Introduction
Hepatocellular carcinoma (HCC) accounts for
approximately 80% of primary liver malignancies, and it is the
third-leading cause of cancer-related deaths worldwide (1). Although surgical resection,
radiofrequency ablation, systemic therapy, and radiotherapy are
available treatment options, patients with advanced-stage HCC face
limited therapeutic choices and poor prognoses (2). Recent advancements in high-precision
radiation techniques, such as stereotactic body radiation therapy
(SBRT), have demonstrated improved local control and survival rates
in patients with HCC (3).
Consequently, the latest 2025 edition of the Japanese guidelines
for HCC now includes radiotherapy as an optional treatment
modality, highlighting its growing importance in HCC management
(4). However, radiotherapy outcomes
vary remarkably among individuals. Notably, the risk factors for
HCC have shifted from viral hepatitis (HBV/HCV) to nonviral
metabolic diseases, particularly those associated with lipid
metabolism abnormalities (5,6).
According to multiple studies, HCC etiology influences the
treatment response, including the efficacy of radiotherapy;
however, the underlying mechanisms remain unclear (7,8).
Therefore, elucidating how lipid metabolism affects
radiosensitivity in HCC cells is of clinical relevance.
Lipids are essential components of cell membranes
and energy metabolism (9). However,
excessive lipid accumulation may induce oxidative stress and lipid
peroxidation, leading to various forms of cell death (10). Oleic acid (OA), a dietary
monounsaturated fatty acid, induces lipid accumulation and
oxidative damage in cells (11–13).
Nonetheless, its impact on cancer cell radiosensitivity remains
poorly understood.
During radiotherapy, nutritional management is
essential to maintain treatment continuity and avoid worsening of
the patient's condition. Clinical guidelines recommend personalized
dietary counseling and the use of oral nutritional supplements to
ensure adequate dietary intake (14). Notably, recent studies have reported
that ketogenic diets, which are rich in lipids and poor in
carbohydrates, can enhance oxidative stress and improve the
therapeutic response to radiotherapy in animal models of lung and
pancreatic cancer (15,16). These findings underscore the
potential role of lipid metabolic status in radiosensitivity
modulation.
This study aimed to evaluate, in vitro, the
effects of dietary lipid uptake on the therapeutic efficacy of
radiotherapy in HCC, using OA to model lipid accumulation. Our
ultimate goal is to optimize radiotherapy by incorporating
considerations of dietary management and lipid metabolism.
Materials and methods
In vitro cell culture
The human HCC cell line Huh7 was obtained from the
RIKEN BioResource Research Center. Cells were cultured in
low-glucose Dulbecco's modified Eagle medium (DMEM; FUJIFILM Wako
Pure Chemical Corporation) supplemented with 10% heat-inactivated
fetal bovine serum (FBS; Japan Bioserum) and 1%
penicillin-streptomycin (Thermo Fisher Scientific, Inc.). Cultures
were maintained at 37°C in a humidified incubator with 5%
CO2 and 95% air. To induce lipid accumulation, Huh7
cells were cultured in low-glucose DMEM supplemented with 5%
heat-inactivated FBS and 1% penicillin-streptomycin and treated
with OA at concentrations ranging from 0.5 to 2 mM. OA was
dissolved in 70% ethanol and added to the medium at the desired
final concentrations. Control cells received an equivalent volume
of 70% ethanol without OA. OA was dissolved in 70% ethanol, and an
equivalent concentration of ethanol was added to all control
groups. Ethanol alone did not significantly affect cell viability,
lipid accumulation, or oxidative stress parameters under the
experimental conditions used.
Exposure to ionizing radiation
(IR)
X-ray irradiation was performed using an X-ray
generator (MBR-1520R-3; Hitachi Medical Co., Ltd.) operated at 150
kVp and 20 mA, with 0.5-mm aluminum and 0.3-mm copper filters. The
source-to-sample distance was set at 45 cm. The delivered dose was
measured using a thimble-type ionization chamber dosimeter placed
adjacent to the samples during irradiation. The dose rate was 1
Gy/min. The 10 Gy dose was selected as a mechanistic in
vitro model to reliably induce oxidative stress and lipid
peroxidation, based on prior radiobiological studies and
preliminary dose-response experiments, rather than to directly
mimic clinical dose regimens.
Cell cycle distribution analysis
Cells were seeded in 35-mm culture dishes (2 ml of
medium per dish) and subjected to X-ray irradiation at a dose of 5
Gy. Following irradiation, the cells were incubated for 3, 6, 12,
18, or 24 h. At each time point, cells were harvested and fixed
with 70% ethanol precooled to −20°C. After fixation, the cells were
treated with RNase I (3.6 µg/ml; Merck KGaA) at 37°C for 20 min in
the dark. Propidium iodide (PI; 37 µg/ml) was then added, and DNA
content was analyzed immediately by flow cytometry using a Cell Lab
Quanta™ SC MPL system (Beckman Coulter, Inc.). The proportions of
cells in the Sub-G1, G0/G1, S, and G2/M phases were calculated
using Kaluza analysis software (version 2.1; Beckman Coulter,
Inc.).
Measurement of lipid accumulation
Intracellular lipid accumulation was quantified
using Nile red stain (FUJIFILM Wako Pure Chemical Corporation). A 1
mM stock solution of Nile red was prepared by dissolving the dye in
DMSO. To assess time-dependent lipid accumulation, cells were
seeded in 60-mm culture dishes (4 ml medium per dish) and treated
the following day with 1 mM OA. Cells were incubated for 3, 6, 12,
18, 24, 36, or 48 h. After incubation, cells were harvested, fixed
with 4% paraformaldehyde, resuspended in PBS, and stored at −20°C.
For measurement, cells were incubated with Nile red at a final
concentration of 0.1 µg/ml in PBS at 37°C for 15 min in the dark.
Fluorescence intensity was measured by flow cytometry using the
Cell Lab Quanta™ SC MPL system (Beckman Coulter, Inc.) and analyzed
using Kaluza analysis software (version 2.1; Beckman Coulter,
Inc.). To assess concentration-dependent lipid accumulation, cells
were similarly seeded in 60-mm dishes and treated the following day
with 0.5, 1, or 2 mM OA for 18 h. Cells were then fixed with 4%
paraformaldehyde, washed with PBS, and stored at −20°C. Staining
and fluorescence measurement were performed as described above.
Measurement of the viable cell
number
To evaluate the cytotoxicity of OA, cell viability
was assessed using the trypan blue exclusion assay. Cells were
collected from the same dishes used in the concentration-dependent
lipid accumulation experiments.
Analysis of cell death
Cell death induced by OA treatment or X-ray
irradiation was analyzed using direct immunofluorescence-based flow
cytometry with the Cell Lab Quanta™ SC MPL system (Beckman Coulter,
Inc.). Cells collected using a single pipette were washed twice
with Annexin V binding buffer (cat. no. 422201; BioLegend, Inc.),
after which they were stained according to the manufacturer's
protocol. Fluorescein isothiocyanate (FITC)-conjugated Annexin V
(cat. no. 640906; BioLegend, Inc.) and PI (MilliporeSigma) were
added to the cell suspension. Fluorescence intensity was quantified
by flow cytometry and analyzed using Kaluza analysis software
(version 2.1; Beckman Coulter, Inc.).
Measurement of intracellular LOOH
levels
Intracellular LOOH levels were measured using
Liperfluo staining solution (Dojindo Laboratories, Inc.). Untreated
cells or cells pretreated with OA and/or exposed to X-ray
irradiation were incubated with 1 µM Liperfluo at 37°C for 30 min
in the dark. After staining, cells were collected and analyzed by
flow cytometry using the Cell Lab Quanta™ SC MPL system (Beckman
Coulter, Inc.). Fluorescence intensity was quantified using Kaluza
analysis software (version 2.1; Beckman Coulter, Inc.).
Measurement of intracellular ROS
levels
Intracellular ROS levels were measured using the
fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate
(DCFH-DA; Dojindo Laboratories, Inc.). Cells were washed twice with
Hanks' balanced salt solution (HBSS) and then incubated with a
working solution of DCFH-DA at 37°C in a humidified atmosphere of
95% air and 5% CO2 for 30 min. After incubation, cells
were washed twice with HBSS and collected. ROS levels were analyzed
by flow cytometry using the Cell Lab Quanta™ SC MPL system (Beckman
Coulter, Inc.). The excitation and emission wavelengths were set at
488 and 530 nm, respectively.
Analysis of mRNA expression
Total RNA was extracted from cultured cells using
the RNeasy® Plus Mini Kit (Qiagen), and RNA
concentrations and purity were determined using a NanoDrop
spectrophotometer (Thermo Fisher Scientific, Inc.). First-strand
complementary DNA (cDNA) was synthesized from total RNA using the
ReverTra Ace® qPCR RT Master Mix (Toyobo Co., Ltd.)
according to the manufacturer's instructions.
Quantitative PCR was performed using the Power SYBR™
Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific)
and the SmartCycler® II system (Takara Bio Inc.).
Thermal cycling was performed as follows: initial denaturation at
95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C
for 1 min. The relative expression levels of glutamate-cysteine
ligase catalytic subunit (GCLC), glutathione peroxidase 4
(GPX4), and ChaC glutathione specific
γ-glutamylcyclotransferase 1 (CHAC1) were normalized to
those of β-actin (ACTB) and calculated using the
2−ΔΔCq method (17).
Gene-specific primers were designed using Primer3
software and synthesized by Eurofins Genomics. Gene sequences were
obtained from the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene/), and the following
accession numbers were used: GCLC (NM_001498.4), GPX4
(NM_002085.5), CHAC1 (NM_024111.6), and ACTB
(NM_001101.5) (Table I).
 | Table I.Sequences of human GCLC, GPX4,
CHAC1 and ACTB quantitative PCR primers. |
Table I.
Sequences of human GCLC, GPX4,
CHAC1 and ACTB quantitative PCR primers.
| Accession
number | Primer name | Primer sequence
(5′-3′) |
|---|
| NM_001498.4 | GCLC
forward |
CATTGATTGTCGCTGGGGAG |
|
| GCLC
reverse |
CTGGGCCAGGAGATGATCAA |
| NM_002085.5 | GPX4
forward |
AGAGATCAAAGAGTTCGCCGC |
|
| GPX4
reverse |
TCTTCATCCACTTCCACAGCG |
| NM_024111.6 | CHAC1
forward |
TGTGGTGACGCTCCTTGAAG |
|
| CHAC1
reverse |
GCCTCTCGCACATTCAGGTAC |
| NM_001101.5 | ACTB
forward |
GGACTTCGAGCAAGAGATGG |
|
| ACTB
reverse |
AGCACTGTGTTGGCGTACAG |
Statistical analysis
All statistical analyses were performed using R
software (version 4.4.1; R Foundation for Statistical Computing).
For comparisons among multiple groups, one-way analysis of variance
(ANOVA) was used, followed by the Tukey-Kramer post hoc test.
Normality and homogeneity of variance were confirmed prior to
ANOVA. Analyses were applied to datasets including cell cycle
distribution, lipid accumulation, cytotoxicity, LOOH levels, ROS
levels, and mRNA expression. A P-value of <0.05 was considered
statistically significant. All experiments were independently
repeated using separate cell cultures (biological replicates). For
each biological replicate, measurements were performed in duplicate
or triplicate as technical replicates where applicable.
Results
Radiation-induced alteration of cell
cycle distribution in Huh7 cells
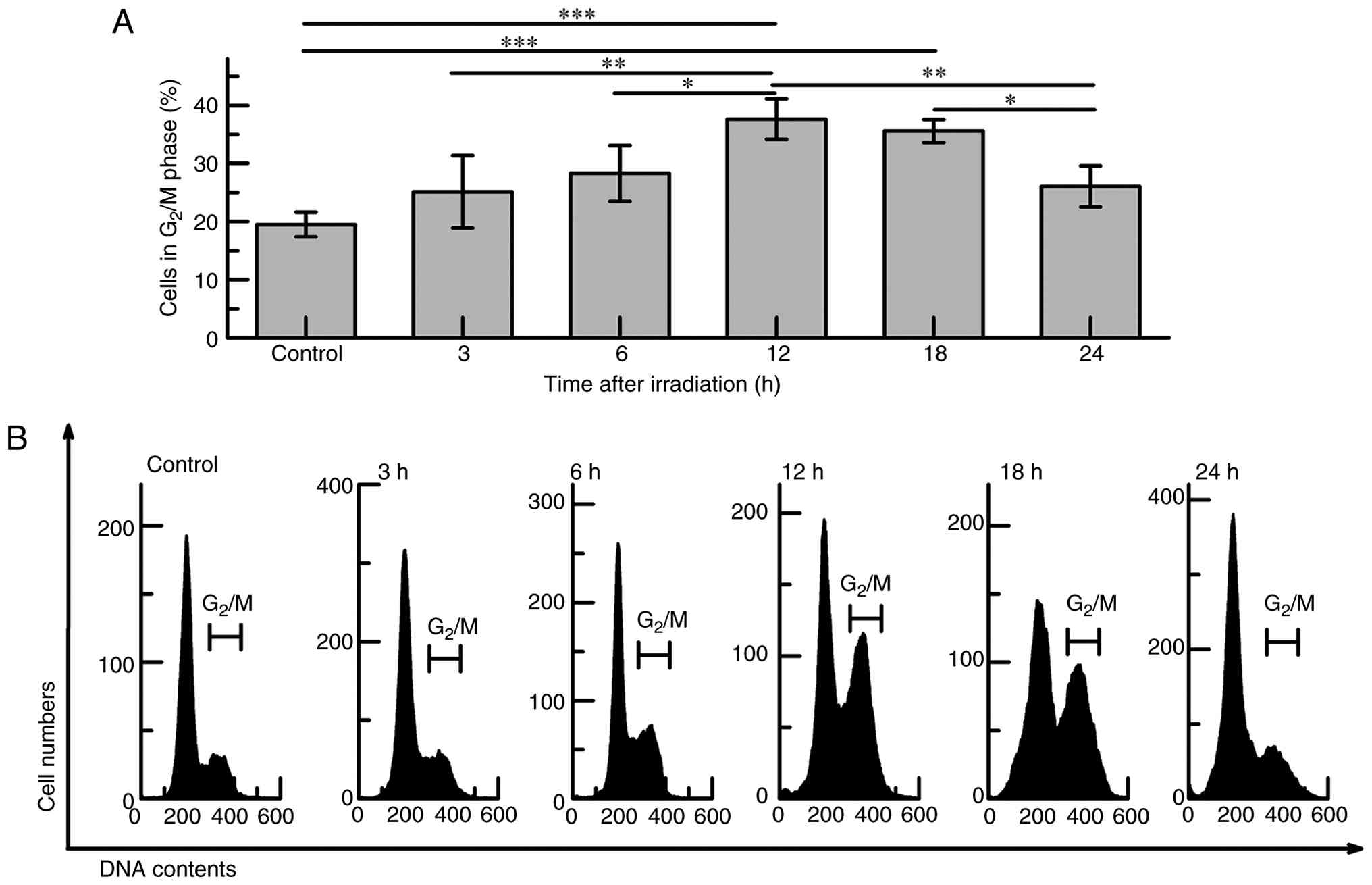
To investigate the effect of IR on cell cycle
progression in HCC cells, Huh7 cells were exposed to 5 Gy X-ray
irradiation, and the cell cycle distribution was assessed at 3, 6,
12, 18, and 24 h post-irradiation using PI staining and flow
cytometry. A marked accumulation of cells in the G2/M phase was
observed at 12 h (37.7±3.5%, P<0.001) and 18 h (35.6±2.0%,
P<0.001) compared with the non-irradiated controls (19.5±2.1%)
(Fig. 1A). In contrast, no
remarkable changes were detected at 3 h or 6 h, whereas a
nonsignificant increase was noted at 24 h. Representative
histograms of PI-stained cells are shown in Fig. 1B. These findings indicate that Huh7
cells undergo a robust G2/M phase arrest following DNA damage,
supporting their use as a reliable in vitro model for
studying radiotherapeutic responses. Based on these results, a
post-irradiation time point of 12 h was selected for downstream
analyses to capture the peak DNA damage response.
OA-induced time- and dose-dependent
lipid accumulation
Huh7 cells were treated with 1 mM OA, and
intracellular lipid accumulation was assessed at 3, 6, 12, 18, 24,
36, and 48 h using Nile red staining and flow cytometry. Compared
to the untreated control (2.9±0.1), OA-treated cells exhibited
remarkably increased lipid levels at all time points (3 h: 6.3±0.4;
6 h: 8.0±0.7; 12 h: 16.5±0.3; 18 h: 23.5±0.7; 24 h: 14.7±0.2; 36 h:
17.6±0.3; 48 h: 13.3±0.6; P<0.001 for all) (Fig. 2A and B). The highest lipid
accumulation was observed at 18 h. Next, cells were exposed to OA
at concentrations of 0.5, 1 or 2 mM for 18 h, and intracellular
lipid levels were analyzed. Cell viability was evaluated using the
trypan blue exclusion assay under the same conditions. A marked
reduction in viability was detected at 2 mM OA (61.2±3.8%) compared
with 0 mM (91.9±2.1%), 0.5 mM (90.0±3.6%) and 1 mM (86.1±6.2%)
(P<0.001) (Fig. 2C). In
addition, a dose-dependent increase in intracellular
lipid-associated Nile red fluorescence intensity was observed (0
mM: 3.04; 0.5 mM: 12.7; 1 mM: 20.5; 2 mM: 29.6) (Fig. 2D and E).
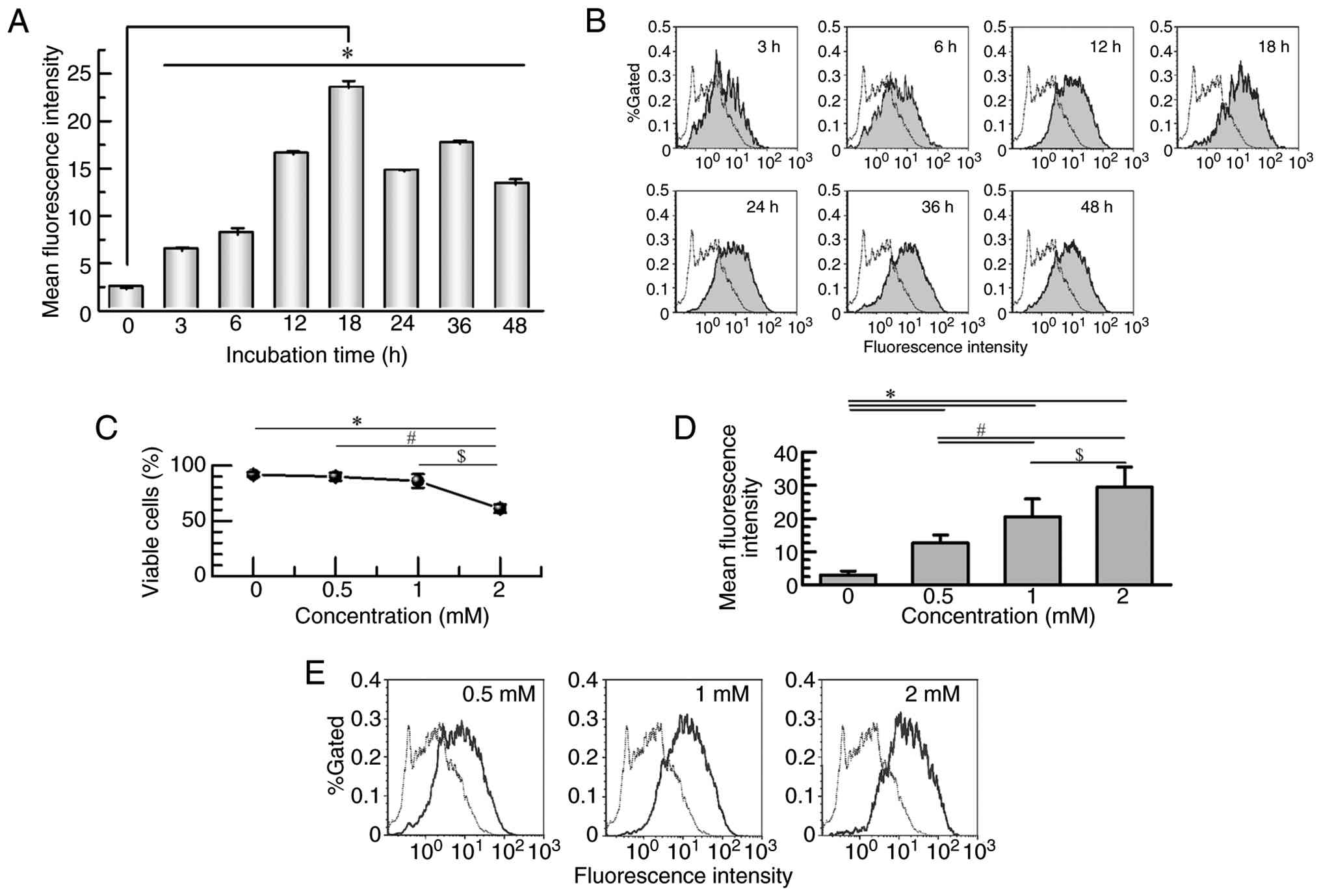 | Figure 2.OA-induced time- and dose-dependent
lipid accumulation in Huh7 cells. (A) Time-course analysis of
intracellular lipid accumulation following treatment with 1 mM OA.
Lipid content was measured at 3, 6, 12, 18, 24, 36 and 48 h using
Nile red staining and flow cytometry. Data are presented as the
mean ± SD from four independent experiments. *P<0.001 vs.
untreated control (0 h). (B) Representative histograms of Nile red
fluorescence showing a time-dependent increase in lipid
accumulation. The dashed line represents the untreated control (0
h). (C) Quantification of cell viability following 18-h treatment
with 0.5, 1 or 2 mM OA, determined using the trypan blue exclusion
assay, in which viable and non-viable cells were manually counted
under a light microscope using a Bürker-Türk hemocytometer.
Continuous data are presented as the mean ± SD of three independent
experiments. *P<0.001 vs. 0 mM, #P<0.001 vs. 0.5
mM and $P<0.001 vs. 1 mM. (D) Quantification of
intracellular lipid content following 18-h treatment with 0.5, 1 or
2 mM OA. Data are presented as the mean ± SD of three independent
experiments. *P<0.001 vs. 0 mM, #P<0.001 vs. 0.5
mM and $P<0.001 vs. 1 mM. Lipid content was assessed
by Nile red staining. (E) Representative histograms of Nile red
fluorescence showing dose-dependent lipid accumulation. The dashed
line represents the untreated control (0 mM). Statistical analysis
was performed using one-way ANOVA followed by the Tukey-Kramer post
hoc test. OA, oleic acid. |
Cytotoxic effect of OA in Huh7
cells
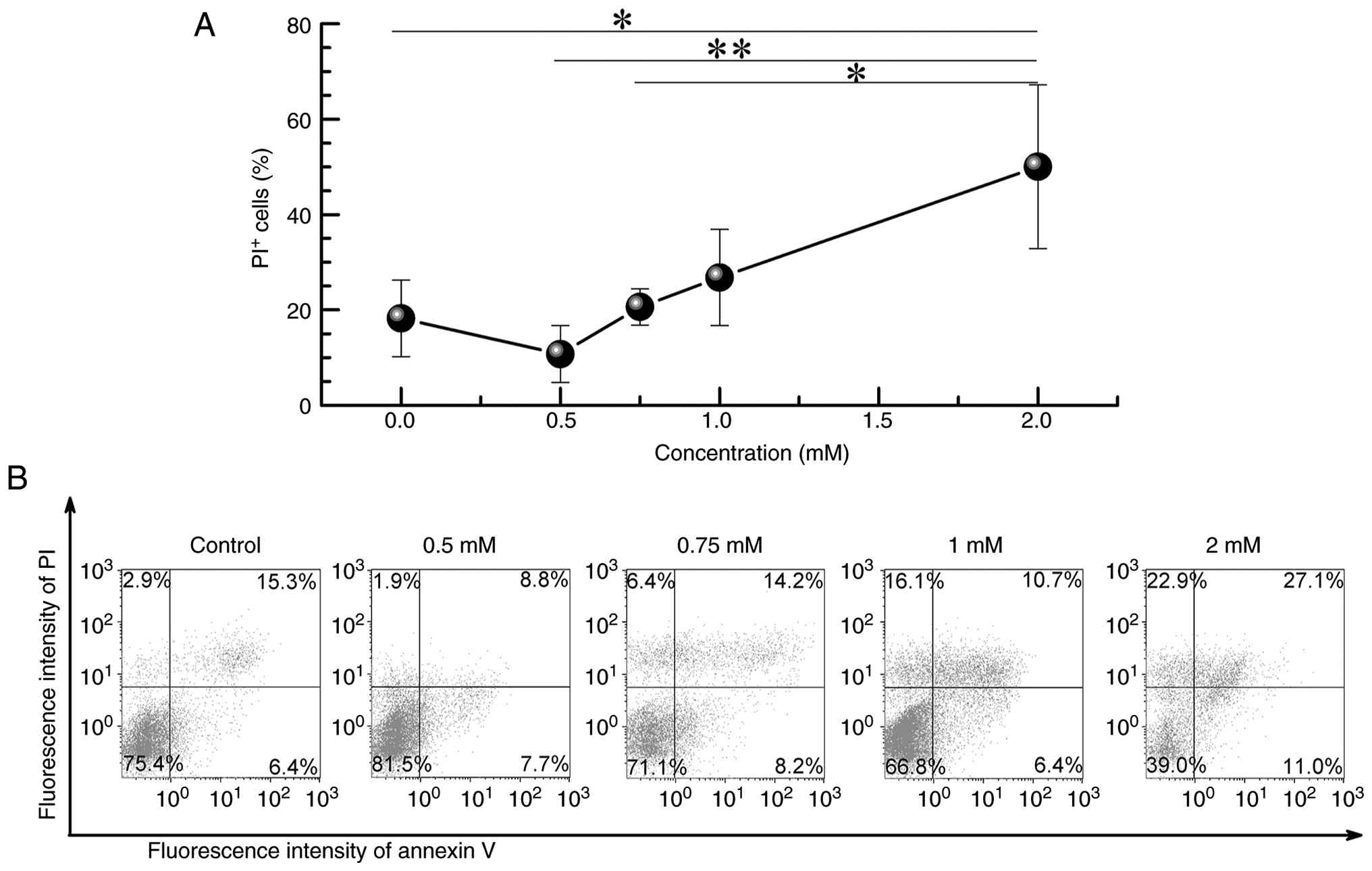
To further assess the cytotoxicity of OA, Huh7 cells
were treated with 0.5, 0.75, 1, or 2 mM OA for 18 h, followed by
annexin V/PI double staining and flow cytometry. A remarkable
increase in the proportion of PI-positive (necrotic/late apoptotic)
cells was observed only in the 2-mM group (50.0±17.1%) compared to
the untreated control group (18.2±8.0%, P<0.05). No
statistically significant differences were detected in the 0.5-mM
(10.8±6.0%), 0.75-mM (20.6±6.8%), or 1-mM (26.8±10.1%) groups
(Fig. 3). These results confirm
that 1 mM OA does not induce substantial cytotoxicity, indicating
its suitability for use as a sublethal dose for subsequent
mechanistic studies.
OA enhances radiation-induced cell
death
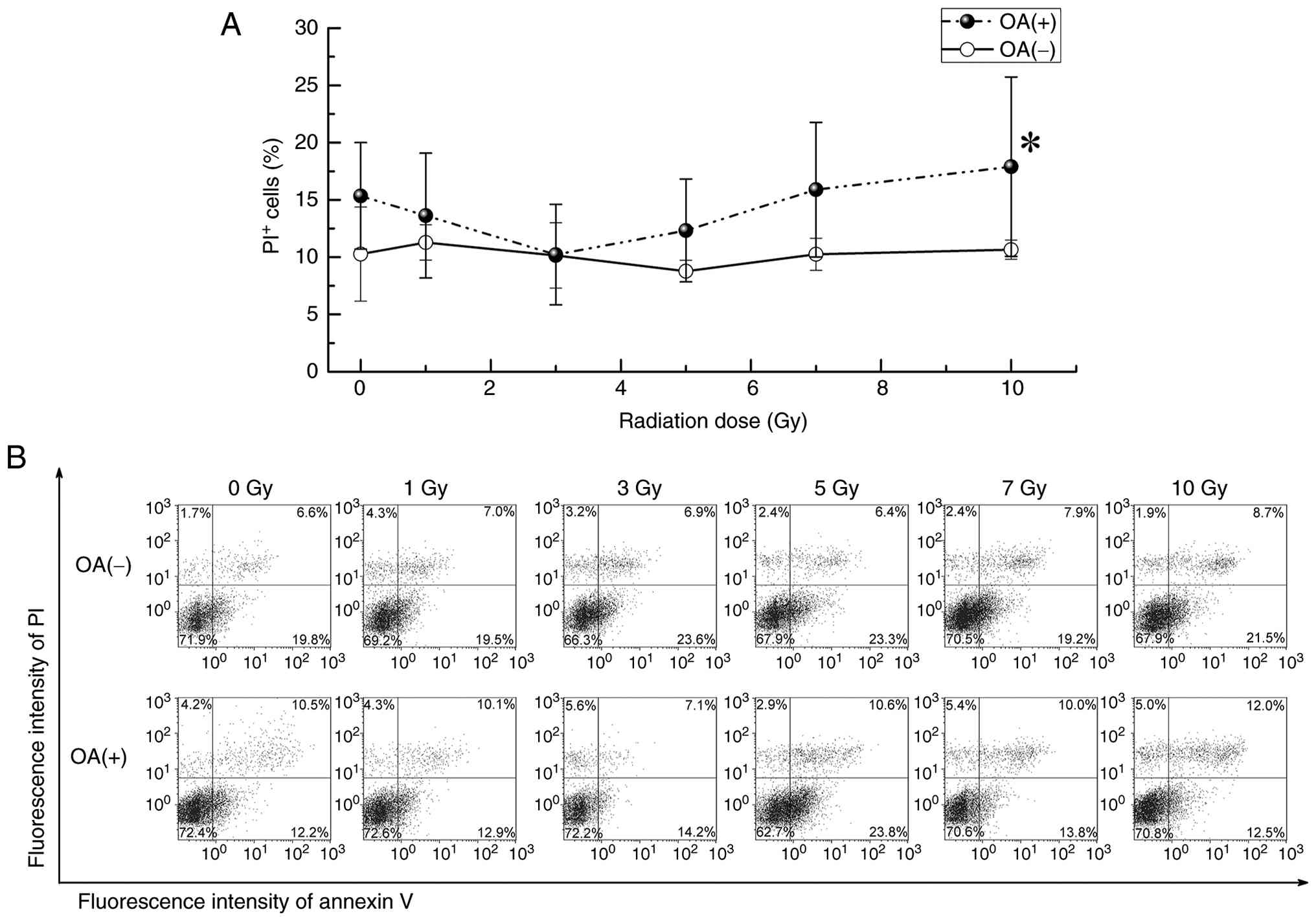
To investigate whether OA-induced lipid accumulation
enhances the cytotoxic effect of ionizing radiation (IR), Huh7
cells were pretreated with 1 mM OA for 18 h prior to X-ray
irradiation at doses ranging from 1 to 10 Gy. Cell death was
assessed 12 h post-irradiation using annexin V/PI staining and flow
cytometry. At 10 Gy, the proportion of PI-positive cells was
remarkably higher in the OA-pretreated group (17.9±7.8%) than in
the IR-only group (10.7±0.8%, P<0.05), indicating enhanced cell
membrane damage (Fig. 4A). No
remarkable differences were observed at lower radiation doses.
These findings suggest that OA-induced lipid accumulation
sensitizes Huh7 cells to radiation-induced cell death in a
dose-dependent manner, particularly at higher radiation doses. The
10-Gy dose was selected for subsequent mechanistic analyses because
a significant increase in PI-positive cells was observed only at
this dose when OA pretreatment was combined with irradiation,
compared with irradiation alone (Fig.
4A and B). In addition, previous in vitro radiobiology
studies have commonly employed single high-dose irradiation to
robustly induce oxidative stress and lipid peroxidation,
facilitating mechanistic evaluation of radiation-induced cell death
pathways (18,19).
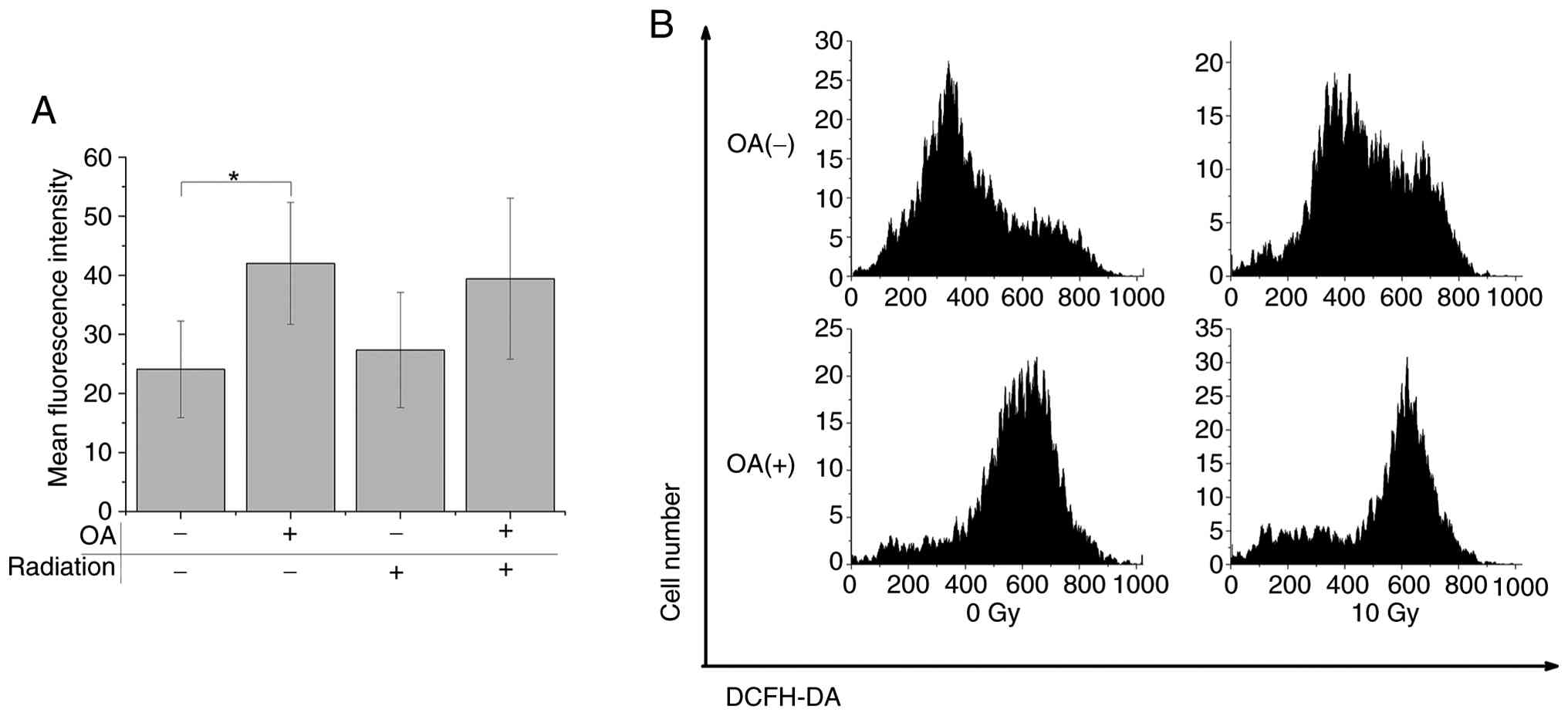
OA augments oxidative stress and lipid
peroxidation after IR exposure
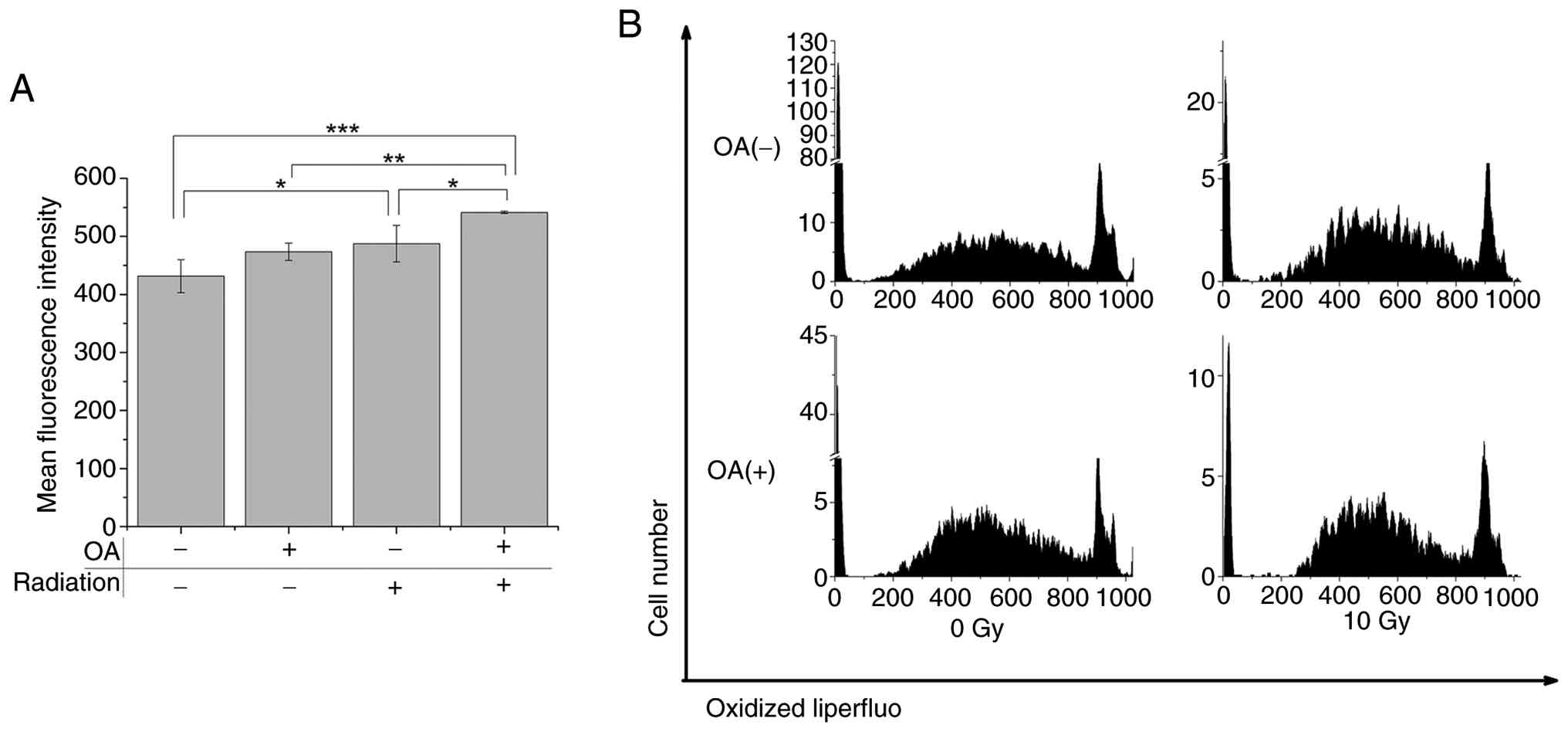
To elucidate the mechanism underlying OA-mediated
radiosensitization, we measured the levels of intracellular
oxidative stress markers, including LOOH and ROS. LOOH levels,
assessed by Liperfluo staining, were not remarkably altered by OA
treatment alone (473.3±14.9) compared to the untreated control. In
contrast, 10 Gy X-ray irradiation remarkably increased LOOH levels
(487.5±31.6, P<0.05). Notably, OA pretreatment combined with 10
Gy irradiation further elevated LOOH accumulation (541.4±2.1,
P<0.05 vs. 10 Gy alone) (Fig.
5A). These findings indicate that OA treatment augments
radiation-induced lipid peroxidation. Moreover, ROS levels were
remarkably increased by OA alone (42.0±10.3) compared to control
(24.1±8.2, P<0.05), as presented in Fig. 6.
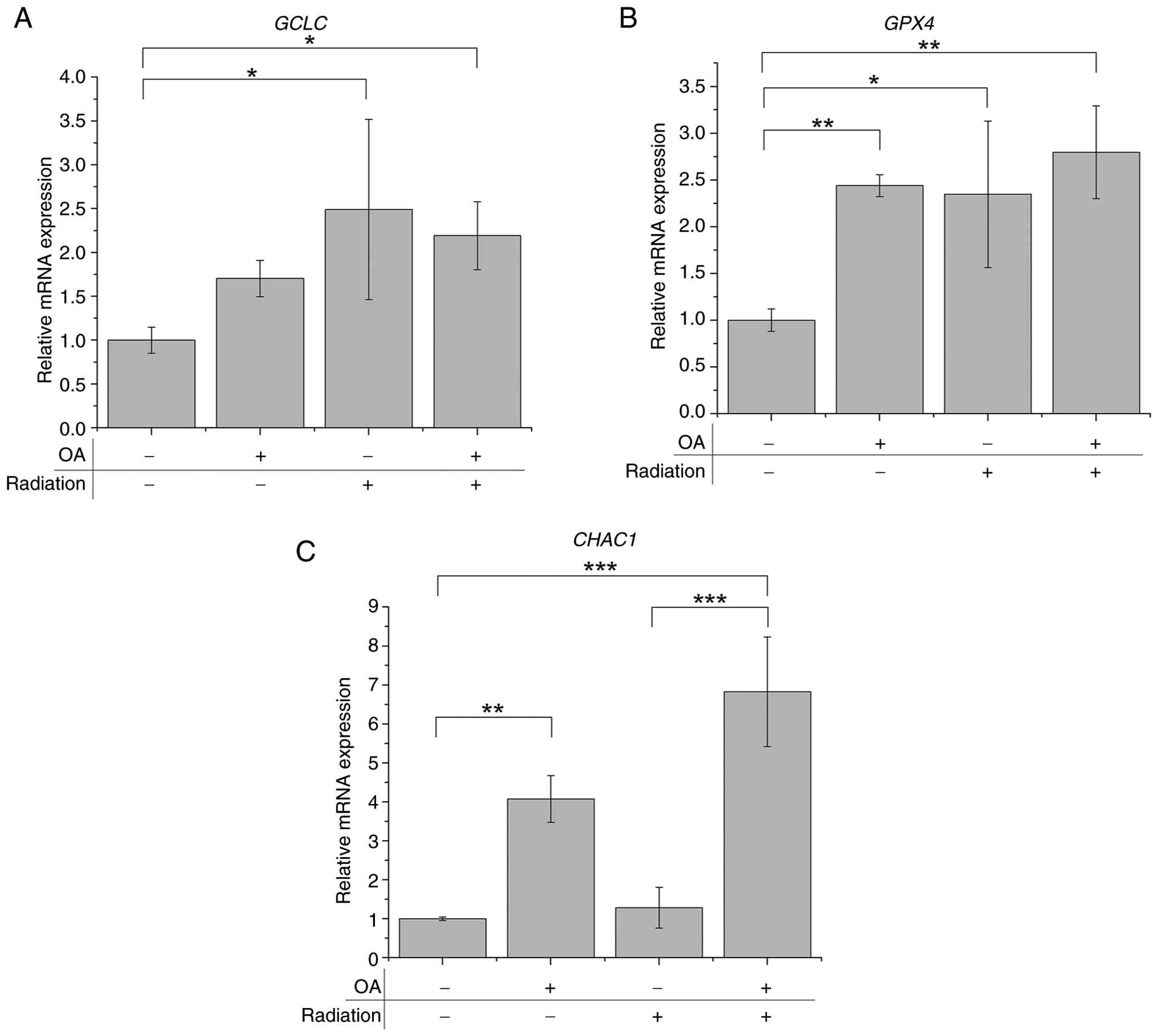
Modulation of oxidative stress-related
gene expression by OA and IR
To further elucidate the molecular mechanisms
underlying OA-mediated radiosensitization, we analyzed the
expression of key oxidative stress-related genes: GCLC,
GPX4, and CHAC1. Irradiation alone remarkably
upregulated GCLC expression (2.5±1.0-fold, P<0.05),
whereas OA administration alone had no statistically significant
effect. The combination of OA and irradiation did not further
increase the GCLC expression beyond that induced by irradiation
alone (2.2±0.4-fold, P<0.05) (Fig.
7A). In contrast, GPX4 expression was remarkably
upregulated in all treatment groups-OA alone (2.4±0.1, P<0.01),
IR alone (2.3±0.8, P<0.05), and the OA + IR combination
(2.8±0.5, P<0.01) (Fig. 7B).
Notably, the levels of CHAC1, a glutathione-degrading enzyme
and marker of ferroptosis, were markedly induced by OA treatment
alone (4.1±0.6-fold, P<0.01) and further elevated in the OA + IR
group (6.8±1.4-fold, P<0.001); however, it was not affected by
IR alone (Fig. 7C). These
transcriptional changes suggest that OA enhances radiation-induced
oxidative stress and may contribute to ferroptosis-related
signaling pathways.
Discussion
In this study, we established an in vitro
model using Huh7 HCC cells and OA, a monounsaturated fatty acid, to
investigate how lipid accumulation affects radiosensitivity. OA
pretreatment at 1 mM for 18 h (resulting in maximal lipid
accumulation without overt cytotoxicity) followed by 10 Gy X-ray
irradiation remarkably increased the proportion of PI-positive
cells and intracellular LOOH compared to radiation alone. These
findings indicate that OA enhances radiation-induced cell death via
additive effects on oxidative membrane damage.
IR primarily targets nuclear DNA and can induce cell
cycle arrest and apoptosis, particularly at the G2/M checkpoint,
which is known to be the most radiosensitive phase (20,21).
In our experiments, a marked accumulation of Huh7 cells in the G2/M
phase was observed 12 h post-irradiation, validating this time
point as optimal for subsequent analyses.
OA and other long-chain fatty acids, such as
palmitic acid, have been widely used in in vitro lipid
metabolism studies. Previous studies have demonstrated that fatty
acid treatment at 0.5–2 mM concentrations for 12–24 h induces lipid
accumulation without affecting viability up to 1 mM (22–24).
Consistent with this, we demonstrated that administration of 1 mM
OA for 18 h maximized lipid accumulation in Huh7 cells without
inducing cytotoxicity, as confirmed by Nile red staining and
annexin V/PI analyses.
Notably, while the number of annexin V-positive
cells (a marker of early apoptosis) was not remarkably increased,
those of PI-positive cells were elevated upon OA + IR co-treatment,
suggesting enhanced necrotic or non-apoptotic cell death. OA
treatment alone also remarkably increased intracellular ROS levels,
consistent with prior findings that OA promotes oxidative stress in
hepatic cells (25). These results
collectively imply that OA may sensitize HCC cells to IR-induced
necrotic cell death via oxidative membrane damage and lipid
peroxidation. Ionizing radiation is also known to activate
inflammation-associated signaling pathways, including
NF-κB-dependent and cytokine-mediated responses, which can
indirectly amplify intracellular oxidative stress through enhanced
ROS generation. Although inflammatory mediators were not directly
assessed in the present study, the observed increase in ROS and
lipid peroxidation following OA pretreatment and irradiation is
consistent with inflammation-associated redox dysregulation
reported in previous radiobiological studies (18,21).
These considerations suggest that inflammatory signaling may
indirectly contribute to oxidative stress amplification under
lipid-rich conditions.
In this context, the potential involvement of lipid
oxidation-related receptors, such as lectin-like oxidized
low-density lipoprotein receptor-1 (LOX-1), warrants consideration.
LOX-1 has been reported to mediate inflammatory and oxidative
responses to oxidized lipids, particularly in immune and vascular
cells, and its deficiency attenuates inflammation-associated tissue
damage in experimental models (26). However, in the present in
vitro system using Huh7 cells, lipid peroxidation is most
likely driven by radiation-induced non-enzymatic oxidative
reactions rather than receptor-mediated uptake of oxidized
lipoproteins. As LOX-1 expression or ox-LDL signaling was not
examined in this study, its involvement remains speculative. Future
studies may clarify whether LOX-1-related pathways contribute to
radiation-induced oxidative stress under lipid-rich conditions in
HCC.
To further elucidate the underlying mechanism, we
evaluated the expression of oxidative stress-related genes.
GCLC expression was upregulated by IR but not by OA, whereas
that of GPX4 was upregulated by both OA and IR. Importantly,
the expression of CHAC1 (a gene associated with glutathione
degradation and ferroptosis) was remarkably induced by OA alone and
further elevated by the combination of OA + IR; however, it was not
induced by IR alone (27,28). These results suggest that OA may
disrupt redox homeostasis by promoting glutathione consumption and
degradation, thereby impairing the cellular antioxidant
defense.
GPX4 requires reduced glutathione (GSH) as a
cofactor to detoxify lipid peroxides such as LOOH. The depletion of
GSH via CHAC1-mediated degradation or ROS-driven consumption
can lead to ferroptotic-like cell death (18). Thus, the observed upregulation of
CHAC1 expression and the increase in LOOH in OA + IR-treated
cells indicate that ferroptosis-related signaling may contribute to
the enhanced radiosensitivity (19). Consistently, Ye et al
(29) demonstrated that
radiation-induced lipid peroxidation triggers ferroptosis and
synergizes with ferroptosis inducers, further supporting the
concept that ferroptotic signaling underlies radiation-enhanced
oxidative damage.
A major limitation of this study is the lack of
functional validation via knockdown or overexpression of key
regulators such as CHAC1 and GPX4. Furthermore,
intracellular GSH levels and GPX4 enzymatic activity were
not directly measured. In addition, the present findings are
derived from a single HCC cell line (Huh7) and an in vitro
experimental system. The absence of in vivo models or
patient-derived samples limits the generalizability of these
results. Future studies employing multiple HCC cell lines, animal
models, and clinically relevant systems will be necessary to
validate the broader relevance of lipid-induced radiosensitization
and its potential translational implications.
Despite these limitations, the present study
provides novel evidence that OA-induced lipid accumulation enhances
HCC cell radiosensitivity by amplifying oxidative stress and lipid
peroxidation. Importantly, OA-induced lipid accumulation alone is
unlikely to be sufficient to determine radiosensitivity, and its
effects become evident primarily in the context of ionizing
radiation-induced oxidative stress. Thus, modulation of lipid
metabolism should be regarded as a potential adjunctive factor
rather than an independent therapeutic strategy. In addition,
validation using multiple hepatocellular carcinoma cell lines with
distinct metabolic characteristics will be required to determine
the generalizability of the present findings. Further validation
using in vivo models and clinical studies will be required
before any translational or clinical implications can be
considered.
Acknowledgements
Not applicable.
Funding
The present study was supported by JSPS KAKENHI, Grants-in-Aid
for Scientific Research (B) (grant no. 21H02861/23K21419),
Grant-in-Aid for Challenging Research (grant no. 25K22722) and
Takeda Science Foundation 2023.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
HT, YM and SM designed the study, prepared the
manuscript draft, and substantively participated in the manuscript
revision. HT and SM analyzed all biological data. YM and SM
supervised the study, critically reviewed the manuscript, and
provided final approval for the version to be submitted and
published. HT, YM and SM confirm the authenticity of all the raw
data. All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Reig M, Forner A, Rimola J, Ferrer-Fàbrega
J, Burrel M, Garcia-Criado Á, Kelley RK, Galle PR, Mazzaferro V,
Salem R, et al: BCLC strategy for prognosis prediction and
treatment recommendation: The 2022 update. J Hepatol. 76:681–693.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kimura T, Fujiwara T, Kameoka T, Adachi Y
and Kariya S: The current role of stereotactic body radiation
therapy (SBRT) in hepatocellular carcinoma (HCC). Cancers (Basel).
14:43832022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
The Japan Society of Hepatology (ed), .
Clinical Practice Guidelines for Hepatocellular Carcinoma 2025. 6th
Edition. Kanehara & Co., Ltd.; Tokyo, Japan: pp. 902025, (In
Japanese).
|
|
5
|
Younossi ZM, Golabi P, Paik JM, Henry A,
Van Dongen C and Henry L: The global epidemiology of nonalcoholic
fatty liver disease (NAFLD) and nonalcoholic steatohepatitis
(NASH): A systematic review. Hepatology. 77:1335–1347. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Toh MR, Wong EYT, Wong SH, Ng AWT, Loo LH,
Chow PK and Ngeow J: Global epidemiology and genetics of
hepatocellular carcinoma. Gastroenterology. 164:766–782. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pfister D, Núñez NG, Pinyol R, Govaere O,
Pinter M, Szydlowska M, Gupta R, Qiu M, Deczkowska A, Weiner A, et
al: NASH limits anti-tumour surveillance in immunotherapy-treated
HCC. Nature. 592:450–456. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimose S, Hiraoka A, Casadei-Gardini A,
Tsutsumi T, Nakano D, Iwamoto H, Tada F, Rimini M, Tanaka M,
Torimura T, et al: The beneficial impact of metabolic
dysfunction-associated fatty liver disease on lenvatinib treatment
in patients with non-viral hepatocellular carcinoma. Hepatol Res.
53:104–115. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bian X, Liu R, Meng Y, Xing D, Xu D and Lu
Z: Lipid metabolism and cancer. J Exp Med. 218:e202016062021.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martin-Perez M, Urdiroz-Urricelqui U,
Bigas C and Benitah SA: The role of lipids in cancer progression
and metastasis. Cell Metab. 34:1675–1699. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li J, Wang T, Liu P, Yang F, Wang X, Zheng
W and Sun W: Hesperetin ameliorates hepatic oxidative stress and
inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic
acid-induced HepG2 cells and a rat model of high-fat diet-induced
NAFLD. Food Funct. 12:3898–3918. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Y, Chen J, Gao Q, Shan X, Wang J and
Lv Z: Study on the attenuated effect of Ginkgolide B on ferroptosis
in high fat diet induced nonalcoholic fatty liver disease.
Toxicology. 445:1525992020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Zhang SD, Wang P, Guo N, Wang W,
Yao LP, Yang Q, Efferth T, Jiao J and Fu YJ: Pinolenic acid
ameliorates oleic acid-induced lipogenesis and oxidative stress via
AMPK/SIRT1 signaling pathway in HepG2 cells. Eur J Pharmacol.
861:1726182019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Muscaritoli M, Arends J, Bachmann P,
Baracos V, Barthelemy N, Bertz H, Bozzetti F, Hütterer E, Isenring
E, Kaasa S, et al: ESPEN practical guideline: Clinical nutrition in
cancer. Clin Nutr. 40:2898–2913. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Allen BG, Bhatia SK, Buatti JM, Brandt KE,
Lindholm KE, Button AM, Szweda LI, Smith BJ, Spitz DR and Fath MA:
Ketogenic diets enhance oxidative stress and radio-chemo-therapy
responses in lung cancer xenografts. Clin Cancer Res. 19:3905–3913.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zahra A, Fath MA, Opat E, Mapuskar KA,
Bhatia SK, Ma DC, Rodman SN III, Snyders TP, Chenard CA,
Eichenberger-Gilmore JM, et al: Consuming a ketogenic diet while
receiving radiation and chemotherapy for locally advanced lung
cancer and pancreatic cancer: The University of Iowa experience of
two phase 1 clinical trials. Radiat Res. 187:743–754. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Z, Tian R, She Z, Cai J and Li H:
Role of oxidative stress in the pathogenesis of nonalcoholic fatty
liver disease. Free Radic Biol Med. 152:116–141. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Verheij M: Clinical biomarkers and imaging
for radiotherapy-induced cell death. Cancer Metastasis Rev.
27:471–480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matt S and Hofmann TG: The DNA
damage-induced cell death response: A roadmap to kill cancer cells.
Cell Mol Life Sci. 73:2829–2850. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Listenberger LL, Han X, Lewis SE, Cases S,
Farese RV Jr, Ory DS and Schaffer JE: Triglyceride accumulation
protects against fatty acid-induced lipotoxicity. Proc Natl Acad
Sci USA. 100:3077–3082. 2023. View Article : Google Scholar
|
|
23
|
Ricchi M, Odoardi MR, Carulli L, Anzivino
C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni
S, et al: Differential effect of oleic and palmitic acid on lipid
accumulation and apoptosis in cultured hepatocytes. J Gastroenterol
Hepatol. 24:830–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Chen C, Chen J, Sang T, Peng H,
Lin X, Zhao Q, Chen S, Eling T and Wang X: Overexpression of
NAG-1/GDF15 prevents hepatic steatosis through inhibiting oxidative
stress-mediated dsDNA release and AIM2 inflammasome activation.
Redox Biol. 52:1023222022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sgonc R and Gruber J: Apoptosis detection:
An overview. Exp Gerontol. 33:525–533. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hashimoto K, Oda Y, Nakagawa K, Ikeda T,
Ohtani K and Akagi M: LOX-1 deficient mice show resistance to
zymosan-induced arthritis. Eur J Histochem. 62:28472018.PubMed/NCBI
|
|
27
|
Balachander GJ, Subramanian S and Ilango
K: Rosmarinic acid attenuates hepatic steatosis by modulating ER
stress and autophagy in oleic acid-induced HepG2 cells. RSC Adv.
8:26656–26663. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JY, Ren C, Wang LX, Yao RQ, Dong N, Wu
Y, Tian YP and Yao YM: Sestrin2 protects dendrite cells against
ferroptosis induced by sepsis. Cell Death Dis. 12:8342021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ye LF, Chaudhary KR, Zandkarimi F, Harken
AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang
Y, et al: Radiation-induced lipid peroxidation triggers ferroptosis
and synergizes with ferroptosis inducers. ACS Chem Biol.
15:469–484. 2020. View Article : Google Scholar : PubMed/NCBI
|