Introduction
Gastric cancer (GC) remains one of the most common
and lethal malignancies worldwide, with persistently high incidence
and mortality rates. According to recent global statistics,
>960,000 new GC cases are diagnosed annually and as estimated in
2024, ~660,000 mortalities occur annually, with men representing
the majority of cases (1). In
China, GC morbidity and mortality also poses a major public health
burden (2); although incidence has
declined moderately across the past decades, this trend is
geographically uneven and population growth may drive an increase
in GC mortalities by 2035 (3). The
development of GC is a complex multistep process, where immune
escape mechanisms serve a major role. Tumor cells can downregulate
antigen expression to escape immune detection, activate
immunosuppressive pathways and recruit suppressive cell populations
to favor growth and metastasis. These methods disarm innate and
adaptive immunity as well as reduce the efficiency of
immunotherapeutics, including immune checkpoint inhibitors (ICIs)
and adoptive cell therapies (4).
The tumor microenvironment (TME) consists of
non-malignant cells and secreted factors which further sustain and
promote GC progression. During early-stage tumor development, the
TME plays a crucial role in inhibiting tumor growth and metastasis
by activating immune cells and enhancing antitumor immune
responses. However, as tumors develop, the TME transforms into an
immunosuppressive niche that further facilitates tumor growth,
metastasis and immune tolerance (5). In GC, the TME provides nutrients and
structural support to tumor cells and, through complex, multi-level
interactions, establishes an immunosuppressive network that impairs
effector immune function and shifts responses toward tolerance
(6).
Immune tolerance refers to natural mechanisms that
maintain the immune system in a state of unresponsiveness or
hyporesponsiveness, thereby preventing autoimmune disease
development and excessive inflammation (7). In cancer, the TME enhances these
mechanisms to inhibit T-cell responses against tumor cells by
increasing regulatory T-cell (Tregs) levels, secreting TGF-β and
IL-10 and upregulating programmed death-ligand 1 (PD-L1). This
creates a tumor-specific immune-tolerant state that induces
effector T-cell exhaustion or apoptosis, markedly impairing immune
clearance (8). Beyond evading
immune surveillance, tumor cells actively acquire immune escape
capabilities through gene mutations, epigenetic alterations and
microenvironmental remodeling (9).
These interconnected processes drive GC progression and influence
immunotherapy outcomes.
Consequently, monotherapies using ICIs or chimeric
antigen receptor T-cells (CAR-T) show limited efficacy in GC, as
some patients develop primary or acquired resistance. However,
these mechanisms may identify clear targets for combination
strategies aimed at TME remodeling (10,11).
The role of the TME in immune escape has attracted
recent attention. A study conducted by He et al (12) demonstrated that stromal modulation
by the tubulin-binding anticancer drugs combretastatin A4 and
eribulin improved tumor perfusion and antitumor immunity. This was
achieved in a tumor mouse model by restoring highly proliferative,
angiogenic pericytes to a quiescent, contractile state, thereby
continuously normalizing the vascular bed and reducing hypoxia. In
addition, Wang et al (13)
explored the role of phosphodiesterase type 5A (PDE5A) in
cancer-associated fibroblasts (CAFs) in shaping an
immunosuppressive TME in GC. The findings identified PDE5A in CAFs
as a key factor influencing TME alterations, such as T cell
exclusion and reduced infiltration of CD8+ cytotoxic T
lymphocytes, which may undermine the effectiveness of GC
immunotherapy. These insights provide both mechanistic
understanding and potential therapeutic implications for improving
GC immunotherapy. Shaibu et al (14) explored how mesenchymal stem cells,
cytokines and ICIs collectively contribute to the formation of an
immunosuppressive GC TME. The study revealed that mesenchymal stem
cells secrete cytokines, including IL-6, TGF-β and IL-10, which
contribute to an immunosuppressive environment, thereby
facilitating tumor progression, immune evasion and resistance to
treatment.
While involvement of the TME in immune escape has
been investigated in the aforementioned studies, the mechanisms
underlying GC immune tolerance remain poorly understood.
Specifically, the roles of cytokines, immune checkpoints and
epigenetic regulation within the TME in driving GC immune tolerance
are not well defined. The present review aimed to address this
knowledge gap, summarizing the role of the TME in GC immune escape
(as reported in the literature) and the mechanisms underpinning GC
immune tolerance. It further explores how immune cells, cytokines
and epigenetic alterations within the TME mediate immune tolerance
and influence immunotherapy efficacy.
In addition, the present review details progress in
overcoming GC immune tolerance and describes innovative
immunotherapeutic strategies for TME remodeling. For example,
emerging ICIs, cell therapies and immunotherapies targeting the
activity of immune cells within the TME represent promising avenues
for future GC immunotherapies.
Composition of the GC TME: The cornerstone
of immune suppression
Cellular components
GC TMEs constitute a complex ecosystem of cellular
and non-cellular components that collectively govern tumor
initiation, progression and immune escape.
Tumor cells
As the central drivers of the TME, tumor cells shape
an immunosuppressive environment through multidimensional
mechanisms. These cells reprogram the TME from an initial antitumor
state to one that favors tumor progression and immune evasion
(15). For example, tumor cells
upregulate PD-L1, which binds to programmed cell death protein 1
(PD-1) on T-cells, to inhibit their activation and effector
functions (16). Furthermore, the
IKAROS family zinc finger 4/non-POU domain-containing
octamer-binding-RAB11 family interacting protein 3 axis reinforces
PD-L1 recycling and surface expression, further suppressing
antitumor immune responses (17).
GC cells also frequently exhibit hypermethylation of the
β2-microglobulin promoter and loss of the transcription factor NLR
family CARD domain containing 5. These alterations inhibit the
synthesis and trafficking of major histocompatibility complex class
I (MHC-I) molecules, hindering effective antigen presentation to
CD8+ T-cells (18).
Zhang et al (19)
demonstrated that tumor cells reprogram neutrophil glucose
metabolism to induce a pro-tumor N2 phenotype and deliver exosomal
microRNA (miR)-4745-5p/miR-3911 to downregulate slit guidance
ligand 2, thereby promoting GC metastasis. This metabolic rewiring
not only sustains tumor growth but also impairs immune cell
function. Antigenic stimulation and metabolic stress drive T-cell
exhaustion, characterized by the co-expression of numerous immune
checkpoints and reduced cytokine secretion. These changes lead to
permanent immune tolerance (20,21).
Immune cells
Immune cells within the TME are key mediators of
immunosuppression and immune escape in GC. Tumor-associated
macrophages (TAMs) secrete IL-10 and TGF-β and express PD-L1 to
inhibit T-cell function. This subsequently suppresses T-cell
activity within the TME, promotes immune escape and is further
reinforced through PI3K-AKT signaling (22,23).
Tumor-associated neutrophils (TANs), recruited through
IL-17/NF-κB/RELB proto-oncogene, NF-kB subunit signaling, adopt a
pro-tumor N2 phenotype and upregulate PD-L1, aiding tumor cells in
evading immune attack (24).
Myeloid-derived suppressor cells (MDSCs) impair the functions of
cytotoxic T-cells and natural killer (NK) cells, facilitating tumor
immune escape (4). Tregs express
forkhead box P3 (FOXP3) and secrete IL-10 within the TME to
suppress effector T-cells, maintain tolerance and drive immune
escape (25). Although NK cells can
recruit CD8+ T-cells through the C-C motif chemokine
ligand (CCL)-3/CCL4/C-C motif chemokine receptor (CCR)-5 axis to
enhance antitumor responses, their cytotoxic activity is often
diminished by the immunosuppressive environment (26).
CAFs
As the predominant stromal cells in the GC TME, CAFs
regulate immunosuppression through a number of signaling axes,
including the TGF-β, IL-6/JAK/STAT and CXCL12/CXCR4 signaling axes.
In GC, CAFs (derived from mesenchymal cells) exhibit high
heterogeneity in their origin, phenotype and function (27), secreting key factors that impair
antitumor components of the immune system.
TGF-β promotes epithelial-mesenchymal transition
(EMT) through the canonical TGF-β/SMAD signaling cascade, enhancing
tumor invasion and dissemination. It also activates STAT3
signaling, a pathway involved in recruiting and polarizing M2
macrophages and Tregs (28). IL-6
sustains a cancer stem-like state through Janus kinase
(JAK)-2/STAT3 activation and impairs dendritic cell (DC) maturation
and antigen presentation, thereby blocking effector T-cell
activation (29). C-X-C motif
chemokine ligand (CXCL)-12 binds to C-X-C motif chemokine receptor
4 on immune cells to recruit Tregs and MDSCs, reinforcing the
immunosuppressive network (30).
Metabolically, CAFs provide tumor cells with substrates, including
lactate, ketone bodies and glutamine through autophagy and aerobic
glycolysis. Meanwhile, tumor-derived by-products exacerbate hypoxia
and nutrient deprivation, inducing metabolic stress in immune cells
and facilitating immune escape (31). Furthermore, increased CAF glycolysis
produces more lactate which both directly damages T-cell metabolism
and cytotoxicity, as well as acidifies the TME to facilitate MDSC
recruitment and activation (32).
Endothelial cells
Endothelial cells in the GC TME enable immune
evasion through abnormal angiogenesis and immunomodulation. GC
cells secrete exosomes and growth factors that activate endothelial
cells to initiate neovascularization (33). In hypoxic environments, tumor
endothelial cells promote an immunosuppressive environment by
upregulating von Willebrand factor and hypoxia-inducible factor
(HIF)-2α (34). Tumor-associated
endothelial cells weaken antitumor immunity by directly inhibiting
T-cell activity through expressing immune checkpoints including
PD-L1 and cytotoxic T-lymphocyte associated protein 4 (CTLA-4)
(35). Endothelial cells also
suppress the effector functions of T-cells, NK cells and DCs
through cytokine secretion (36). A
study demonstrated that endothelial-derived chemokines, including
IL-8 and CXCL1, selectively recruit immunosuppressive neutrophils
to perivascular regions, further reinforcing the local
immunosuppressive environment (37). Notably, endothelial cells and CAFs
engage in reciprocal interactions through the Notch signaling
pathway, cooperatively driving vascular abnormalities and
maintaining the immune barrier (38).
Non-cellular components
Extracellular matrix (ECM)
ECM, the primary non-cellular component of the GC
TME, contributes to establishing an immunosuppressive environment.
GC-associated ECM is heterogeneous and undergoes dynamic
remodeling, which modulates tumor cell proliferation, migration and
invasion, as well as immune cell infiltration and function
(39). Sustained stimulation by
CAFs triggers extensive fibrotic remodeling of the GC ECM whereby
the dense structure transmits mechanical stress to tumor and immune
cells, disrupting the T-cell cytoskeleton and impairing motility
(40,41).
The ECM serves an important role in
immunosuppression through a number of mechanisms. For example,
excessive ECM deposition elevates interstitial fluid pressure,
which restricts immune cell infiltration and drug penetration. In
addition, specific ECM components including serpin family E member
1 and collagen IV-α1 enhance immune tolerance and tumor progression
by upregulating TGF-β1 and matrix metalloproteinases (42,43).
The stiffness and porosity of the ECM also regulates cancer and
stromal cell behavior. Increased matrix stiffness activates
intracellular mechanotransduction pathways to enhance tumor cell
proliferation and survival, induce EMT and enhance invasion and
metastasis (44).
Soluble factors
Soluble factors including cytokines, chemokines,
growth factors and metabolites, collectively drive GC progression
and immune evasion by modulating immune cell function, promoting
tumor growth and metastasis and remodeling the stromal compartment.
Among these, IL-6 and TGF-β are key immunosuppressive cytokines.
IL-6 activates the JAK/STAT3 pathway to promote tumor cell
proliferation and survival while inhibiting CD8+ T-cell
function (45). TGF-β exacerbates
immunosuppression by inducing Treg differentiation and suppressing
effector T-cell activity (46).
The chemokine network orchestrates immune cell
recruitment. CCL2 recruits M2 TAMs, CXCL12 attracts Tregs and MDSCs
and CXCL8 (also known as IL-8) recruits neutrophils, together
establishing an immunosuppressive cellular microenvironment
(47). Growth factors, particularly
vascular endothelial growth factor (VEGF), not only drive
angiogenesis but also impair DC maturation and function, thereby
weakening antitumor immunity (48).
Metabolites further reinforce immunosuppression in
the GC TME. For example, lactate produced by tumor glycolysis
lowers extracellular pH, inhibiting the function of T-cells and NK
cells while promoting Treg and MDSC activity (49). Similarly, adenosine activates the
A2A receptor to suppress effector T-cell responses and facilitate
Treg differentiation (50).
Physicochemical conditions
Physicochemical properties of GC TMEs are markedly
dysregulated, contributing to immunosuppression by altering immune
cell metabolism and function. Acidification, primarily driven by
lactic acid accumulation from tumor glycolysis, directly impairs
T-cell and NK cell activity, diminishing antigen presentation and
cytokine secretion to weaken antitumor immunity. Moreover, acidic
conditions polarize TAMs toward an M2 phenotype, further
reinforcing immunosuppression (49). Hypoxia, resulting from aberrant
angiogenesis and inadequate perfusion, promotes tumor cell
survival, invasion and metastasis through HIF-1α signaling, while
also facilitating the recruitment and activation of
immunosuppressive cells (51).
Elevated interstitial fluid pressure, a key component of the TME,
impedes immune cell infiltration into the tumor core, confining
effector cells to the periphery and limiting their cytotoxic
capacity (52).
In summary, the GC TME represents a heterogeneous
‘ecosystem’ in which cellular and non-cellular components interact
to establish an immunosuppressive network (Fig. 1). Table
I summarizes the principal components of the GC TME and their
key immunosuppressive mechanisms, providing a framework for
subsequent analyses and therapeutic strategy design.
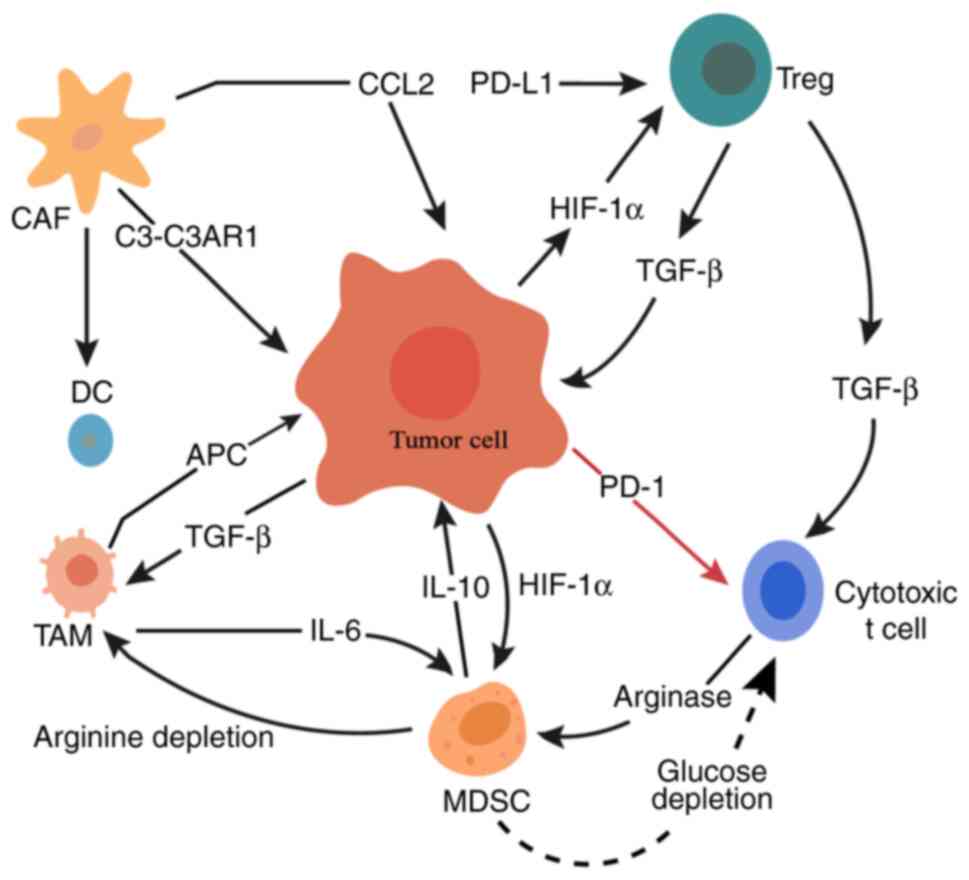 | Figure 1.Schematic diagram of immune
regulatory mechanisms in the tumor microenvironment of gastric
cancer. MDSCs suppress the activity of immune effector cells
through metabolic pathways. This leads to increased immune energy
depletion and enhanced immunosuppression within the tumor
microenvironment. The red arrow indicates inhibition, the black
arrows indicate promotion, and the dotted arrow represents indirect
metabolic signaling. MDSC, myeloid-derived suppressor cell; CAF,
cancer-associated fibroblast; DC, dendritic cell; C3-C3AR1,
complement C3-complement C3a receptor 1; CCL2, C-C motif chemokine
ligand 2; APC, antigen-presenting cell; TAM, tumor-associated
macrophage; HIF-1α, hypoxia-inducible factor 1α; PD-1, programmed
cell death protein 1; Treg, regulatory T-cell; PD-L1, programmed
death-ligand 1. |
 | Table I.Major cellular and non-cellular
components of the GC TME and their immunosuppressive
mechanisms. |
Table I.
Major cellular and non-cellular
components of the GC TME and their immunosuppressive
mechanisms.
| Component | Key factors and
pathways | Mechanisms of
immune suppression and impact |
|---|
| Tumor cells | PD-L1/PD-1
pathway | Inhibit T-cell
activation, impair antigen presentation and |
|
|
IKZF4/NONO-RAB11FIP3 axis | induce immune
tolerance and stress in immune cells |
|
| β2-microglobulin
hypermethylation |
|
|
| NLRC5 loss |
|
|
| Metabolic
reprogramming |
|
| Immune cells | IL-10 | Suppress T-cell
function, maintain immune tolerance and |
|
| TGF-β | promote immune
escape |
|
| PD-L1 |
|
|
| IL-17/NF-κB/RelB
signaling |
|
|
| PI3K-AKT
pathway |
|
|
| FOXP3 |
|
|
| CCL3/CCL4-CCR5
axis |
|
|
Cancer-associated | TGF-β/SMAD
signaling | Promote tumor
invasion, inhibit T-cell activation and |
| fibroblasts | IL-6/JAK2/STAT3
signaling | increase metabolic
stress, |
|
| CXCL12/CXCR4 | aiding immune
escape |
|
| Autophagy |
|
|
| Aerobic
glycolysis |
|
| Endothelial
cells | HIF-2α | Inhibit T-cell and
NK-cell activity, recruit |
|
| Von Willebrand
factor | immunosuppressive
cells and maintain immune barrier |
|
| PD-L1 and
CTLA-4 |
|
|
| IL-8 and CXCL1 |
|
|
| Notch
signaling |
|
| Extracellular
matrix | SERPINE1 | Restrict immune
cell access, enhance tumor progression |
|
| COL4A1 | and limit drug
penetration |
|
| TGF-β1 |
|
|
| MMPs |
|
|
| ECM stiffness and
porosity |
|
| Soluble
factors | IL-6 | Inhibit T-cell
function, recruit immunosuppressive cells |
|
| TGF-β | and impair immune
responses |
|
| CCL2 |
|
|
| CXCL12 |
|
|
| CXCL8 (IL-8) |
|
|
| VEGF |
|
|
| Lactate |
|
|
| Adenosine |
|
|
Physicochemical | Lactic acid | Inhibit T-cell and
NK cell activity, promote tumor survival |
| conditions | HIF-1α | and limit immune
cell infiltration |
|
| Interstitial fluid
pressure |
|
Molecular mechanisms of immune
tolerance
Impairment of antigen presentation and
recognition
GC cells employ multiple strategies to downregulate
MHC-I expression, which hampers the presentation of
tumor-associated antigens to CD8+ T-cells, facilitating
immune evasion (53). Concurrently,
persistent hypoxia in the TME increases HIF-1α levels. HIF-1α
inhibits the function of antigen-processing enzymes and impairs
peptide loading onto MHC-I molecules (54,55).
In addition, the DCs within the TME exhibit limited maturation and
activation, reduced expression of costimulatory molecules
(CD80/CD86) and impaired antigen uptake and processing. This
deprives T-cells of adequate activation molecules, namely signal II
and signal III cytokines. This dual impairment markedly reduces the
primary T-cell response, establishing an initial barrier to
antitumor immunity and promoting immune tolerance (56,57).
Activation of immune checkpoint
signaling
Immune checkpoints act as key negative regulators of
T-cell activity. In GC, PD-L1s on tumor cells bind to PD-1 on
T-cells, recruiting the Src homology-2 protein tyrosine
phosphatase, blocking the PI3K/AKT pathway, reducing T-cell
metabolism, impairing cytotoxicity and promoting cell cycle arrest
(58). CTLA-4, also upregulated in
the GC microenvironment, has a higher affinity for CD80/CD86
compared with CD28, thus inhibiting costimulatory signals and
suppressing naive T-cell activation and proliferation (59).
Furthermore, CD8+ T-cells infiltrating GC
upregulate both T-cell immunoglobulin and mucin domain 3 (TIM-3).
The interaction of galectin-9 with its ligand (TIM-3) induces the
apoptosis of T-cells and also exacerbates T-cell exhaustion,
sustaining an immunosuppressive microenvironment, even in the
context of PD-1 blockade, thus driving therapeutic resistance
(60).
Immunosuppressive cytokines and
receptor networks
Inhibitory cytokines within the GC TME establish an
autocrine-paracrine suppressive network. TGF-β directly inhibits
the proliferation and cytokine secretion of effector T-cells. In
addition, TGF-β drives the differentiation of naive CD4+
T-cells into Tregs by upregulating FOXP3 (61). IL-10 activates STAT3 to polarize DCs
and macrophages toward a tolerogenic phenotype. It also reduces the
production of proinflammatory cytokines (such as IL-12 and TNF-α),
thereby suppressing effector T-cell activation (62). VEGF impairs DC maturation through
VEGFR-2 and reduces antigen presentation, further downregulating
endothelial adhesion molecules, restricting the infiltration of
CD8+ T cells and NK cells into the tumor parenchyma
(63,64).
Metabolic reprogramming-mediated
immune suppression
GC cells and tumor-associated DCs upregulate
indoleamine 2,3-dioxygenase 1 and tryptophan 2,3-dioxygenase, which
catalyze the conversion of tryptophan to kynurenine (Kyn).
Accumulated Kyn engages the aryl hydrocarbon receptor to drive
naive T-cell differentiation into Tregs, establishing an
immunosuppressive environment (65,66).
Arginase 1 (ARG1), abundant in TAMs and MDSCs, depletes arginine,
downregulates the CD3ζ chain expression and attenuates T-cell
receptor (TCR) signaling and cytotoxicity (67). Lipid droplets accumulate in TAMs and
tumor cells, generating oxidized lipids that activate the NOD-like
receptor pyrin domain-containing 3 inflammasome through endoplasmic
reticulum stress or reactive oxygen species (ROS), further
reinforcing immunosuppression (68). Furthermore, excess cholesterol and
its derivatives (such as bile acids, steroid hormones and
oxysterols) bind to fatty acid-binding proteins in Tregs and MDSCs,
regulating their lipid metabolism to enhance survival and
suppressive function (69).
Epigenetic and transcriptional
regulation
GC cells utilize DNA methyltransferases and enhancer
of zeste homolog 2 (EZH2) to silence proinflammatory genes (such as
IFN-γ and TNF-α) while upregulating immunosuppressive loci
(70,71). In T-cells, epigenetic modifications
govern the balance between exhaustion and memory phenotypes
(72). In TAMs, the downregulation
of miR-155 relieves the repression of suppressor of cytokine
signaling 1, limiting M1 polarization, as restoring miR-155
reinstates M1 characteristics and enhances antitumor inflammatory
responses (73,74). In addition, the long non-coding RNA
PVT1 binds to EZH2 to prevent its degradation, sustaining the
histone H3 lysine 27 trimethyltransferase activity of EZH2 in
Tregs. This upregulates FOXP3 expression, preserving Treg-mediated
immunosuppression (75,76).
Information transfer mediated by
exosomes and microvesicles
Exosomes secreted by tumor cells carry miR-21, which
targets antigen presentation transcription factors and MHC class II
(MHC-II) molecules in DCs. This interaction impairs DC maturation
and antigen presentation capabilities, thereby suppressing the
activation of CD4+ and CD8+ T-cells (77). Tumor-derived exosomes (TDEs) can
accumulate in distant organs and modulate the local TME, promoting
immunosuppressive stromal remodeling and establishing a
pre-metastatic niche conducive to tumor cell colonization (78). A study has shown that inhibiting the
expression of neutral sphingomyelinase 2 or Ras-related protein
Rab27a, markedly reduces exosome production and secretion, thereby
enhancing immune cell function and improving the efficacy of
antitumor therapies (79).
Infiltration and interaction of
immunosuppressive cells
CCL17 and CCL22 are chemokines highly expressed in
the GC TME, which serve key roles in shaping the tumor immune
microenvironment (80). In patients
with GC, MDSC levels are notably elevated, as these cells
overexpress ARG1 and inducible nitric oxide synthase (iNOS; which
produces reactive nitrogen species). This depletion of arginine in
the TME hinders the proliferation of effector T-cells and reduces
the expression of the TCR ζ-chain (81).
Macrophages undergo M2 polarization in response to
colony stimulating factor (CSF)-1/CSF-1R and IL-4/IL-13 signals.
M2-polarized TAMs secrete CCL18 and activate the ERK1/2-NF-κB
pathway to promote tumor cell invasion. Concurrently, M2 TAMs
upregulate VEGF-A, which drives aberrant angiogenesis and impedes
effector T-cell infiltration (82,83).
Overall, these molecular mechanisms of immune
tolerance involve numerous pathways, including impaired antigen
presentation, immune checkpoint activation, cytokine signaling,
metabolic reprogramming, epigenetic regulation, exosomal transfer
and immunosuppressive cell infiltration. These mechanisms act both
independently and collaboratively through cross-regulatory
networks, collectively forming the molecular basis for GC immune
escape.
Core immune cells driving immune tolerance
in the GC TME and their interactions
MDSCs
MDSCs are key immune regulators that induce immune
tolerance in the TME. Under physiological conditions, MDSCs
differentiate into macrophages, granulocytes or DCs upon
maturation. However, persistent inflammatory cues in the TME
prevent their own differentiation into mature cells and promote
their expansion. As a result, MDSCs subvert T-cell function through
increased accumulation in local tumor tissue and peripheral
circulation (84).
MDSCs suppress antitumor immune responses through a
number of mechanisms. These cells produce high levels of ROS and
excessive peroxynitrite, which oxidize and nitrate T-cell surface
receptors and co-stimulatory molecules, directly impairing T-cell
recognition and cytotoxic functions (85,86).
Additionally, MDSCs secrete immunosuppressive enzymes including
ARG1 and iNOS, further inhibiting T-cell function and weakening
antitumor immunity (87). In GC,
MDSC accumulation is markedly associated with CD8+
T-cell dysfunction, facilitating tumor progression. These cells
suppress CD8+ T-cell activity and proliferation either
through direct cell-cell interactions or through releasing
inhibitory factors (88). MDSCs
also exacerbate immunosuppression by promoting the increase of
Tregs (89). Tumor cells recruit
MDSCs into the TME through TDEs or chemokines including CXCL12 and
CXCL5. Notably, miR-107 secreted by GC cells is transferred to
MDSCs through exosomes, where it downregulates the expression of
the dicer 1, ribonuclease III gene as well as phosphatase and
tensin homolog. This promotes the increase and activation of MDSC,
intensifying immunosuppression within the TME (90). Furthermore, in the GC TME, MDSC
accumulation is associated with elevated levels of inflammatory
cytokines such as IL-6, TNF-α and IL-22. These cytokines not only
enhance MDSC recruitment and activation but also aggravate immune
suppression by inducing the differentiation of T helper (Th)-22 and
Th17 cells (91).
In summary, MDSCs serve a key role in promoting
immune tolerance in the GC TME by inhibiting T-cell function,
facilitating Treg recruitment and interacting with cytokines
amongst other immune cells.
TAMs
TAMs are among the most abundant immune cell types
in the TME. Their functional plasticity and interactions with other
cells markedly suppress antitumor immune responses and promote
tumor progression. TAMs are highly plastic, polarizing into
pro-inflammatory M1 or immunosuppressive M2 phenotypes. The
majority of M2-polarized TAMs in GC contribute to immune tolerance
by secreting immunosuppressive cytokines (including IL-10 and
TGF-β) and promoting Treg infiltration (92). Cytokines within the TME enhance Treg
recruitment and differentiation while inhibiting effector T-cell
function (93). M2 TAMs also
increase ARG1 levels, which deplete local arginine stores and
impair T-cell function (94).
Interactions between TAMs and other immune cells
further exacerbates immune tolerance in GC. TAMs promote Treg
activation and infiltration through TGF-β secretion, thereby
suppressing CD8+ T-cell activity. Additionally, the
CCL5-CCR5 signaling axis between TAMs and NK cells contribute to a
chemotherapy-induced immunosuppressive TME (95). TAMs also regulate MDSCs, as through
shared metabolic inhibitory pathways and cytokine networks
(including IL-6 and TGF-β), TAMs and MDSCs jointly consume key
nutrients for T-cell activation and cooperatively inhibit DC
maturation, leading to impaired antigen presentation and reduced
T-cell-mediated immunity (96,97).
In addition, TAMs directly suppress NK cell function by
downregulating NK group 2 member D receptor expression and
inhibiting the release of perforin and granzyme (through IL-10
secretion or human leukocyte antigen G expression), thereby
reducing NK cell cytotoxicity against GC cells (98). Notably, IL-10 and TGF-β secreted by
TAMs induce the differentiation of naive CD4+ T-cells
into FOXP3+ Tregs and support their functional
stability. Conversely, IL-35 secreted by Tregs further promotes TAM
polarization toward the immunosuppressive M2 phenotype,
establishing a TAM-Treg positive feedback loop that reinforces
immunosuppression (99,100).
In summary, TAMs in the GC TME orchestrate a complex
immunosuppressive network through synergistic interactions with
other immune cells and cytokines, ultimately hindering the
initiation of effective antitumor immune responses.
Tregs
Tregs serve a key role in maintaining immune
homeostasis by preventing autoimmunity. Within the GC TME, Tregs
employ multiple mechanisms to suppress immune attacks and support
tumor growth. Tregs upregulate CTLA-4, which competitively binds to
CD80/CD86 on effector T-cells and antigen-presenting cells (APCs)
to block co-stimulatory signals. This inhibits CD8+
T-cell activation and proliferation (101). In addition, Tregs suppress
effector T-cell function through both the release of
immunosuppressive cytokines and direct cell-cell contact (102). Treg infiltration is associated
with GC progression and a poor prognosis. Tregs sustain an
immunosuppressive TME, supporting tumor cell proliferation,
invasion and metastasis (103).
Tregs also interact closely with other immune cells in the TME, as
they secrete TGF-β and other factors to polarize TAMs toward the M2
phenotype, enhancing immunosuppression (95). Tregs block DC maturation and antigen
presentation, hampering the initiation of antitumor immunity
(104). Tregs also secrete IL-10
and TGF-β, which weaken NK cell cytotoxicity and negatively
regulate NK cell-mediated tumor cell death (105).
Notably, studies have demonstrated a bidirectional
regulatory relationship between Tregs, MDSCs and TAMs. MDSCs
secrete chemokines such as CCL5 to recruit Tregs into tumor tissue.
In turn, TGF-β released by Tregs further enhances MDSC
differentiation into a suppressive state. These interactions
establish an immunosuppressive loop that reinforces immune
tolerance in the TME (84,106). Overall, Treg function is tailored
to the TME, enabling them to establish an efficient,
self-sustaining immunosuppressive network in the GC TME. Thus,
Tregs are key drivers of tumor immune tolerance.
DCs
As key APCs, DCs serve as important regulators of
immune responses within the TME. In GC, DC function is frequently
impaired, driving immune tolerance and promoting tumor progression
and metastasis. This dysfunction is closely linked to aberrant
activation of the PI3K/AKT and EMT signaling pathways. Notably,
these pathways not only enhance the immunosuppressive properties of
DCs but also facilitate tumor invasion and metastasis.
Metabolic dysregulation in the TME profoundly
modulates DC function, as dysregulated glucose metabolism
diminishes their antigen-presenting capacity, while abnormal lipid
metabolism promotes the development of an immunosuppressive DC
phenotype (107). Furthermore,
TDEs and immunosuppressive cytokines in the GC TME suppress DC
function through paracrine mechanisms. These factors impair DC
maturation while sustaining the proliferation of other
immunosuppressive cell populations (108,109).
Within the GC TME, DCs exhibit downregulated surface
expression of co-stimulatory molecules, as well as MHC-I and MHC-II
molecules, markedly reducing antigen presentation efficiency. This
leads to insufficient infiltration of tumor-specific
CD8+ T-cells and their subsequent dysfunction (110). In addition, aberrantly activated
plasmacytoid DCs (pDCs) in GC tissues promote the expansion of
FOXP3+ Tregs through the inducible co-stimulator
(ICOS)-ligand/ICOS axis. High levels of indoleamine 2,3-dioxygenase
secreted by pDCs further contribute to Treg induction and the
maintenance of their suppressive function (104,111).
Functional defects in DCs also impair NK cell
activity, as in the GC TME, DCs fail to adequately activate NK
cells, resulting in reduced antibody-dependent cellular
cytotoxicity and diminished tumor cell clearance (112).
Intercellular interactions in the
immune suppression network
In the GC TME, immunosuppressive cells do not act in
isolation. Instead, they cooperate through intricate intercellular
interactions and molecular signaling networks to establish a highly
integrated immunosuppressive system. A key mechanism underlying
this cooperation involves chemokine-receptor interactions, which
are key in recruiting and localizing inhibitory immune cells. Tumor
and stromal cells secrete large quantities of chemokines through
chemokine receptor-mediated pathways, facilitating the recruitment
of MDSCs, TAMs and Tregs to the tumor site (113,114).
For example, CAFs produce CCL2 to attract TAMs and
activate them through the JAK/STAT3 signaling pathway (115). CAFs also recruit and polarize TAMs
through the complement C3 (C3)-complement C3a receptor 1 (C3AR1)
axis, helping to establish an immunosuppressive microenvironment
(116). Importantly, TAM-derived
TGF-β and IL-6 further sustain CAF activation, creating a positive
feedback loop that amplifies immunosuppression (117).
Beyond chemokine signaling, direct cell-cell
interactions, metabolic crosstalk and cytokine networks
collectively strengthen the suppressive microenvironment. Tregs
inhibit APCs by competitively binding to B7 molecules through
CTLA-4, blocking CD28-mediated co-stimulatory signaling. Tregs also
secrete TGF-β, which disrupts the metabolic reprogramming of
effector T-cells, leading to functional exhaustion and diminished
cytotoxicity (118,119). Lysosomal associated membrane
protein 3+ DCs interact with tumor-associated stromal
cells through the nectin cell adhesion molecule 2-T-cell
immunoglobulin and ITIM domain axis, suppressing CD8+
T-cell activity and promoting Treg enrichment to promote immune
tolerance (120).
MDSCs consume arginine through their arginase
activity, impairing T cell function. TAMs further enhance MDSC
recruitment and activation by consuming lipids released by tumor
cells (121,122). Tumor cells also engage in
metabolic competition to suppress immunity. Excess glucose
consumption by tumor cells (which exhibit a high glycolytic
phenotype) deprives infiltrating T-cells of energy, inducing their
functional exhaustion (123).
Meanwhile, the metabolic microenvironment sustains Treg survival
and suppressive capacity by regulating their fatty acid metabolism,
preserving their immunosuppressive phenotype (124).
Tumor cells actively shape the immunosuppressive
network by releasing signaling molecules, including VEGF,
prostaglandin E2 (PGE2) and HIF-1α. VEGF inhibits DC
differentiation and maturation through VEGFR binding, reducing
antigen presentation and leading to the formation of a ‘cold’ tumor
microenvironment (125). Elevated
PGE2 levels upregulate p50, NF-κB and inhibitory gene expression in
MDSCs, enhancing their suppressive activity. PGE2 also promotes M2
polarization of TAMs, increasing the secretion of immunosuppressive
cytokines including IL-10 and TGF-β (126). HIF-1α directly binds to the
promoter region of the PD-L1 gene, inducing its expression in tumor
cells and MDSCs. Concurrently, HIF-1α upregulates chemokines such
as CCL28 and CXCL12, facilitating the selective recruitment of
Tregs and MDSCs to hypoxic tumor regions and intensifying
immunosuppression (127,128).
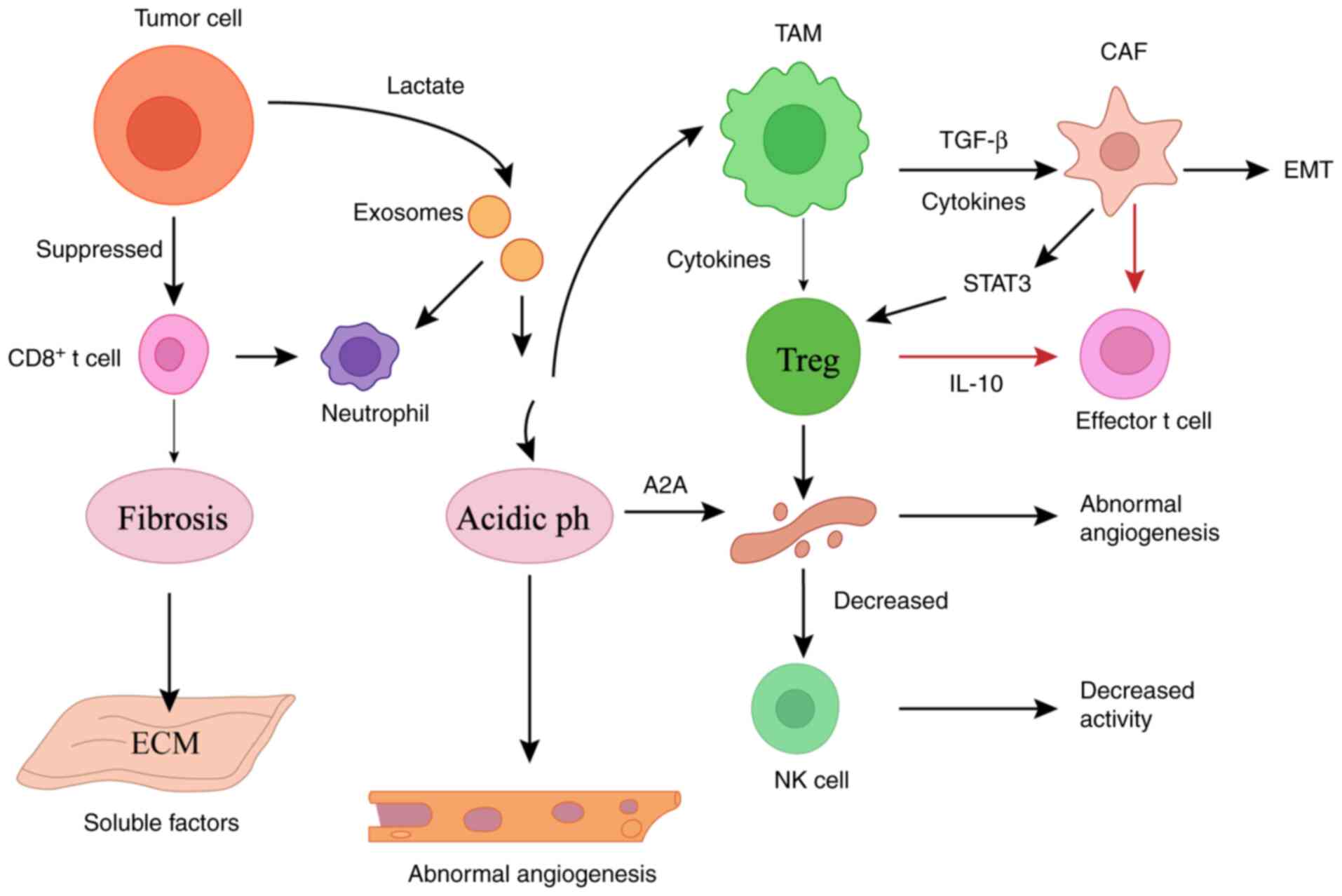
In summary, immune tolerance in the GC TME is not an
isolated trait of individual cells but rather a highly dynamic and
plastic inhibitory network established through complex interactions
among principal immune cell populations (Fig. 2).
Treatment strategies targeting immune
tolerance in the TME: Challenges and opportunities
ICIs
ICIs represent a transformative therapeutic strategy
that targets immunosuppressive mechanisms in the TME, reshaping
cancer treatment paradigms. PD-1, a co-inhibitory receptor that
binds to its ligand PD-L1, is frequently expressed on GC cells,
where it inhibits T-cell function to enable immune evasion. PD-1
inhibitors (nivolumab, pembrolizumab and camrelizumab) restore
T-cell function and alleviate the immunosuppressive state of the GC
TME (129,130).
CheckMate-649 (131) is a pivotal global phase III
clinical trial to assess the efficacy and safety of nivolumab
combined with chemotherapy as first-line treatment for patients
with advanced GC, gastroesophageal junction cancer (GEJC) and
esophageal adenocarcinoma (EAC). The trial enrolled 1,581
treatment-naive patients with advanced or metastatic
non-HER2+ GC, GEJC or EAC, who were randomly assigned to
either the nivolumab + chemotherapy group or the chemotherapy-alone
group. Primary endpoints included overall survival (OS) and
progression-free survival (PFS), with secondary endpoints such as
health-related quality of life also assessed. The results showed
that nivolumab combined with chemotherapy notably prolonged both OS
and PFS, with more pronounced efficacy in patients with a PD-L1
combined positive score (CPS) ≥5.
KEYNOTE-062, a phase III trial, compared
pembrolizumab monotherapy, pembrolizumab + chemotherapy and
chemotherapy alone in patients with untreated advanced GC or
gastroesophageal junction (G/GEJ) cancer. In Asian patients,
pembrolizumab monotherapy resulted in a longer OS in those with a
PD-L1 CPS ≥1 (22.7 months) and CPS ≥10 (28.5 months), with
favorable tolerability (132).
Additional analyses of KEYNOTE-062 demonstrated that pembrolizumab
monotherapy was non-inferior compared with chemotherapy in patients
with untreated advanced GC/G/GEJ cancer and exhibited an improved
safety profile (133).
KEYNOTE-590 (134),
another important trial, primarily assessed pembrolizumab combined
with chemotherapy for esophagogastric junction tumors. Among
patients with esophageal squamous cell carcinoma (ESCC) and a PD-L1
CPS ≥10, the median PFS and OS were 7.3 and 13.9 months,
respectively. In all patients with ESCC, these values were 6.3 and
12.6 months. These findings demonstrate that pembrolizumab combined
with chemotherapy markedly delays disease progression and improves
patient survival.
While ICIs have improved GC treatment, not all
patients derive benefit. Identifying appropriate patient
populations and developing more effective combination regimens to
broaden clinical benefit remain key challenges, largely due to
notable heterogeneity in treatment responses. PD-L1 has been used
as a biomarker to predict immunotherapy responses in clinical
trials, but its sensitivity and specificity remain unresolved.
Current research is also exploring inhibitors targeting other
immune co-inhibitory molecules, with the goal of overcoming the
limitations of single-agent therapies through multi-target
combination strategies. Furthermore, future studies should aim to
focus on overcoming tumor adaptive resistance through combination
strategies, such as pairing ICIs with TME remodeling
approaches.
Cell therapy
In recent years, CAR-T immunotherapy has emerged as
a promising advancement in cancer immunotherapy. CAR-T therapy
involves engineering T-cells to express a CAR, fusing a tumor
antigen-specific single-chain variable fragment with intracellular
activation domains, endowing T-cells with MHC-independent tumor
cell recognition and cytotoxic capacity.
CT041, a CAR-T therapy targeting Claudin18.2
(CLDN18.2), has shown promise in GC treatment. An ongoing phase I
trial (trial ID no. NCT03874897) is assessing the safety and
efficacy of CT041 in patients with CLDN18.2-positive digestive
system cancers (135). Data from
37 treated patients showed an overall response rate (ORR) of 48.6%
and a disease control rate (DCR) of 73.0%. In patients with GC, the
ORR was 57.1% while DCR was 75.0% and the 6-month OS rate was
81.2%. These results suggest CT041 is effective and well tolerated
in GC.
Beyond CLDN18.2, CAR-T research in GC has focused on
other targets such as mucin 1 (cell surface associated), HER2 and
CEA. For example, clinical trials are underway for TCR-based
therapies targeting MAGE family member A4 (MAGE-A4). While MAGE-A4
expression in GC is relatively low (9%), MAGE-A4-targeted TCR
therapy is also under investigation in other solid tumors (136). Meyer et al (137) assessed the safety and preliminary
efficacy of AFP-targeted TCR-T cells (ADP-A2AFP) in a phase I trial
(trial ID no. NCT03132792) for patients with gastric hepatoid
carcinoma. Results showed this therapy induced complete and partial
remissions in some patients, with manageable toxicity.
NK cells, either expanded in vitro or derived
from induced pluripotent stem cells, can bypass immune evasion by
recognizing tumor cells with reduced MHC-I expression. A study has
detailed that activated NK cells exhibit enhanced cytotoxicity
against GC cells (138).
Currently, numerous GC-related cell therapy studies
are in phase I/II trials, which primarily evaluate safety and early
efficacy, and have not yet advanced to phase III registration
studies. Phase I trials typically investigate safe dosages, toxic
reactions and cellular kinetics, CAR-T cells can proliferate within
the body and effectively target tumor cells in the short term;
however, their long-term persistence is limited. These cells may be
eliminated by the immune system or lose their functionality due to
immune evasion mechanisms, such as the inhibitory effects of the
TME. Phase II trials are initiated to explore combination regimens,
and early data suggests these combinations may modestly improve
efficacy but at the cost of increased adverse events. The lack of
phase III studies is mainly attributed to patient heterogeneity,
complex manufacturing processes and cost considerations.
Cell therapy for GC also faces ongoing challenges,
including tumor heterogeneity and difficulties in target selection.
GC lacks highly specific antigen targets and single-target
heterogeneity can lead to antigen escape (139). The short lifespan of CAR-T cells
also hinders the establishing of long-term immune memory.
Furthermore, CAR-T cell overload can cause high fever and organ
dysfunction, highlighting the importance of managing adverse events
(140,141). Most notably, clinical validation
systems remain incomplete and large-scale multi-center randomized
controlled phase III trials to confirm efficacy are still
lacking.
While cell therapy holds promise for GC
immunotherapy, its application is limited by the complex regulation
of the TME, tumor heterogeneity and immunosuppressive states. The
majority of current studies are still in phase I/II stages, as
while encouraging efficacy signals exist, they are insufficient to
support widespread clinical implementation. To enhance cell therapy
effectiveness, future efforts should focus on ensuring each target
elicits appropriate immune responses and establishing triggered
self-destruction mechanisms to mitigate adverse effects.
Antibody-drug conjugates (ADCs)
Antibody-drug conjugates consist of a monoclonal
antibody targeting tumor-associated antigens, a cleavable linker
and a highly potent cytotoxic payload. They integrate the
specificity of monoclonal antibodies with the same efficacy as
small-molecule drugs at inducing cell death. Trastuzumab
deruxtecan, a novel anti-HER2 ADC, has exhibited manageable safety
profiles and robust tumor activity in patients with
HER2+ advanced GC (142). This agent comprises a humanized
anti-HER2 monoclonal antibody, an enzyme-cleavable peptide linker
and the topoisomerase I inhibitor deruxtecan, with a high
drug-to-antibody ratio of 8:1 and offers the additional advantage
of a bystander effect (143).
Notably, disitamab vedotin (RC48) demonstrated
promising efficacy in phase I/II clinical trials involving patients
with HER2+ GEJC who had received at least two prior
lines of therapy. These patients achieved a median PFS of 3.8–4.1
months and a median OS of 6.8–7.9 months. The majority of adverse
events were grade 1 or 2, confirming manageable safety (144). Ongoing studies are evaluating the
combination of RC48 with PD-1 inhibitors, with preliminary findings
indicating enhanced antitumor activity and acceptable toxicity,
suggesting that pairing ADCs with ICIs may further improve
therapeutic efficacy (145).
However, numerous barriers limit ADC effectiveness.
Dense stroma and high endocytosis rates restrict ADC penetration
and distribution within the tumor parenchyma. In addition, elevated
lysosomal enzyme activity or the use of non-cleavable linkers can
impair intracellular drug release efficiency (146). Tumor cells can also develop
resistance to ADCs by downregulating target antigens, upregulating
ATP-binding cassette transporters to promote drug efflux or
altering endocytic and lysosomal trafficking (147). In the future, combining liquid
biopsy-based biomarkers (such as circulating tumor cells and
cell-free tumor DNA) with multi-omics approaches may facilitate
real-time monitoring of treatment responses and resistance
patterns, guiding the personalized use of ADCs (148).
Overall, ADCs represent an innovative therapeutic
platform that merges targeted delivery with potent cytotoxicity,
demonstrating efficacy in overcoming immune tolerance in GC. Future
advancements in linker design, multi-targeting strategies and
combinations with immunotherapies are expected to yield more
precise, effective and safer therapies for patients.
Biologics
Biologics hold notable promise for reversing immune
tolerance in the TME, owing to their specificity and diverse
immunomodulatory effects. Bispecific antibodies (BsAbs), a distinct
class of biologics, are engineered to bind simultaneously to two
distinct antigens or two epitopes on the same antigen. The core
mechanism of BsAbs involves enhancing antitumor immune responses by
co-targeting tumor cells and immune cells, such as T-cells. For
example, CD3 BsAbs direct T-cells to the tumor microenvironment,
activate them and induce tumor cell cytotoxicity (149,150). Furthermore, BsAbs can reshape the
TME and boost antitumor responses by blocking immunosuppressive
signals (such as PD-L1 and TGF-β) or activating co-stimulatory
signals (including CD28 and 4-1BB) (151,152).
Additional TME-modulating agents, such as
anti-TGF-β antibodies and VEGFR inhibitors, can modulate
immunosuppressive factors in the TME, thereby enhancing immune
cell-mediated tumor recognition and inducing cell death. Anti-TGF-β
therapy blocks the TGF-β signaling pathway, inhibits its protumor
effects and strengthens the antitumor response of the immune system
(153). A separate study found
that anti-TGF-β therapy can reprogram TANs from a protumor
phenotype to an antitumor phenotype, suppressing tumor growth and
metastasis (154). Anti-VEGFR
inhibitors enhance immune cell antitumor responses by inhibiting
tumor angiogenesis and increasing immune cell infiltration into the
tumor (155).
Jung et al (156) developed an oncolytic adenovirus
designed to express IL-12p35, IL-12p40, granulocyte macrophage-CSF
and relaxin. This construct efficiently degrades collagen networks,
modifies matrix density and enhances CD8+ T-cell
infiltration across a number of tumor types (including GC and
pancreatic cancer), markedly improving the efficacy of PD-1
blockade therapy. Additionally, eganelisib (IPI-549), an oral
selective inhibitor of phosphoinositide 3-kinase γ, restores the
function of TAMs and MDSCs to enhance immune effector activity. By
inhibiting PI3K signaling in both the TME and immune cells, IPI-549
enhances the efficacy of PD-1 and PD-L1 checkpoint inhibitors
(157).
Despite their promise, biologics face challenges in
clinical application. Accurate targeting is required to minimize
off-target toxicity, as immune responses against nanoparticle-based
carriers remain a concern and tumor cells can develop adaptive
resistance mechanisms (158,159).
In summary, biologics represent promising
therapeutic strategies for overcoming tumor immune tolerance by
precisely modulating the TME. Despite ongoing challenges, their
potential to reverse immunosuppression and enhance antitumor immune
responses remains notable. With continued technological innovation
and interdisciplinary collaboration, biologics may serve an
increasingly important role in cancer immunotherapy.
Overall, treatment strategies targeting immune
tolerance within the GC TME are moving away from single target
approaches toward multi-dimensional, coordinated regulation, with
the TME immunosuppressive network disruption at their core.
Although technical barriers persist, advances in understanding TME
heterogeneity and optimizing combination therapies are expected to
improve immunotherapy response rates and deliver meaningful
clinical benefits to patients with GC. Table II summarizes current
immunotherapeutic approaches for overcoming immune tolerance in GC,
highlighting the distinct mechanisms, clinical progress and
limitations of ICIs, cell-based therapies, antibody-drug conjugates
and biologics.
| Table II.Emerging immunotherapeutic strategies
for overcoming immune tolerance in GC. |
Table II.
Emerging immunotherapeutic strategies
for overcoming immune tolerance in GC.
| Therapy | Mechanism | Key results | Challenges and
future directions |
|---|
| Immune checkpoint
inhibitors | PD-1 inhibitors
(such as nivolumab and pembrolizumab) | Prolonged survival
in GC with PD-L1 CPS ≥5 | Heterogeneous
responses and PD-L1 biomarker controversy |
| Cell-based
therapy | CAR-T targeting
tumor antigens (such as CT041 and CLDN18.2) | 57.1% ORR, 75.0%
DCR in GC and 81.2% 6-month survival | Limited
persistence, antigen escape and severe toxicity |
| Antibody-drug
conjugates | Monoclonal
antibodies and cytotoxic drugs (such as trastuzumab and
deruxtecan) | Effective in
HER2+ advanced GC and GEJC | Limited tumor
penetration and resistance mechanisms |
| Biological
agents | Bispecific
antibodies (such as CD3 bispecific antibodies) | Enhances T-cell
activation and tumor cytotoxicity | Off-target toxicity
and adaptive resistance |
Differences between preclinical and clinical
evidence and their importance
In recent years, numerous studies have shown that
TME serves a key role in establishing and sustaining immune
tolerance in GC. To improve scientific rigor, the present review
distinguishes between preclinical and clinical evidence to avoid
conflating experimental results with clinical outcomes.
A preclinical study using animal models and in
vitro systems has shown that the TME regulates GC immune
tolerance through the activation of key signaling pathways
(160). For example, TAMs and
MDSCs jointly deplete nutrients important for T-cell activation and
alter DC maturation through shared metabolic inhibitory pathways
and cytokine networks. These mechanisms disrupt antigen
presentation, thereby diminishing T cell-mediated antitumor
responses. Additionally, the CAF-mediated C3-C3AR1 axis promotes
the recruitment and functional polarization of TAMs, establishing
an immunosuppressive microenvironment. These findings advance the
understanding of cellular mechanisms underlying TME-mediated immune
regulation.
However, preclinical outcomes do not directly
translate to clinical efficacy. Animal and cell-based models have
limitations in recapitulating the complexity of the human GC immune
microenvironment, including discrepancies in immune cell ratios,
simplified cytokine networks and the absence of a fully functional
human immune system. Thus, while preclinical studies provide
valuable clues for mechanistic investigation, their conclusions
require validation with large-scale clinical trials and real-world
data to demonstrate clinical relevance.
In clinical trials, analyses of patient tissue
samples, immunohistochemistry and transcriptomic data have further
confirmed the role of the TME in GC immune escape. For example, in
phase III trials such as CheckMate-649 and KEYNOTE-062, patients
with high PD-L1 expression and elevated immunosuppressive cell
infiltration often exhibit poor responses to immunotherapy. This
clinical evidence indicates that TME features are associated with
the efficacy of ICIs, reflecting the pathological state of tumors
and their response to treatment. However, fully validating the
precise mechanisms driving GC immune tolerance remains challenging
due to limitations in sample size, inadequate patient
stratification and short follow-up periods.
Overall, preclinical studies provide molecular and
cellular insights into TME-mediated immune tolerance in GC, while
clinical studies demonstrate the relevance and therapeutic
importance of these mechanisms in patients. By distinguishing
between these two types of evidence, the present review not only
enhances scientific integrity but also reduces the risk of
artefactual misinterpretation of experimental outcomes as clinical
conclusions. To advance TME-targeted immunotherapy for GC, future
research should aim to prioritize the translational validation of
preclinical findings.
Challenges and limitations of TME-targeted
therapeutic strategies
Although TME-targeted approaches have emerged as
promising strategies for GC immunotherapy recently, their clinical
efficacy has been limited in numerous aspects.
GC is a highly heterogeneous malignancy, with
notable variations in genomic profiles, immune cell composition and
microenvironmental features between patients and even across
different regions of the same tumor. This extensive heterogeneity
limits the effectiveness of single-agent TME-targeted therapies,
highlighting the need for personalized treatment strategies.
The TME is not just a ‘supportive environment’ for
tumor cells but a dynamic system that rapidly adapts to therapeutic
pressures. During immunotherapy, tumor cells often develop adaptive
resistance through multiple mechanisms. For example, while ICIs may
initially activate T-cells, immunosuppressive cells within the TME,
such as Tregs and TAMs, may reverse immune tolerance by modulating
immune responses. This resistance reduces the long-term efficacy of
single-target therapies.
PD-L1 expression and tumor-infiltrating lymphocytes
have been proposed as predictive indicators of immunotherapy
efficacy, but their utility in GC remains inconsistent. The
predictive value of these biomarkers across different GC subtypes
has not been fully validated and they lack sufficient specificity
and sensitivity. Thus, identifying novel and more reliable
biomarkers remains a major barrier to advancing TME-targeted
immunotherapy.
To address these limitations and improve the
efficacy of GC immunotherapy, future research directions may focus
on the following methods. Firstly, combining ICIs, as immune cell
reprogramming and tumor angiogenesis inhibitors may yield more
effective strategies for TME modification and overcoming immune
tolerance. For example, pairing anti-PD-1/PD-L1 antibodies with
anti-CTLA-4 antibodies can enhance T-cell activation and rewire the
immunosuppressive microenvironment.
Beyond classical ICIs, targeted therapies directed
against other immunomodulatory factors in the TME are promising.
Targeting immunosuppressive cells (such as Tregs and TAMs) that
actively suppress antitumor immunity, can enhance immunotherapy
efficacy. Identifying such new targets may contribute to
advancements in GC immunotherapy.
The current inadequacy of biomarkers for predicting
responses to TME-targeted therapies underscores the need for more
accurate tools. Studying dynamic changes in TME immune cells,
phenotypic features of tumor-infiltrating immune cells and tumor
immune metabolic states may enable the development of more precise
biomarkers for individualized treatment. The screening and
validation of these biomarkers could provide more reliable guidance
for the clinical application of immunotherapy.
Summary and outlook
GC cell growth is associated with TME-induced
immune tolerance, as tumor cells cannot be identified by the immune
system through a complex regulatory network. The present review
exhibits a systematic investigation of cellular and non-cellular
composition of the GC TME, as well as their mechanisms for
orchestrating immune tolerance.
From a compositional perspective, dynamic
interaction of cellular and non-cellular components of TME
establishes an immunosuppressive environment. At the molecular
level, the immune responses are suppressed by impaired antigen
presentation, immune checkpoint activation, imbalance of cytokine
networks, metabolic reprogramming, epigenetic regulation and
exosome-mediated information transfer. All components act
synergistically through numerous pathways. Furthermore, core immune
cells, including MDSCs, TAMs, Tregs and DCs interact dynamically to
form a stable immunosuppressive network, exacerbating the immune
tolerance state in GC.
Consequently, novel therapies targeting TME-induced
immune tolerance exhibit promise. ICIs enhance immune response and
tumor immunity. Similarly, cell therapies modify immune effector
cells to enhance anti-tumor immunity. ADCs enable precise targeting
of tumors and infusion of immune effector cells. Finally, biologics
induce immunization and regulate TME composition and function.
The immune cells and cytokines in the TME work in
conjunction to adaptively suppress the host antitumor immune
response within the tumor and eventually help in evading host
antitumor recognition. However, current research still has exhibits
gaps and limitations. Effective biomarkers to predict immunotherapy
responses remain lacking. While PD-L1 expression and
tumor-infiltrating lymphocytes have been explored as potential
biomarkers for GC, their predictive value is often
subtype-dependent and has not been accurately verified. This
underscores the need for new biomarkers to optimize the clinical
application of immunotherapy. Also, the heterogeneity of immune
responses in GC is unknown. The immune tolerance mechanisms may
vary across subtypes. Some subtypes may evade immune surveillance
by suppressing T-cell activity, while others may rely on immune
cell reprogramming or exploitation of immune checkpoint pathways.
Current understanding of this heterogeneity remains limited, so
multidimensional analyses are needed to uncover disparities in
immune responses across subtypes. In addition, existing preclinical
models have notable limitations. The majority of studies on
TME-mediated immune escape use mouse xenograft models, which
provide useful insights, but capacity to shed light on GC is
limited. Therefore, more animal models are urgently required to
accurately evaluate the efficacy of a number of immunotherapeutic
strategies for GC.
Ethical and societal considerations regarding
TME-targeted therapies require complex genetic testing and
precision medication assistance, which places additional strain on
healthcare resource allocation and limits patient access.
Exorbitant costs of testing and medications prevent some patients
from accessing these treatments. Furthermore, TME-targeted
immunotherapies are generally costly and long-term data
demonstrating improvements in survival and quality of life remain
scarce.
Additionally, TME modulation strategies typically
bypass immune tolerance through widespread immune activation, which
can trigger non-specific immune-related adverse events such as
immune enteritis, pneumonia, hepatitis and endocrine dysfunction.
Although the side effects of monoclonal antibodies can be partially
managed with corticosteroids and immunosuppressants in clinical
practice, long-term risks (including disease recurrence and chronic
tissue damage) remain a concern for multi-target combination
therapies. From a health economics perspective, the
cost-effectiveness and cost-benefit ratio of immunotherapy in
comparison with traditional chemotherapy or targeted chemotherapy
will largely determine their inclusion in clinical guidelines and
health insurance coverage.
In conclusion, while notable progress has been made
in understanding the role of the TME in GC immune tolerance,
numerous challenges remain before clinical advancements can be
achieved. The complexity and dynamism of the TME hinder the
translation of experimental findings into clinical practice. Future
studies should aim to use system immunomics and multi-omics
approaches and high-throughput spatial transcriptomics to
facilitate the translation from mechanism to clinical practice.
Only through rigorous validation and careful assessment of
immunomodulatory strategies, can the goals of precise GC treatment
be achieved.
Acknowledgements
Not applicable.
Funding
The authors declare that financial support was received for the
research, authorship and/or publication of this article. The
present review was supported by the Natural Science Foundation of
Heilongjiang Province (grant no. LH2023H059).
Availability of data and materials
Not applicable.
Authors' contributions
ZG and LH conceived the present review, conducted
the literature review and wrote the manuscript. BC, YM and QN
assisted with the literature review and manuscript revision. ZW and
JY contributed to manuscript writing and interpretation. ZL
provided critical feedback and helped with manuscript revision.
Data authentication is not applicable. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tan N, Wu H, Cao M, Yang F, Yan X, He S,
Cao M, Zhang S, Teng Y, Li Q, et al: Global, regional, and national
burden of early-onset gastric cancer. Cancer Biol Med. 21:667–678.
2024.PubMed/NCBI
|
|
2
|
Yu WY, Li X, Zhu J, Ding YM, Tao HQ and Du
LB: Epidemiological characteristics of gastric cancer in China and
worldwide. Zhonghua Zhong Liu Za Zhi. 47:468–476. 2025.(In
Chinese). PubMed/NCBI
|
|
3
|
Lin JL, Lin JX, Lin GT, Huang CM, Zheng
CH, Xie JW, Wang JB, Lu J, Chen QY and Li P: Global incidence and
mortality trends of gastric cancer and predicted mortality of
gastric cancer by 2035. BMC Public Health. 24:17632024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma ES, Wang ZX, Zhu MQ and Zhao J: Immune
evasion mechanisms and therapeutic strategies in gastric cancer.
World J Gastrointest Oncol. 14:216–229. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y, Li C, Lu Y, Liu C and Yang W: Tumor
microenvironment-mediated immune tolerance in development and
treatment of gastric cancer. Front Immunol. 13:10168172022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yasuda T and Wang YA: Gastric cancer
immunosuppressive microenvironment heterogeneity: Implications for
therapy development. Trends Cancer. 10:627–642. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Romagnani S: Immunological tolerance and
autoimmunity. Intern Emerg Med. 1:187–196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Dai Z, Wu W, Wang Z, Zhang N,
Zhang L, Zeng WJ, Liu Z and Cheng Q: Regulatory mechanisms of
immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer
Res. 40:1842021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J, Liu T, Huang T, Shang M and Wang
X: The mechanisms on evasion of anti-tumor immune responses in
gastric cancer. Front Oncol. 12:9438062022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi AN, Zhou YB and Wang GH:
Immunotherapy: Progress and challenges of a revolutionary treatment
for gastric cancer. Zhonghua Wai Ke Za Zhi. 63:563–567. 2025.(In
Chinese). PubMed/NCBI
|
|
11
|
Zhao Y, Bai Y, Shen M and Li Y:
Therapeutic strategies for gastric cancer targeting immune cells:
Future directions. Front Immunol. 13:9927622022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He B, Wood KH, Li ZJ, Ermer JA, Li J,
Bastow ER, Sakaram S, Darcy PK, Spalding LJ, Redfern CT, et al:
Selective tubulin-binding drugs induce pericyte phenotype switching
and anti-cancer immunity. EMBO Mol Med. 17:1071–1100. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang K, Xie CJ, Ding Z, Shan T, Zhong Z,
Yuan FL, Wu JJ, Yuan ZD, Qian C, Yu L, et al: PDE5A+
cancer-associated fibroblasts enhance immune suppression in gastric
cancer. Gut. October 20–2025.(Epub ahead of print). View Article : Google Scholar
|
|
14
|
Shaibu Z, Danbala IA, Chen Z and Zhu W:
Role of mesenchymal stem cells in modulating cytokine networks and
immune checkpoints in gastric cancer therapy. Biochim Biophys Acta
Rev Cancer. 1880:1894332025. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He X, Guan XY and Li Y: Clinical
significance of the tumor microenvironment on immune tolerance in
gastric cancer. Front Immunol. 16:15326052025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lv Y, Zhao Y, Wang X, Chen N, Mao F, Teng
Y, Wang T, Peng L, Zhang J, Cheng P, et al: Increased intratumoral
mast cells foster immune suppression and gastric cancer progression
through TNF-α-PD-L1 pathway. J Immunother Cancer. 7:542019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weng N, Zhou C, Zhou Y, Zhong Y, Jia Z,
Rao X, Qiu H, Zeng G, Jin X, Zhang J, et al: IKZF4/NONO-RAB11FIP3
axis promotes immune evasion in gastric cancer via facilitating
PD-L1 endosome recycling. Cancer Lett. 584:2166182024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shukla A, Cloutier M, Appiya Santharam M,
Ramanathan S and Ilangumaran S: The MHC Class-I Transactivator
NLRC5: Implications to cancer immunology and potential applications
to cancer immunotherapy. Int J Mol Sci. 22:19642021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Yu D, Ji C, Wang M, Fu M, Qian Y
and Zhang X, Ji R, Li C, Gu J and Zhang X: Exosomal
miR-4745-5p/3911 from N2-polarized tumor-associated neutrophils
promotes gastric cancer metastasis by regulating SLIT2. Mol Cancer.
23:1982024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nair R, Somasundaram V, Kuriakose A,
Krishn SR, Raben D, Salazar R and Nair P: Deciphering T-cell
exhaustion in the tumor microenvironment: Paving the way for
innovative solid tumor therapies. Front Immunol. 16:15482342025.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chow A, Perica K, Klebanoff CA and Wolchok
JD: Clinical implications of T cell exhaustion for cancer
immunotherapy. Nat Rev Clin Oncol. 19:775–790. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu K, Cao Y, Zhang Z, Wang L, Gu Y, Xu T,
Zhang X, Guo X, Shen Z and Qin J: Blockade of CLEVER-1 restrains
immune evasion and enhances anti-PD-1 immunotherapy in gastric
cancer. J Immunother Cancer. 13:e0110802025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi T, Zhang Y, Wang Y, Song X, Wang H,
Zhou X, Liang K, Luo Y, Che K, Wang X, et al: DKK1 promotes tumor
immune evasion and impedes Anti-PD-1 treatment by inducing
immunosuppressive macrophages in gastric cancer. Cancer Immunol
Res. 10:1506–1524. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang T, Li Y, Zhai E, Zhao R, Qian Y,
Huang Z, Liu Y, Zhao Z, Xu X, Liu J, et al: Intratumoral
fusobacterium nucleatum recruits Tumor-associated neutrophils to
promote gastric cancer progression and immune evasion. Cancer Res.
85:1819–1841. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Z, Zhang W, Bai J, Li J and Li H:
Emerging role of helicobacter pylori in the immune evasion
mechanism of gastric cancer: An insight into tumor
microenvironment-Pathogen interaction. Front Oncol. 12:8624622022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang L, Zhao X, Li Y, Hu Y, Sun Y, Liu S,
Zhang Z, Li Y, Feng X, Yuan J, et al: The tumor immune
microenvironment remodeling and response to HER2-targeted therapy
in HER2-positive advanced gastric cancer. IUBMB Life. 76:420–436.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun H, Wang X, Wang X, Xu M and Sheng W:
The role of cancer-associated fibroblasts in tumorigenesis of
gastric cancer. Cell Death Dis. 13:8742022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Allam A, Yakou M, Pang L, Ernst M and
Huynh J: Exploiting the STAT3 nexus in cancer-associated
fibroblasts to improve cancer therapy. Front Immunol.
12:7679392021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X,
Li J, Li C, Yan M, Zhu Z, et al: IL-6 secreted by cancer-associated
fibroblasts promotes epithelial-mesenchymal transition and
metastasis of gastric cancer via JAK2/STAT3 signaling pathway.
Oncotarget. 8:20741–20750. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hilmi M, Nicolle R, Bousquet C and
Neuzillet C: Cancer-associated fibroblasts: Accomplices in the
tumor immune evasion. Cancers (Basel). 12:29692020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Avagliano A, Granato G, Ruocco MR, Romano
V, Belviso I, Carfora A, Montagnani S and Arcucci A: Metabolic
reprogramming of cancer associated fibroblasts: The slavery of
stromal fibroblasts. Biomed Res Int. 2018:60754032018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kitamura F, Semba T, Yasuda-Yoshihara N,
Yamada K, Nishimura A, Yamasaki J, Nagano O, Yasuda T, Yonemura A,
Tong Y, et al: Cancer-associated fibroblasts reuse cancer-derived
lactate to maintain a fibrotic and immunosuppressive
microenvironment in pancreatic cancer. JCI Insight. 8:e1630222023.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iha K, Sato A, Tsai HY, Sonoda H, Watabe
S, Yoshimura T, Lin MW and Ito E: Gastric cancer cell-derived
exosomal GRP78 enhances angiogenesis upon stimulation of vascular
endothelial cells. Curr Issues Mol Biol. 44:6145–6157. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang R, Liu G, Wang K, Pan Z, Pei Z and Hu
X: Hypoxia signature derived from tumor-associated endothelial
cells predict prognosis in gastric cancer. Front Cell Dev Biol.
13:15156812025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fang J, Lu Y, Zheng J, Jiang X, Shen H,
Shang X, Lu Y and Fu P: Exploring the crosstalk between endothelial
cells, immune cells, and immune checkpoints in the tumor
microenvironment: New insights and therapeutic implications. Cell
Death Dis. 14:5862023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Q, Yu M, Lin Z, Wu L, Xia P, Zhu M,
Huang B, Wu W, Zhang R, Li K, et al: COL1A1-positive endothelial
cells promote gastric cancer progression via the ANGPTL4-SDC4 axis
driven by endothelial-to-mesenchymal transition. Cancer Lett.
623:2177312025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu Y, Xiang M, Brulois KF, Lazarus NH,
Pan J and Butcher EC: Endothelial cell Notch signaling programs
cancer-associated fibroblasts to promote tumor immune evasion. Res
Sq. Jun 11–2024.doi: 10.21203/rs.3.rs-4538031/v1.
|
|
38
|
Xu Y, Miller CP, Tykodi SS, Akilesh S and
Warren EH: Signaling crosstalk between tumor endothelial cells and
immune cells in the microenvironment of solid tumors. Front Cell
Dev Biol. 12:13871982024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang Y, Xu X, Lu Y, Sun Q, Zhang L, Shao
J, Chen D, Chang Y, Sun X, Zhuo W and Zhou T: The phase separation
of extracellular matrix protein matrilin-3 from cancer-associated
fibroblasts contributes to gastric cancer invasion. FASEB J.
38:e234062024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ji L, Peng J, Lin Y, Zhong Y, Ni B, Zhu C
and Zhang Z: High extracellular matrix stiffness upregulates TNNT1
to awaken dormant tumor cells in liver metastatic niches of gastric
cancer. Cell Oncol (Dordr). 48:815–834. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li S, Sampson C, Liu C, Piao HL and Liu
HX: Integrin signaling in cancer: Bidirectional mechanisms and
therapeutic opportunities. Cell Commun Signal. 21:2662023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zaheer J, Shanmugiah J, Kim S, Kim H and
Kim JS: Tumor microenvironment modulation by SERPINE1 increases
radioimmunotherapy in murine model of gastric cancer. Sci Rep.
15:164492025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qian X, Jia W, Li Y, Chen J, Zhang J and
Sun Y: COL4A1 promotes gastric cancer progression by regulating
tumor invasion, tumor microenvironment and drug sensitivity. Curr
Med Chem. Apr 7–2025.doi: 10.2174/0109298673351943250314074632
(Epub ahead of print). View Article : Google Scholar
|
|
44
|
Moreira AM, Pereira J, Melo S, Fernandes
MS, Carneiro P, Seruca R and Figueiredo J: The extracellular
matrix: An accomplice in gastric cancer development and
progression. Cells. 9:3942020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Raja UM, Gopal G, Shirley S, Ramakrishnan
AS and Rajkumar T: Immunohistochemical expression and localization
of cytokines/chemokines/growth factors in gastric cancer. Cytokine.
89:82–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chung HW and Lim JB: Role of the tumor
microenvironment in the pathogenesis of gastric carcinoma. World J
Gastroenterol. 20:1667–1680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tian Y, Xie Y, Yi G, Wu F, Dang X, Bai F,
Wang J and Zhang D: Prognostic value and therapeutic significance
of CCL chemokines in gastric cancer. Curr Med Chem. 31:7043–7058.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oya Y, Hayakawa Y and Koike K: Tumor
microenvironment in gastric cancers. Cancer Sci. 111:2696–2707.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhu M, Wang Z, He Y, Zhang B, Wu L, Liu C,
Fei Y, Gao P, Cai J and Zuo X: Acidic tumor
microenvironment-modulated nanoparticle potentiates gastric cancer
photoimmunotherapy. J Adv Res. June 6–2025.(Epub ahead of
print).
|
|
50
|
Liu HY, Wang FH, Liang JM, Xiang YY, Liu
SH, Zhang SW, Zhu CM, He YL and Zhang CH: Targeting NAD metabolism
regulates extracellular adenosine levels to improve the
cytotoxicity of CD8+ effector T cells in the tumor microenvironment
of gastric cancer. J Cancer Res Clin Oncol. 149:2743–2756. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rojas A, Araya P, Gonzalez I and Morales
E: Gastric tumor microenvironment. Adv Exp Med Biol. 1226:23–35.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lin K, Lin X and Luo F: IGF2BP3 boosts
lactate generation to accelerate gastric cancer immune evasion.
Apoptosis. 29:2147–2160. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cornel AM, Mimpen IL and Nierkens S: MHC
Class I downregulation in cancer: Underlying mechanisms and
potential targets for cancer immunotherapy. Cancers (Basel).
12:17602020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Isobe T, Aoyagi K, Koufuji K, Shirouzu K,
Kawahara A, Taira T and Kage M: Clinicopathological significance of
hypoxia-inducible factor-1 alpha (HIF-1α) expression in gastric
cancer. Int J Clin Oncol. 18:293–304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Balasubramanian A, John T and
Asselin-Labat ML: Regulation of the antigen presentation machinery
in cancer and its implication for immune surveillance. Biochem Soc
Trans. 50:825–837. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zong J, Keskinov AA, Shurin GV and Shurin
MR: Tumor-derived factors modulating dendritic cell function.
Cancer Immunol Immunother. 65:821–833. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schmidt SV, Nino-Castro AC and Schultze
JL: Regulatory dendritic cells: There is more than just immune
activation. Front Immunol. 3:2742012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sodir NM, Pathria G, Adamkewicz JI, Kelley
EH, Sudhamsu J, Merchant M, Chiarle R and Maddalo D: SHP2: A
pleiotropic target at the interface of cancer and its
microenvironment. Cancer Discov. 13:2339–2355. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sobhani N, Tardiel-Cyril DR, Davtyan A,
Generali D, Roudi R and Li Y: CTLA-4 in regulatory T cells for
cancer immunotherapy. Cancers (Basel). 13:14402021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kandel S, Adhikary P, Li G and Cheng K:
The TIM3/Gal9 signaling pathway: An emerging target for cancer
immunotherapy. Cancer Lett. 510:67–78. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yuan XL, Chen L, Zhang TT, Ma YH, Zhou YL,
Zhao Y, Wang WW, Dong P, Yu L, Zhang YY and Shen LS: Gastric cancer
cells induce human CD4+Foxp3+ regulatory T cells through the
production of TGF-β1. World J Gastroenterol. 17:2019–2027. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen L, Shi Y, Zhu X, Guo W, Zhang M, Che
Y, Tang L, Yang X, You Q and Liu Z: IL-10 secreted by
cancer-associated macrophages regulates proliferation and invasion
in gastric cancer cells via c-Met/STAT3 signaling. Oncol Rep.
42:595–604. 2019.PubMed/NCBI
|
|
63
|
Bourhis M, Palle J, Galy-Fauroux I and
Terme M: Direct and indirect modulation of T Cells by VEGF-A
counteracted by Anti-angiogenic treatment. Front Immunol.
12:6168372021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lee WS, Yang H, Chon HJ and Kim C:
Combination of Anti-angiogenic therapy and immune checkpoint
blockade normalizes vascular-immune crosstalk to potentiate cancer
immunity. Exp Mol Med. 52:1475–1485. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ye Z, Yue L, Shi J, Shao M and Wu T: Role
of IDO and TDO in cancers and related diseases and the therapeutic
implications. J Cancer. 10:2771–2782. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Platten M, von Knebel Doeberitz N, Oezen
I, Wick W and Ochs K: Cancer Immunotherapy by Targeting IDO1/TDO
and their downstream effectors. Front Immunol. 5:6732014.PubMed/NCBI
|
|
67
|
Sosnowska A, Chlebowska-Tuz J, Matryba P,
Pilch Z, Greig A, Wolny A, Grzywa TM, Rydzynska Z, Sokolowska O,
Rygiel TP, et al: Inhibition of arginase modulates T-cell response
in the tumor microenvironment of lung carcinoma. Oncoimmunology.
10:19561432021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liang JJ, Fraser IDC and Bryant CE: Lipid
regulation of NLRP3 inflammasome activity through organelle stress.
Trends Immunol. 42:807–823. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lagunas-Rangel FA: Cholesterol effects on
the tumor immune microenvironment: From fundamental concepts to
mechanisms and implications. Front Oncol. 15:15790542025.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ji Y, Xiao C, Fan T, Deng Z, Wang D, Cai
W, Li J, Liao T, Li C and He J: The epigenetic hallmarks of immune
cells in cancer. Mol Cancer. 24:662025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Emran AA, Chatterjee A, Rodger EJ, Tiffen
JC, Gallagher SJ, Eccles MR and Hersey P: Targeting DNA Methylation
and EZH2 activity to overcome melanoma resistance to immunotherapy.
Trends Immunol. 40:328–344. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Belk JA, Daniel B and Satpathy AT:
Epigenetic regulation of T cell exhaustion. Nat Immunol.
23:848–860. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang C, Wang X, Zhang D, Sun X, Wu Y, Wang
J, Li Q and Jiang G: The macrophage polarization by miRNAs and its
potential role in the treatment of tumor and inflammation (Review).
Oncol Rep. 50:1902023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang
CY and Zen K: Re-polarization of tumor-associated macrophages to
pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol.
4:341–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wozniak M and Czyz M: lncRNAs-EZH2
interaction as promising therapeutic target in cutaneous melanoma.
Front Mol Biosci. 10:11700262023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Su Y, Bai Q, Zhang W, Xu B and Hu T: The
role of Long Non-coding RNAs in modulating the immune
microenvironment of Triple-negative breast cancer: Mechanistic
insights and therapeutic potential. Biomolecules. 15:4542025.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xu Z, Chen Y, Ma L, Chen Y, Liu J, Guo Y,
Yu T, Zhang L, Zhu L and Shu Y: Role of exosomal non-coding RNAs
from tumor cells and tumor-associated macrophages in the tumor
microenvironment. Mol Ther. 30:3133–3154. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yang X, Zhang Y, Zhang Y, Zhang S, Qiu L,
Zhuang Z, Wei M, Deng X, Wang Z and Han J: The key role of exosomes
on the pre-metastatic niche formation in tumors. Front Mol Biosci.
8:7036402021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Han QF, Li WJ, Hu KS, Gao J, Zhai WL, Yang
JH and Zhang SJ: Exosome biogenesis: Machinery, regulation, and
therapeutic implications in cancer. Mol Cancer. 21:2072022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Mizukami Y, Kono K, Kawaguchi Y, Akaike H,
Kamimura K, Sugai H and Fujii H: CCL17 and CCL22 chemokines within
tumor microenvironment are related to accumulation of Foxp3+
regulatory T cells in gastric cancer. Int J Cancer. 122:2286–2293.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ren W, Zhang X, Li W, Feng Q, Feng H, Tong
Y, Rong H, Wang W, Zhang D, Zhang Z and Tu S: Circulating and
tumor-infiltrating arginase 1-expressing cells in gastric
adenocarcinoma patients were mainly immature and monocytic
Myeloid-derived suppressor cells. Sci Rep. 10:80562020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang J, Hu C, Zhang R, Xu J, Zhang Y,
Yuan L, Zhang S, Pan S, Cao M, Qin J, et al: The role of
macrophages in gastric cancer. Front Immunol. 14:12821762023.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Baradaran A, Asadzadeh Z, Hemmat N,
Baghbanzadeh A, Shadbad MA, Khosravi N, Derakhshani A, Alemohammad
H, Afrashteh Nour M, Safarpour H, et al: The cross-talk between
tumor-associated macrophages and tumor endothelium: Recent advances
in macrophage-based cancer immunotherapy. Biomed Pharmacother.
146:1125882022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gabrilovich DI and Nagaraj S:
Myeloid-derived suppressor cells as regulators of the immune
system. Nat Rev Immunol. 9:162–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Corzo CA, Cotter MJ, Cheng P, Cheng F,
Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC and
Gabrilovich DI: Mechanism regulating reactive oxygen species in
tumor-induced myeloid-derived suppressor cells. J Immunol.
182:5693–5701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Nagaraj S, Gupta K, Pisarev V, Kinarsky L,
Sherman S, Kang L, Herber DL, Schneck J and Gabrilovich DI: Altered
recognition of antigen is a mechanism of CD8+ T cell tolerance in
cancer. Nat Med. 13:828–835. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yamaguchi T, Fushida S, Kinoshita J,
Okazaki M, Ishikawa S, Ohbatake Y, Terai S, Okamoto K, Nakanuma S,
Makino I, et al: Extravasated platelet aggregation contributes to
tumor progression via the accumulation of myeloid-derived
suppressor cells in gastric cancer with peritoneal metastasis.
Oncol Lett. 20:1879–1887. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhou Y and Guo F: A selective
sphingosine-1-phosphate receptor 1 agonist SEW-2871 aggravates
gastric cancer by recruiting myeloid-derived suppressor cells. J
Biochem. 163:77–83. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kusmartsev S, Su Z, Heiser A, Dannull J,
Eruslanov E, Kübler H, Yancey D, Dahm P and Vieweg J: Reversal of
myeloid cell-mediated immunosuppression in patients with metastatic
renal cell carcinoma. Clin Cancer Res. 14:8270–8278. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ren W, Zhang X, Li W, Feng Q, Feng H, Tong
Y, Rong H, Wang W, Zhang D, Zhang Z, et al: Exosomal miRNA-107
induces myeloid-derived suppressor cell expansion in gastric
cancer. Cancer Manag Res. 11:4023–4040. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Chen X, Wang Y, Wang J, Wen J, Jia X, Wang
X and Zhang H: Accumulation of T-helper 22 cells, interleukin-22
and myeloid-derived suppressor cells promotes gastric cancer
progression in elderly patients. Oncol Lett. 16:253–261.
2018.PubMed/NCBI
|
|
92
|
Gordon S and Martinez FO: Alternative
activation of macrophages: Mechanism and functions. Immunity.
32:593–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bronte V and Zanovello P: Regulation of
immune responses by L-arginine metabolism. Nat Rev Immunol.
5:641–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tsutsumi C, Ohuchida K, Yamada Y, Shimada
Y, Imamura M, Son K, Mochida Y, Katayama N, Iwamoto C, Torata N, et
al: Claudin18.2-positive gastric cancer-specific changes in
neoadjuvant chemotherapy-driven immunosuppressive tumor
microenvironment. Br J Cancer. 132:793–804. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Aizaz M, Khan A, Khan F, Khan M, Musad
Saleh EA, Nisar M and Baran N: The cross-talk between macrophages
and tumor cells as a target for cancer treatment. Front Oncol.
13:12590342023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
He S, Zheng L and Qi C: Myeloid-derived
suppressor cells (MDSCs) in the tumor microenvironment and their
targeting in cancer therapy. Mol Cancer. 24:52025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Masmoudi D, Villalba M and Alix-Panabières
C: Natural killer cells: The immune frontline against circulating
tumor cells. J Exp Clin Cancer Res. 44:1182025. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wang J, Huang H, Lu J, Bi P, Wang F, Liu
X, Zhang B, Luo Y and Li X: Tumor cells induced-M2 macrophage
favors accumulation of Treg in nasopharyngeal carcinoma. Int J Clin
Exp Pathol. 10:8389–8401. 2017.PubMed/NCBI
|
|
100
|
Mirlekar B: Tumor promoting roles of
IL-10, TGF-β, IL-4, and IL-35: Its implications in cancer
immunotherapy. SAGE Open Med. 10:205031212110690122022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Sakaguchi S, Yamaguchi T, Nomura T and Ono
M: Regulatory T cells and immune tolerance. Cell. 133:775–787.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Shaopeng Z, Yang Z, Yuan F, Chen H and
Zhengjun Q: Regulation of regulatory T cells and tumor-associated
macrophages in gastric cancer tumor microenvironment. Cancer Med.
13:e69592024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wang B, Zhang Z, Liu W and Tan B:
Targeting regulatory T cells in gastric cancer: Pathogenesis,
immunotherapy, and prognosis. Biomed Pharmacother. 158:1141802023.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ling Z, Shao L and Liu X, Cheng Y, Yan C,
Mei Y, Ji F and Liu X: Regulatory T cells and plasmacytoid
dendritic cells within the tumor microenvironment in gastric cancer
are correlated with gastric microbiota dysbiosis: A preliminary
study. Front Immunol. 10:5332019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lin Y, Jing X, Chen Z, Pan X, Xu D, Yu X,
Zhong F, Zhao L, Yang C, Wang B, et al: Histone
deacetylase-mediated tumor microenvironment characteristics and
synergistic immunotherapy in gastric cancer. Theranostics.
13:4574–4600. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hoechst B, Ormandy LA, Ballmaier M, Lehner
F, Krüger C, Manns MP, Greten TF and Korangy F: A new population of
myeloid-derived suppressor cells in hepatocellular carcinoma
patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology.
135:234–243. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Peng X, He Y, Huang J, Tao Y and Liu S:
Metabolism of dendritic cells in tumor microenvironment: For
immunotherapy. Front Immunol. 12:6134922021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Chen J, Duan Y, Che J and Zhu J:
Dysfunction of dendritic cells in tumor microenvironment and
immunotherapy. Cancer Commun (Lond). 44:1047–1070. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang B, Gu B, Gao L, Ma C, Li X, Wang Y,
Hu J, Wang N, Xiang L, Yu Y, et al: SERPINE1 facilitates metastasis
in gastric cancer through anoikis resistance and tumor
microenvironment remodeling. Small. 21:e25001362025. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Fu C and Jiang A: Dendritic cells and CD8
T cell immunity in tumor microenvironment. Front Immunol.
9:30592018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ni L: Advances in human dendritic
Cell-based immunotherapy against gastrointestinal cancer. Front
Immunol. 13:8871892022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ghasemi M, Abbasi L, Ghanbari Naeini L,
Kokabian P, Nameh Goshay Fard N and Givtaj N: Dendritic cells and
natural killer cells: The road to a successful oncolytic
virotherapy. Front Immunol. 13:9500792022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zou W: Regulatory T cells, tumour immunity
and immunotherapy. Nat Rev Immunol. 6:295–307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jin J, Lin J, Xu A, Lou J, Qian C, Li X,
Wang Y, Yu W and Tao H: CCL2: An important mediator between tumor
cells and host cells in tumor microenvironment. Front Oncol.
11:7229162021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Li Y, Zheng Y, Huang J, Nie RC, Wu QN, Zuo
Z, Yuan S, Yu K, Liang CC, Pan YQ, et al: CAF-macrophage crosstalk
in tumour microenvironments governs the response to immune
checkpoint blockade in gastric cancer peritoneal metastases. Gut.
74:350–363. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wan S, Zhao E, Kryczek I, Vatan L,
Sadovskaya A, Ludema G, Simeone DM, Zou W and Welling TH:
Tumor-associated macrophages produce interleukin 6 and signal via
STAT3 to promote expansion of human hepatocellular carcinoma stem
cells. Gastroenterology. 147:1393–1404. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
McRitchie BR and Akkaya B: Exhaust the
exhausters: Targeting regulatory T cells in the tumor
microenvironment. Front Immunol. 13:9400522022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Patsoukis N, Bardhan K, Chatterjee P, Sari
D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al:
PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis
and promoting lipolysis and fatty acid oxidation. Nat Commun.
6:66922015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wang Z, Ji X, Zhang Y, Yang F, Su H, Zhang
H, Li Z, Zhang W and Sun W: Interactions between LAMP3+ dendritic
cells and T-cell subpopulations promote immune evasion in papillary
thyroid carcinoma. J Immunother Cancer. 12:e0089832024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Raber P, Ochoa AC and Rodríguez PC:
Metabolism of L-arginine by myeloid-derived suppressor cells in
cancer: Mechanisms of T cell suppression and therapeutic
perspectives. Immunol Invest. 41:614–634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Jin HR, Wang J, Wang ZJ, Xi MJ, Xia BH,
Deng K and Yang JL: Lipid metabolic reprogramming in tumor
microenvironment: From mechanisms to therapeutics. J Hematol Oncol.
16:1032023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Cascone T, McKenzie JA, Mbofung RM, Punt
S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al:
Increased tumor glycolysis characterizes immune resistance to
adoptive T cell therapy. Cell Metab. 27:977–987.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhang S, Lv K, Liu Z, Zhao R and Li F:
Fatty acid metabolism of immune cells: A new target of tumour
immunotherapy. Cell Death Discov. 10:392024. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Osada T, Chong G, Tansik R, Hong T,
Spector N, Kumar R, Hurwitz HI, Dev I, Nixon AB, Lyerly HK, et al:
The effect of anti-VEGF therapy on immature myeloid cell and
dendritic cells in cancer patients. Cancer Immunol Immunother.
57:1115–1124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Porta C, Consonni FM, Morlacchi S,
Sangaletti S, Bleve A, Totaro MG, Larghi P, Rimoldi M, Tripodo C,
Strauss L, et al: Tumor-derived prostaglandin E2 promotes p50
NF-κB-Dependent differentiation of monocytic MDSCs. Cancer Res.
80:2874–2888. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK,
Li LL, Koh HY, Tsang FH, Wei LL, Wong CM, et al: Hypoxia inducible
factor HIF-1 promotes myeloid-derived suppressor cells accumulation
through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun.
8:5172017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Bailey CM, Liu Y, Liu M, Du X, Devenport
M, Zheng P, Liu Y and Wang Y: Targeting HIF-1α abrogates
PD-L1-mediated immune evasion in tumor microenvironment but
promotes tolerance in normal tissues. J Clin Invest.
132:e1508462022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Mou P, Ge QH, Sheng R, Zhu TF, Liu Y and
Ding K: Research progress on the immune microenvironment and
immunotherapy in gastric cancer. Front Immunol. 14:12911172023.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Kang YK, Chen LT, Ryu MH, Oh DY, Oh SC,
Chung HC, Lee KW, Omori T, Shitara K, Sakuramoto S, et al:
Nivolumab plus chemotherapy versus placebo plus chemotherapy in
patients with HER2-negative, untreated, unresectable advanced or
recurrent gastric or gastro-oesophageal junction cancer
(ATTRACTION-4): A randomised, multicentre, double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 23:234–247. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Moehler M, Xiao H, Blum SI, Elimova E,
Cella D, Shitara K, Ajani JA, Janjigian YY, Garrido M, Shen L, et
al: Health-related quality of life with nivolumab plus chemotherapy
versus chemotherapy in patients with advanced
Gastric/Gastroesophageal junction cancer or esophageal
adenocarcinoma from CheckMate 649. J Clin Oncol. 41:5388–5399.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Satake H, Lee KW, Chung HC, Lee J,
Yamaguchi K, Chen JS, Yoshikawa T, Amagai K, Yeh KH, Goto M, et al:
Pembrolizumab or pembrolizumab plus chemotherapy versus standard of
care chemotherapy in patients with advanced gastric or
gastroesophageal junction adenocarcinoma: Asian subgroup analysis
of KEYNOTE-062. Jpn J Clin Oncol. 53:221–229. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Shitara K, Van Cutsem E, Bang YJ, Fuchs C,
Wyrwicz L, Lee KW, Kudaba I, Garrido M, Chung HC, Lee J, et al:
Efficacy and safety of pembrolizumab or pembrolizumab plus
chemotherapy vs chemotherapy alone for patients with First-line,
advanced gastric cancer: The KEYNOTE-062 Phase 3 randomized
clinical trial. JAMA Oncol. 6:1571–1580. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Sun JM, Shen L, Shah MA, Enzinger P,
Adenis A, Doi T, Kojima T, Metges JP, Li Z, Kim SB, et al:
Pembrolizumab plus chemotherapy versus chemotherapy alone for
first-line treatment of advanced oesophageal cancer (KEYNOTE-590):
A randomised, placebo-controlled, phase 3 study. Lancet.
398:759–771. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Qi C, Gong J, Li J, Liu D, Qin Y, Ge S,
Zhang M, Peng Z, Zhou J, Cao Y, et al: Claudin18.2-specific CAR T
cells in gastrointestinal cancers: Phase 1 trial interim results.
Nat Med. 28:1189–1198. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Wang T, Navenot JM, Rafail S, Kurtis C,
Carroll M, Van Kerckhoven M, Van Rossom S, Schats K, Avraam K,
Broad R, et al: Identifying MAGE-A4-positive tumors for TCR T cell
therapies in HLA-A*02-eligible patients. Mol Ther Methods Clin Dev.
32:1012652024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Meyer T, Finn RS, Borad M, Mahipal A,
Edeline J, Houot R, Hausner PF, Hollebecque A, Goyal L, Frigault M,
et al: Phase I trial of ADP-A2AFP TCR T-cell therapy in patients
with advanced hepatocellular or gastric hepatoid carcinoma. J
Hepatol. 84:113–121. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Wang F, Lau JKC and Yu J: The role of
natural killer cell in gastrointestinal cancer: Killer or helper.
Oncogene. 40:717–730. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Lee HJ, Hwang SJ, Jeong EH and Chang MH:
Genetically engineered CLDN18.2 CAR-T cells expressing synthetic
PD1/CD28 fusion receptors produced using a lentiviral vector. J
Microbiol. 62:555–568. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Cai F, Zhang J, Gao H and Shen H: Tumor
microenvironment and CAR-T cell immunotherapy in B-cell lymphoma.
Eur J Haematol. 112:223–235. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Frey N and Porter D: Cytokine release
syndrome with chimeric antigen receptor T cell therapy. Biol Blood
Marrow Transplant. 25:e123–e127. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Shitara K, Bang YJ, Iwasa S, Sugimoto N,
Ryu MH, Sakai D, Chung HC, Kawakami H, Yabusaki H, Lee J, et al:
Trastuzumab deruxtecan in previously treated HER2-Positive gastric
cancer. N Engl J Med. 382:2419–2430. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Jang JY, Kim D, Lee NK, Im E and Kim ND:
Antibody-drug conjugates powered by deruxtecan: Innovations and
challenges in oncology. Int J Mol Sci. 26:65232025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Dong S, Wei C, Wang X, Yang X, Shen W, Li
S, Xu J, Ma Y, Bie L, Yu W and Li N: A retrospective multicenter
study on the efficacy and safety of disitamab vedotin monotherapy
versus combination with anti-PD-1 immunotherapy in advanced gastric
cancer. Sci Rep. 15:22322025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wang Y, Gong J, Wang A, Wei J, Peng Z,
Wang X, Zhou J, Qi C, Liu D, Li J, et al: Disitamab vedotin (RC48)
plus toripalimab for HER2-expressing advanced gastric or
gastroesophageal junction and other solid tumours: A multicentre,
open label, dose escalation and expansion phase 1 trial.
EClinicalMedicine. 68:1024152024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yasunaga M, Manabe S, Tsuji A, Furuta M,
Ogata K, Koga Y, Saga T and Matsumura Y: Development of
Antibody-drug conjugates using DDS and molecular imaging.
Bioengineering (Basel). 4:782017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Roth JS, Guo H, Chen L, Shen M, Gbadegesin
O, Robey RW, Gottesman MM and Hall MD: Identification of
antibody-drug conjugate payloads which are substrates of
ATP-binding cassette drug efflux transporters. bioRxiv. Jul
18–2025.doi: 10.1101/2025.05.22.651305.
|
|
148
|
Moon GY, Dalkiran B, Park HS, Shin D, Son
C, Choi JH, Bang S, Lee H, Doh I, Kim DH, et al: Dual biomarker
strategies for liquid biopsy: Integrating circulating tumor cells
and circulating tumor DNA for enhanced tumor monitoring. Biosensors
(Basel). 15:742025. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Braun T, Rade M, Merz M, Klepzig H, Große
F, Fandrei D, Pham NN, Kreuz M, Kuhn CK, Kuschel F, et al:
Multiomic profiling of T cell lymphoma after therapy with anti-BCMA
CAR T cells and GPRC5D-directed bispecific antibody. Nat Med.
31:1145–1153. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Middelburg J, Schaap G, Sluijter M, Lloyd
K, Ovcinnikovs V, Schuurman J, van der Burg SH, Kemper K and van
Hall T: Cancer vaccines compensate for the insufficient induction
of protective tumor-specific immunity of CD3 bispecific antibody
therapy. J Immunother Cancer. 13:e0103312025. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Liu X, Le Gall C, Alexander RK, Borgman E,
Balligand T and Ploegh HL: Nanobody-based bispecific antibody
engagers targeting CTLA-4 or PD-L1 for cancer immunotherapy. Nat
Biomed Eng. Jul 16–2025.doi: 10.1038/s41551-025-01447-z (Epub ahead
of print). View Article : Google Scholar
|
|
152
|
Niu M, Yi M, Wu Y, Lyu L, He Q, Yang R,
Zeng L, Shi J, Zhang J, Zhou P, et al: Synergistic efficacy of
simultaneous anti-TGF-β/VEGF bispecific antibody and PD-1 blockade
in cancer therapy. J Hematol Oncol. 16:942023. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Scialpi R, Espinosa-Sotelo R, Bertran E,
Dituri F, Giannelli G and Fabregat I: New Hepatocellular carcinoma
(HCC) Primary cell cultures as models for exploring personalized
Anti-TGF-β therapies based on tumor characteristics. Int J Mol Sci.
26:24302025. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Qin F, Liu X, Chen J, Huang S, Wei W, Zou
Y, Liu X, Deng K, Mo S, Chen J, et al: Anti-TGF-β attenuates tumor
growth via polarization of tumor associated neutrophils towards an
anti-tumor phenotype in colorectal cancer. J Cancer. 11:2580–2592.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Liu Y, Li Y, Wang Y, Lin C, Zhang D, Chen
J, Ouyang L, Wu F, Zhang J and Chen L: Recent progress on vascular
endothelial growth factor receptor inhibitors with dual targeting
capabilities for tumor therapy. J Hematol Oncol. 15:892022.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Jung BK, Ko HY, Kang H, Hong J, Ahn HM, Na
Y, Kim H, Kim JS and Yun CO: Relaxin-expressing oncolytic
adenovirus induces remodeling of physical and immunological aspects
of cold tumor to potentiate PD-1 blockade. J Immunother Cancer.
8:e0007632020. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Hong DS, Postow M, Chmielowski B, Sullivan
R, Patnaik A, Cohen EEW, Shapiro G, Steuer C, Gutierrez M,
Yeckes-Rodin H, et al: Eganelisib, a First-in-Class PI3Kγ
Inhibitor, in Patients with Advanced Solid Tumors: Results of the
Phase 1/1b MARIO-1 Trial. Clin Cancer Res. 29:2210–2219. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Sabit H, Pawlik TM, Radwan F, Abdel-Hakeem
M, Abdel-Ghany S, Wadan AS, Elzawahri M, El-Hashash A and Arneth B:
Precision nanomedicine: Navigating the tumor microenvironment for
enhanced cancer immunotherapy and targeted drug delivery. Mol
Cancer. 24:1602025. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Kundu M, Butti R, Panda VK, Malhotra D,
Das S, Mitra T, Kapse P, Gosavi SW and Kundu GC: Modulation of the
tumor microenvironment and mechanism of Immunotherapy-based drug
resistance in breast cancer. Mol Cancer. 23:922024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Fujiwara Y, Kinoshita J, Shimada M, Saito
H, Tsuji T, Yamamoto D, Moriyama H, Horii M, Nomura S, Matsushita
T, et al: Regulatory B cells drive immune evasion in the tumor
microenvironment and are involved peritoneal metastasis in gastric
cancer. Sci Rep. 15:274992025. View Article : Google Scholar : PubMed/NCBI
|