Introduction
Extraskeletal myxoid chondrosarcoma (EMC) is a rare,
clinicopathologically distinct soft-tissue sarcoma and a subtype of
chondrosarcoma, and it represents a malignant mesenchymal neoplasm
of uncertain differentiation and exceptional rarity, accounting for
<3% of all soft-tissue sarcomas, with an annual incidence of ~1
per million individuals. EMC is characterized histologically by
bland-appearing spindle to stellate cells arranged in cords,
reticular or lace-like patterns within abundant myxoid stroma and
typically, a lack of overt hyaline cartilage differentiation
(1). EMC most commonly arises in
the deep soft tissues of the proximal limbs in middle-aged to older
adults, predominantly affecting middle-aged adults (median age, 50
years) and showing a male-to-female ratio of ~2:1, with a
predilection for the thigh and popliteal fossa, and less frequent
involvement of the trunk (2).
Despite its deceptively bland cytology, EMC has been associated
with substantial risks of local recurrence and distant metastasis,
most often to the lungs and soft tissues. Recurrent chromosomal
rearrangements involving the nuclear receptor subfamily 4 group A
member 3 (NR4A3) gene on chromosome 9q22 have emerged as
highly characteristic and diagnostically informative hallmarks. The
classic translocation is t(9;22)(q22;q12), which fuses Ewing
sarcoma breakpoint region 1 (EWSR1) with nuclear receptor
subfamily 4 group A member 3 (NR4A3). Other variant partners
include TATA-box binding protein associated factor 15
(TAF15), rearranged through t(9;17)(q22;q11), and less
commonly, transcription factor 12 (TCF12), involved in
t(9;15)(q22;q21) (3,4). By contrast, primary intracranial EMC
is exceedingly rare and has been reported to arise from the
meninges or, less commonly, the brain parenchyma; its clinical and
radiological features frequently overlap with meningioma, glioma
and other sarcomas, complicating the formation of an accurate
diagnosis (5). In the absence of
large series and standardized treatment protocols, evaluation
relies on integrated assessment combining imaging, histopathology
and, where available, molecular genetics. The present study reports
a case of adolescent intracranial EMC with high-grade features and
outlines the diagnostic pitfalls and management considerations
pertinent to this uncommon entity.
Case report
In April 2025, a 17-year-old male patient presented
to the Department of Neurosurgery at Yantai Yuhuangding Hospital
(Yantai, China) with a 1-week history of a progressive headache
that had acutely worsened over the preceding 24 h, accompanied by
nausea and emesis. A neurological examination showed impaired
orientation, calculation and memory. The pupils measured 3 mm
bilaterally, limb strength was Medical Research Council grade 5-
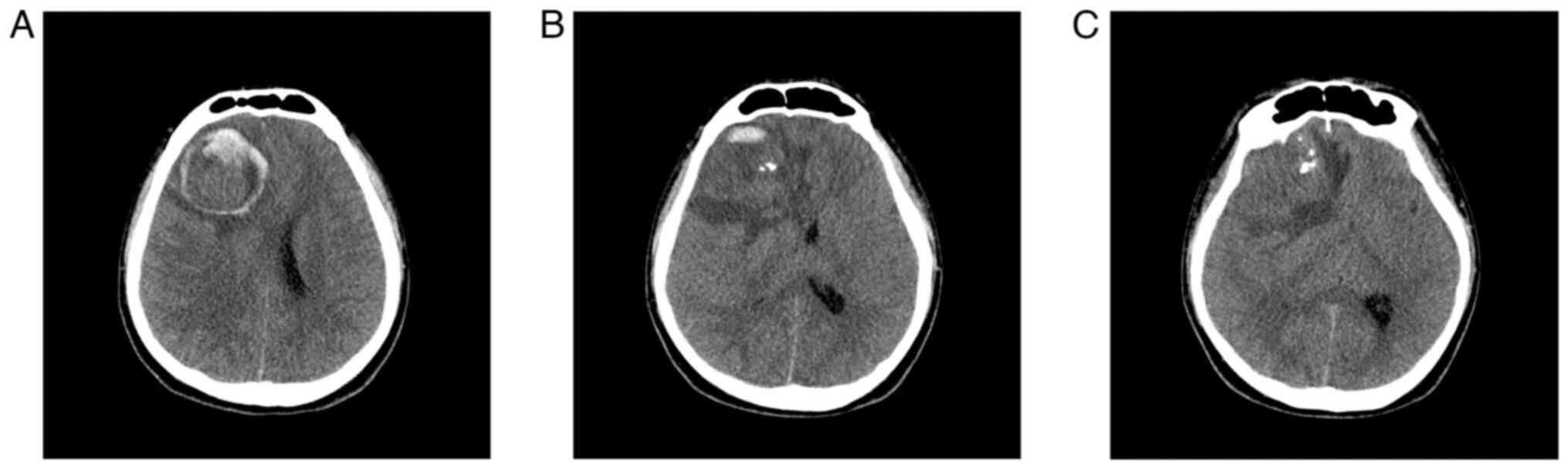
and no pathological reflexes were elicited (6). Non-contrast head computed tomography
(CT) scans taken in an external hospital revealed a right frontal
space-occupying lesion, with intracranial neoplasm and stroke
considered in the initial differential.
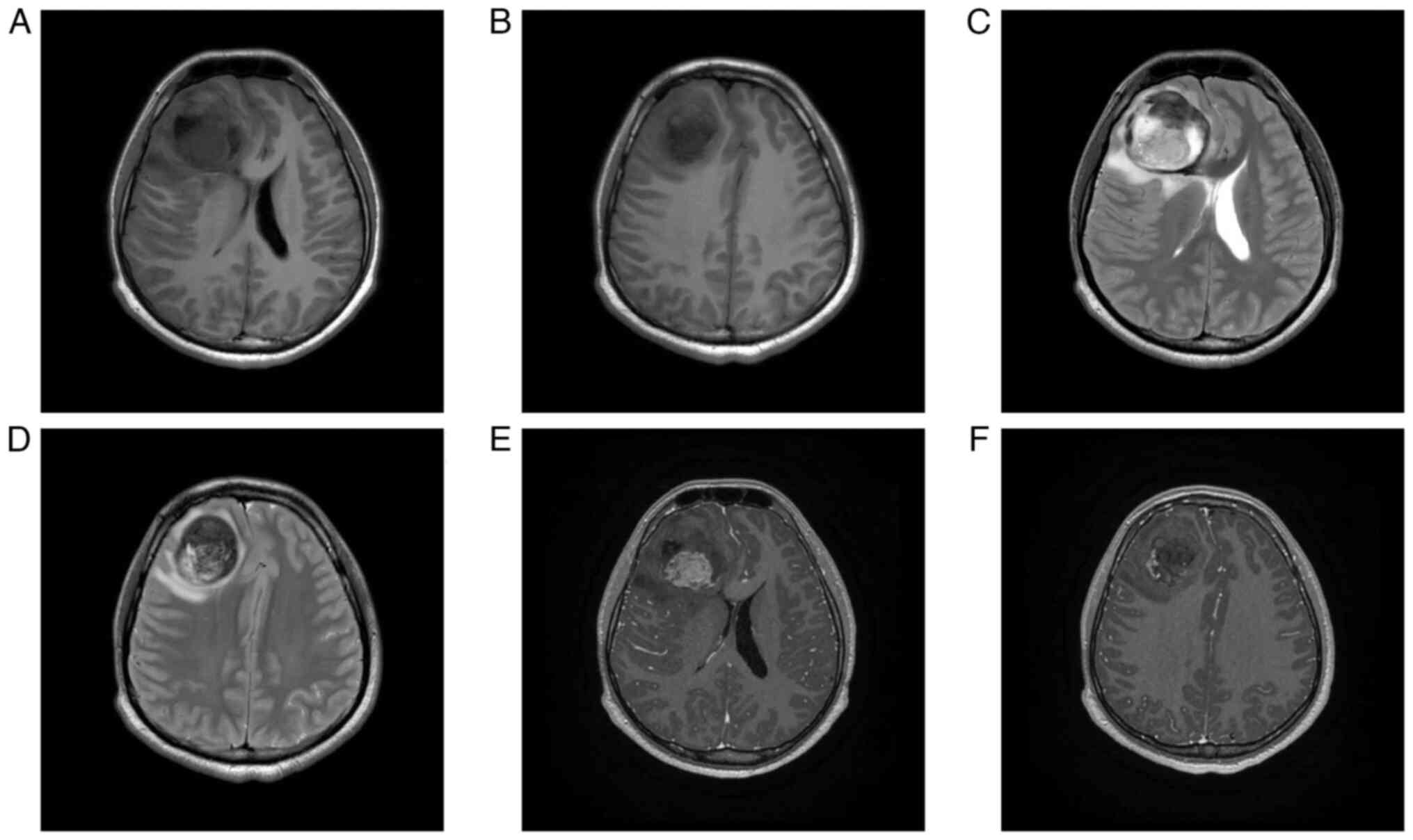
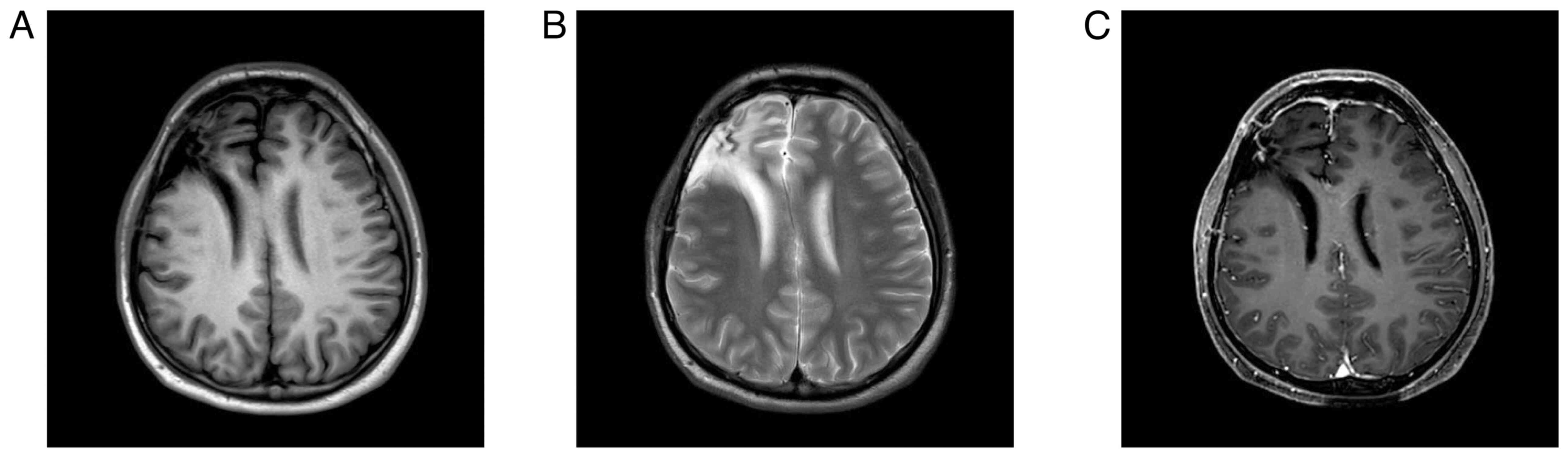
On hospital day 2, contrast-enhanced 3T magnetic
resonance imaging (MRI) demonstrated a 7.2×5.2-cm right frontal
mass with intralesional hemorrhage and mass effect displacing the
anterior and middle cerebral arteries (Fig. 1). CT angiography additionally
suggested anterior skull-base involvement with adjacent dural
contact (Fig. 2). A working
diagnosis of a tumor-related stroke was considered;
oligodendroglioma and meningioma were included in the
differential.
On hospital day 5, after exclusion of surgical
contraindications, the patient underwent a craniotomy for tumor
resection. Intraoperatively, a solid, firm, hypervascular mass
without a discrete capsule was identified with invasion of the
anterior skull-base dura. Fluorescein sodium aided delineation of
the tumor-brain interface, and a gross total resection was
achieved. The cut surface was soft and gelatinous. Dural defects
were repaired with artificial dura. Postoperatively, the patient
was transferred to the intensive care unit for ventilatory
support.
At 24 h post-surgery, the patient was somnolent with
a Glasgow Coma Scale score of E3V5M6 and proximal limb strength of
5- (6,7). Follow-up CT demonstrated a hematoma
with progressive fluid accumulation within the resection cavity
when compared with immediate postoperative imaging. By
postoperative day 3, after discontinuation of therapeutic
hypothermia, the patient's temperature peaked at 37.5°C and the
clinical status stabilized, permitting transfer to the general
ward.
On postoperative day 5, a high-grade fever and
incisional discharge prompted a lumbar puncture, which showed
elevated opening pressure and marked cerebrospinal-fluid
pleocytosis. Culture of the fluid grew methicillin-resistant
Staphylococcus aureus, confirming postoperative bacterial
meningitis. The patient received intravenous meropenem (1 g every 8
h) and vancomycin (1 g every 12 h); defervescence and wound
resolution were achieved after 6 days.
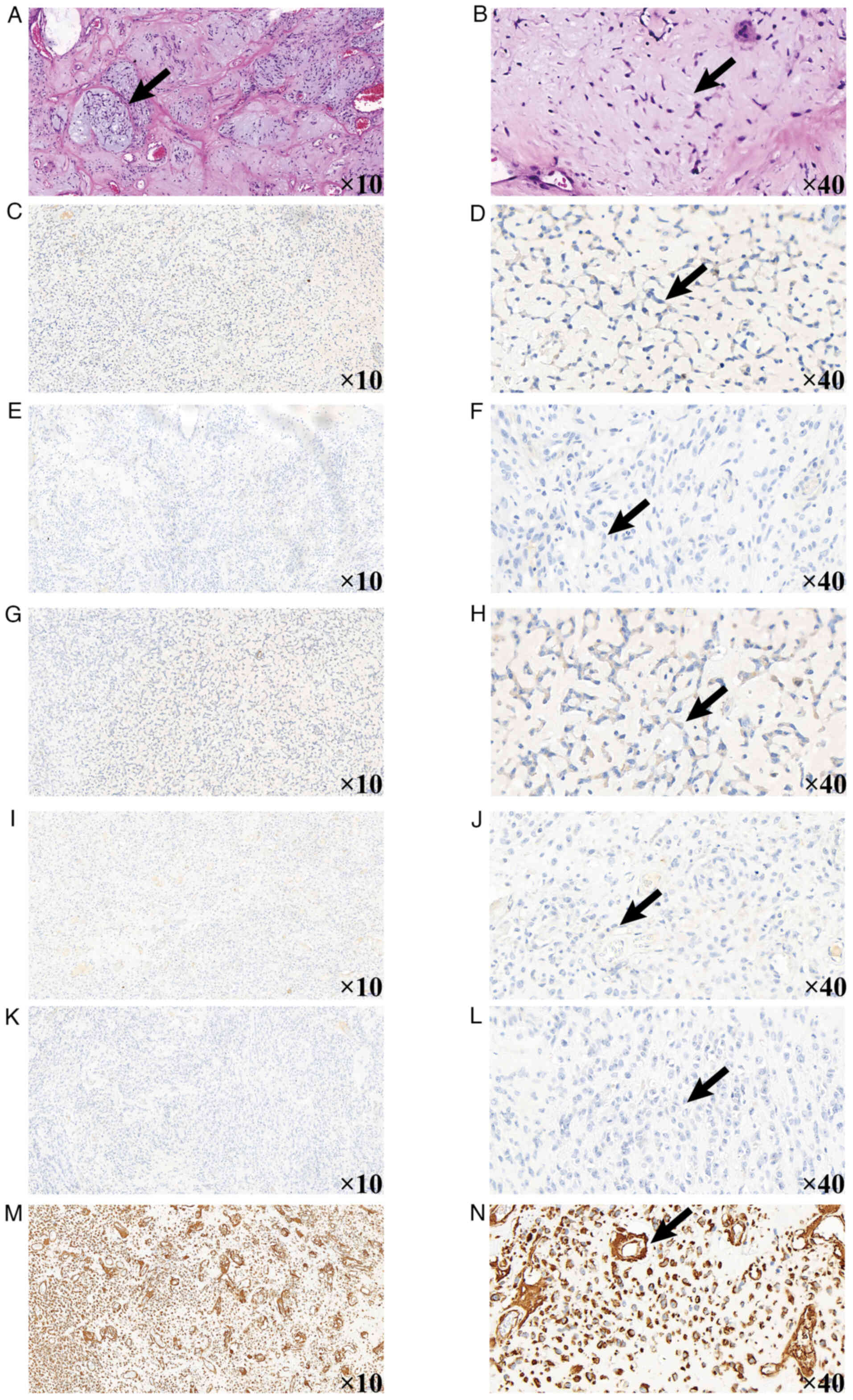
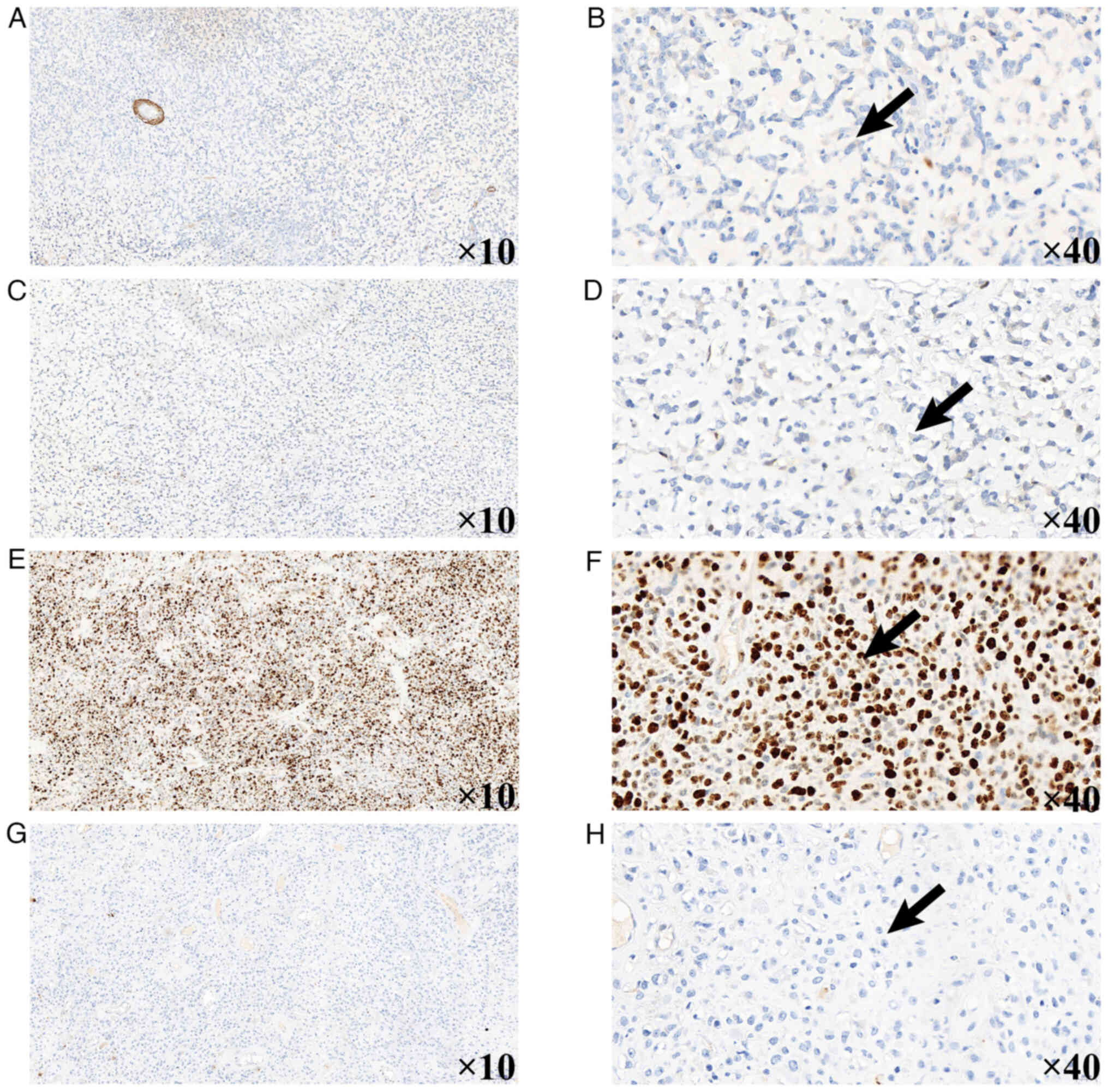
On postoperative day 16, histopathological analysis
established a diagnosis of EMC. Tissue samples were fixed in 4%
Paraformaldehyde Fix Solution (cat. no. P0099-500 ml; Beyotime
Biotechnology) at room temperature for 24 h, followed by routine
dehydration and embedding in paraffin. Sections were cut at a
thickness of 3 µm and mounted on slides. To minimize non-specific
background, sections were blocked with 3% bovine serum albumin in
PBS for 1 h at room temperature. The Roche BenchMark automated
staining platform was used according to the manufacturer's
instructions, with the following ready-to-use primary antibodies,
incubated at room temperature for 32 min: p53 (cat. no. 61209507;
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.), vimentin (cat.
no. ZM-0260; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.),
Ki-67 (Gene Tech Co., Ltd.; cat. no. GT209407), CK(Pan) (GeneTech
Co., Ltd.; cat. no. GM351507), INI1 (Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd.; cat. no. ZA-0696), S100 (Beijing Zhongshan
Jinqiao Biotechnology Co., Ltd.; cat. no. ZA-0225), Desmin
(GeneTech Co., Ltd.; cat. no. GT225207), EMA (Thermo Fisher
Scientific, Inc.; cat. no. 24 h-0095), GFAP (cat. no. ZM-0118;
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.), synaptophysin
(cat. no. ZM-0246; Beijing Zhongshan Jinqiao Biotechnology Co.,
Ltd.), CD34 (GeneTech Co., Ltd.; cat. no. GM716507), IDH1 (Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.; cat. no. ZM-0447), SOX10
(cat. no. ZA-0624; Beijing Zhongshan Jinqiao Biotechnology Co.,
Ltd.), STAT6 (cat. no. ZA-0647; Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd.), NKX2.2 (cat. no. ZA-0655; Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.) and SMA (cat. no.
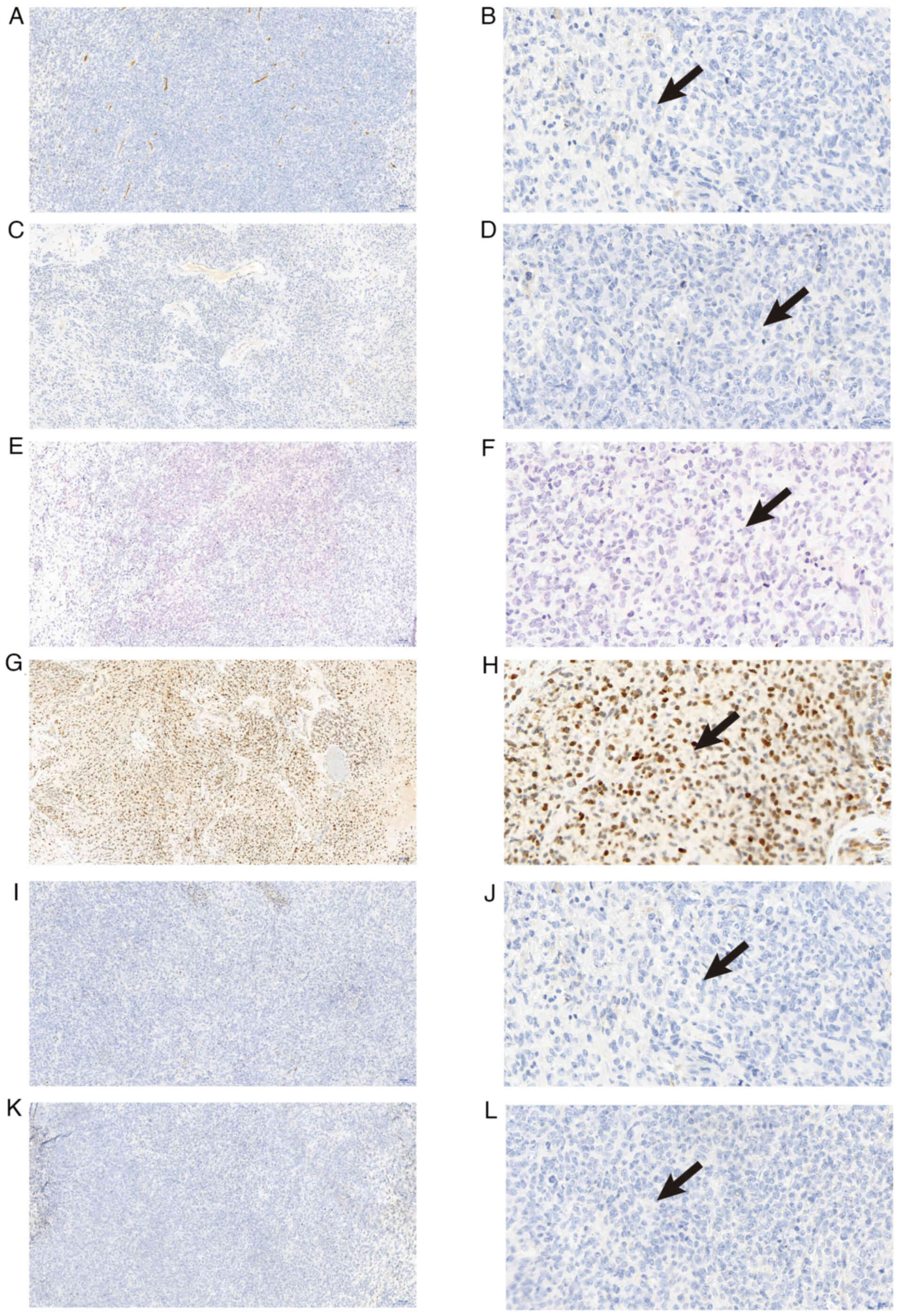
Kit-0006/MAB-0890; Abcam). Immunohistochemical analysis showed the
following results: S-100(−), integrase interactor 1 (INI1)(−),
cytokeratin (CK)(−), smooth muscle actin (SMA)(−), desmin(−),
CD34(−), STAT6(−), homeobox protein Nkx-2.2 (NKX2.2)(−),
transcription factor SOX-10 (SOX10)(−), synaptophysin(−),
epithelial membrane antigen (EMA)(−), glial fibrillary acidic
protein (GFAP)(−), diffuse vimentin(+), p53(+, 60%), isocitrate
dehydrogenase 1 (IDH-1)(−) and a Ki-67 labeling index of 80%
(Fig. 3, Fig. 4, Fig.
5). NR4A3 fluorescence in situ hybridization (FISH) was
recommended to confirm the molecular subtype, but was declined due
to financial constraints and concerns about invasive testing.
After discontinuation of intensive antimicrobial
therapy, the patient remained afebrile. Following a 23-day
hospitalization period, the patient was discharged with baseline
neurological function restored. Discharge medications included 30
mg oral idebenone three times daily and 0.2 g oral citicoline
sodium three times daily; neurosurgical follow-up was scheduled for
surveillance.
Approximately 1 month postoperatively, the patient
was readmitted for adjuvant radiotherapy (RT). Baseline
hematological parameters were within reference ranges: Red blood
cells, 4.71×10¹2/l (normal range,
4.0–5.5×10¹2/l); hemoglobin, 142 g/l (normal range,
130–175 g/l); platelets, 285×109/l (normal range,
125–350×109/l); and leukocytes, 8.36×109/l
(normal range, 4.0–10.0×109/l).
Simulation CT (3-mm slice thickness) was acquired
with the patient in a supine position and immobilized in a
thermoplastic mask. The tumor bed and contrast-enhancing region
were contoured as the gross tumor volume with a 3-mm margin to
generate the planning gross tumor volume (59.40 Gy in 33
fractions). Preoperative edema and enhancing dura defined the
clinical target volume; a further 3-mm margin produced the planning
target volume (PTV) (66 Gy in 33 fractions). Treatment-planning
verification confirmed ≥95% isodose coverage of the PTV with
organ-at-risk doses within institutional constraints.
Hematological indices were monitored throughout RT
and showed a mild downward trend consistent with non-actionable
myelosuppression (week 7: hemoglobin, 140 g/l; platelets,
238×109/l; and leukocytes, 4.83×109/l). No
laboratory threshold for intervention was reached.
During the course of radiotherapy, adjunctive
osmotherapy with intravenous 20% mannitol (125 ml every 8 h) was
administered to manage perilesional edema. The patient's Eastern
Cooperative Oncology Group performance status remained at 1
(8), and no radiation-induced
dermatitis or neurological toxicity was observed.
Post-RT MRI demonstrated substantial resolution of
the postoperative hematoma and associated fluid collections
(Fig. 6). Given the high-risk
pathological features (INI1 loss and Ki-67 at 80%) and the limited
responsiveness of EMC to conventional chemotherapy reported in the
literature, a multidisciplinary team concluded that adjuvant
chemotherapy would provide no clear clinical benefit; systemic
therapy was therefore not pursued, and the patient was discharged.
The patient's long-term prognosis remains uncertain, and imaging
surveillance is planned at 3- to 6-month intervals for 2 years and
annually thereafter.
Discussion
EMC is a rare soft-tissue sarcoma, accounting for
2.5–3% of all soft-tissue sarcomas. EMC is classified by the World
Health Organization as a ‘mesenchymal tumor of uncertain
differentiation’ (9).
Pathologically, EMC exhibits a multinodular architecture with a
mucin-rich matrix and chondroid cells showing malignant cytological
features (10). Although initially
regarded as a low-grade neoplasm, subsequent studies have shown a
notable propensity for local recurrence and distant metastasis
(11,12), particularly to the lungs (13). Intracranial EMC is hypothesized to
arise from embryonic remnants within cranial bones or from
pluripotent mesenchymal cells in the dura mater (14). EMC most often affects middle-aged
men and typically presents as a slowly enlarging, painless,
deep-seated mass in the proximal limbs (9). By contrast, intracranial lesions
usually have an insidious onset with non-specific symptoms,
frequently delaying the diagnosis.
The present study reports a rare case of
intracranial EMC in a 17-year-old male patient; to the best of our
knowledge, only 16 comparable cases have been reported worldwide
(Table I). The present case adds to
the spectrum of primary intracranial EMC, a review of which is
summarized in Table I (5,14–28).
The current patient presented with an acute headache, in contrast
to the classically indolent course of EMC. Imaging disclosed a
large right frontal mass (7.2×5.2 cm) with intralesional hemorrhage
and invasion of the anterior cranial-fossa dura. Notably, the
supratentorial location challenges the conventional expectation
that intracranial EMCs predominantly arise in the posterior cranial
fossa; fewer than 5 supratentorial cases have been documented
(17,28). Postoperative histopathology
confirmed EMC with loss of INI1 expression and a high Ki-67
labeling index (80%), indicating an aggressive phenotype. The
distinctiveness of this case lies in its adolescent onset (typical
onset, 50–60 years), right frontal location and high proliferative
activity with INI1 loss. The postoperative course was further
complicated by bacterial meningitis, adding to the management
complexity.
 | Table I.Reported cases of primary
intracranial extraskeletal myxoid chondrosarcoma. |
Table I.
Reported cases of primary
intracranial extraskeletal myxoid chondrosarcoma.
| First author/s,
year | Sex | Age, years | Clinical
symptoms | Size, cm | Locations of
tumor | Treatment | Recurrence | Metastasis | (Refs.) |
|---|
| Scott et al,
1976 | M | 39 | Headache, nausea
and vomiting | NA | 4th ventricle | PR | N | N | (15) |
| Smith and Davidson,
1981 | M | 12 | Headache, nausea,
vomiting and difficulty ambulating | 3.5×1.0×0.5 | Left
cerebellum | GTR | N | N | (16) |
| Salcman et
al, 1992 | F | 28 | Headache, slow
speech and right limb weakness | 7.0×5.0×4.0 | Left parafalcine
region | GTR | Y | N | (17) |
| Sato et al,
1993 | F | 43 | Blurred vision and
gait disturbance | NA | Pineal region | PR, RT (60 Gy),
chemotherapy | Y | Y | (18) |
| Chaskis et
al, 2002 | F | 17 | Headache and status
epilepticus | NA | Right
frontal-parietal lobe | GTR | Y | N | (14) |
| González-Lois et
al, 2002 | M | 69 | Headache, dizziness
and behavior change | NA | Right frontal
lobe | GTR | N | N | (19) |
| Im et al,
2003 | M | 43 | Headache, nausea
and vomit | 2.0 | Left parietal
lobe | GTR, RT (59.4
Gy) | N | N | (20) |
| Cummings et
al, 2004 | M | 63 | Hearing loss and
gait disturbance | 2.4×1.8×2.4 | Right jugular
foramen, right Cerebellopontine Angle (CPA) | GTR | NA | NA | (21) |
| Sorimachi et
al, 2008 | F | 37 | Headache, nausea,
vomit and upward gaze palsy | NA | Pineal region | GTR, RT (59.4
Gy) | N | N | (22) |
| O'Brien et
al, 2008 | F | 26 | Headache, nausea
and seizure | 2.5 | Left CPA | STR proton
therapy | N | N | (23) |
| Arpino et
al, 2011 | F | 54 | Headache and left
ophthalmoplegia | NA | Sellar and
parasellar area | GTR | NA | NA | (24) |
| Dulou et al,
2012 | F | 70 | Behavior change and
difficulty in walking | NA | Left frontal
lobe | GTR, preoperative
RT (60 Gy) | Y | N | (25) |
| Park et al,
2012 | F | 21 | Headache, right
limb weakness, bilateral hearing loss and bilateral vision
loss | 3.2×6.3×4.9 | Left lateral
ventricle | GTR, RT (60.8
Gy) | NA | NA | (26) |
| Qin et al,
2017 | F | 41 | Headache and
vomiting | 3.0×3.0×3.0 | Left
cerebellum | GTR, two stage RT
(56 Gy/50 Gy), chemo | N | N | (27) |
| Akakin et
al, 2018 | F | 75 | Right limb
weakness | NA | Left parafalcine
region | GTR | Y | N | (28) |
| Zhu et al,
2022 | M | 52 | Headache, dizzy and
nausea | 3.4×3.0 | Left cavernous
sinus | GTR, RT (45
Gy) | N | N | (5) |
| Present case | M | 17 | Headache, nausea
and vomiting | 7.2×5.2 | Right frontal
lobe | GTR, RT (59.4
Gy) | N | N |
|
The present case underscores the pronounced clinical
and pathological heterogeneity of EMC. An accurate diagnosis
requires an integrated, multimodal assessment.
Immunohistochemically, the tumor showed diffuse vimentin
positivity, consistent with mesenchymal differentiation, and was
negative for S-100, aligning with the frequently low rate of S-100
expression reported in intracranial EMCs (28,29)
(Table II). The
immunohistochemical profile of the present case aligns with
reported intracranial EMCs, with a detailed comparison provided in
Table II (5,16–20,22,23,25–28).
| Table II.Immunohistochemical features of
intracranial myxoid chondrosarcomas in reported cases. |
Table II.
Immunohistochemical features of
intracranial myxoid chondrosarcomas in reported cases.
| First author,
year | GFAP | Cytokeratin | EMA | S-100 | Vimentin | NSE | Synaptophysin | Chromogranin A | (Refs.) |
|---|
| Smith and Davidson,
1981 | (−) |
|
|
|
|
|
|
| (16) |
| Salcman et
al, 1992 | (−) | (−) | (−) | (−) | (++) |
|
|
| (17) |
| Sato et al,
1993 | (−) |
|
| (++) | (++) |
|
|
| (18) |
| González-Lois et
al, 2002 | (+) | (−) | (−) | (+), focal | (+) | (−) |
|
| (19) |
| Im et al,
2003 | (−) | (−) | (−) | (+), weak | (+) |
|
|
| (20) |
| Sorimachi et
al, 2008 | (−) | (−) | (−) | (−) | (+) | (−) | (−) | (−) | (22) |
| O'Brien et
al, 2008 | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (23) |
| Park et al,
2012 | (−) | (+), focal
strong | (+) | (+) |
|
|
|
| (26) |
| Dulou et al,
2012 | (+) | (+), strong | (+), weak |
|
|
|
|
| (25) |
| Qin et al,
2017 | (−) | (−) | (−) | (+) |
|
|
|
| (27) |
| Akakin et
al, 2018 | (−) | (−) | (+) | (+) | (+) | ND | (−) | (−) | (28) |
| Zhu et al,
2022 | (−) | (−) | (−) | (+) | (+) | ND | ND | ND | (5) |
| Present case | (−) | (−) | (−) | (−) | (+) | ND | (−) | ND |
|
Histopathological evaluation of hematoxylin and
eosin (H&E)-stained sections revealed a hypercellular neoplasm
with a multinodular architecture within an abundant myxoid stroma.
Tumor cells were predominantly eosinophilic, ranging from
epithelioid to plump spindle forms, arranged in cords, nests and
reticular patterns. There was marked nuclear atypia, elevated
mitotic activity and foci of hemorrhagic change. A panel of
negative immunomarkers supported systematic exclusion of mimics:
GFAP and SOX10 negativity argued against gliomas and schwannomas
(30); CK and EMA negativity argued
against epithelial tumors and meningiomas (31); synaptophysin, desmin and SMA
negativity argued against neuroendocrine and myogenic neoplasms
(32); and the absence of CD34 and
STAT6 expression did not support a pericytic/solitary fibrous tumor
lineage (33). Furthermore, while
CD99 is a valuable marker in the differential diagnosis of small
round cell tumors, particularly for excluding Ewing sarcoma, it was
not performed in the present retrospective case as the
comprehensive diagnostic workup was already achieved through the
extensive immunohistochemical panel and characteristic
morphological features. However, based on its established role in
differential diagnosis, its inclusion is recommended in a routine
panel. The hemorrhagic transformation may reflect aberrant vascular
proliferation, concordant with the high Ki-67 index (up to 80%) and
strong p53 immunoreactivity (>60%), collectively suggesting
activated angiogenesis-related pathways.
Loss of INI1 expression is infrequently observed in
conventional intracranial EMCs (reported in <10% of cases) but
has been described in high-grade morphological variants with
pronounced atypia and brisk mitotic activity (34). The combination of characteristic
H&E features, multinodular myxoid stroma with corded growth and
the specific immunoprofile is strongly supportive of EMC. Diffuse
vimentin positivity with absent synaptophysin/EMA expression
effectively excludes malignant rhabdoid tumors, while CK negativity
argues against chordoma. In addition, negative NKX2.2 and IDH-1
staining disfavors Ewing's sarcoma and IDH-mutant gliomas,
respectively (35).
The immunoprofile in the present case, namely strong
vimentin positivity, S-100 negativity, synaptophysin negativity and
the absence of lineage-defining markers, closely mirrors reported
intracranial EMCs, including those associated with the TAF15-NR4A3
fusion subtype; in the absence of molecular confirmation, however,
this remains inferential. This fusion subtype accounts for ~20% of
EMCs and has been linked to aggressive clinical behavior and
resistance to pazopanib (36).
Molecular confirmation remains rare in the literature, with
definitive subtyping reported in only 1 of 16 documented
intracranial cases (5). Thus, while
the diagnosis is well supported by histomorphology and
immunohistochemistry, definitive classification would require
genetic confirmation, which was not performed in the present case
(5).
A definitive diagnosis typically requires molecular
detection of NR4A3 rearrangements, most commonly confirmation of
the EWSR1-NR4A3 fusion (37). The
patient's refusal to undergo NR4A3 testing imposed two critical
limitations: First, the TAF15-NR4A3 subtype, which confers relative
resistance to pazopanib, could not be distinguished from the
EWSR1-NR4A3 subtype, precluding accurate molecular subtyping; and
second, prognostication may be less reliable, as patients with
TAF15 fusions have significantly lower 5-year survival rates than
those with EWSR1 fusions.
Given the high-risk pathological features,
management raised three major challenges. First, postoperative
bacterial meningitis, reflected by a cerebrospinal-fluid white-cell
count of 836×106/l, may necessitate delaying RT, thereby
increasing the risk of recurrence. Second, invasion of the
skull-base dura complicates margin assessment and favors the
judicious use of neuronavigation. Third, in this high-risk context,
systemic therapies such as temozolomide or pazopanib merit
consideration; temozolomide penetrates the blood-brain barrier
effectively, whereas pazopanib may be more suitable for patients
lacking the TAF15 fusion. At the 3-month postoperative follow-up,
no recurrence was detected in the present case. Nonetheless,
vigilant long-term surveillance is warranted, as distant metastases
often emerge years after surgery. Proton-beam therapy may offer
dosimetric advantages over conventional RT, but the optimal
strategy requires further evaluation (37).
Imaging findings are pivotal for differential
diagnosis and require synthesis across modalities. CT typically
shows a well-circumscribed hypodense mass, with calcification in
~12.5% of cases (5).
Contrast-enhanced CT often demonstrates mild or absent enhancement
(2). MRI provides greater
diagnostic resolution: Lesions are commonly iso- to hypointense on
T1-weighted sequences, with hemorrhagic foci appearing hyperintense
(38). On T2-weighted imaging, they
are frequently markedly hyperintense with hypointense fibrous
septa, often producing a multiloculated cystic appearance (2,39).
Post-contrast enhancement is typically septal, ring-like or
lobulated; ~10% of cases show minimal or no enhancement (38). The MRI characteristics in the
present case align closely with these descriptors (Table III). The imaging findings in the
present case are consistent with the spectrum of MRI features
summarized in Table III (5,14–28).
| Table III.MRI features of primary intracranial
extraskeletal myxoid chondrosarcoma case. |
Table III.
MRI features of primary intracranial
extraskeletal myxoid chondrosarcoma case.
| First author,
year | T1WI | T2WI | Enhanced MRI | (Refs.) |
|---|
| Scott et al,
1976 | NA | NA | NA | (15) |
| Smith and Davidson,
1981 | NA | NA | NA | (16) |
| Salcman et
al, 1992 | Well-defined,
hyperintensity | Homogeneous
hyperintensity | NA | (17) |
| Sato et al,
1993 | NA | NA | NA | (18) |
| Chaskis et
al, 2002 | Hypointensity | NA | Heterogeneous
enhancement | (14) |
| González-Lois et
al, 2002 | NA | NA | Significantly
homogeneous enhancement | (19) |
| Im et al,
2003 | Unclear-defined,
hypointensity | Hyperintensity | Homogeneously well
enhanced | (20) |
| Cummings et
al, 2004 | NA | NA | Heterogeneous
enhancement | (21) |
| Sorimachi et
al, 2008 | Mixed signal
intensity, hyperintensity (hemorrhage) | NA | Heterogeneous
enhancement | (22) |
| O'Brien et
al, 2008 | Hypointensity | Hyperintensity | NA | (23) |
| Arpino et
al, 2011 | Hypointensity | Hyperintensity | Heterogeneously
peripheral enhancement | (24) |
| Dulou et al,
2012 | NA | Hyperintensity,
peritumor edema | Heterogeneously
ring-like enhancement | (25) |
| Park et al,
2012 | Homogeneous
iso-intensity | Heterogeneous
hyperintensity, Peritumor edema | Heterogeneously
lobulated enhancement | (26) |
| Qin et al,
2017 | NA | NA | NA | (27) |
| Akakin et
al, 2018 | NA | Heterogeneous
hyperintensity | Heterogeneously
rim-like enhancement, DWI showed intratumor calcification | (28) |
| Zhu et al,
2022 | Homogeneous
hypointensity | Heterogeneous
hyperintensity | Heterogeneously
well enhanced | (5) |
| Present case | Hypointensity | Hyperintensity | Heterogeneously
well enhanced |
|
Key differential diagnoses of EMC include chondroid
meningioma, which often exhibits calcification and a characteristic
dural-tail sign (1), ependymoma,
which commonly arises in the fourth ventricle and may disseminate
along cerebrospinal-fluid pathways (38), cavernous hemangioma, which shows a
‘popcorn-like’ mixed T2 signal with a surrounding hemosiderin rim,
and metastatic tumors, which are frequently multiple and typically
associated with substantial vasogenic edema (5).
For clinical practice, heightened vigilance is
warranted when encountering intracranial mucin-rich tumors with
hemorrhage and skull-base invasion in adolescents. A routine
immunohistochemical panel should include vimentin, S-100, CD99 and
INI1, with strong consideration of NR4A3 assessment. Molecular
confirmation is recommended using FISH or RNA sequencing.
Therapeutically, meticulous control of surgical
boundaries at the skull base and timely initiation of RT, at a
recommended dose of 50–54 Gy, are crucial. Systemic options may
include temozolomide or pazopanib. For patients with a relevant
family history, genetic screening should be considered.
The present study has several limitations, including
the absence of NR4A3 fusion testing, a short postoperative
follow-up of only 3 months and the lack of mechanistic validation.
Given that EMC can metastasize years after surgery, long-term
standardized follow-up is essential. Future work should prioritize
establishing a dedicated registry for adolescent EMCs and
investigating age-specific mechanisms, including functional
interrogation of the LSM14A-NR4A3 fusion, the consequences of INI1
loss and strategies for combination immunotherapy.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HW designed and conducted the study, collected the
case report data, and analyzed and interpreted the data. HZ
acquired the pathological data. FW contributed to the analysis and
revision of the discussion section and prepared the pathological
images. HW wrote the manuscript, and all authors analyzed the
results and revised the manuscript. HZ made significant
contributions to the conception and design of the study. HW, HZ,
and FW confirm the authenticity of all the raw data. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Yantai Yuhuangding Hospital (Yantai, China; approval no.
2025-561).
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of any potentially identifiable images
or data included in this article.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript or
to generate images, and subsequently, the authors revised and
edited the content produced by the AI tools as necessary, taking
full responsibility for the ultimate content of the present
manuscript.
Glossary
Abbreviations
Abbreviations:
|
EMC
|
extraskeletal myxoid
chondrosarcoma
|
|
MRI
|
magnetic resonance imaging
|
|
RT
|
radiotherapy
|
References
|
1
|
Stacchiotti S, Baldi GG, Morosi C, Gronchi
A and Maestro R: Extraskeletal myxoid chondrosarcoma: State of the
art and current research on biology and clinical management.
Cancers (Basel). 12:27032020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang L, Wang R, Xu R, Qin G and Yang L:
Extraskeletal myxoid chondrosarcoma: A comparative study of imaging
and pathology. Biomed Res Int. 2018:96842682018.PubMed/NCBI
|
|
3
|
Noguchi H, Mitsuhashi T, Seki K, Tochigi
N, Tsuji M, Shimoda T and Hasegawa T: Fluorescence in situ
hybridization analysis of extraskeletal myxoid chondrosarcomas
using EWSR1 and NR4A3 probes. Hum Pathol. 41:336–342. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei S, Pei J, von Mehren M, Abraham JA,
Patchefsky AS and Cooper HS: SMARCA2-NR4A3 is a novel fusion gene
of extraskeletal myxoid chondrosarcoma identified by RNA
next-generation sequencing. Genes Chromosomes Cancer. 60:709–712.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu ZY, Wang YB, Li HY and Wu XM: Primary
intracranial extraskeletal myxoid chondrosarcoma: A case report and
review of literature. World J Clin Cases. 10:4301–4313. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nodal S, Khalafallah AM, Sanghera BS,
Garcia Barreto M, Errante EL, Levi AD, Ray WZ and Burks SS: A
meta-analysis and systematic review: Association of timing and
muscle strength after nerve transfer in upper trunk palsy.
Neurosurg Rev. 48:5032025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mehta R and Chinthapalli K: Glasgow coma
scale explained. BMJ. 365:l12962019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oken MM, Creech RH, Tormey DC, Horton J,
Davis TE, McFadden ET and Carbone PP: Toxicity and response
criteria of the eastern cooperative oncology group. Am J Clin
Oncol. 5:649–655. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kandoussi AE, Hung YP, Tung EL, Bauer F,
Vicentini JRT, Lozano-Calderon S and Chang CY: Clinical, imaging
and pathological features of extraskeletal myxoid chondrosarcoma.
Skeletal Radiol. 54:959–966. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okamoto S, Hisaoka M, Ishida T, Imamura T,
Kanda H, Shimajiri S and Hashimoto H: Extraskeletal myxoid
chondrosarcoma: A clinicopathologic, immunohistochemical, and
molecular analysis of 18 cases. Hum Pathol. 32:1116–1124. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giner F, López-Guerrero JA, Machado I,
Rubio-Martínez LA, Espino M, Navarro S, Agra-Pujol C, Ferrández A
and Llombart-Bosch A: Extraskeletal myxoid chondrosarcoma: p53 and
Ki-67 offer prognostic value for clinical outcome-an
immunohistochemical and molecular analysis of 31 cases. Virchows
Arch. 482:407–417. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ulucaköy C, Atalay İB, Yapar A, Kaptan AY,
Bingöl İ, Doğan M and Ekşioğlu MF: Surgical outcomes of
extraskeletal myxoid chondrosarcoma. Turk J Med Sci. 52:1183–1189.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fice MP, Lee L, Kottamasu P, Almajnooni A,
Cohn MR, Gusho CA, Gitelis S and Blank AT: Extraskeletal myxoid
chondrosarcoma: A case series and review of the literature. Rare
Tumors. 14:203636132210797542022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chaskis C, Michotte A, Goossens A, Stadnik
T, Koerts G and D'Haens J: Primary intracerebral myxoid
chondrosarcoma. Case illustration. J Neurosurg. 97:2282002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scott RM, Dickersin R, Wolpert SM and
Twitchell T: Myxochondrosarcoma of the fourth ventricle. Case
report. J Neurosurg. 44:386–389. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smith TW and Davidson RI: Primary
meningeal myxochondrosarcoma presenting as a cerebellar mass: Case
report. Neurosurgery. 8:577–581. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salcman M, Scholtz H, Kristt D and
Numaguchi Y: Extraskeletal myxoid chondrosarcoma of the falx.
Neurosurgery. 31:344–348. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sato K, Kubota T, Yoshida K and Murata H:
Intracranial extraskeletal myxoid chondrosarcoma with special
reference to lamellar inclusions in the rough endoplasmic
reticulum. Acta Neuropathol. 86:525–528. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
González-Lois C, Cuevas C, Abdullah O and
Ricoy JR: Intracranial extraskeletal myxoid chondrosarcoma: Case
report and review of the literature. Acta Neurochir (Wien).
144:735–740. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Im SH, Kim DG, Park IA and Chi JG: Primary
intracranial myxoid chondrosarcoma: Report of a case and review of
the literature. J Korean Med Sci. 18:301–307. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cummings TJ, Bridge JA and Fukushima T:
Extraskeletal myxoid chondrosarcoma of the jugular foramen. Clin
Neuropathol. 23:232–237. 2004.PubMed/NCBI
|
|
22
|
Sorimachi T, Sasaki O, Nakazato S, Koike T
and Shibuya H: Myxoid chondrosarcoma in the pineal region. J
Neurosurg. 109:904–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O'Brien J, Thornton J, Cawley D, Farrell
M, Keohane K, Kaar G, McEvoy L and O'Brien DF: Extraskeletal myxoid
chondrosarcoma of the cerebellopontine angle presenting during
pregnancy. Br J Neurosurg. 22:429–432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arpino L, Capuano C, Gravina M and Franco
A: Parasellar myxoid chondrosarcoma: A rare variant of cranial
chondrosarcoma. J Neurosurg Sci. 55:387–389. 2011.PubMed/NCBI
|
|
25
|
Dulou R, Chargari C, Dagain A, Teriitehau
C, Goasguen O, Jeanjean O and Védrine L: Primary intracranial
extraskeletal myxoid chondrosarcoma. Neurol Neurochir Pol.
46:76–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park JH, Kim MJ, Kim CJ and Kim JH:
Intracranial extraskeletal myxoid chondrosarcoma: Case report and
literature review. J Korean Neurosurg Soc. 52:246–249. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qin Y, Zhang HB, Ke CS, Huang J, Wu B, Wan
C, Yang CS and Yang KY: Primary extraskeletal myxoid chondrosarcoma
in cerebellum: A case report with literature review. Medicine
(Baltimore). 96:e86842017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Akakın A, Urgun K, Ekşi M, Yılmaz B,
Yapıcıer Ö, Mestanoğlu M, Toktaş ZO, Demir MK and Kılıç T: Falcine
myxoid chondrosarcoma: A rare aggressive case. Asian J Neurosurg.
13:68–71. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kallen ME and Hornick JL: The 2020 WHO
classification: What's new in soft tissue tumor pathology? Am J
Surg Pathol. 45:e1–e23. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tekavec K, Švara T, Knific T, Gombač M and
Cantile C: Histopathological and immunohistochemical evaluation of
canine nerve sheath tumors and proposal for an updated
classification. Vet Sci. 9:2042022.PubMed/NCBI
|
|
31
|
Sarathy D, Snyder MH, Ampie L, Berry D and
Syed HR: Dural convexity chondroma mimicking meningioma in a young
female. Cureus. 13:e207152021.PubMed/NCBI
|
|
32
|
Bellizzi AM: An algorithmic
immunohistochemical approach to define tumor type and assign site
of origin. Adv Anat Pathol. 27:114–163. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Almaghrabi A, Almaghrabi N and Al-Maghrabi
H: Glomangioma of the kidney: A rare case of glomus tumor and
review of the literature. Case Rep Pathol.
2017:74236422017.PubMed/NCBI
|
|
34
|
Velz J, Agaimy A, Frontzek K, Neidert MC,
Bozinov O, Wagner U, Fritz C, Coras R, Hofer S, Bode-Lesniewska B
and Rushing E: Molecular and clinicopathologic heterogeneity of
intracranial tumors mimicking extraskeletal myxoid chondrosarcoma.
J Neuropathol Exp Neurol. 77:727–735. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu Z, Bian J, Yang Y, Wei D and Qi S:
Ewing sarcoma of the pancreas: A pediatric case report and
narrative literature review. Front Oncol. 14:13685642024.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dulken BW, Kingsley L, Zdravkovic S,
Cespedes O, Qian X, Suster DI and Charville GW: CHRNA6 RNA in situ
hybridization is a useful tool for the diagnosis of extraskeletal
myxoid chondrosarcoma. Mod Pathol. 37:1004642024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ngo C, Verret B, Vibert J, Cotteret S,
Levy A, Pechoux CL, Haddag-Miliani L, Honore C, Faron M, Quinquis
F, et al: A novel fusion variant LSM14A::NR4A3 in extraskeletal
myxoid chondrosarcoma. Genes Chromosomes Cancer. 62:52–56. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wilbur HC, Robinson DR, Wu YM, Kumar-Sinha
C, Chinnaiyan AM and Chugh R: Identification of novel PGR-NR4A3
fusion in extraskeletal myxoid chondrosarcoma and resultant patient
benefit from tamoxifen therapy. JCO Precis Oncol. 6:e22000392022.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hong YG, Yoo J, Kim SH and Chang JH:
Intracranial extraskeletal myxoid chondrosarcoma in fourth
ventricle. Brain Tumor Res Treat. 9:75–80. 2021. View Article : Google Scholar : PubMed/NCBI
|