Introduction
Chromophobe renal cell carcinoma (ChRCC) is a rare
type of RCC, accounting for 5–7% of RCCs cases (1). ChRCC has a favorable prognosis
compared with other RCC subtypes, including clear-cell RCC and
papillary RCC (1). ChRCC originates
from intercalated cells of the renal cortex, but sarcomatoid
changes can occur in 2–8% of ChRCC cases. The presence of
sarcomatoid changes in ChRCC significantly alters its clinical
behavior, transforming it into a highly aggressive variant with a
poor prognosis (2).
Sarcomatoid changes may occur in any RCC subtype,
leading to the appearance of spindle-shaped pleomorphic cells that
resemble sarcomas. These histological transformations are
associated with rapid disease progression, increased metastatic
potential and resistance to conventional therapies. While
sarcomatoid changes are well documented in clear-cell and papillary
RCC, their occurrence in ChRCC is rare and not fully understood
(3).
The present study reports a case of ChRCC with
sarcomatoid changes, highlighting distinct histopathological,
genetic and immunohistochemical features. Through detailed
next-generation sequencing (NGS) analysis, we aimed to identify the
molecular changes responsible for this transformation. Analyzing
the genetic alterations and immune profiles of sarcomatoid
transformation in ChRCC may enhance therapeutic strategies and
improve outcomes in affected patients.
Case report
A 54-year-old woman with no significant medical or
family history presented to the Urology Department (Jeonbuk
National University Hospital, Jeonju, South Korea) in February 2023
for evaluation of an incidentally detected left renal mass.
Laboratory results were within normal ranges: Serum tumor markers,
α-fetoprotein at 2.72 ng/ml (normal <7.0 ng/ml),
carcinoembryonic antigen at 1.8 ng/ml (normal <5.2 ng/ml),
carbohydrate antigen (CA) 19-9 at 9.0 U/ml (normal <34.0 U/ml)
and CA125 at 28.4 U/ml (normal <35.0 U/ml). Computed tomography
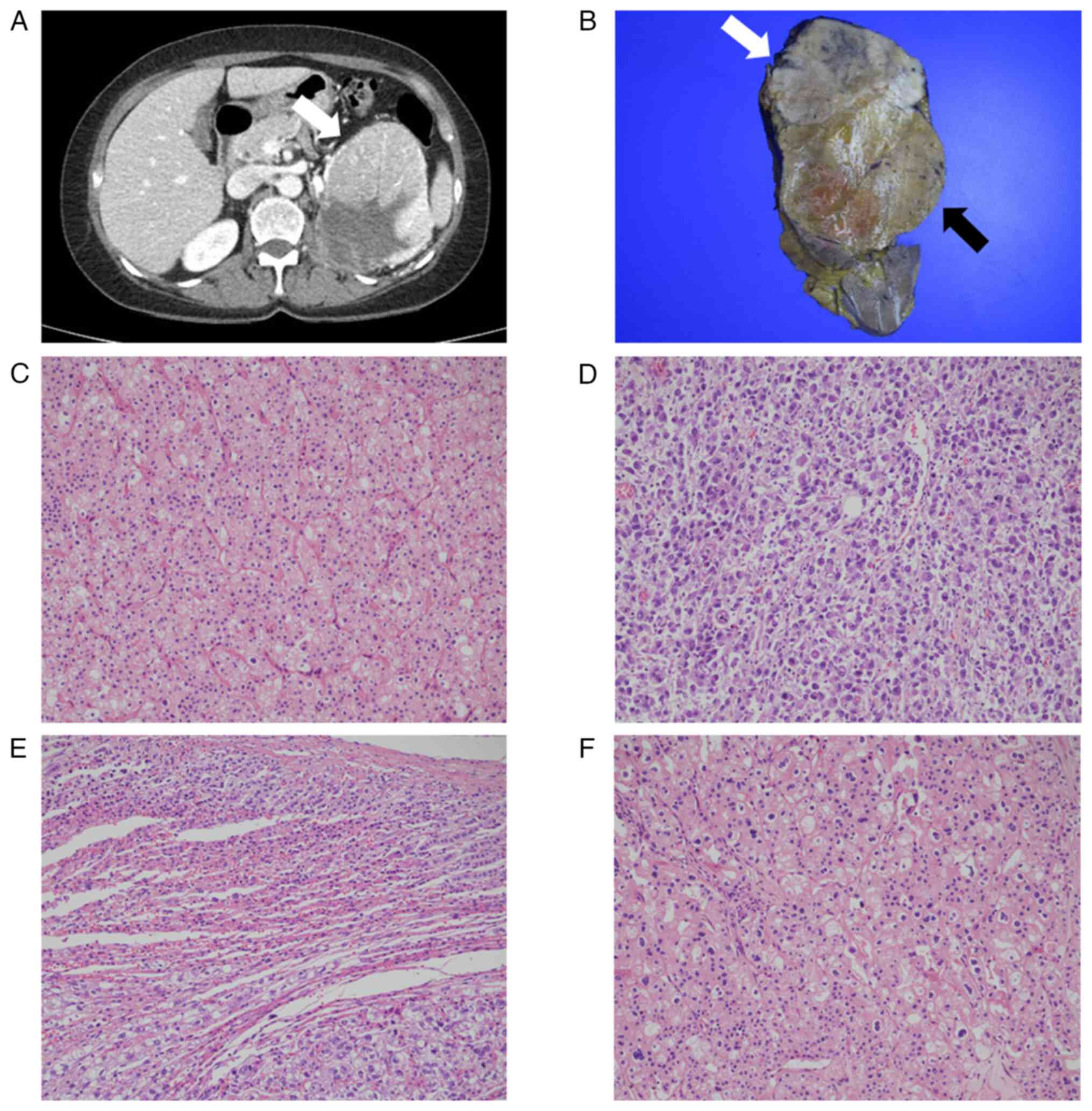
of the abdomen revealed a large left renal mass measuring 13×10 cm
in maximum dimensions, with heterogeneous enhancement and irregular
margins (Fig. 1A). The patient
underwent an open radical nephrectomy for diagnosis and
treatment.
Gross examination of the specimen revealed a tumor
measuring 14.2×9.8 cm with two distinct components: A yellowish
hard lesion (Fig. 1B; white arrow)
and a white-grayish soft lesion (Fig.
1B; black arrow). The tumor exhibited an irregular margin and
revealed a possible invasion into perinephric fat and adrenal
tissue. For histopathological analysis, the resected specimen was
fixed in 10% neutral buffered formalin at room temperature for 24
h. After fixation, the specimen was processed and embedded in
paraffin. Each section was cut at 4 µm thickness using a microtome
and mounted on glass slides. Hematoxylin and eosin (H&E)
staining was performed with hematoxylin staining for 3 min at room
temperature, followed by eosin staining for 7 min at room
temperature. All stained sections were observed under a light
microscope. On histology, the yellowish hard lesion showed
morphological features typical of ChRCC (Fig. 1C), while the white-grayish soft
lesion showed large cells with marked nuclear pleomorphism and
bizarre mitotic figures (Fig. 1D).
The tumor cells had invaded the perinephric fat and adrenal glands
(Fig. 1E). In a limited area, some
foci showed a transition between lesion 1 and lesion 2 (Fig. 1F). The conventional and poorly
differentiated ChRCC components exhibited contrasting
immunohistochemical and special staining patterns. Staining was
performed using an automated immunostainer (BenchMark ULTRA;
Ventana Medical Systems, Inc.) according to the manufacturer's
protocol. Tissue sections were fixed in 10% neutral buffered
formalin at room temperature for 24 h, processed routinely and
embedded in paraffin. Sections were cut at a thickness of 4 µm and
mounted on glass slides. All primary antibodies used were
ready-to-use products provided by the manufacturer (Roche);
therefore, no dilution was required. All staining steps, including
deparaffinization, antigen retrieval, blocking, antibody incubation
and detection, were carried out automatically under pre-optimized
and standardized conditions according to the manufacturer's
instructions. Hematoxylin was applied as a counterstain for 5 min
at room temperature. All stained slides were examined using a light
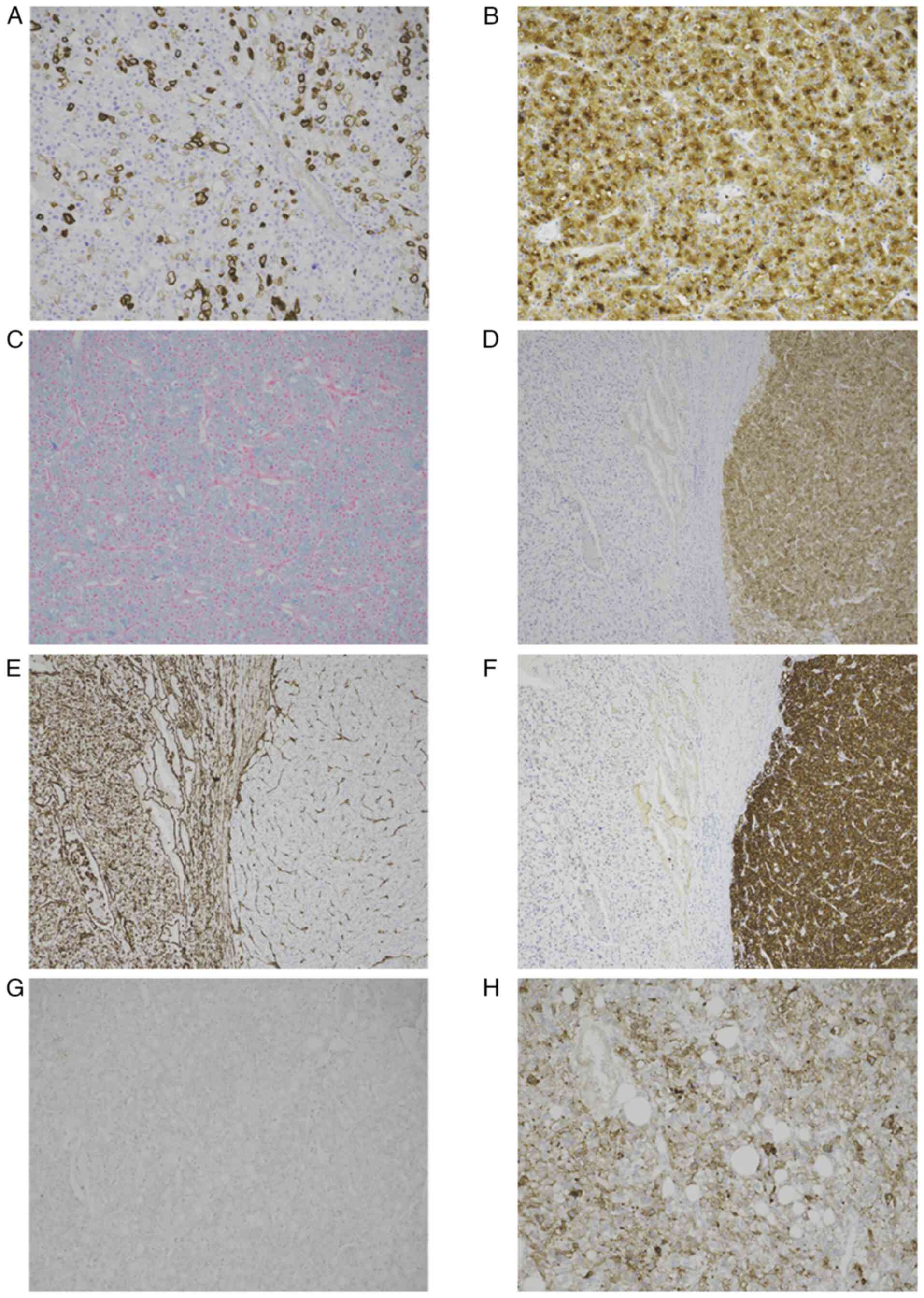
microscope. The conventional ChRCC component was positive for
Keratin-7 (KRT-7; cat. no. 790-4462; Roche Tissue Diagnostics), KIT
(CD117; cat. no. 790-7061; Roche Tissue Diagnostics) and
pan-cytokeratin (cat. no. 760-2595; Roche Tissue Diagnostics) and
negative for vimentin (cat. no. 760-2917; Roche Tissue
Diagnostics), with positive staining for Hale's colloidal iron
(cat. no. 860-009; Roche Tissue Diagnostics). All
immunohistochemical staining was performed using a Ventana
BenchMark ULTRA immunostainer (Roche Tissue Diagnostics) according
to the manufacturer's protocols. Chromogenic detection was
performed using the OptiView DAB IHC Detection Kit (cat. no.
760-700; Roche Tissue Diagnostics), which contains biotinylated
secondary antibody, streptavidin-HRP conjugate and DAB substrate.
By contrast, the poorly differentiated components displayed the
opposite staining pattern. However, focal KRT expression was
observed in certain tumor cells within the poorly differentiated
component (Fig. 2A-F). Targeted
NGS-based genomic profiling was performed for the two components.
Targeted NGS was performed using formalin-fixed, paraffin-embedded
(FFPE) tumor tissues. H&E-stained slides were reviewed and the
tumor area with sufficient viable tumor cells was marked for use as
a guide for macrodissection. Tumor areas with >50% tumor cells
were used for molecular examination. In brief, total nucleic acid
was extracted from FFPE tissue using RecoverAll™ Total
Nucleic Acid Isolation Kit (Ambion; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Library preparation
for the Oncomine® Comprehensive Assay Plus (Thermo
Fisher Scientific, Inc.), which covers 2,737 amplicons (2,530 DNA +
207 RNA) within 143 cancer-related genes, was performed. An
IonTorrent S5 XL platform was used for sequencing according the
manufacturer's specifications. The percentage of covered amplicons
was 95%. Reads were aligned to the hg19 reference genome and
variants with allele frequencies <3% were excluded. Both lesions
shared an insertion-deletion (indel) variant, RNF46
(c.349_350delCGinsA, p.Arg117ThrfsTer41). Copy number analysis
identified identical chromosomal losses in lesions 1, 2, 6, 10, 13,
15, 17, 21 and X. Based on these findings, the pathological
diagnosis confirmed ChRCC with sarcomatoid changes.
While the two components shared some genetic
alterations, there were significant differences. The sarcomatoid
component displayed distinct genetic features absent in the
conventional ChRCC component. Copy number analysis identified
chromosomal gains on chromosomes 1, 2, 6, 10, 13, 15, 17, 21 and X,
contrasting with the chromosomal losses found in the ChRCC
component (Fig. 3A and B).
Additional genomic alterations included single nucleotide variants
and indels in TP53 (c.1176_1179delAGAC, p.Ter394IlefsTer27),
TSC2 (c.1675G>A, p.Asp559Asn), NF1 (c.60+1G>A,
p.?), CDKN1B (c.384_385insAG, p.His129SerfsTer17) and
MET-CAPZA2 fusion mutation. These data have been uploaded to
Jeonbuk National University Hospital repository.
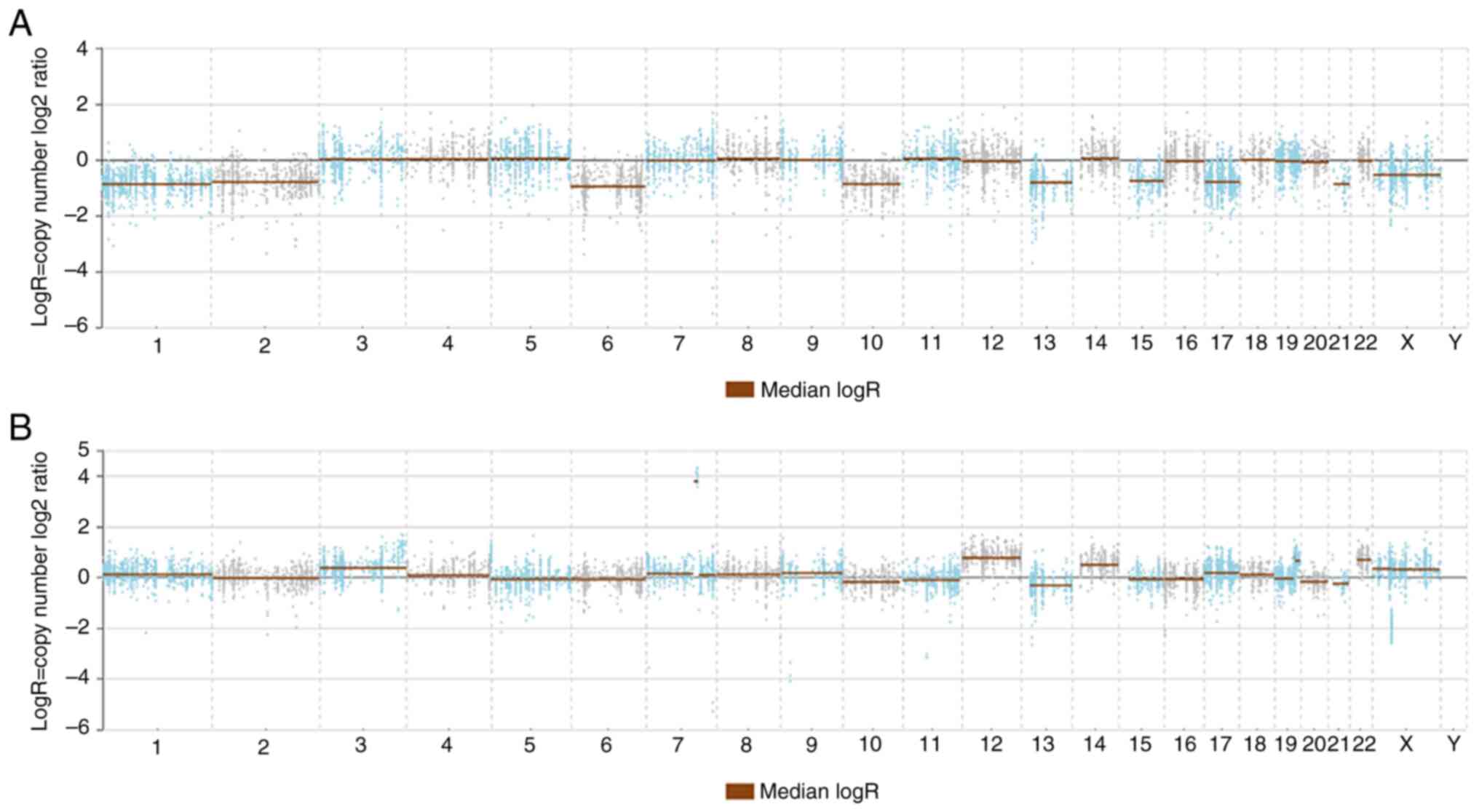 | Figure 3.Next-generation sequencing of
chromophobe renal cell carcinoma with sarcomatoid differentiation.
(A) In the chromophobe renal cell carcinoma component, copy number
analysis identified chromosomal loss in chromosomes 1, 2, 6, 10,
13, 15, 17, 21 and X. (B) The sarcomatoid differentiation component
exhibited distinct genetic features not seen in the chromophobe
renal cell carcinoma component. Copy number analysis identified
chromosomal gains in chromosomes 1, 2, 6, 10, 13, 15, 17, 21 and
X. |
To assess the immunotherapeutic potential,
immunohistochemical staining for programmed cell death ligand 1
(PD-L1; cat. no. 790-4905; Roche Tissue Diagnostics) was performed.
Chromogenic detection was performed using the OptiView DAB IHC
Detection Kit (cat. no. 760-700; Roche Tissue Diagnostics), which
contains biotinylated secondary antibody, streptavidin-HRP
conjugate and DAB substrate. Immunohistochemical staining for PD-L1
was performed using an automated immunostainer (BenchMark ULTRA;
Ventana Medical Systems, Inc.) according to the manufacturer's
protocol. Tissue sections were fixed in 10% neutral buffered
formalin at room temperature for 24 h, processed routinely and
embedded in paraffin. Sections were cut at a thickness of 4 µm and
mounted on glass slides. All primary antibodies used were
ready-to-use products provided by the manufacturer (Roche);
therefore, no dilution was required. All staining steps, including
deparaffinization, antigen retrieval, blocking, antibody incubation
and detection, were carried out automatically under pre-optimized
and standardized conditions according to the manufacturer's
instructions. Hematoxylin was applied as a counterstain for 5 min
at room temperature. All stained slides were examined using a light
microscope. The ChRCC component was negative for PD-L1, whereas the
area showed ≥10% positivity (Fig. 2G
and H).
Following diagnosis, the patient underwent
chemotherapy with nivolumab and cabozantinib. The patient received
chemotherapy with cabozantinib 40 mg (orally) and nivolumab 240 mg
(orally). After treatment for 2 consecutive days, the patient
exhibited symptoms of colitis and immune-mediated pneumonitis.
Despite adjuvant chemotherapy, the tumor progressed and
metastasized to multiple organs, including the lungs, liver, bones
and lymph nodes. The patient's condition rapidly deteriorated due
to hypercalcemia caused by multiple metastasis, and the patient
died in April 2023.
Discussion
ChRCC was first identified as a distinctive subtype
of RCC by Thoenes in 1985 (4).
ChRCC is uncommon subtype with generally favorable prognosis
compared to clear-cell RCC or papillary RCC, exhibiting a 5-year
survival rate of 78–100% and a 10-year survival rate of 80–90%
(5). However, sarcomatoid
differentiation significantly alters its clinical course
transforming ChRCC into an aggressive variant with poor outcomes
(2). Although documented across all
RCC subtypes, sarcomatoid transformation remains poorly understood
in ChRCC because of its rarity. The present case highlights the
histopathological, genetic and immunohistochemical differences
between conventional ChRCC and its sarcomatoid counterparts and
provides insights into the molecular changes that drive sarcomatoid
transformation.
In the present case, the coexistence of two distinct
tumor components, one conventional ChRCC and the other sarcomatoid,
along with the presence of a transitional area, suggested that
sarcomatoid transformation arose from pre-existing ChRCC cells by
acquiring additional molecular alterations that drive aggressive
behavior. NGS analysis revealed that both components shared certain
genetic changes such as the RNF46 variant and chromosomal
loss, whereas the sarcomatoid component exhibited additional
distinct genetic alterations. The conventional ChRCC component
exhibited loss of chromosomes 1, 2, 6, 10, 13, 15, 17, 21 and X.
Among these, the loss of chromosomes 1, 2, 6, 10 and 17 are
recognized as hallmarks of ChRCC (6,7). The
NGS analysis revealed that additional alterations in the
sarcomatoid component were chromosomal gains detected in
chromosomes 3, 5, 7, 9, 12, 13, 14, 19, 22 and X. These chromosomal
gains have also been reported in previous literature on sarcomatoid
ChRCC, suggesting that they may contribute to the sarcomatoid
transformation of ChRCC (7).
Additionally, in the sarcomatoid component, mutations in key tumor
suppressor genes such as TP53, TSC2 and NF1 were
observed, along with the detection of a MET-CAPZA2
fusion. These mutations could open possibilities for the
sarcomatoid differentiation of ChRCC component.
The MET gene, encoding a tyrosine kinase
receptor in the hepatocyte growth factor/scatter factor pathway, is
implicated in various oncogenic processes, including cell
proliferation, angiogenesis, invasion and metastasis (8,9). Among
MET alteration, MET fusions have been identified as
oncogenic drivers in various cancers, such as lung cancer, glioma,
colorectal cancer and cholangiocarcinoma (10). In RCC, MET alterations,
particularly MET exon 14 skipping mutations and MET
amplifications, have been associated with sarcomatoid
transformation and poor prognosis (11). However, to the best of our
knowledge, no studies have reported MET fusion in the
sarcomatoid transformation of ChRCC at present. The present study's
identification of the MET-CAPZA2 fusion in the sarcomatoid
component suggests that aberrant MET activation may play a
key role in driving the aggressive phenotype observed in this
case.
In addition, in the present case, the TP53
mutation was exclusively detected in the sarcomatoid component.
TP53 is a well-known tumor suppressor gene that regulates
cell cycle arrest, DNA repair and apoptosis. Loss-of-function
TP53 mutations are commonly associated with high-grade,
undifferentiated and aggressive tumors, as they lead to increased
genomic instability, uncontrolled cell proliferation and resistance
to apoptosis (12). In RCC,
TP53 mutations are frequently observed in sarcomatoid
dedifferentiation and are associated with poor prognosis and
resistance to immune checkpoint inhibitors (13,14).
The sarcomatoid component in the present case
harbored additional alterations in TSC2 and NF1, both
of which are tumor suppressor genes involved in the mTOR and RAS
signaling pathways, respectively (15,16).
TSC2 mutations can lead to constitutive mTOR activation,
driving cellular proliferation and metabolic reprogramming, which
contribute to an aggressive tumor phenotype (15). Meanwhile, NF1 mutations
disrupt the negative regulation of the RAS-MAPK pathway, promoting
tumor growth and invasion. Loss of NF1 has also been
associated with resistance to targeted therapies, further
highlighting the treatment challenges in sarcomatoid RCC (16).
Studies have demonstrated that targeting MET
fusions with selective MET tyrosine kinase inhibitors (TKI)
such as savolitinib and crizotinib can be effective in various
cancers, including non-small cell lung cancer and other solid
tumors (17). For example, patients
with MET fusion-positive tumors, including lung and
hepatobiliary cancers, respond to crizotinib in clinical trials
(10,18). These findings support the potential
of MET TKIs in the treatment of MET fusion-driven
cancers and reinforce their role in precision medicine. Management
of ChRCC with sarcomatoid changes based on genetic alterations has
not yet been standardized. Although MET fusion was
identified in the present case, we were unable to use TKIs for
treatment. However, we hypothesize that targeted therapies against
MET should be considered as future treatment strategies for
sarcomatoid ChRCC, although such approaches need to be fully
explored in clinical trials. The heterogeneity of MET
alterations influences sensitivity to MET TKI and also serve
as a predictive biomarker for improving patient selection. A
previous study highlighted the pivotal role of the MET
pathway in RCC and provide a comprehensive review of clinical data
on multiple drugs targeting the MET pathway. Combination
strategies of MET TKI and immune checkpoint inhibitors could
lead to sustained and deep responses in RCC (19).
Another important finding in the present case was
the differential expression of PD-L1 between conventional ChRCC and
the sarcomatoid components. PD-L1 positivity in >10% of
sarcomatoid tumor cells suggests that immune checkpoint inhibitors
such as nivolumab may be potential therapeutic options (20). Aberrant PD-L1 expression is
associated with high-grade histology such as sarcomatoid
differentiation. A previous study has shown that sarcomatoid
components exhibit higher PD-L1 expression than non-sarcomatoid
components (21). The present case
is similar to those of previous studies (20,21).
In the present case, the ChRCC component was negative for PD-L1,
whereas the sarcomatoid comoponent showed ≥10% positivity. Aberrant
PD-L1 expression in the sarcomatoid component could indicate the
biological distinctiveness of sarcomatoid differentiation and could
have potential therapeutic implications. Because previous studies
suggest that higher PD-L1 expression is associated with a higher
histological grade in RCC, it is important to compare the immune
checkpoint marker such as PD-L1 between the classical RCC component
and the sarcomatoid component specifically (22). However, in the present study,
despite the use of nivolumab, the patient's tumor continued to
progress, leading to multiple organ metastases and death within 1
month. This rapid progression highlights the aggressive nature of
sarcomatoid ChRCC and suggests that while immune checkpoint
inhibitors may offer some benefits, they may not be sufficient as a
monotherapy, particularly in advanced or metastatic cases.
The current patient's poor response to combination
therapy with nivolumab and cabozantinib underscores the need for
further research on more effective treatment strategies for
sarcomatoid ChRCC. A previous review article reported that
combination therapy using cabozantinib and nivolumab can make
treatment more complex by causing an immunosuppressive state and
drug toxicity (23). This could be
possible explanation for the present patient's poor response to
combination therapy. Due to the molecular heterogeneity observed
between the conventional and sarcomatoid components, a more
personalized therapeutic approach targeting specific genetic
alterations and immune profiles may be necessary to improve
outcomes in patients with this aggressive variant of RCC. For
instance, the combination of immune checkpoint inhibitors with
targeted therapies against MET, TP53 and other genetic
alterations identified in sarcomatoid ChRCC should be explored in
future studies.
In conclusion, the present case highlights the
unique molecular and immunohistochemical characteristics of ChRCC
with sarcomatoid transformation. The distinct genetic differences
between the two components emphasize the complex molecular
landscape of this tumor and highlight the challenges in treating
sarcomatoid ChRCC. Further research is needed to better understand
the mechanisms driving sarcomatoid transformation and to develop
more effective and tailored therapeutic strategies for patients
with this rare and aggressive form of RCC.
Acknowledgements
Not applicable.
Funding
This paper was supported by the Fund of Biomedical Research
Institute, Jeonbuk National University Hospital.
Availability of data and materials
The NGS data generated in the present study may be
found in the Ion Reporter under the following URL: https://figshare.com/articles/dataset/4_S23_3641_A4_2_study_v1_b1dcd489-a2c3-4355-b6c7-42e61ad47e55_2023-04-04_21-51-44-090_All_zip/29002592?file=54388502.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
ARA and KMK conceptualized the study and wrote the
original manuscript. ARA and SJN searched the literature and
obtained case-related data. KMK and YBJ analyzed data and relevant
literature. KMK reviewed and edited the final draft. KMK and SJN
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
This case report was approved by Jeonbuk National
University Hospital Institutional Review Board (approval no. IRB
2023-07-004). This case report was conducted in accordance with the
Declaration of Helsinki of 1975.
Patient consent for publication
Patient consent was obtained for publication of
images and data.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ChRCC
|
chromophobe renal cell carcinoma
|
|
NGS
|
next-generation sequencing
|
|
CA
|
carbohydrate antigen
|
|
H&E
|
hematoxylin and eosin
|
|
KRT
|
keratin
|
|
FFPE
|
formalin-fixed paraffin-embedded
|
|
Indel
|
insertion-deletion
|
|
PD-L1
|
programmed cell death ligand 1
|
|
TKI
|
tyrosine kinase inhibitor
|
References
|
1
|
Moch H, Amin MB, Berney DM, Compérat EM,
Gill AJ, Hartmann A, Menon S, Raspollini MR, Rubin MA, Srigley JR,
et al: The 2022 World Health Organization classification of tumours
of the urinary system and male genital organs-part A: Renal,
penile, and testicular tumours. Eur Urol. 82:458–468. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lobo J, Ohashi R, Amin M-B, Berney DM,
Compérat EM, Cree IA, Gill AJ, Hartmann A, Menon S, Netto GJ, et
al: WHO 2022 landscape of papillary and chromophobe renal cell
carcinoma. Histopathology. 81:426–438. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shuch B, Bratslavsky G, Linehan WM and
Srinivasan R: Sarcomatoid renal cell carcinoma: A comprehensive
review of the biology and current treatment strategies. Oncologist.
17:46–54. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thoenes W, Störkel S and Rumpelt HJ: Human
chromophobe cell renal carcinoma. Virchows Arch B Cell Pathol Incl
Mol Pathol. 48:207–217. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bian L, Duan J, Wang X, Yang Y, Zhang X
and Xiao S: Sarcomatoid chromophobe renal cell carcinoma: A case
report and review of the literature. Am J Case Rep. 20:1225–1230.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brunelli M, Gobbo S, Cossu-Rocca P, Cheng
L, Hes O, Delahunt B, Pea M, Bonetti F, Mina MM, Ficarra V, et al:
Chromosomal gains in the sarcomatoid transformation of chromophobe
renal cell carcinoma. Mod Pathol. 20:303–309. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brunelli M, Eble JN, Zhang S, Martignoni
G, Delahunt B and Cheng L: Eosinophilic and classic chromophobe
renal cell carcinomas have similar frequent losses of multiple
chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this
pattern of genetic abnormality is not present in renal oncocytoma,
and this pattern of genetic abnormality is not present in renal
oncocytoma. Mod Pathol. 18:161–169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo R, Luo J, Chang J, Rekhtman N, Arcila
M and Drilon A: MET-dependent solid tumours-molecular diagnosis and
targeted therapy. Nat Rev Clin Oncol. 17:569–587. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rhoades Smith KE and Bilen MA: A review of
papillary renal cell carcinoma and MET inhibitors. Kidney Cancer.
3:151–161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Turpin A, Descarpentries C, Grégoire V,
Farchi O, Cortot AB and Jamme P: Response to capmatinib in a MET
fusion-positive cholangiocarcinoma. Oncologist. 28:80–83. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu XW, Chen XR, Rong YM, Lyu N, Xu CW,
Wang F, Sun WY, Fang SG, Yuan JP, Wang HJ, et al: MET exon 14
skipping mutation, amplification and overexpression in pulmonary
sarcomatoid carcinoma: A multi-center study. Tranl Oncol.
13:1008682020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Amendolare A, Marzano F, Petruzzella V,
Vacca RA, Guerrini L, Pesole G, Sbisà E and Tullo A: The
underestimated role of the p53 pathway in renal cancer. Cancers
(Basel). 14:57332022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bi M, Zhao S, Said JW, Merino MJ, Adeniran
AJ, Xie Z, Nawaf CB, Choi J, Belldegrun AS, Pantuck AJ, et al:
Genomic characterization of sarcomatoid transformation in clear
cell renal cell carcinoma. Proc Natl Acad Sci USA. 113:2170–2175.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carlsen L, Zhang S, Tian X, De La Cruz A,
George A, Arnoff TE and El-Deiry WS: The role of p53 in anti-tumor
immunity and response to immunotherapy. Front Mol Biosci.
1:11483892023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwiatkowski DJ, Choueiri TK, Fay AP, Rini
BI, Thorner AR, de Velasco G, Tyburczy ME, Hamieh L, Albiges L,
Agarwal N, et al: Mutations in TSC1, TSC2, and MTOR are associated
with response to rapalogs in patients with metastatic renal cell
carcinoma. Clin Cancer Res. 22:2445–2452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giraud JS, Bièche I, Pasmant É and
Tlemsani C: NF1 alterations in cancers: Therapeutic implications in
precision medicine. Expert Opin Investing Drugs. 32:941–957. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Recondo G, Che J, Jänne PA and Awad MM:
Targeting MET dysregulation in cancer. Cancer Discov. 10:922–934.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng W, Xu T, Yang L, Yan N, Yang J and
Fang S: Dramatic response to crizotinib through MET phosphorylation
inhibition in rare TFG-MET fusion advanced squamous cell lung
cancer. Oncologist. 30:oyae1662025. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nandagopal L, Sonpavde GP and Agarwal N:
Investigational MET inhibitors to treat renal cell carcinoma.
Expert Opin Investig Drugs. 28:851–860. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goodman AM, Piccioni D, Kato S, Boichard
A, Wang HY, Frampton G, Lippman SM, Connelly C, Fabrizio D, Miller
V, et al: Prevalence of PDL1 amplification and preliminary response
to immune checkpoint blockade in solid tumors. JAMA Oncol.
4:1237–1244. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thompson RH, Kuntz SM, Leibovich BC, Dong
H, Lohse CM, Webster WS, Sengupta S, Frank I, Parker AS, Zincke H,
et al: Tumor B7-H1 is associated with poor prognosis in renal cell
carcinoma patients with long-term follow-up. Cancer Res.
66:3381–3385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kawakami F, Sircar K, Rodriguez-Canales J,
Fellman BM, Urbauer DL, Tamboli P, Tannir NM, Jonasch E, Wistuba
II, Wood CG and Karam JA: Programmed cell death ligand 1 and
tumor-infiltrating lymphocyte status in patients with renal cell
carcinoma and sarcomatoid dedifferentiation. Cancer. 123:4823–4831.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McGregor B, Mortazavi A, Cordes L, Salabao
C, Vandlik S and Apolo AB: Management of adverse events associated
with cabozantinib plus nivolumab in renal cell carcinoma: A review.
Cancer Treat Rev. 103:1023332022. View Article : Google Scholar : PubMed/NCBI
|