas and Chondrosarcomas of the Skull Base and Spine. 2nd
Edition. Elsevier Inc.; pp. 67–78. 2018, View Article : Google Scholar
|
|
11
|
Farsad K, Kattapuram SV, Sacknoff R, Ono J
and Nielsen GP: Sacral chordoma. Radiographics. 29:1525–1530. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loken EK and Huang RY: Advanced meningioma
imaging. Neurosurg Clin N Am. 34:335–345. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sangoi AR, Dulai MS, Beck AH, Brat DJ and
Vogel H: Distinguishing chordoid meningiomas from their histologic
mimics: An immunohistochemical evaluation. Am J Surg Pathol.
33:669–681. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lehtonen E, Stefanovic V and Saraga-Babic
M: Changes in the expression of intermediate filaments and
desmoplakins during development of human notochord.
Differentiation. 59:43–49. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Folpe AL, Agoff SN, Willis J and Weiss SW:
Parachordoma is immunohistochemically and cytogenetically distinct
from axial chordoma and extraskeletal myxoid chondrosarcoma. Am J
Surg Pathol. 23:1059–1067. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vujovic S, Henderson S, Presneau N, Odell
E, Jacques TS, Tirabosco R, Boshoff C and Flanagan AM: Brachyury, a
crucial regulator of notochordal development, is a novel biomarker
for chordomas. J Pathol. 209:157–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dridi M, Boutonnat J, Dumollard JM, Peoc'h
M and Karpathiou G: Patterns of brachyury expression in chordomas.
Ann Diagn Pathol. 53:1517602021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miettinen M, Wang Z, Lasota J, Heery C,
Schlom J and Palena C: Nuclear brachyury expression is consistent
in chordoma, common in germ cell tumors and small cell carcinomas,
and rare in other carcinomas and sarcomas: An immunohistochemical
study of 5229 cases. Am J Surg Pathol. 39:1305–1312. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scheil S, Brüderlein S, Liehr T, Starke H,
Herms J, Schulte M and Möller P: Genome-wide analysis of sixteen
chordomas by comparative genomic hybridization and cytogenetics of
the first human chordoma cell line, U-CH1. Genes Chromosomes
Cancer. 32:203–211. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hallor KH, Staaf J, Jönsson G, Heidenblad
M, Vult von Steyern F, Bauer HC, Ijszenga M, Hogendoorn PC, Mandahl
N, Szuhai K and Mertens F: Frequent deletion of the CDKN2A locus in
chordoma: Analysis of chromosomal imbalances using array
comparative genomic hybridisation. Br J Cancer. 98:434–442. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuźniacka A, Mertens F, Strömbeck B,
Wiegant J and Mandahl N: Combined binary ratio labeling
fluorescence in situ hybridization analysis of chordoma. Cancer
Genet Cytogenet. 151:178–181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roberts CW and Biegel JA: The role of
SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther.
8:412–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim KH and Roberts CWM: Targeting EZH2 in
cancer. Nat Med. 22:128–134. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Horbinski C, Oakley GJ, Cieply K, Mantha
GS, Nikiforova MN, Dacic S and Seethala RR: The prognostic value of
Ki-67, p53, epidermal growth factor receptor, 1p36, 9p21, 10q23,
and 17p13 in skull base chordomas. Arch Pathol Lab Med.
134:1170–1176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei J, Wu J, Yin Z, Li X, Liu Y, Wang Y,
Wang Z, Xu C and Fan L: Low expression of H3K27me3 is associated
with poor prognosis in conventional chordoma. Front Oncol.
12:10484822022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Margueron R and Reinberg D: The polycomb
complex PRC2 and its mark in life. Nature. 469:343–349. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karele EN and Paze AN: Chordoma: To know
means to recognize. Biochim Biophys Acta Rev Cancer.
1877:1887962022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heery CR, Palena C, McMahon S, Donahue RN,
Lepone LM, Grenga I, Dirmeier U, Cordes L, Marté J, Dahut W, et al:
Phase I study of a poxviral TRICOM-based vaccine directed against
the transcription factor brachyury. Clin Cancer Res. 23:6833–6845.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yurter A, Sciubba D, Gokaslan Z and
Kaloostian PJJN: Spinal Chordomas: Current Medical and Surgical
Management: JSM Neurosurgery and Spine. 2014.
|
|
30
|
Pan Y, Lu L, Chen J, Zhong Y and Dai Z:
Analysis of prognostic factors for survival in patients with
primary spinal chordoma using the SEER Registry from 1973 to 2014.
J Orthop Surg Res. 13:762018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Daoud EV, Zhu K, Mickey B, Mohamed H, Wen
M, Delorenzo M, Tran I, Serrano J, Hatanpaa KJ, Raisanen JM, et al:
Epigenetic and genomic profiling of chordoid meningioma:
Implications for clinical management. Acta Neuropathol Commun.
10:562022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tahta A, Genc B, Cakir A and Sekerci Z:
Chordoid meningioma: Report of 5 cases and review of the
literature. Br J Neurosurg. 37:41–44. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ren L, Hua L, Deng J, Cheng H, Wang D,
Chen J, Xie Q, Wakimoto H and Gong Y: Favorable long-term outcomes
of chordoid meningioma compared with the other WHO grade 2
meningioma subtypes. Neurosurgery. 92:745–755. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rekhi B and Karmarkar S:
Clinicocytopathological spectrum, including uncommon forms, of nine
cases of chordomas with immunohistochemical results, including
brachyury immunostaining: A single institutional experience.
Cytopathology. 30:229–235. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murali R and Ng T: Chordoid meningioma
masquerading as chordoma. Pathology. 36:198–201. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Presneau N, Shalaby A, Ye H, Pillay N,
Halai D, Idowu B, Tirabosco R, Whitwell D, Jacques TS, Kindblom LG,
et al: Role of the transcription factor T (brachyury) in the
pathogenesis of sporadic chordoma: A genetic and functional-based
study. J Pathol. 223:327–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goode B, Mondal G, Hyun M, et al: A
recurrent kinase domain mutation in PRKCA defines chordoid glioma
of the third ventricle. Nat Commuml. 9:8102018. View Article : Google Scholar
|
|
38
|
Reifenberger G, Weber T, Weber RG, Wolter
M, Brandis A, Kuchelmeister K, Pilz P, Reusche E, Lichter P and
Wiestler OD: Chordoid glioma of the third ventricle:
Immunohistochemical and molecular genetic characterization of a
novel tumor entity. Brain Pathol. 9:617–626. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ogura K, Fujiwara T, Beppu Y, Chuman H,
Yoshida A, Kawano H and Kawai A: Extraskeletal myxoid
chondrosarcoma: A review of 23 patients treated at a single
referral center with long-term follow-up. Arch Orthop Trauma Surg.
132:1379–1386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Flucke U, Tops BBJ, Verdijk MAJ, van Cleef
PJH, van Zwam PH, Slootweg PJ, Bovée JVMG, Riedl RG, Creytens DH,
Suurmeijer AJH and Mentzel T: NR4A3 rearrangement reliably
distinguishes between the clinicopathologically overlapping
entities myoepithelial carcinoma of soft tissue and cellular
extraskeletal myxoid chondrosarcoma. Virchows Archiv. 460:621–628.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Amary MF, Bacsi K, Maggiani F, Damato S,
Halai D, Berisha F, Pollock R, O'Donnell P, Grigoriadis A, Diss T,
et al: IDH1 and IDH2 mutations are frequent events in central
chondrosarcoma and central and periosteal chondromas but not in
other mesenchymal tumours. J Pathol. 224:334–343. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pansuriya TC, van Eijk R, d'Adamo P, van
Ruler MAJH, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk
JG, Verbeke SLJ, Meijer D, et al: Somatic mosaic IDH1 and IDH2
mutations are associated with enchondroma and spindle cell
hemangioma in Ollier disease and Maffucci syndrome. Nat Genet.
43:1256–1261. 2011. View Article : Google Scholar : PubMed/NCBI
|