Introduction
Sarcoma is frequently misdiagnosed in ~30% of cases
due to its challenging morphological features, which can cause
delays in receiving appropriate treatment (1). Liposarcoma (LPS) is one of the most
common types of soft-tissue sarcoma (STS), accounting for 15–20% of
all instances (2,3). LPS tumors are characterized by notable
clinical and pathological diversity, which complicate diagnosis and
treatment strategies.
The classification of LPS is based on
histopathological analysis, immunohistochemistry (IHC) and
molecular profiling, which categorizes them into four distinct
subtypes, namely, well-differentiated LPS (WDL), dedifferentiated
LPS (DDL), myxoid LPS (ML) and pleomorphic LPS (PL) (3). Each subtype has unique morphological
and molecular characteristics that affect disease progression and
treatment responsiveness.
Challenges in aligning morphological findings with
underlying molecular mechanisms persist despite advancements in
understanding LPS tumors. Key genetic changes, such as MDM2
proto-oncogene (MDM2) and cyclin-dependent kinase 4 (CDK4)
amplifications, have been noted in the well-differentiated and
dedifferentiated subtypes (4).
However, the role of genetic changes in disease variability and
treatment resistance is unclear. Additionally, the interactions of
molecular pathways in more aggressive subtypes, namely, myxoid and
pleomorphic, raise key questions that could improve prognostic
accuracy and treatment efficacy (3–5).
Systemic therapies that target specific molecular
pathways (MDM2, CDK4) demonstrate potential in overcoming the
limitations of traditional treatments (surgical), especially for
advanced and high-grade LPS (3–5). The
histopathological images included in the present review were
obtained from Dr Sardjito General Hospital (Yogyakarta, Indonesia).
The selected representative slides, collected between 2021 and
2024, were chosen due to the rarity of the cases and their
relevance to the discussion. The histopathological images were used
solely for educational and illustrative purposes, without the
inclusion of any identifiable patient data. The present review aims
to provide a detailed overview of the morphological and molecular
features of LPS, emphasizing how these factors may contribute to
improved diagnostic precision, prognostic assessments, innovative
therapeutic strategies and novel approaches for molecular targeting
in the future.
WDL
WDL is the most common LPS subtype, accounting for
31–33% of all liposarcomas (1,5). WDL
is typically found in the deep soft tissues of the limbs,
especially the thigh and retroperitoneum, but can also appear in
the chest wall, head and neck (5).
WDL grows gradually but is often asymptomatic. However,
retroperitoneal tumors may cause symptoms such as abdominal pain
and bloating due to organ compression. Larger tumors may appear as
bulges or swelling. WDL develops slowly and has a low risk of
metastasis, but local recurrence is common, complicating complete
surgical resection (1,5). WDL primarily affects adults aged 50–60
years (5). Imaging studies (MRI or
CT) demonstrate WDL as a fatty mass with thickened septa or nodular
elements that could indicate malignancy. The diagnosis involves
imaging, biopsy and molecular testing for MDM2 or CDK4
amplification (3).
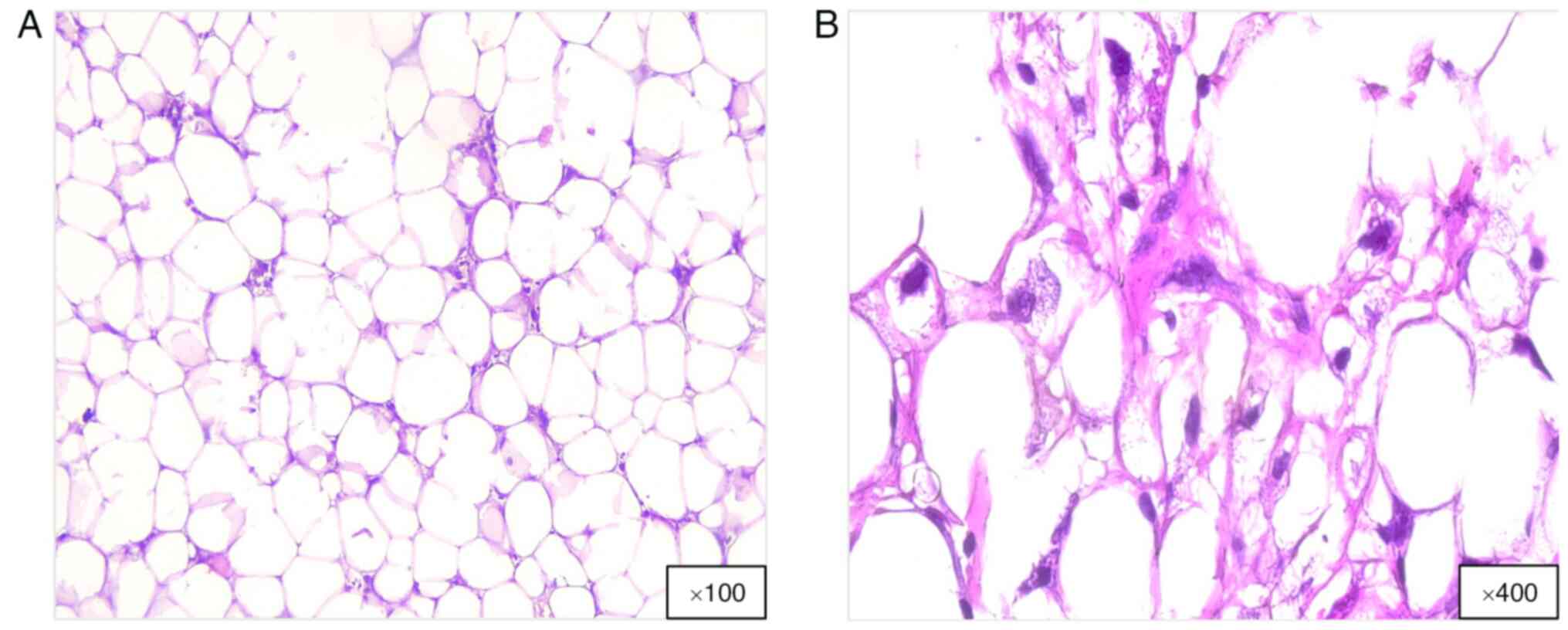
Histologically, WDL consists of mature adipocytes,
atypical stromal cells and a few lipoblasts (Fig. 1). Moreover, MDM2 and CDK4 oncogenes,
which facilitate the differentiation of WDL from benign lipomas
through IHC or molecular testing, are often upregulated (4,6). WDL
mainly arises from genetic alterations, especially amplification of
the 12q13-15 region. This abnormality causes supernumerary rings or
giant rod chromosomes, which are characteristic of WDL (4,6).
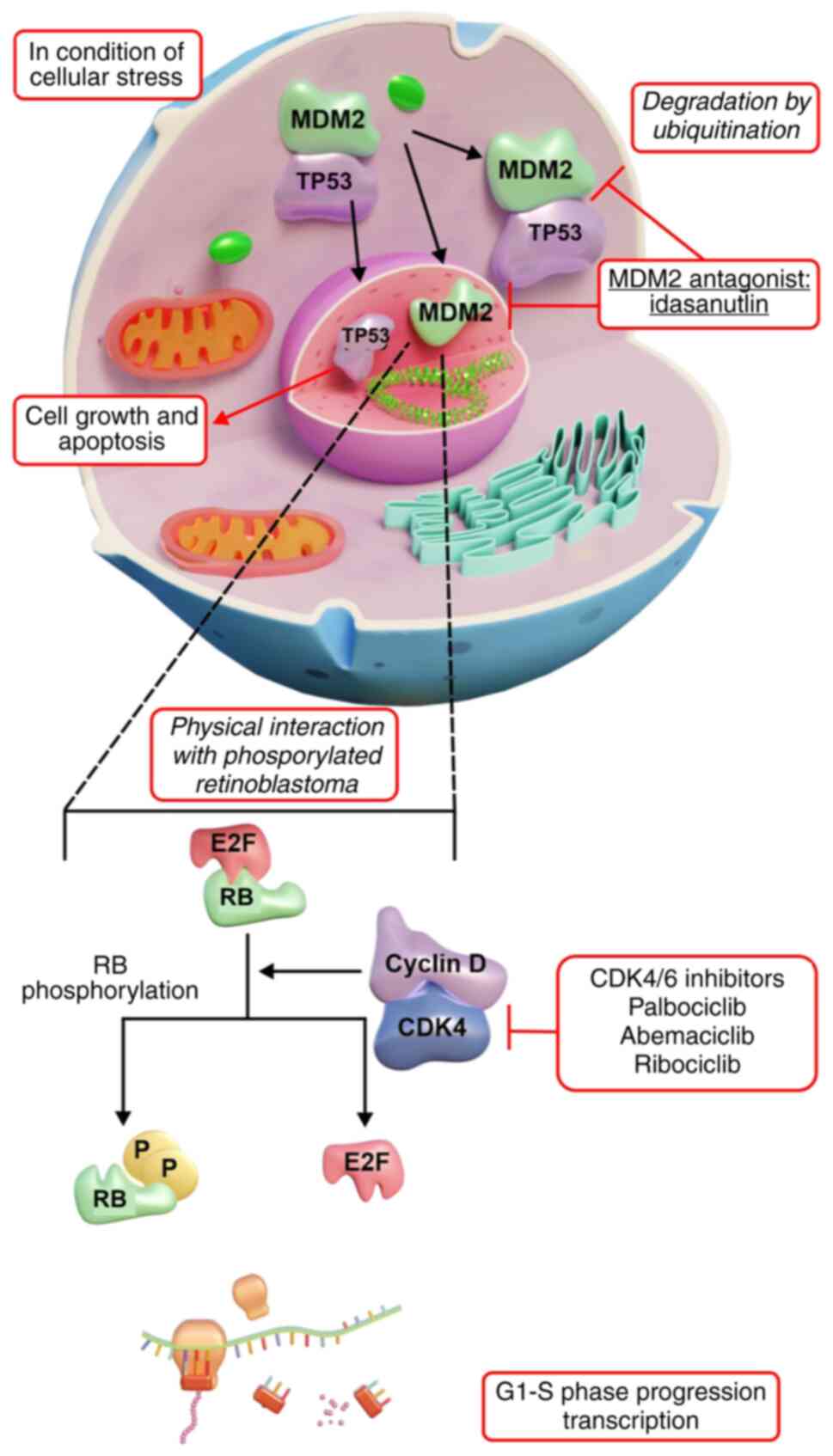
Amplified genes, such as MDM2 and CDK4, are key in WDL and DDL.
CDK4 activates D-type cyclins for hyperphosphorylation of
retinoblastoma protein (RB) (3,5,7).
Phosphorylated RB does not suppress E2F transcription factor 1,
which is a key transcription factor for the G1 to S
phase transition, resulting in unregulated cell proliferation. CDK4
is amplified in ≤90% of LPS cases, causing persistent RB
inactivation and tumor cells to bypass the G1/S
checkpoint, facilitating tumor growth (7).
In most WDL and DDL cases, the MDM2-p53 pathway is
altered due to MDM2 gene amplification (3,5,8). MDM2,
an E3 ubiquitin-protein ligase, inhibits p53, which is a key tumor
suppressor. MDM2 typically promotes p53 degradation, keeping its
levels low. However, stress phosphorylates p53, stabilizing it to
regulate genomic stability and apoptosis (3,5,8). In
WDL, increased MDM2 expression levels cause rapid p53 degradation,
neutralizing its tumor-suppressive effects and promoting
uncontrolled growth. This dysregulation emphasizes the malignant
potential of WDL and identifies CDK4 and MDM2 as vital therapeutic
targets (8).
The main treatment for WDL is surgery aimed at
complete excision with clear margins (9). Recurrence rates depend on the tumor
size and location, with retroperitoneal tumors having a higher
recurrence compares with the limbs, retroperitoneum, paratesticular
region, mediastinum and head and neck region (8,9).
Radiation and chemotherapy are usually ineffective for WDL
(9). The prognosis for extremity
WDL is generally favorable with 74% overall survival rates;
however, retroperitoneal WDL has a greater risk of progressing to
more aggressive DDL (9).
WDL often exhibits an increase in the MDM2 oncogene,
which inhibits p53, positioning MDM2 as a notable therapeutic
target. For instance, idasanutlin, a small-molecule MDM2
antagonist, has demonstrated potential in preclinical and early
clinical studies (3,8,9).
Furthermore, CDK4 expression, often elevated in WDL, is key in
cell-cycle regulation. Currently, palbociclib, a CDK4/6 inhibitor,
is under study for its efficacy in tumor management (3,8,9).
Therapeutic strategies that target and disrupt the PI3K/AKT/mTOR
pathway have proven to be effective in LPS. Everolimus, an mTOR
inhibitor, has been explored for tumor growth inhibition (10). Anti-angiogenic agents have been
investigated in LPS due to a dependence on blood supply. Pazopanib,
a multitarget tyrosine kinase inhibitor, blocks VEGFR and related
pathways. Immune checkpoint inhibitors (ICIs) such as pembrolizumab
[anti-programmed cell death protein 1 (PD-1)] were evaluated;
however, their efficacy in WDL remains to be elucidated (5). Previous studies have explored
epigenetic modulation with therapeutic agents such as histone
deacetylase (HDAC) inhibitors (8).
Vorinostat has demonstrated potential in the treatment of other
sarcoma subtypes, such as synovial sarcoma and chondrosarcoma, it
is undergoing clinical investigations for its efficacy in LPS
(3,5,9).
LPS presents treatment challenges due to the varying
immune microenvironments in each subtype, which affects prognosis
and therapy response. DDL exhibits higher tumor-infiltrating
lymphocyte (TIL) levels compared with WDL and ML, which is
associated with improved overall survival (OS) and progression-free
survival (PFS), emphasizing the potential of immunotherapy
(11,12). DDL is key to sarcoma immune classes
C, D and E, with sarcoma immune class E demonstrating the highest
immune reactivity characterized by tertiary lymphoid structures
(TLSs), which contain B and CD8+ T cells that enhance
antitumor immunity. Nonetheless, the tumor microenvironment of DDL
remains immunosuppressive due to M2-like macrophages that impede
antitumor responses. Conversely, ML contains fewer TILs compared
with DDL, which makes ML less suitable for immunotherapy (11). Immunotherapy notably improves LPS,
particularly in immune-active subtypes such as DDL. PD-1 and
programmed death-ligand 1 (PD-L1) expression levels, which regulate
immune evasion, vary across LPS subtypes and influence responses to
ICIs. Previous studies have indicated that 100% of DDL cases have
PD-1-positive lymphocyte infiltration, whereas only 10% of ML cases
have PD-1-positive lymphocyte infiltration, demonstrating the
differing immune checkpoint activity in LPS (11,13).
Additionally, the presence of TLSs in retroperitoneal LPS is
associated with greater immune infiltration, indicating that tumors
rich in TLSs may demonstrate an improved response to ICIs. Current
clinical trials on ICIs, such as anti-PD1 antibody pembrolizumab
(NCT02301039) and anti-PD-L1 antibody atezolizumab (NCT03474094),
have yielded mixed outcomes across STS subtypes, especially in WDL
(11). Further research is
warranted to enhance immunotherapies, identify biomarkers and
optimize patient selection for improved treatment outcomes in
patients with WDL (11).
WDL has a 5-year OS rate of 75–98%, depending on the
tumor location, with extremity tumors exhibiting the highest OS
(14). Treatment of WDL with more
aggressive methods, such as surgery and radiotherapy (RT), resulted
in lower local recurrence rates compared with surgery alone,
although it did not markedly alter disease-specific survival (DSS)
(15). The DSS for patients with
WDL post-surgery is ~98.3% (16).
In a previous study, treatment of WDL with radiation alone
demonstrated a higher mortality rate compared with WDL treated with
surgery alone, surgery combined with radiation or no treatment at
all (17).
DDL
DDL often presents as a growing, initially painless
mass (4). As it enlarges, it can
cause discomfort by pressing on nearby structures. DDL typically
originates from a well-differentiated subtype (4). Depending on the location of the tumor,
symptoms include abdominal pain, bloating, bowel obstruction and
urinary retention. DDL is typically diagnosed at a larger size
owing to deep anatomical positioning (4,18). In
the limbs, DDL may appear as a solid mass causing functional issues
or localized pain. In trunk soft tissues, DDL can present as a
palpable, non-tender mass (18).
DDL exhibits rapid growth and higher recurrence rate compared with
WDL, and can spread to the lungs or liver. Advanced stages may
cause pain, weight loss and functional impairments. Furthermore,
DDL commonly affects adults aged 50–70 years and is slightly more
prevalent in men (18). Several
cases of DDL have arisen from dedifferentiating previously
diagnosed or undiagnosed WDL (4,18). CT
or MRI indicate a heterogeneous mass with non-lipomatous areas, and
calcifications or necrotic zones may indicate dedifferentiation
(4,18).
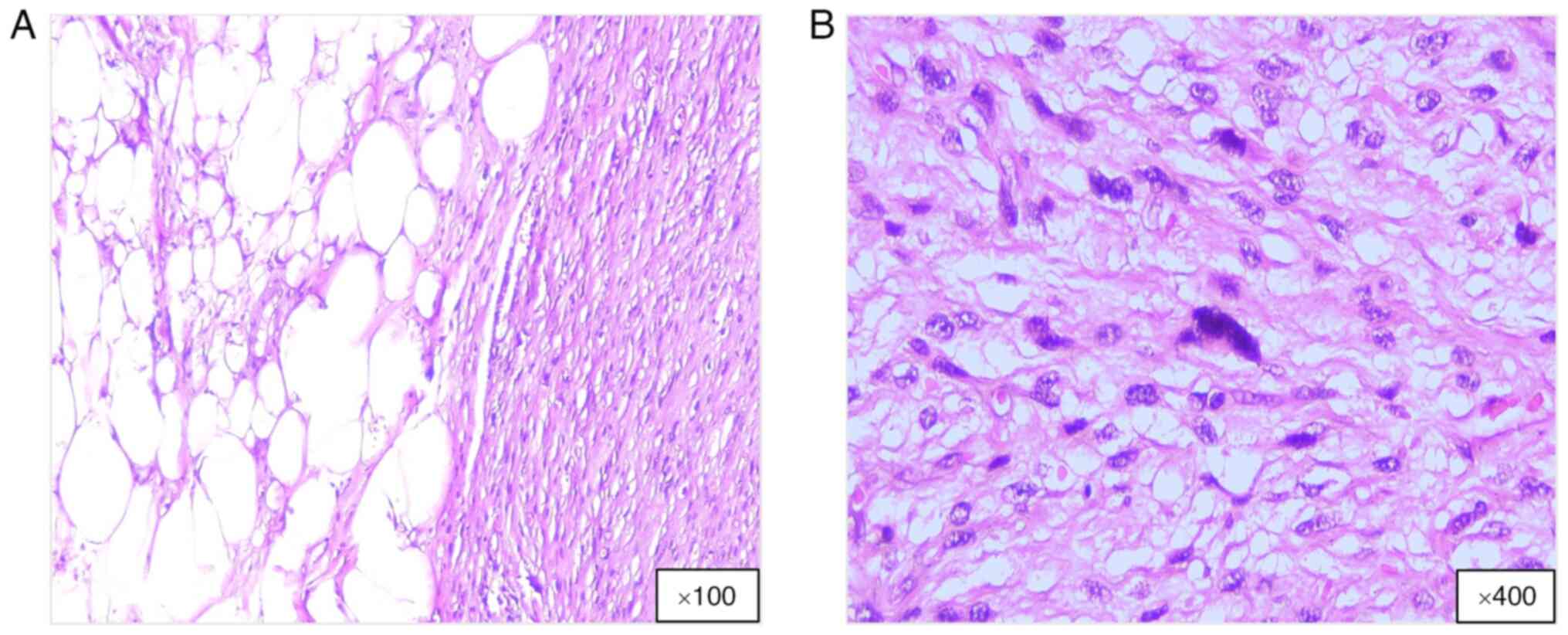
DDL has unique histopathological characteristics
that mark its evolution from a WDL; it includes differentiated
adipose tissue combined with fibrous elements, and dedifferentiated
areas may appear grayish-white and denser compared with fatty
regions. Larger tumors may exhibit necrosis or hemorrhage, whereas
tumors in the retroperitoneum often appear as large, multinodular
masses (3,4). Microscopic analysis demonstrates a
biphasic structure: A well-differentiated component with mature
adipocytes and atypical stromal cells with hyperchromatic nuclei,
and a dedifferentiated portion containing high-grade sarcomatous
segments with spindle cells, hyperchromasia, mitotic figures and
pleomorphism, resembling undifferentiated pleomorphic sarcomas or
other high-grade sarcomas, namely, fibrosarcoma or myxofibrosarcoma
(Fig. 2). In the dedifferentiated
area, atypical spindle cells are arranged in a fascicular pattern
with few lipoblasts. DDLs exhibit fibrous and myxoid traits and may
contain fibrous, myxoid or cartilaginous materials, with rare cases
demonstrating heterologous differentiation, including osseous or
rhabdomyoblastic changes (4,18).
IHC is key for the identification of diagnostic
markers. MDM2 amplification and upregulation can be detected by IHC
or fluorescence in situ hybridization (FISH). Moreover, CDK4
exhibits amplification and upregulation, which is beneficial in
distinguishing DDL from other sarcomas. Other markers, such as
protein S100, indicate adipocytic differentiation under certain
conditions (4). Previous genetic
studies have reported amplification in the 12q13-15 region,
particularly MDM2 and CDK4 oncogenes (3,5,8).
Prognostic features demonstrate that increased mitotic activity in
dedifferentiated areas is associated with aggressive behavior and
worse outcomes. Necrosis often manifests a higher tumor grade and
metastasis risk (4). Previous
molecular studies have indicated that ~90% of WDL/DDL cases have
MDM2 and CDK4 amplifications as primary oncogenic drivers (3,4).
However, DDL has a more aggressive molecular profile compared with
WDL, which is attributed to greater genomic complexity and
instability (19). Furthermore, WDL
is primarily defined by 12q13-15 region amplification, whereas DDL
displays additional chromosomal aberrations, gene fusions and
rearrangements, which contribute to the increased malignancy of DDL
(19). A key feature that
distinguishes DDL from WDL is its extensive genomic instability,
which includes 11q23 loss, 6q23 or 1q32 amplification and gene
fusions such as C15orf7::CBX3, CTDSP1::DNM3OS and CTDSP2::DNM3OS.
By contrast, MDM2-CDK4 amplification remains as the primary
oncogenic alteration of WDL (20).
DDL is defined by upregulated pathways for cell
proliferation and survival, particularly PI3K/AKT/mTOR and DNA
damage response, which are more pronounced in DDL compared with
those in WDL (21). The
PI3K/AKT/mTOR pathway in DDL promotes tumor growth and apoptosis
resistance. Conversely, WDL relies on MDM2 and CDK4 for cell cycle
dysregulation, which causes RB hyperphosphorylation and p53
suppression (Fig. 3). Additionally,
DDL demonstrates more disruptions in cell-cycle regulation,
especially at the G2/M checkpoint and E2F target genes,
which lead to faster tumor cell proliferation (21). Aurora A kinase is upregulated in
retroperitoneal DDL and markedly contributes to metastasis and
recurrence, which is less common in WDL (22). A notable difference is the higher
somatic copy number alterations (SCNAs) in DDL. A previous study
has linked specific SCNA clusters, such as 12q15 amplification, to
a poor prognosis and reduced PFS (23). SCNAs can appear in WDL; however,
they lack the same prognostic significance as in DDL (23). Therefore, although both WDL and DDL
exhibit MDM2 and CDK4 amplification, DDL has greater genomic
complexity, additional activated oncogenic pathways and enhanced
proliferation, which marks DDL as a more aggressive LPS subtype.
Understanding these molecular distinctions is key in the
development of targeted therapies to potentially improve treatment
outcomes and the survival of patients in the future.
The treatment of DDL focuses on local tumor control,
the prevention of recurrence and the management of systemic
disease. The aggressive nature and high recurrence rate of DDL
presents notable treatment challenges. Surgical treatments involve
wide local excision for complete resection and negative margins
(5). However, the achievement of
clear margins in retroperitoneal DDL is difficult due to the
proximity of vital organs, which increases the recurrence risk.
Complex cases may require a multidisciplinary approach. Recurrence
often needs repeat surgeries, especially for retroperitoneal
tumors. Radiation therapy can serve as an adjuvant or neoadjuvant
treatment, which potentially reduces tumor size and improves
resectability (5,24). Although effective for local control,
radiation has a limited impact on distant metastases (5). Systemic therapies, including
chemotherapy, have limited effectiveness against DDL, with standard
drugs, such as doxorubicin and ifosfamide, typically used for
unresectable or metastatic cases. Novel agents, including
trabectedin and eribulin, demonstrate potential in the treatment of
advanced stages (5,24). Targeted therapies, for example, MDM2
inhibitors (idasanutlin), are being clinically evaluated (8). CDK4 inhibitors (for example,
palbociclib) may reduce tumor growth in MDM2/CDK4-amplified tumors.
Studies of alternative pathways continue for effective targeted
treatments (5,24). Palliative care aims to enhance
quality of life through pain management and support, whereas
radiation therapy alleviates symptoms from mass effect metastases
(5,9,24).
DDL exhibits a 48–51.5% 5-year OS rate, which has
been linked to tumor location and the quality of the treatment
facility. Local recurrence occurs in 62.4% of cases. Surgical
resection is the standard therapy, with complete resection
preferred over marginal resection. Radiation is advisable for
high-grade DDL in the extremity for improved local control, and
chemotherapy is an option for DDL with a high recurrence risk
(16,25–27).
ML
ML is a distinct subtype of LPS that represents
20–30% of all LPS cases and is characterized by its unique clinical
behavior, histopathological features and molecular profile. ML is a
slow-growing, painless mass that predominantly affects adults aged
30–60 years, with the most common site being the deep soft tissue
of the extremities (28,29). ML is classified into two types based
on the 5% cut-off of the round cell component, namely, low-grade ML
(also called pure ML) and high-grade ML (30,31).
Despite being radiosensitive and occasionally recurrent locally, ML
tends to metastasize to unusual sites, including the soft tissues
and bones (32,33). ML diagnosis and staging are
challenging owing to the broad spectrum of clinical presentations.
Staging of ML is usually performed by whole-body MRI and chest CT
(3).
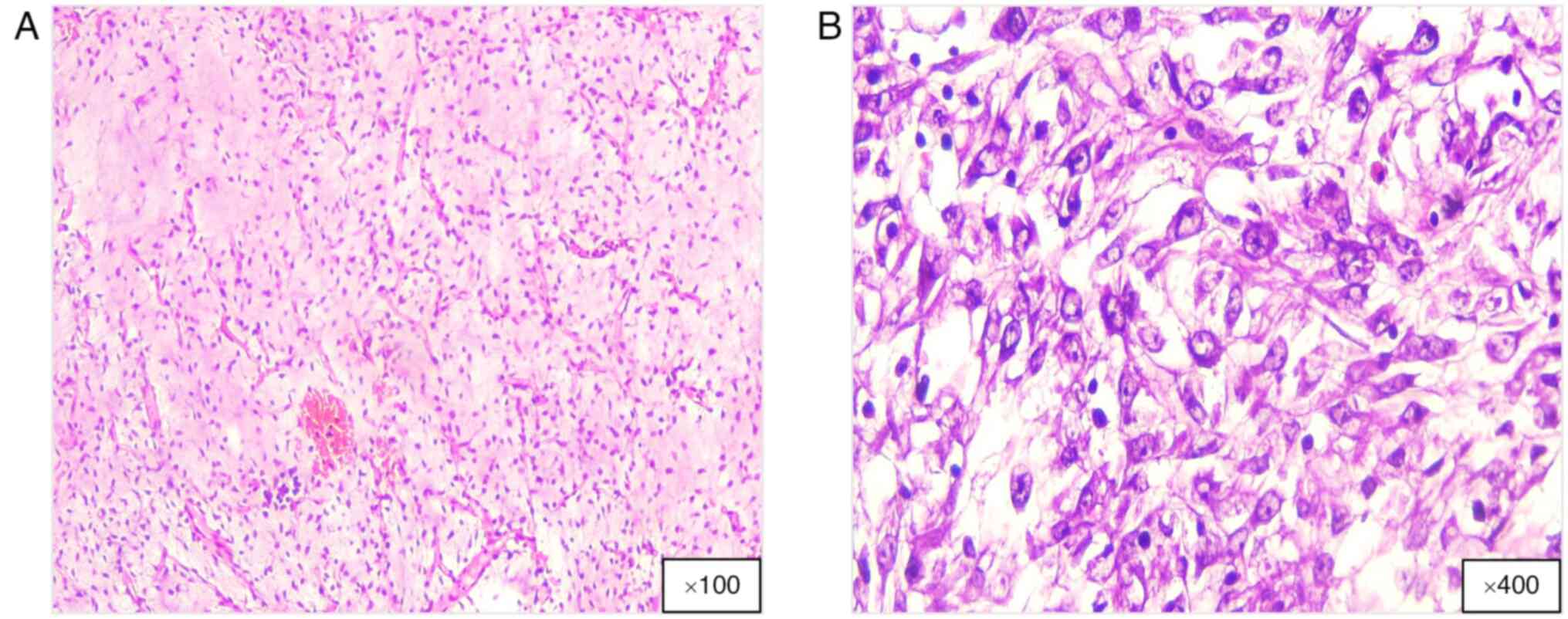
Histologically, ML exhibits a distinctive feature
from WDL and DDL, and is marked by a unique gelatinous myxoid
stroma interspersed with lipoblasts and a prominent capillary
network (Fig. 4). High-grade ML
frequently exhibits necrosis, hemorrhage and elevated mitotic
activity, which are associated with a worse prognosis (3,34).
Additionally, IHC serves a key role in the diagnosis of ML. DNA
damage-inducible transcript 3 protein (DDIT3) is a sensitive marker
for ML and is highly specific when diffusely present among tumor
cells (35). In a previous study,
the histological grade of ML was assessed using IHC with
phospho-histone H3 and was determined to be associated with ML
prognosis (36).
In most ML cases (>90%), a specific chromosomal
translocation, t(12;16) (q13;p11), characterizes them and results
in the formation of the FUS::DDIT3 fusion protein (37). In some cases, translocation
t(12;22)(q13;q12) may occur, leading to the EWSR1::DDIT3 fusion
protein. These protein fusions can be identified by FISH (38,39).
Moreover, their presence may lead to the upregulation of oncogenic
genes such as MET proto-oncogene, receptor tyrosine kinase (MET),
ret proto-oncogene (RET) and phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit α (PIK3CA) (40). Upregulation and/or activation
through RTKs, including MET, RET and VEGFRs, contribute to the high
activity level of the PI3K pathway in ML (Fig. 5). Furthermore, the FUS-DDIT fusion
is associated with the IGF-IR/PI3K/AKT and mTOR signaling pathways
(41–43). In addition, a telomerase reverse
transcriptase promoter mutation is commonly observed in most ML
cases, serving a key role in tumorigenesis (44,45).
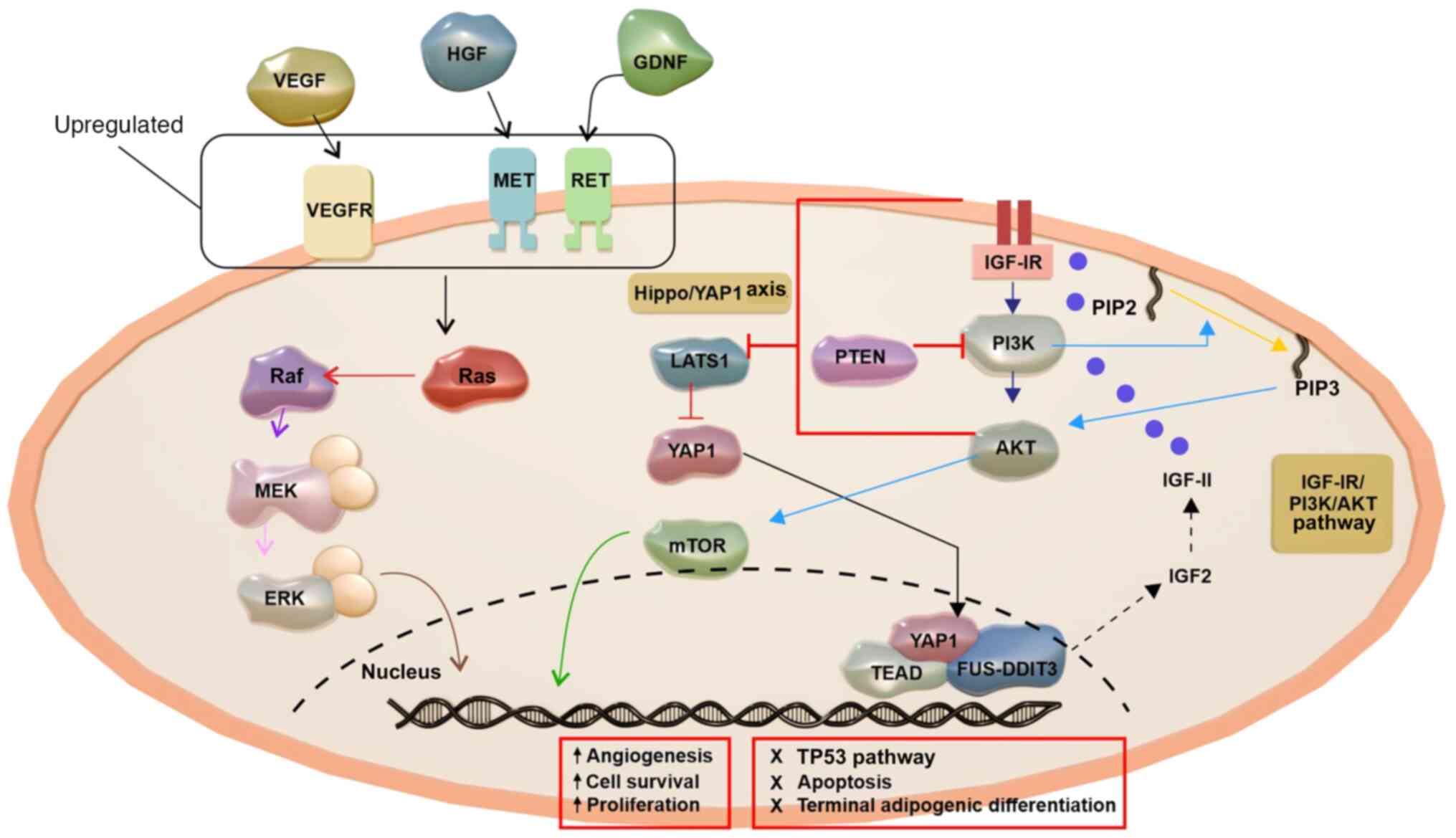 | Figure 5.The pathogenesis of ML. ML
pathogenesis involves the upregulation and activation of RTKs,
including MET, RET and VEGFRs, leading to enhanced PI3K pathway
activity. Growth factors that bind to RTKs, such as IGF-IR,
activate PI3K, transforming PIP2 into PIP3 and triggering AKT
signaling and phosphorylating downstream targets. PTEN inhibits
PI3K signaling by conversion of PIP3 back to PIP2. Moreover, RTKs
activate genes linked to angiogenesis, proliferation and survival
by Ras and the PI3K/AKT pathway. ML is associated with AKT
activation and PIK3CA and PTEN alterations. The oncogenic circuit
includes FUS-DDIT3, the IGF-IR/PI3K/AKT pathway and the Hippo/YAP1
axis. FUS-DDIT3 induces IGF-2 expression, forming an autocrine
IGF-II/IGF-IR signaling loop that activates the IGF-IR/PI3K/AKT
pathway. Signals from IGF-IR and PI3K inhibit Hippo kinase LATS1,
enabling nuclear accumulation of YAP1. FUS-DDIT3 interacts with
YAP1/TEAD in the nucleus, regulating oncogenic gene programs
involved in apoptosis, adipogenesis, the cell cycle and
proliferation. ML, myxoid liposarcoma; RTK, receptor tyrosine
kinase; MET, MET proto-oncogene receptor tyrosine kinase; RET, ret
proto-oncogene; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3,
phosphatidylinositol (3–5)-trisphosphate; PIK3CA,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α;
IGF, insulin-like growth factor; IGF-IR, IGF-1 receptor; FUS-DDIT3,
fused in liposarcoma-DNA damage-inducible transcript 3; YAP1,
Yes-associated protein 1; LATS1, large tumor suppressor kinase 1;
TEAD, transcriptional enhanced associate domain; HGF, hepatocyte
growth factor; GDNF, glial cell line-derived neurotrophic
factor. |
Current management approaches involve a combination
of surgical resection, adjuvant RT and chemotherapy. If the tumor
location is deep, preoperative RT should be considered before
surgical resection. Patients with metastatic ML should receive
neoadjuvant chemotherapy (46). ML
has a higher 5-year OS rate compared with DDL, ranging from 81 to
84% (15,47). ML is radiation-sensitive; thus,
surgical resection with radiation is the primary treatment
(48). Preoperative radiation is
recommended for deep or large tumors before surgical resection, and
chemotherapy is used for patients with a positive-margin resection
and metastatic ML (46). Treatment
with radiation alone is not recommended, as it has been associated
with a higher risk of mortality (17).
PL
PL is the rarest and most aggressive subtype of LPS,
accounting for <10% of cases and mainly affecting older adults
aged 50–70 years. Owing to its rarity in young adults, PL diagnosis
in this group should be differentiated from that of other LPSs
(49). PL is histologically
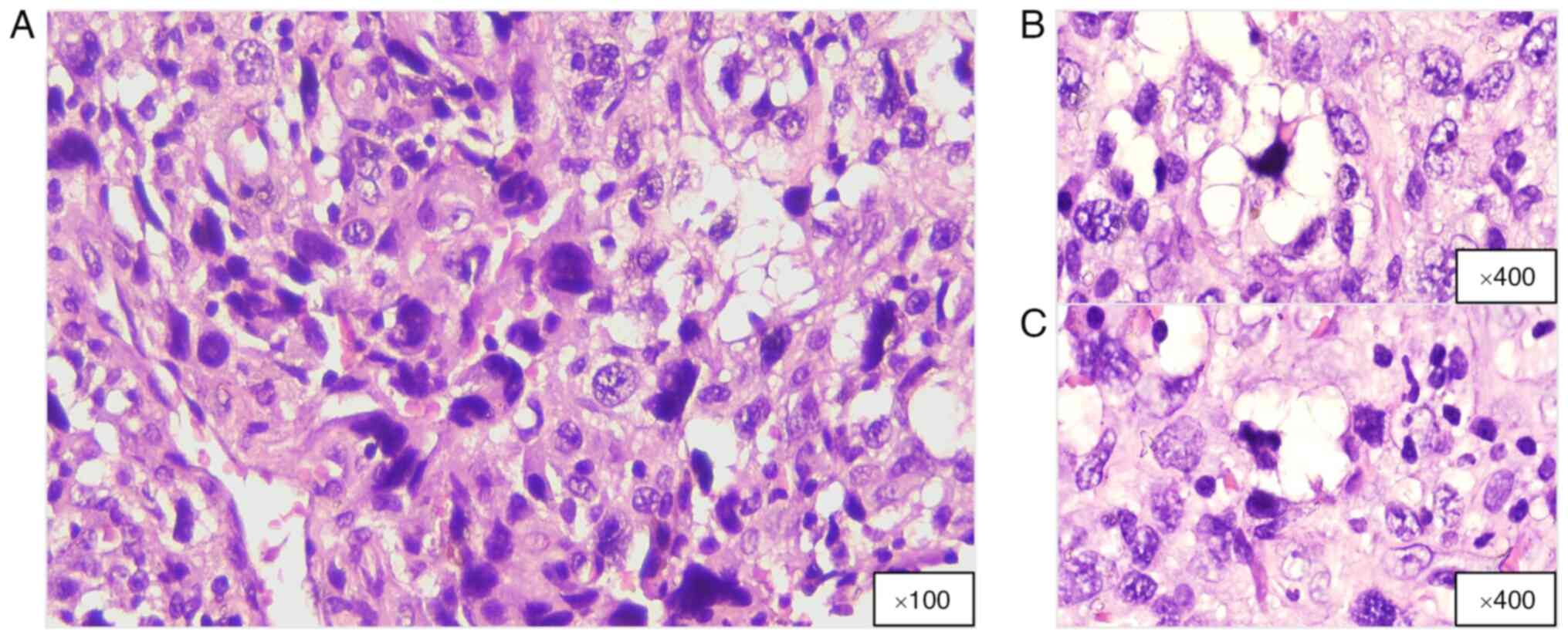
characterized by a pleomorphic cellular structure (Fig. 6), including bizarre, multinucleated
tumor cells and pleomorphic lipoblasts (50). Most patients with PL present with a
rapidly growing, painless mass, predominantly in the extremities
(28,51). Although PL frequently develops in
the deep soft tissues, it can also occur in the retroperitoneum and
in superficial tissues such as the subcutis or dermis (52,53).
PL pathogenesis involves complex genetic and molecular changes
notable for the lack of unifying molecular alterations, which is
frequently observed in STS with intricate karyotypes. Unlike other
LPS subtypes, such as WDL and DDL, PL does not exhibit MDM2 or CDK4
amplification. This genetic complexity indicates that distinct
dominant molecular abnormalities are unlikely to be the basis of PL
tumorigenesis and progression (54).
PL exhibits molecular profiles that align more
closely with other pleomorphic sarcomas than with atypical
lipomatous tumors, WDL, DDL or ML. Molecular investigations of PL
are hindered due to the rarity of PL. Genetic abnormalities in PL
include complex karyotypic alterations, with TP53 mutations
observed in 60% of cases and NF1 mutations in 5% of all PL cases
(55). Tumors typically exhibit
intricate patterns, demonstrating gains in regions 1p, 1q21-q32,
2q, 3p, 3q, 5p12-p15, 5q, 6p21, 7p and 7q22 (55). A previous study has reported losses
in the areas of 1q, 2q, 3p, 4q, 10q, 11q, 12p13, 13q14, 13q21-qter
and 13q23-24 (55). Other studies
have demonstrated that 60% of PL cases feature a deletion at
13q14.2-q14.3, which encompasses the tumor suppressor RB1 (55–57).
Amplified in PL, the mitotic arrest-deficient gene (MAD2) may serve
a notable role (57). A previous
study with a small sample size of PL cases reported a 13-fold
upregulation of MAD2 compared with normal fat samples (57). Additionally, deletions in PL,
specifically at 17p13 and 17q11.2, where the TP53 gene and
sarcoma-associated tumor suppressor neurofibromatosis type 1
reside, have been observed (56,57).
The diagnosis of PL requires a multidisciplinary
approach. MRI and CT are key for tumor staging. In PL, 60% of the
cases appear as well-defined masses with heterogeneous features,
which indicate necrosis and hemorrhage (51). Histopathological evaluation is the
gold standard, often enhanced by IHC to differentiate PL from other
high-grade sarcomas. The presence of spindle epitheloid pleomorphic
cells and pleomorphic lipoblasts are characteristic of PL. IHC has
limited value in diagnosing PL due to its non-specific
immunoprofile and variable positivity for SMA, desmin and CD34,
usually showing focal positivity. Other IHC markers, such as MDM2
and CDK4, are generally negative in PL, allowing differentiation
from other LPS subtypes (50,58–60).
Managing PL is challenging due to its aggressive
nature and high metastatic potential. Even with optimal surgical
management, local and distant recurrence risks remain elevated.
Preoperative RT may help reduce tumor size for improved surgical
margins. PL prognosis is poorer compared with that of other LPS
types, with stagnant survival rates of >20 years. Previously,
doxorubicin combined with ifosfamide demonstrated some efficacy and
neoadjuvant/adjuvant therapies were associated with improved
survival. Trabectedin and eribulin are therapeutic options for
advanced cases of PL. Ongoing efforts are key to developing new
treatments; however, the identification of targetable aberrations,
particularly the loss of p53 and RB pathway proteins, are difficult
to utilize therapeutically (61–65).
PL exhibits local recurrence and metastasis rates
that range from 30 to 50%. The overall 5-year survival rate is
~60%. Metastatic spread predominantly impacts the lungs and pleura.
Various factors, including central tumor location, increased depth,
larger size and a higher mitotic count, are associated with a less
favorable prognosis (51,66).
Table I summarizes
the genomic alterations in the LPS subtypes, detailing the
demographics, morphology, immunophenotype, growth rates,
recurrence, metastasis and therapy responses. WDL and DDL exhibit
MDM2 and CDK4 amplifications on chromosome 12q13-15 (3,5,8–10).
MDM2 inhibits p53, promoting unchecked cellular growth; thus,
inhibitors such as idasanutlin are undergoing investigations to
restore p53 function and induce apoptosis. CDK4 amplification
disrupts cell-cycle regulation, which makes CDK4/6 inhibitors, such
as palbociclib, a potential therapeutic agent for tumor control.
Resistance to chemotherapy and RT is one of the key challenges in
the treatment of WDL and DDL. Multitargeted tyrosine kinase
inhibitors, including pazopanib, demonstrate antitumor effects.
Current research on HDAC inhibitors, such as vorinostat, along with
ICIs, including pembrolizumab, has demonstrated potential in the
treatment of WDL (3,5,8–10). ML
features chromosomal translocations such as t(12;16) and t(12;22),
which result in FUS-DDIT 3 and EWSR1-DDIT 3 fusions (3,5,8). ML
responds well to chemotherapy, especially anthracycline regimens.
Novel immunotherapeutic strategies targeting cancer-testis antigens
(for example, New York esophageal squamous cell carcinoma 1)
demonstrate potential with vaccine-based approaches. Checkpoint
inhibitors, such as atezolizumab, are evaluated with other
treatments against ML tumors (3,5,8). PL is
the most aggressive LPS subtype, lacking MDM2 and CDK 4
amplifications, and characterized by high pleomorphic histology
(3,5,58,61).
Treatment options are limited. However, doxorubicin and ifosfamide
are effective, and eribulin and trabectedin are alternatives for
advanced disease. Molecular studies point to VEGFR-2 as a target,
prompting investigations into tyrosine kinase inhibitors such as
apatinib (3,5,58,61).
 | Table I.Summary of the genomic biomarkers of
LPS. |
Table I.
Summary of the genomic biomarkers of
LPS.
| LPS subtype | Age range,
years | Predilection | Morphological
features |
Immuno-phenotype | Molecular
markers/genomics alterations | Growth rate | Likelihood of
metastasis | Treatment | Recurrence | Therapy response
(radiotherapy and chemotherapy) | Targeted
therapy | (Refs.) |
|---|
| WDL | 50-60 | Extremities,
retroperitoneum | Mature adipocytes,
atypical stromal cells and a limited number of lipoblasts | MDM2(+), CDK(+),
DDIT3(−), S100(+), CD34(−). p16(+), p53 wild-type | MDM2 and CDK4,
12q13-15 amplification | Slow | Low | Surgical resection
with negative margins | Low | Poor | Idasanutlin, MDM2
antagonist; palbociclib, CDK4/6 inhibitor; everolimus, mTOR
inhibitor; pazopanib, multitarget tyrosine kinase inhibitor;
pembrolizumab (anti-PD-1, immune checkpoint inhibitors; vorinostat,
histone deacetylase inhibitors) | (3,5,8–10) |
| DDL | 50-70 | Extremities,
retroperitoneum | Biphasic structure
with i) WD component: Mature adipocytes intermingled with atypical
stromal cells and hyperchromatic nuclei; and ii) DD component:
Spindle cells, hyperchromasia, mitotic figures and
pleomorphism | MDM2(+), CDK4(+),
DDIT3(−), S100(+), CD34(−), p16(+), p53 wild-type or mutant | MDM2 and CDK4,
12q13-15 amplification and other chromosomal abnormalities | Rapid | High | Surgical resection,
radiotherapy and chemotherapy/targeted therapy | High | Poor | Idasanutlin, MDM2
antagonist; palbociclib, CDK4/6 inhibitor | (3,5,8,9) |
| ML | 30-60 | Thigh or other
proximal extremities | Unique gelatinous
myxoid stroma interspersed with lipoblasts and a prominent
capillary network; occasionally exhibits necrosis, hemorrhage and
elevated mitotic activity | MDM2(−), CDK4(−),
DDIT3(+), PHH3(+), S100(+), CD34(−), p16(−), p53 wild type | t(12;16) with
FUS-DDIT3 fusion t(12;22) with EWSR1-DDIT3 fusion | Slow | Low to medium,
depends on the degree of differentiation | Surgical resection,
radiotherapy and chemotherapy/targeted therapies | High | Commonly
sensitive | CMB305,
immuno-therapeutic for NY-ESO-1; atezolizumab, anti PD-L1 | (3,5,8) |
| PL | 50-70 | Lower and/or upper
limbs | Pleomorphic
cellular architecture, bizarre, multinucleated tumor cells and
pleomorphic lipoblasts | MDM2(−), CDK4(−),
DDIT3(−), S100(+), SMA(+), desmin(+), CD34(+), p16(+), p53 mutant
hyper-expression | No MDM2 or CDK4
amplification; complex karyotype and lack of specificity | Rapid | High | Surgical resection,
radiotherapy and chemotherapy | High | Poor | Apatinib, VEGFR-2
tyrosine kinase inhibitor | (3,5,58,61) |
Conclusion
Numerous studies have established the use of
molecular biomarkers in elucidating the characteristics of sarcoma
and predicting its prognosis. Specific genetic alterations, such as
MDM2 and CDK4, along with 12q13-15 amplification in WDL and DDL,
are associated with a poor therapeutic response. By contrast,
chromosomal translocations including t(12;16) with FUS-DDIT3 fusion
and t(12;22) with EWSR1-DDIT3 fusion in ML are linked to
sensitivity to therapy, although they confer higher recurrence
rates. Furthermore, the complex karyotype and lack of molecular
specificity in PL is associated with an unfavorable response to
treatment and increased recurrence. MDM2 and CDK4 amplification
serve a key role in diagnosis, prediction of tumor recurrence and
prognosis. A comprehensive diagnosis of sarcoma requires further
study into each molecular alteration and the current World Health
Organization classification (28).
Surgical intervention facilitates tumor removal and provides
samples for subsequent molecular analysis. Further research is
warranted to explore the integration of molecular biomarkers with
the WHO grading system to enhance treatment decision-making, which
may potentially lead to a more accurate diagnosis, prognosis and
therapeutic strategies for the treatment of sarcoma in the
future.
Acknowledgements
The authors would like to thank Dr Auliana Hayu
Kusumastuti and Dr Thea Saphira Mugiarto (Department of Anatomical
Pathology, Faculty of Medicine, Public Health and Nursing, Gadjah
Mada University, Yogyakarta, Indonesia) for their assistance in
managing the administrative tasks related to this study.
Funding
The present review was supported by the Academic Excellence
Grant (Program Peningkatan Academic Excellence 2024) provided by
the Universitas Gadjah Mada (grant no.
6243/UN1/FKKMK/PPKE/PT/2024).
Availability of data and materials
Not applicable.
Authors' contributions
ED conceptualized the article and wrote the first
draft of the manuscript. RM, YP, SA, RB and AS collected materials
(literature). RB and AS prepared the table and figures. IW
contributed with pathological expertise. ED, RM, YP, SA, RB, AS and
IW made significant contributions to the critical review, editing,
revision and final decision to submit the manuscript for
publication. All authors read and approved the final manuscript.
Data authentication is not applicable.
Ethics approval and consent to
participate
The present review received ethical approval from
the Medical and Health Research Ethics Committee of the Faculty of
Medicine, Public Health and Nursing, Gadjah Mada University
(Yogyakarta, Indonesia; approval no. KE/FK/1510/EC/2024).
Patient consent for publication
The requirement to obtain informed consent for the
publication of histopathological images was waived by the Ethics
Committee of the Faculty of Medicine, Public Health and Nursing,
Gadjah Mada University (Yogyakarta, Indonesia) in accordance with
institutional guidelines for the use of anonymized, retrospective
data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jonczak E, Grossman J, Alessandrino F,
Seldon Taswell C, Velez-Torres JM and Trent J: Liposarcoma: A
journey into a rare tumor's epidemiology, diagnosis,
pathophysiology, and limitations of current therapies. Cancers
(Basel). 16:38582024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ducimetière F, Lurkin A, Ranchère-Vince D,
Decouvelaere AV, Péoc'h M, Istier L, Chalabreysse P, Muller C,
Alberti L, Bringuier PP, et al: Incidence of sarcoma histotypes and
molecular subtypes in a prospective epidemiological study with
central pathology review and molecular testing. PLoS One.
6:e202942011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee ATJ, Thway K, Huang PH and Jones RL:
Clinical and molecular spectrum of liposarcoma. J Clin Oncol.
36:151–159. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fletcher C, Unni K and Mertens F: World
Health Organization Classification of Tumours. Pathology and
genetics of tumours of soft tissue and bone. 3rd edition. IARC
Press; 2002
|
|
5
|
Zhou XP, Xing JP, Sun LB, Tian SQ, Luo R,
Liu WH, Song XY and Gao SH: Molecular characteristics and systemic
treatment options of liposarcoma: A systematic review. Biomed
Pharmacother. 178:1172042024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yee EJ, Stewart CL, Clay MR and McCarter
MM: Lipoma and its doppelganger: The atypical lipomatous
tumor/well-differentiated liposarcoma. Surg Clin North Am.
102:637–656. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu J, Wood D, Ingley E, Koks S and Wong D:
Update on genomic and molecular landscapes of well-differentiated
liposarcoma and dedifferentiated liposarcoma. Mol Biol Rep.
48:3637–3647. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Somaiah N and Tap W: MDM2-p53 in
liposarcoma: The need for targeted therapies with novel mechanisms
of action. Cancer Treat Rev. 122:1026682024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thway K: Well-differentiated liposarcoma
and dedifferentiated liposarcoma: An updated review. Semin Diagn
Pathol. 36:112–121. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bouyahya A, El Allam A, Aboulaghras S,
Bakrim S, El Menyiy N, Alshahrani MM, Al Awadh AA, Benali T, Lee
LH, El Omari N, et al: Targeting mTOR as a cancer therapy: Recent
advances in natural bioactive compounds and immunotherapy. Cancers
(Basel). 14:55202022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Z, Lu L, Gao Z, Zhou Q, Wang Z, Sun L
and Zhou Y: Immunotherapy for liposarcoma: Emerging opportunities
and challenges. Future Oncol. 18:3449–3461. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dancsok AR, Setsu N, Gao D, Blay JY,
Thomas D, Maki RG, Nielsen TO and Demicco EG: Expression of
lymphocyte immunoregulatory biomarkers in bone and soft-tissue
sarcomas. Mod Pathol. 32:1772–1785. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S,
Kim KM, Park HS, Lee H, Moon WS, Chung MJ, et al: Tumor
infiltrating PD1-positive lymphocytes and the expression of PD-L1
predict poor prognosis of soft tissue sarcomas. PLoS One.
8:e828702013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smith CA, Martinez SR, Tseng WH, Tamurian
RM, Bold RJ, Borys D and Canter RJ: Predicting survival for
well-differentiated liposarcoma: The importance of tumor location.
J Surg Res. 175:12–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vos M, Koseła-Paterczyk H, Rutkowski P,
van Leenders GJLH, Normantowicz M, Lecyk A, Sleijfer S, Verhoef C
and Grünhagen DJ: Differences in recurrence and survival of
extremity liposarcoma subtypes. Eur J Surg Oncol. 44:1391–1397.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vos M, Grünhagen DJ, Koseła-Paterczyk H,
Rutkowski P, Sleijfer S and Verhoef C: Natural history of
well-differentiated liposarcoma of the extremity compared to
patients treated with surgery. Surg Oncol. 29:84–89. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amer KM, Congiusta DV, Thomson JE, Elsamna
S, Chaudhry I, Bozzo A, Amer R, Siracuse B, Ghert M and Beebe KS:
Epidemiology and survival of liposarcoma and its subtypes: A dual
database analysis. J Clin Orthop Trauma. 11 (Suppl 4):S479–S484.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nascimento AG: Dedifferentiated
liposarcoma. Semin Diagn Pathol. 18:263–266. 2001.PubMed/NCBI
|
|
19
|
Haddox CL and Riedel RF: Recent advances
in the understanding and management of liposarcoma. Fac Rev.
10:12021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mantilla JG, Ricciotti RW, Chen EY, Liu YJ
and Hoch BL: Amplification of DNA damage-inducible transcript 3
(DDIT3) is associated with myxoid liposarcoma-like morphology and
homologous lipoblastic differentiation in dedifferentiated
liposarcoma. Mod Pathol. 32:585–592. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Wang X, Liu L, Yan B, Qiu F and
Zhou B: Targeting liposarcoma: Unveiling molecular pathways and
therapeutic opportunities. Front Oncol. 14:14840272024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Casadei L, de Faria FCC, Lopez-Aguiar A,
Pollock RE and Grignol V: Targetable pathways in the treatment of
retroperitoneal liposarcoma. Cancers (Basel). 14:13622022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hirata M, Asano N, Katayama K, Yoshida A,
Tsuda Y, Sekimizu M, Mitani S, Kobayashi E, Komiyama M, Fujimoto H,
et al: Integrated exome and RNA sequencing of dedifferentiated
liposarcoma. Nat Commun. 10:56832019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gahvari Z and Parkes A: Dedifferentiated
liposarcoma: Systemic therapy options. Curr Treat Options Oncol.
21:152020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haddox CL, Hornick JL, Roland CL, Baldini
EH, Keedy VL and Riedel RF: Diagnosis and management of
dedifferentiated liposarcoma: A multidisciplinary position
statement. Cancer Treat Rev. 131:1028462024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao J, Du W, Tao X, Li A, Li Y and Zhang
S: Survival and prognostic factors among different types of
liposarcomas based on SEER database. Sci Rep. 15:17902025.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gootee JM, Curtin CE, Aurit SJ, Randhawa
SE, Kang BY and Silberstein PT: Treatment facility: An important
prognostic factor for dedifferentiated liposarcoma survival. Fed
Pract. 36 (Suppl 5):S34–S41. 2019.PubMed/NCBI
|
|
28
|
Fletcher C, Bridge J, Hogendoorn P and
Mertens F: WHO Classification of Tumours of Soft Tissue and Bone.
4th edition. IARC Press; 2013
|
|
29
|
Yang L, Chen S, Luo P, Yan W and Wang C:
Liposarcoma: Advances in cellular and molecular genetics
alterations and corresponding clinical treatment. J Cancer.
11:100–107. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moreau LC, Turcotte R, Ferguson P, Wunder
J, Clarkson P, Masri B, Isler M, Dion N, Werier J, Ghert M, et al:
Myxoid\round cell liposarcoma (MRCLS) revisited: An analysis of 418
primarily managed cases. Ann Surg Oncol. 19:1081–1088. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Haniball J, Sumathi VP, Kindblom LG, Abudu
A, Carter SR, Tillman RM, Jeys L, Spooner D, Peake D and Grimer RJ:
Prognostic factors and metastatic patterns in primary
myxoid/round-cell liposarcoma. Sarcoma. 2011:5380852011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chung PWM, Deheshi BM, Ferguson PC, Wunder
JS, Griffin AM, Catton CN, Bell RS, White LM, Kandel RA and
O'Sullivan B: Radiosensitivity translates into excellent local
control in extremity myxoid liposarcoma: A comparison with other
soft tissue sarcomas. Cancer. 115:3254–3261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ho TP: Myxoid liposarcoma: How to stage
and follow. Curr Treat Options Oncol. 24:292–299. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fiore M, Grosso F, Lo Vullo S,
Pennacchioli E, Stacchiotti S, Ferrari A, Collini P, Lozza L,
Mariani L, Casali PG and Gronchi A: Myxoid/round cell and
pleomorphic liposarcomas: Prognostic factors and survival in a
series of patients treated at a single institution. Cancer.
109:2522–2531. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scapa JV, Cloutier JM, Raghavan SS,
Peters-Schulze G, Varma S and Charville GW: DDIT3
immunohistochemistry is a useful tool for the diagnosis of myxoid
liposarcoma. Am J Surg Pathol. 45:230–239. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takazawa A, Yoshimura Y, Okamoto M, Tanaka
A, Kito M, Aoki K, Uehara T, Takahashi J, Kato H and Nakayama J:
The usefulness of immunohistochemistry for phosphohistone H3 as a
prognostic factor in myxoid liposarcoma. Sci Rep. 13:47332023.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Powers MP, Wang WL, Hernandez VS, Patel
KS, Lev DC, Lazar AJ and López-Terrada DH: Detection of myxoid
liposarcoma-associated FUS-DDIT3 rearrangement variants including a
newly identified breakpoint using an optimized RT-PCR assay. Mod
Pathol. 23:1307–1315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Crozat A, Åman P, Mandahl N and Ron D:
Fusion of CHOP to a novel RNA-binding protein in human myxoid
liposarcoma. Nature. 363:640–644. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dal Cin P, Sciot R, Panagopoulos I, Aman
P, Samson I, Mandahl N, Mitelman F, Van den Berghe H and Fletcher
CD: Additional evidence of a variant translocation t(12;22) with
EWS/CHOP fusion in myxoid liposarcoma: Clinicopathological
features. J Pathol. 182:437–441. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Negri T, Virdis E, Brich S, Bozzi F,
Tamborini E, Tarantino E, Jocollè G, Cassinelli G, Grosso F,
Sanfilippo R, et al: Functional mapping of receptor tyrosine
kinases in myxoid liposarcoma. Clin Cancer Res. 16:3581–3593. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Trautmann M, Cyra M, Isfort I, Jeiler B,
Krüger A, Grünewald I, Steinestel K, Altvater B, Rossig C, Hafner
S, et al: Phosphatidylinositol-3-kinase (PI3K)/Akt signaling is
functionally essential in myxoid liposarcoma. Mol Cancer Ther.
18:834–844. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Trautmann M, Menzel J, Bertling C, Cyra M,
Isfort I, Steinestel K, Elges S, Grünewald I, Altvater B, Rossig C,
et al: FUS-DDIT3 fusion protein-driven IGF-IR signaling is a
therapeutic target in myxoid liposarcoma. Clin Cancer Res.
23:6227–6238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Demicco EG, Torres KE, Ghadimi MP, Colombo
C, Bolshakov S, Hoffman A, Peng T, Bovée JV, Wang WL, Lev D and
Lazar AJ: Involvement of the PI3K/Akt pathway in myxoid/round cell
liposarcoma. Mod Pathol. 25:212–221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Koelsche C, Renner M, Hartmann W, Brandt
R, Lehner B, Waldburger N, Alldinger I, Schmitt T, Egerer G, Penzel
R, et al: TERT promoter hotspot mutations are recurrent in myxoid
liposarcomas but rare in other soft tissue sarcoma entities. J Exp
Clin Cancer Res. 33:332014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kunieda J, Yamashita K, Togashi Y, Baba S,
Sakata S, Inamura K, Ae K, Matsumoto S, Machinami R, Kitagawa M and
Takeuchi K: High prevalence of TERT aberrations in myxoid
liposarcoma: TERT reactivation may play a crucial role in
tumorigenesis. Cancer Sci. 113:1078–1089. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tfayli Y, Baydoun A, Naja AS and Saghieh
S: Management of myxoid liposarcoma of the extremity. Oncol Lett.
22:5962021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dürr HR, Rauh J, Baur-Melnyk A, Knösel T,
Lindner L, Roeder F, Jansson V and Klein A: Myxoid liposarcoma:
Local relapse and metastatic pattern in 43 patients. BMC Cancer.
18:3042018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zheng K, Yu XC, Xu M and Yang Y: Surgical
outcomes and prognostic factors of myxoid liposarcoma in
extremities: A retrospective study. Orthop Surg. 11:1020–1028.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Alaggio R, Coffin CM, Weiss SW, Bridge JA,
Issakov J, Oliveira AM and Folpe AL: Liposarcomas in young
patients: A study of 82 cases occurring in patients younger than 22
years of age. Am J Surg Pathol. 33:645–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Downes KA, Goldblum JR, Montgomery EA and
Fisher C: Pleomorphic liposarcoma: A clinicopathologic analysis of
19 cases. Mod Pathol. 14:179–184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hornick JL, Bosenberg MW, Mentzel T,
McMenamin ME, Oliveira AM and Fletcher CDM: Pleomorphic
liposarcoma: Clinicopathologic analysis of 57 cases. Am J Surg
Pathol. 28:1257–1267. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gardner JM, Dandekar M, Thomas D, Goldblum
JR, Weiss SW, Billings SD, Lucas DR, McHugh JB and Patel RM:
Cutaneous and subcutaneous pleomorphic liposarcoma: A
clinicopathologic study of 29 cases with evaluation of MDM2 gene
amplification in 26. Am J Surg Pathol. 36:1047–1051. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang L, Luo R, Xiong Z, Xu J and Fang D:
Pleomorphic liposarcoma: An analysis of 6 case reports and
literature review. Medicine (Baltimore). 97:e99862018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ghadimi MP, Liu P, Peng T, Bolshakov S,
Young ED, Torres KE, Colombo C, Hoffman A, Broccoli D, Hornick JL,
et al: Pleomorphic liposarcoma: Clinical observations and molecular
variables. Cancer. 117:5359–5369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Conyers R, Young S and Thomas DM:
Liposarcoma: Molecular genetics and therapeutics. Sarcoma.
2011:4831542011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Taylor BS, Barretina J, Socci ND,
Decarolis P, Ladanyi M, Meyerson M, Singer S and Sander C:
Functional copy-number alterations in cancer. PLoS One.
3:e31792008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Singer S, Socci ND, Ambrosini G, Sambol E,
Decarolis P, Wu Y, O'Connor R, Maki R, Viale A, Sander C, et al:
Gene expression profiling of liposarcoma identifies distinct
biological types/subtypes and potential therapeutic targets in
well-differentiated and dedifferentiated liposarcoma. Cancer Res.
67:6626–6636. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wei S, Henderson-Jackson E, Qian X and Bui
MM: Soft tissue tumor immunohistochemistry update: Illustrative
examples of diagnostic pearls to avoid pitfalls. Arch Pathol Lab
Med. 141:1072–1091. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Oliveira AM and Nascimento AG: Pleomorphic
liposarcoma. Semin Diagn Pathol. 18:274–285. 2001.PubMed/NCBI
|
|
60
|
Anderson WJ and Jo VY: Pleomorphic
liposarcoma: Updates and current differential diagnosis. Semin
Diagn Pathol. 36:122–128. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Assi T, Ngo C, Faron M, Verret B, Lévy A,
Honoré C, Hénon C, Le Péchoux C, Bahleda R and Le Cesne A: Systemic
therapy in advanced pleomorphic liposarcoma: A comprehensive
review. Curr Treat Options Oncol. 24:1598–1613. 2023.PubMed/NCBI
|
|
62
|
Wan L, Tu C, Qi L and Li Z: Survivorship
and prognostic factors for pleomorphic liposarcoma: A
population-based study. J Orthop Surg Res. 16:1752021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Eilber FC, Eilber FR, Eckardt J, Rosen G,
Riedel E, Maki RG, Brennan MF and Singer S: The impact of
chemotherapy on the survival of patients with high-grade primary
extremity liposarcoma. Ann Surg. 240:686–697. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Crago AM and Dickson MA: Liposarcoma:
Multimodality management and future targeted therapies. Surg Oncol
Clin N Am. 25:761–773. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ebey BN, Naouar S, Faidi B, Lahouar R, Ben
Khalifa B and El Kamel R: Pleomorphic spermatic cord liposarcoma: A
case report and review of management. Int J Surg Case Rep.
81:1057252021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gebhard S, Coindre JM, Michels JJ, Terrier
P, Bertrand G, Trassard M, Taylor S, Château MC, Marquès B, Picot V
and Guillou L: Pleomorphic liposarcoma: Clinicopathologic,
immunohistochemical, and follow-up analysis of 63 cases: A study
from the French federation of cancer centers sarcoma group. Am J
Surg Pathol. 26:601–616. 2002. View Article : Google Scholar : PubMed/NCBI
|