Introduction
Epidemiology and treatment challenges of hepatocellular carcinoma (HCC)
HCC ranks sixth globally in terms of incidence among malignant tumors and is the third leading cause of cancer-related mortalities, with particular prevalence in Asia (1). Current treatment modalities for HCC primarily include surgical resection, liver transplantation, radiotherapy, local ablation and targeted therapy, with the latter typically focusing on key oncogenic signaling pathways, such as the PI3K/AKT pathway (2). Due to the insidious presentation of symptoms during the early stages, the majority of HCC cases are first diagnosed already at an advanced stage, limiting curative treatment options. For such patients, systemic therapy remains the mainstay, yet survival rates remain low. HCC exhibits high recurrence rates, metastatic potential and drug resistance, with particularly high postoperative recurrence rates (3). Early recurrence indicates a poor prognosis, which is frequently associated with numerous factors, including microvascular invasion (MVI), albumin-bilirubin classification, satellite lesions and a high tumor burden score (4). Therefore, further investigations into the molecular mechanisms and influencing factors of HCC development are key in identifying novel diagnostic biomarkers and therapeutic targets.
Overview of the biological properties of stomatin-like protein 2 (STOML2). STOML2 is a member of the stomatin superfamily and a membrane-associated protein. It is primarily localized to the inner mitochondrial membrane; however, it was initially discovered on the surfaces of red blood cell membranes (4). The protein contains a number of conserved domains, including the characteristic stomatin/prohibitin/flotillin/HflK/C domain (also known as the band 7 domain) and a coiled-coil domain, and functions as a scaffold protein that regulates membrane protein localization, mainly influencing mitochondrial dynamics (5). In normal cells, STOML2 participates in various physiological processes, including maintaining cell membrane stability and regulating ion channel activity (6).
Research background regarding STOML2 in tumors
In tumor cells, STOML2 expression levels frequently undergo abnormal changes, which are closely associated with tumorigenesis and progression (7). Extensive evidence has indicated that STOML2, as a lipid raft-associated protein, exhibits markedly elevated expression in a variety of malignancies, including esophageal carcinoma, breast cancer and other solid tumors, as well as hematological malignancies. Its expression levels are positively associated with tumor staging, metastasis and poor prognosis (8,9). For the present review, a systematic survival analysis was conducted to assess the prognostic value of STOML2 expression across different cancer types using clinical survival data from the Human Protein Atlas (HPA) database (version 23.0; http://www.proteinatlas.org/). Standardized survival datasets for multiple cancer types, including breast cancer, liver hepatocellular carcinoma (LIHC) and renal cell carcinoma, were downloaded from the HPA. Data processing and visualization were performed using R software (version 4.3.1; R Foundation for Statistical Computing; http://www.R-project.org/) (10,11) with relevant bioinformatics packages, such as survival (https://cran.r-project.org/package=survival) and survminer (https://cran.r-project.org/package=survminer). Kaplan-Meier survival curves were plotted, and differences between the groups were compared using the log-rank test. For each cancer type, samples were dichotomized into STOML2-high and STOML2-low expression groups based on the median expression value of STOML2 across the respective cohort. The analysis of overall survival revealed that high STOML2 expression was significantly associated with poor prognosis in both breast cancer (P<0.001) and LIHC (P<0.001). By contrast, high STOML2 expression exhibited a potentially favorable prognostic trend in renal cell carcinoma (P<0.001). Throughout the current review, P<0.05 was considered to indicate a statistically significant difference. These clinical phenotype data support the rationale for investigating the mechanism by which STOML2 promotes HCC progression in the current review.
Central role of the PI3K/AKT signaling pathway in HCC
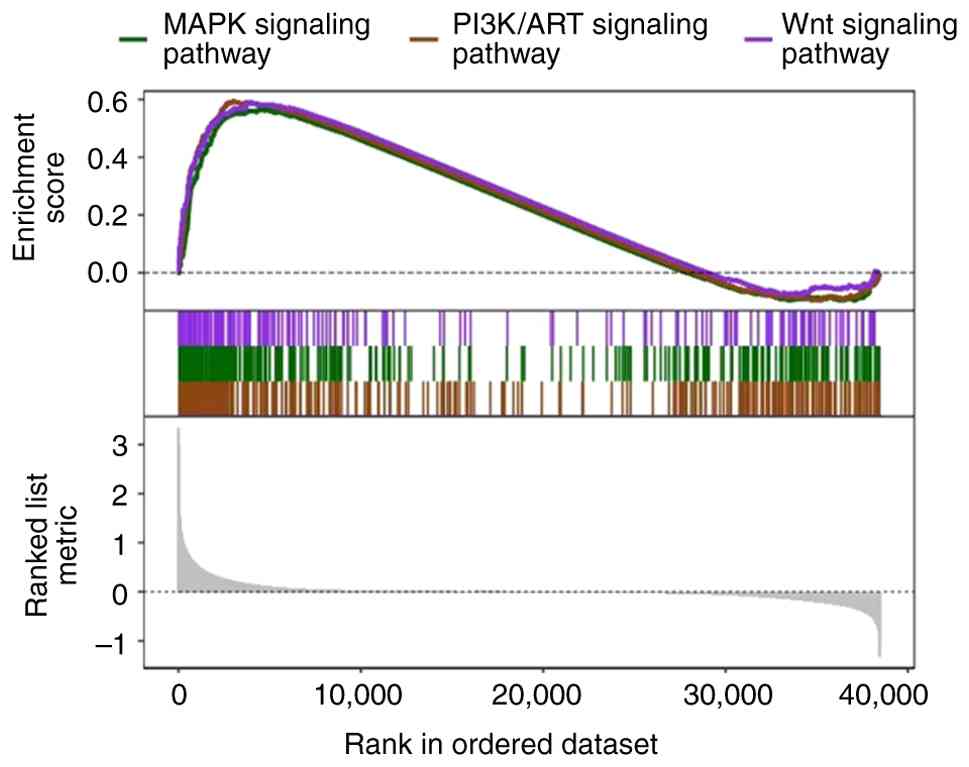
As a pivotal signaling hub, the PI3K/AKT pathway serves as a regulator of cellular survival and proliferation. In HCC, this pathway exhibits abnormal activation in >50% of cases, making it a core driver of HCC initiation and progression (12). Upon activation, PI3K catalyzes the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 further recruits AKT to the cell membrane and induces its phosphorylation; activated AKT then phosphorylates various downstream substrates, such as mTOR and GSK-3β, extensively regulating a plethora of biological processes in HCC, including cell cycle progression, protein synthesis, glucose metabolism, apoptosis resistance, epithelial-mesenchymal transition (EMT) and metastasis (13). At present, targeting the PI3K/AKT pathway has become a key therapeutic direction for HCC management (14). Gene Set Enrichment Analysis (GSEA) was performed to investigate signaling pathway enrichment in samples with differential STOML2 expression. The analysis utilized The Cancer Genome Atlas (TCGA)-LIHC cohort dataset, which was accessed and downloaded from the Genomic Data Commons portal (https://portal.gdc.cancer.gov/). Gene expression data (RNA-Seq by Expectation-Maximization normalized counts) were log2-transformed. Samples were dichotomized into ‘STOML2-high’ and ‘STOML2-low’ expression groups based on the median expression value of STOML2 across the entire cohort. GSEA was conducted using the GSEA software (version 4.3.2; Broad Institute) (15), available from https://www.gsea-msigdb.org/gsea/index.jsp, with the canonical signaling pathway gene sets (c2.cp.v2023.2.Hs.symbols.gmt) obtained from the Molecular Signatures Database (16) (https://www.gsea-msigdb.org/gsea/msigdb). Enrichment analysis was run with 1,000 permutations. A gene set was considered significantly enriched if it met the following thresholds: |normalized enrichment score (NES)|>1, nominal P<0.05, and false discovery rate (FDR) q-value <0.25. The analysis demonstrated significant enrichment of the PI3K/AKT signaling pathway in STOML2-high expression samples [enrichment score (ES)=0.594; NES=1.733; FDR q-value=2.004×10−7]. Concurrently, the MAPK pathway (ES=0.566; NES=1.625; FDR q-value=5.299×10−6) and Wnt pathway (ES=0.592; NES=1.637; FDR q-value=4.204×10−4) were also markedly enriched (Fig. 1).
Expression characteristics and clinical importance of STOML2 in HCC
High expression of STOML2 in HCC tissues and cell lines
Tissue level
At the tissue level, numerous studies have demonstrated that both STOML2 mRNA transcripts and STOML2 protein levels are notably higher in HCC tissues compared with those in adjacent normal liver tissues, as evidenced by techniques including reverse transcription-quantitative PCR, western blotting and immunohistochemistry (17,18). In the present study, a paired comparative analysis of STOML2 transcript levels was performed using the aforementioned TCGA-LIHC cohort. As shown in Fig. 2, the log2(transcripts per million +1) value of STOML2 was significantly higher in HCC tumor tissues compared with that in matched adjacent non-cancerous tissues from the same patients (P<0.001). The statistical significance was determined using a paired two-tailed Student's t-test, which is appropriate for comparing the means of two related (paired) samples. This finding, derived from analysis of TCGA-LIHC dataset, further demonstrated the abnormal upregulation of STOML2 in HCC at the transcriptional level.
Cell line level
STOML2 is commonly upregulated in established liver cancer cell lines (including Hep3B, Huh-7, MHCC97-H cells) relative to normal hepatic counterparts, as supported by comparative expression analyses (19).
Subcellular localization
While STOML2 is primarily localized to the inner mitochondrial membrane under physiological conditions, it undergoes relocalization in HCC cells. In the context of HCC, STOML2 is found primarily in the cytoplasm and cell membrane, particularly in lipid rafts, which is consistent with its altered biological functions in promoting tumor progression (20).
Association between STOML2 expression and clinicopathological characteristics of HCC
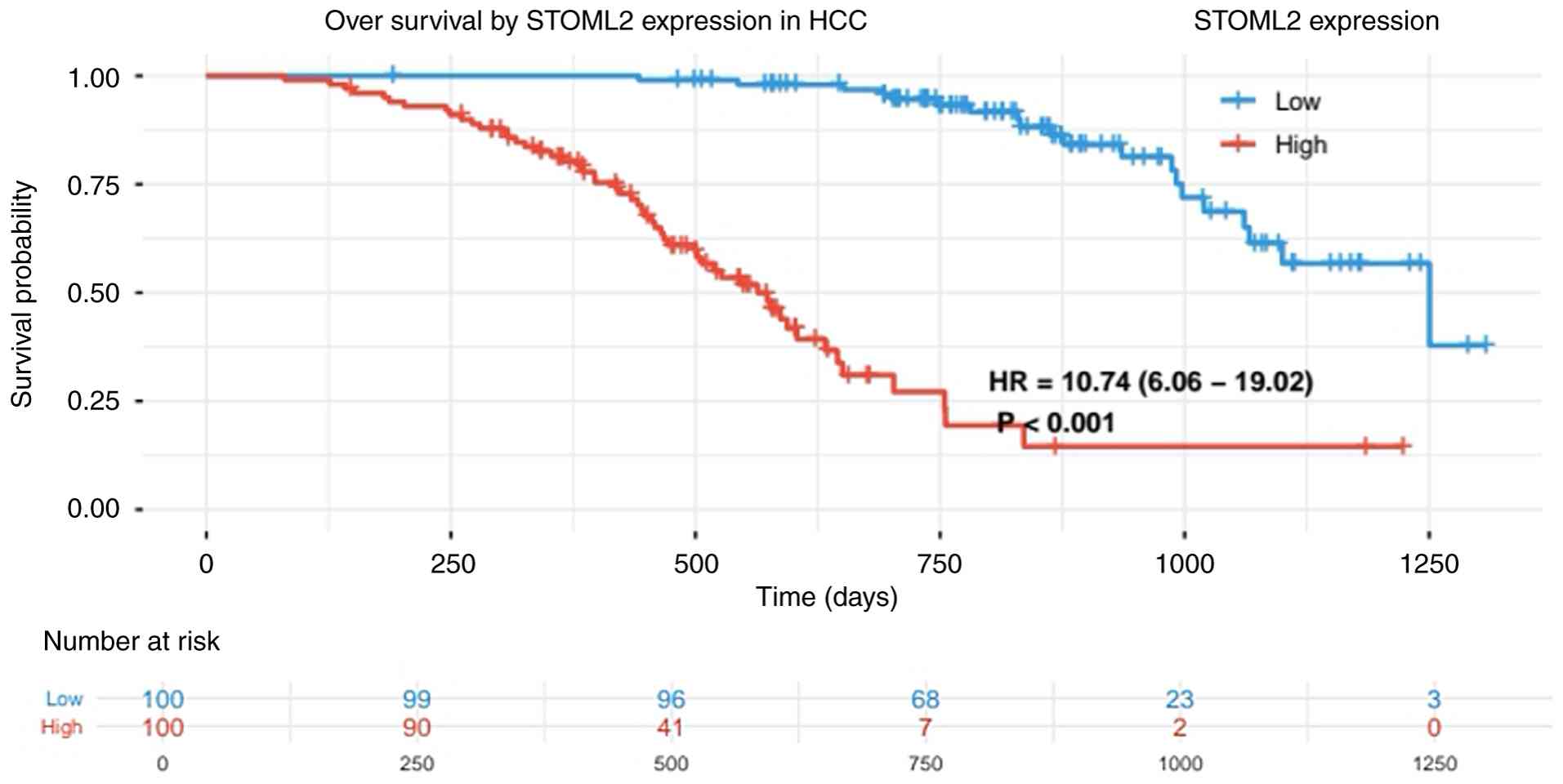
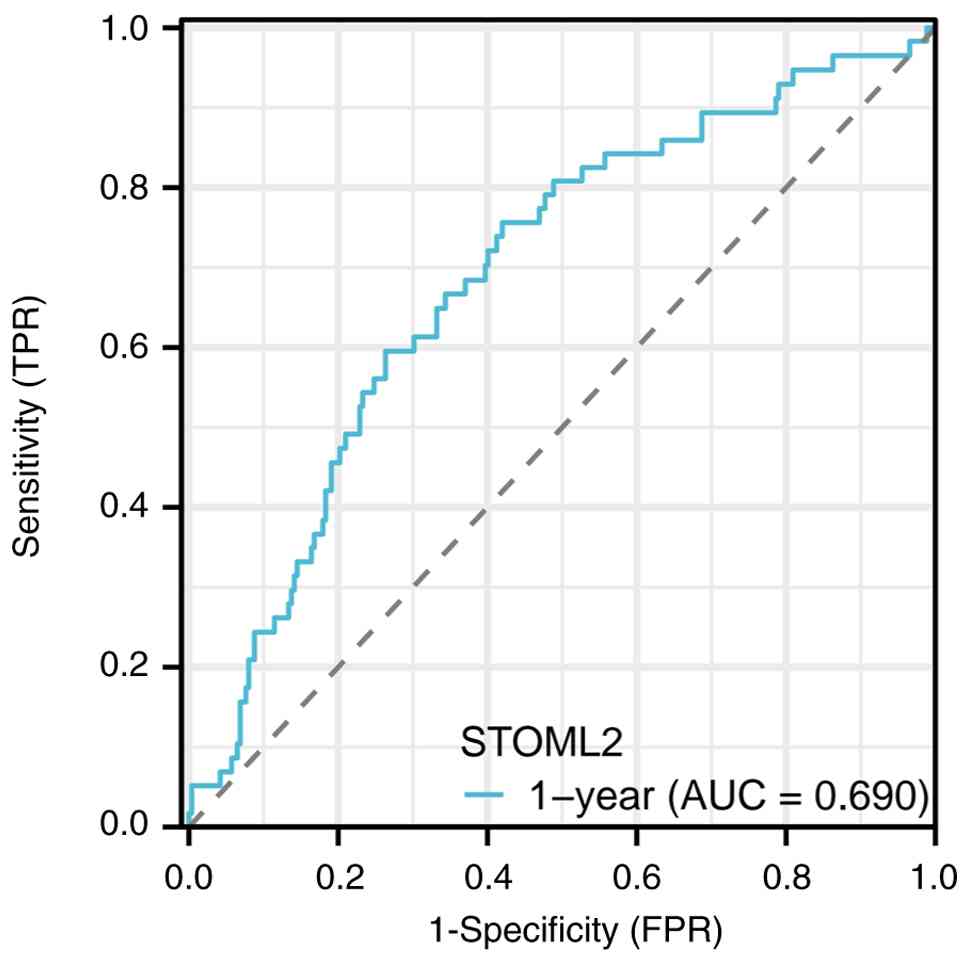
High STOML2 expression is markedly associated with multiple adverse clinical and pathological characteristics in patients with HCC. Patients with elevated STOML2 expression tend to exhibit larger tumor volumes, where its expression levels progressively increase with advancing TNM staging, particularly in patients with stage III–IV disease (21). In terms of histological differentiation, STOML2 expression is generally higher in poorly differentiated HCC tissues compared with that in moderately or well-differentiated groups, suggesting that its expression level is associated with the degree of tumor malignancy (22). Notable upregulation of STOML2 has been observed in patients with HCC and MVI or portal vein tumor thrombus, further indicating the potential involvement of this protein in HCC invasion (23). High STOML2 expression is also associated with an increased risk of lymph node metastasis, since its expression levels have been shown to be markedly higher in cases with extrahepatic metastasis compared with those in patients who are non-metastatic. In one previous study, the STOML2 protein exhibited a significantly higher positive expression rate in the distant metastasis group compared with that in the non-metastatic group (94.4 vs. 58.3; P<0.05), suggesting a potential positive association between STOML2 and distant tumor metastasis (24). To specifically evaluate the prognostic impact of STOML2 in HCC, an independent survival analysis was conducted using TCGA-LIHC cohort. The analysis utilized the full dataset from TCGA-LIHC project, comprising RNA-seq and clinical data for 371 patients. As in previous analyses, patients were dichotomized into STOML2-high and STOML2-low expression groups based on the median expression value of STOML2 mRNA across the cohort. Survival curves were generated using the Kaplan-Meier method, and differences in overall survival distributions between groups were compared using the Renyi test (a variant of the Cramér-von Mises test). This rank-based test is sensitive to early differences in survival, and was applied with the standard parameter setting (ρ=0.5) to weight such early deviations. The analysis demonstrated that STOML2 expression was significantly associated with the overall survival of patients with HCC. As shown in Fig. 3, the overall survival of patients in the STOML2-high expression group was significantly lower compared with that in the low-expression group (P<0.001), with a hazard ratio of 10.74 (95% CI, 6.06–19.02). This indicates that high STOML2 expression is an independent adverse prognostic factor for HCC. Collectively, these findings suggest that STOML2 serves a key role in the progression and metastasis of HCC. To further evaluate the dynamic prognostic accuracy of STOML2, a time-dependent receiver operating characteristic (ROC) curve analysis was conducted. This analysis utilized the same TCGA-LIHC cohort (comprising RNA-seq and clinical data for 371 patients with HCC), ensuring consistency with the preceding survival analysis. STOML2 mRNA expression levels, represented as continuous log2 (transcripts per million + 1) values, served as the sole predictive variable. The primary outcome was 1-year overall survival. The analysis was performed using R software (version 4.3.1); specifically, the timeROC package (version 0.4) was employed to compute the time-dependent ROC curves and the corresponding area under the curve (AUC) value at the 1-year time point. The timeROC package implements a nonparametric inverse probability of censoring weighting estimator for calculating time-dependent sensitivity and specificity, which properly accounts for right-censored survival data. The package is publicly available from the Comprehensive R Archive Network at https://CRAN.R-project.org/package=timeROC. As shown in Fig. 4, the AUC value of STOML2 for predicting 1-year overall survival was 0.690. An AUC value exceeding the chance-level threshold of 0.5 indicates predictive capacity, with the observed value of 0.690 suggesting a moderate predictive efficiency of STOML2 for the short-term prognosis of HCC (25).
Mechanism underlying the STOML2 activation of the PI3K/AKT signaling pathway
Direct interaction and regulation
Interaction between STOML2 and p85
p85 is a regulatory subunit of PI3K. STOML2 may bind specifically to p85 through specific domains, such as the sterile α-motif. This interaction enhances PI3K activity by promoting the formation of a stable p85-p110 heterodimer between p85 and the catalytic subunit p110, thereby preventing degradation or conformational inactivation of free p110 (26). It may also directly enhance PI3K catalytic efficiency by relieving the self-inhibition of p110 by p85 through conformational regulation, thereby promoting the conversion of PIP2 to PIP3 (27).
Stabilization of the PI3K complex
As a scaffold protein, STOML2 may enable the PI3K complex (consisting of the p85 regulatory subunit and p110 catalytic subunit) to specifically bind with other upstream activators, such as receptor tyrosine kinases (RTKs; for example, EGFR or IGF-1R), or localize to specific membrane domains, such as lipid rafts or plasma membrane invaginations. This process enhances signal proximity, improves signaling efficiency and PI3K complex stability, and also ensures directed transmission of downstream signaling (28).
Facilitating AKT membrane recruitment and phosphorylation activation
PIP3 serves as an important anchoring site for AKT recruitment to the plasma membrane. STOML2 may enhance AKT activation through the following two mechanisms: i) Its localization in lipid rafts maintains a locally enriched PIP3 microenvironment on the membrane, providing a more stable binding site for AKT; or ii) STOML2 may indirectly assist AKT phosphorylation by recruiting 3-phosphoinositide-dependent protein kinase 1 and mTOR complex (mTORC)2 to the membrane vicinity, thereby promoting their respective phosphorylation of AKT at Thr308 and Ser473 sites, ultimately enhancing the stability of the active conformation of AKT (29).
Indirect regulation of upstream signals
STOML2 interacts with RTKs
STOML2 resides in lipid rafts, which serve as platforms for the signal transduction of multiple RTKs. STOML2 may stabilize RTK localization on the membrane by affecting lipid raft integrity or directly interacting with specific RTKs (such as EGFR), thereby inhibiting their endocytic degradation (30). These actions enhance ligand-induced RTK activation, such as EGFR phosphorylation upon binding to EGF, which further promotes the downstream recruitment and activation of PI3K, ultimately upregulating the PI3K/AKT pathway (31).
Negative regulation of PTEN
PTEN serves as a key negative regulator of the PI3K/AKT pathway by directly inhibiting AKT activation through the dephosphorylation of PIP3 (32). STOML2 may relieve the inhibitory effect of PTEN on the PI3K/AKT pathway by affecting PTEN stability, subcellular localization or through directly/indirectly inhibiting its phosphatase activity. Notably, this regulatory pattern has been inferred by previous studies in other malignancies (33,34). Tripartite motif-containing 37 has been shown to accelerate PTEN ubiquitination and degradation through their interaction, thereby activating the PI3K/AKT pathway and promoting T-cell acute lymphoblastic leukemia proliferation (35). However, the specific molecular mechanisms by which STOML2 regulates PTEN in HCC remain unvalidated and require further experimental verification in HCC cell lines or animal models.
Synergistic effects with other positive regulators of the PI3K/AKT pathway
Activation of the PI3K/AKT pathway typically relies on multifactorial cooperation. STOML2 may interact with, (or function synergistically with,) known positive regulators of this pathway. STOML2 has been demonstrated to amplify the activation effect on the PI3K/AKT pathway by enhancing the binding efficiency between Ras and PI3K or by cooperating with GRB2-associated binding protein 1 to recruit PI3K to the membrane vicinity (36). Additionally, STOML2 can participate in cross-talk between the PI3K/AKT pathway and MAPK/Wnt pathway through interactions with molecules, such as platelet derived growth factor receptor-β (37). GSEA revealed co-enrichment of these three pathways in STOML2-high expression samples, suggesting they can synergistically amplify the regulatory effects of STOML2 on malignant phenotypes in HCC. However, the specific molecular mechanisms underlying this cross-regulation require further validation.
Downstream effects driven by the STOML2/PI3K/AKT axis and its pro-cancer mechanisms
Promotion of cell proliferation and inhibition of apoptosis
A previous study conducted in hepatocellular carcinoma cell lines has indicated that knocking out STOML2 can suppress the expression of the PI3K/AKT/mTOR signaling pathway, leading to increased apoptosis and excessive activation of autophagy (38). Consequently, STOML2 upregulation in HCC cells may activate the PI3K/AKT signaling pathway to promote proliferation. AKT inhibits GSK-3β activity through phosphorylation, blocking its mediation of cyclin D1 degradation. This leads to the intracellular accumulation of cyclin D1, facilitating the transition from G1 to S phase (39). In renal carcinoma cells, 6-gingerol can induce G1 arrest by inhibiting the AKT/GSK-3β/cyclin D1 signaling pathway. Conversely, AKT regulates the FOXO family of transcription factors by promoting their cytoplasmic retention and inactivation, thereby downregulating the expression of cell cycle inhibitors p21Cip1 and p27Kip1. This promotes the action of their inhibitory effects on cyclin-dependent kinases, accelerating cell cycle progression (40). This mechanism has been observed across numerous cancer types. In prostate cancer, AKT activation has been shown to suppress FOXO transcription factors, reducing p27 expression and promoting cell proliferation (41). Similarly, in breast cancer, constitutive AKT signaling leads to phosphorylation and cytoplasmic sequestration of FOXO transcription factors, resulting in decreased nuclear p27Kip1 levels and accelerated G1/S phase progression (42). In addition, AKT can phosphorylate the pro-apoptotic protein Bad, causing it to bind to 14–3-3 proteins and lose activity, thereby failing to antagonize the anti-apoptotic functions of Bcl-2/Bcl-xL (43). AKT can also directly phosphorylate caspase-9 precursors, inhibiting their activation process and blocking the initiation of the apoptotic cascade. In non-small cell lung cancer, AKT regulates the alternative splicing of caspase-9 by phosphorylating the splicing factor SRp30a (also known as SRSF1). Phosphorylation of SRp30a by AKT enhances its activity and promotes the inclusion of a specific exonic sequence in the CASP9 pre-mRNA. This alternative splicing event leads to the predominant production of the anti-apoptotic isoform caspase-9b, which lacks the catalytic domain, instead of the pro-apoptotic isoform caspase-9a. Consequently, the pro-apoptotic cascade cannot be initiated, thereby promoting tumor cell survival (44). In endometrial carcinoma, silencing the S100A8 gene has been shown to inhibit AKT phosphorylation, leading to caspase-9 activation and subsequent apoptosis induction (45). These studies collectively demonstrate the important role of the AKT/caspase-9 signaling pathway in tumorigenesis and progression. AKT also promotes mouse double minute 2 homolog nuclear translocation through phosphorylation, accelerating p53 ubiquitination and degradation, thereby weakening p53-mediated apoptosis pathways in various cancer cells, including HCC and glioblastoma cells (46).
Enhancing cell invasion and migration
Inducing EMT
EMT is an important biological process through which tumor cells of epithelial origin acquire invasive and migratory capabilities. This transition enables tumor cells to escape the constraints of intercellular junctions, enhance their ability to invade the extracellular matrix and acquire stem cell-like properties (47). The PI3K/AKT pathway serves as the core regulator of EMT. STOML2 can activate AKT, which phosphorylates and inhibits GSK-3β activity. This release of regulatory control over GSK-3β allows EMT transcription factors to escape phosphorylation-mediated degradation and become stabilized (48). This subsequently downregulates epithelial markers, such as E-cadherin and claudin-1, while upregulating mesenchymal markers, such as N-cadherin, vimentin and fibronectin. Consequently, HCC cells lose epithelial polarity and acquire the migratory and invasive properties that are characteristic of mesenchymal cells (49).
Regulation of cytoskeletal remodeling and pseudopodia formation
AKT can activate the Rho family of GTPases by phosphorylating their guanine exchange factors (or by inhibiting their GTPase-activating proteins), thereby regulating the activity states of these GTPases, such as Rac1 and Cdc42. This promotes the reorganization of the actin cytoskeleton, the dynamic assembly and dissociation of focal adhesions, and the formation of migration-related pseudopodia, such as pseudopodia and filopodia, providing structural support and propulsion for the directed migration of HCC cells (50).
Promotion of extracellular matrix
As a key component of the cellular microenvironment, the extracellular matrix not only provides structural scaffolding for cells but also regulates cell adhesion, migration and survival signaling through interactions with receptors, such as integrins (51). Under normal conditions, synthesis and degradation of extracellular matrix components are maintained at a dynamic equilibrium, which tumor cells can disrupt to promote invasion and metastasis. AKT can upregulate the transcriptional expression and secretion of MMPs, such as MMP-2 and MMP-9, by activating mTORC1 or the NF-κB pathway. These MMPs specifically degrade extracellular matrix components, such as collagen and laminin, clearing physical barriers for HCC cells to breach the basement membrane and invade surrounding tissues (52). Therefore, it may be inferred that STOML2 upregulation can notably enhance the secretory activity of MMPs in HCC cells, thereby promoting their invasion and migration through this mechanism.
Promoting angiogenesis
Angiogenesis is a key process that enables tumors to obtain nutrients and oxygen after exceeding a certain size. This process is initiated when tumor cells, often under hypoxic stress, secrete pro-angiogenic factors such as vascular endothelial growth factor (VEGF). VEGF signaling activates nearby endothelial cells, prompting their proliferation, migration and eventual tube formation to create new, often disorganized, blood vessels. These newly formed blood vessels not only supply the growing tumor but also serve as a key prerequisite for distant metastasis. Tumor cells at the invasive front can intravasate into these immature, leaky vessels, enter the systemic circulation, and subsequently spread to distant tissues and organs where they may extravasate and form secondary tumors (53). Although the role of STOML2 in HCC angiogenesis has not been directly validated, the PI3K/AKT pathway, a key regulator of VEGF expression, may represent a potential mechanism (54). STOML2 activates AKT, initiating specific cascade reactions. AKT can phosphorylate and inhibit prolyl hydroxylase activity, reducing the hydroxylation of hypoxia-inducible factor-1α (HIF-1α). This inhibits its ubiquitination and degradation, enhancing its accumulation in the nucleus (55). As a notable transcription factor, HIF-1α directly binds to the hypoxia response element on the VEGF gene promoter, markedly upregulating VEGF transcription and secretion. AKT may also synergistically promote the expression of VEGF and other proangiogenic factors by activating various transcription factors, such as c-Myc and NF-κB (56). These factors exert paracrine effects on vascular endothelial cells within the tumor microenvironment, activating their proliferation, migration and luminal formation capabilities (57). However, direct evidence of STOML2-mediated angiogenesis in HCC is currently lacking and requires further validation.
Regulation of tumor metabolic reprogramming (Warburg effect)
Tumor metabolic reprogramming is a hallmark adaptation of cancer cells to rapid proliferation demands, with the Warburg effect (the preferential use of glycolysis for energy production even under aerobic conditions) being one of its most prominent metabolic phenotypes (58). This metabolic reprogramming not only fuels rapid cancer cell proliferation, but also promotes immune evasion and tumor progression by altering the tumor microenvironment (59). The PI3K/AKT pathway, as a central regulatory hub of cellular metabolism, serves a notable role in initiating and sustaining the Warburg effect. In addition, STOML2 may participate in HCC cell metabolic reprogramming by activating this pathway (60). AKT promotes the recruitment of glucose transporter type 1 (GLUT1) to the cell membrane through phosphorylation, markedly enhancing cellular glucose uptake capacity. Simultaneously, AKT directly phosphorylates and activates hexokinase, particularly hexokinase 2 (which binds to mitochondria to prioritize glucose utilization), phosphofructokinase, pyruvate kinase and other key rate-limiting enzymes of glycolysis, accelerating glucose breakdown into pyruvate through glycolysis. Furthermore, AKT upregulates GLUT1 expression by activating HIF-1α, with this regulation being particularly pronounced under hypoxic conditions (61). Furthermore, AKT inhibits GSK-3β activity, thereby releasing its suppression of glycogen synthase and promoting glucose storage as glycogen, providing cells with stable energy reserves (62). These combined regulatory actions enhance glycolytic flux in HCC cells, not only rapidly generating ATP to meet energy demands, but also supplying intermediates for the synthesis of biomolecules, such as nucleotides and amino acids, thereby sustaining the continuous proliferation of HCC cells (63). The precise mechanism by which STOML2 regulates the Warburg effect in HCC cells through PI3K/AKT pathway activation, including whether it influences lactate dehydrogenase activity or metabolic diversion of pyruvate, remains to be fully elucidated in HCC models.
Promoting chemotherapy resistance
Chemotherapy resistance is one of the primary causes of clinical treatment failure in HCC and sustained activation of the PI3K/AKT pathway is a key mechanism underlying this resistance to specific anticancer chemotherapies (64). Previous studies have revealed that phosphatidylinositol-binding clathrin assembly protein-interacting mitotic regulator can enhance HCC cell resistance to sorafenib by activating the PI3K/AKT pathway (65), whereas syndecan-1 can promote HCC cell resistance to cisplatin through the PI3K/AKT pathway (66). STOML2 notably reduces HCC cell sensitivity to conventional chemotherapeutic agents (such as sorafenib, cisplatin, doxorubicin and 5-fluorouracil) by activating AKT-mediated anti-apoptotic and pro-survival signaling, in addition to enhancing DNA damage repair capacity. By contrast, STOML2 knockdown or combined treatment with the PI3K/AKT pathway inhibitor LY294002 can effectively reverse this resistance phenotype, restoring cellular response to chemotherapy. This discovery identified a potential therapeutic target for overcoming chemotherapy resistance in HCC (67).
Collectively, the STOML2/PI3K/AKT axis drives multiple hallmarks of HCC progression, including sustained proliferation, evasion of apoptosis, enhanced invasion and metastasis, and therapy resistance. A schematic summary of these mechanisms, distinguishing between validated pathways in HCC and hypotheses extrapolated from other types of cancer, is presented in Fig. 5.
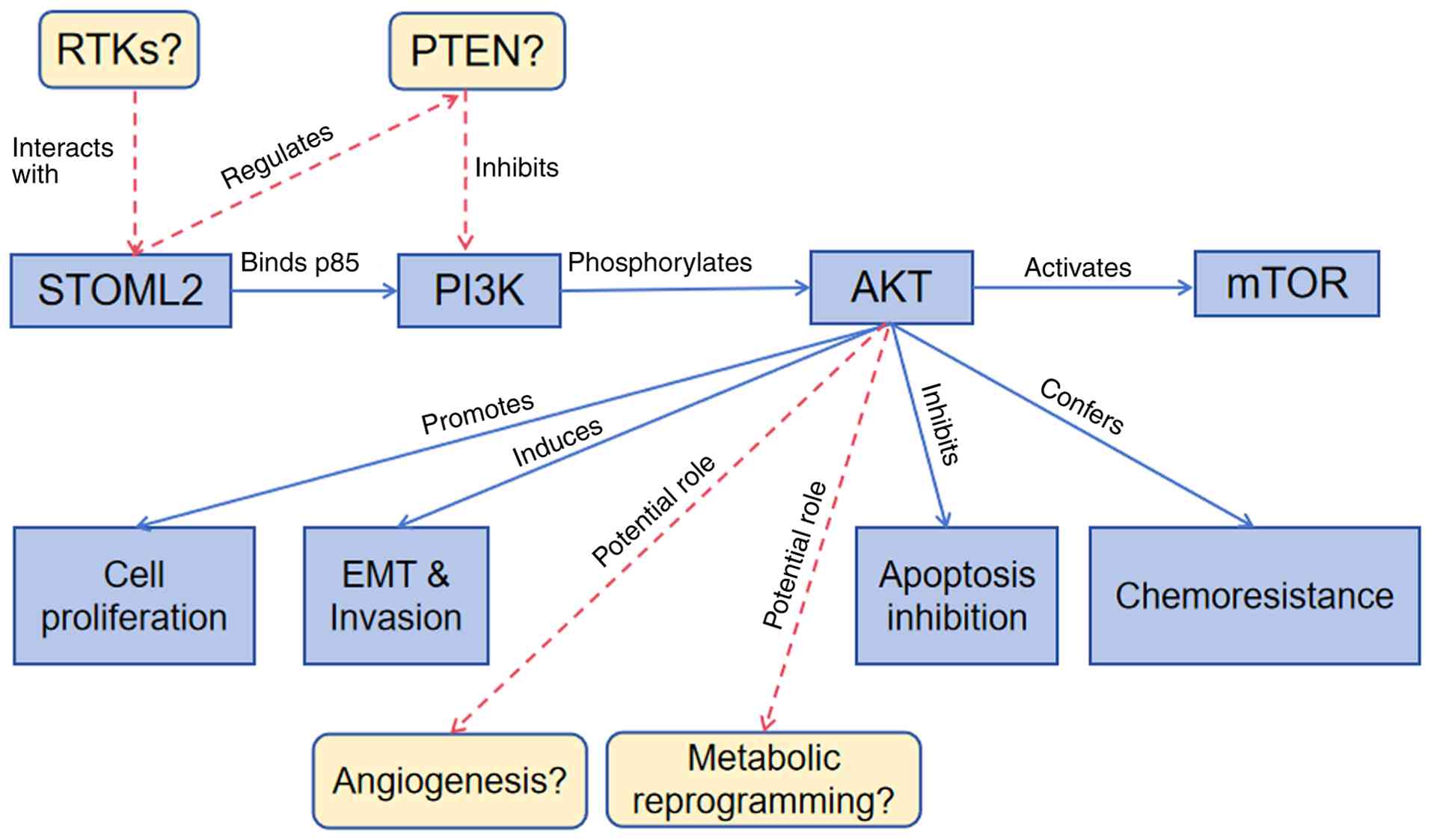 |
Figure 5.
Schematic illustration of STOML2-mediated activation of the PI3K/AKT signaling pathway and its regulation of malignant phenotypes in HCC. Solid blue lines represent mechanisms experimentally validated in HCC: i) STOML2 (primarily localized in the cytoplasm and lipid rafts of HCC cells) directly binds to the p85 regulatory subunit of PI3K, promoting the formation of a stable p85-p110 heterodimer and enhancing PI3K catalytic activity. ii) Activated PI3K converts phosphatidylinositol-4,5-bisphosphate to phosphatidylinositol-3,4,5-trisphosphate, which recruits AKT to the cell membrane and facilitates its phosphorylation (at Thr308 by phosphoinositide-dependent kinase-1 and Ser473 by mTOR complex 2). iii) Phosphorylated AKT further activates downstream mTOR signaling to promote cell proliferation, phosphorylates GSK-3β to stabilize EMT transcription factors (Snail, Slug and Twist), thereby downregulating epithelial markers (E-cadherin and claudin-1) and upregulating mesenchymal markers (N-cadherin and vimentin), and inhibits apoptosis by phosphorylating Bad and caspase-9. Dashed red lines indicate hypothetical mechanisms extrapolated from other malignancies: i) STOML2 may interact with RTKs to maintain RTK membrane localization and inhibit endocytic degradation. ii) STOML2 may promote the ubiquitin-mediated degradation of PTEN (a negative regulator of PI3K/AKT) or block its membrane targeting. iii) Activated AKT could upregulate VEGF expression through hypoxia-inducible factor-1α stabilization to facilitate angiogenesis. iv) AKT may enhance glucose transporter 1 membrane recruitment and activate glycolytic enzymes to induce the Warburg effect (metabolic reprogramming). Notably, these hypothetical mechanisms have not been demonstrated by HCC-specific experiments and require further validation. Boxes denote the final malignant phenotypes of HCC regulated by the STOML2/PI3K/AKT axis, including cell proliferation, invasion/migration, apoptosis inhibition and chemoresistance (to sorafenib, cisplatin and 5-fluorouracil). This figure was created using Adobe Illustrator (version 27.0; Adobe Inc.). HCC, hepatocellular carcinoma; STOML2, stomatin-like protein 2; EMT, epithelial-mesenchymal transition; RTKs, receptor tyrosine kinases.
|
Upstream regulatory mechanisms of STOML2 expression
Abnormal upregulation of STOML2 in HCC serves as a prerequisite for its activation of the PI3K/AKT pathway and subsequent promotion of tumor progression. Therefore, understanding the reasons behind this abnormal increase of STOML2 in HCC is important.
Transcriptional level regulation
Activation of transcription factors
Abnormally activated oncogenic transcription factors in HCC can directly target and regulate STOML2 gene transcription (68). The Wnt/β-catenin pathway, which is frequently abnormally activated in HCC, initiates transcription by binding to the T-cell factor/lymphoid-enhancer factor binding element on the STOML2 gene promoter through the β-catenin/transcription factor 4 complex (69). Additionally, transcription factors frequently upregulated in HCC, such as c-Myc, STAT3 and NF-κB, may synergistically promote STOML2 transcription activation by binding to specific sites within its promoter or enhancer regions (70).
Epigenetic regulation of STOML2 expression
i) DNA methylation. Methylation of CpG islands in gene promoter regions typically suppresses transcription. The hypomethylation observed in the STOML2 promoter region may contribute to its elevated expression in HCC. Downregulation of methyltransferases or inhibition of their activity in this region reduces methylation levels, thereby releasing the suppression of transcription initiation (71); however, the specific methylation sites and regulatory patterns in HCC require further validation.
ii) Histone modifications. Histone modifications regulate gene expression by altering chromatin accessibility. In HCC, the STOML2 promoter region is hypothesized to accumulate activating histone modifications such as H3K27ac, mediated by activating histone acetyltransferases, while simultaneously reducing repressive modifications. While specific histone modifications at the STOML2 promoter in HCC require experimental validation, general dysregulation of histone modifiers (e.g., acetyltransferases and deacetylases) contributing to oncogene activation has been documented in HCC. This creates an open chromatin state, facilitating transcription factor binding and initiating transcription (72).
iii) Non-coding RNA regulation. Certain tumor-suppressing microRNAs (miRNAs/miRs), such as miR-101, miR-139-5p or miR-485-5p, can directly induce STOML2 mRNA degradation or inhibit translation by specifically binding to its 3′-untranslated region (UTR), thereby limiting STOML2 expression under normal physiological conditions. However, in HCC, these tumor-suppressing miRNAs are frequently downregulated due to gene segment deletions, abnormal promoter methylation or dysfunction in upstream regulatory pathways, markedly weakening their inhibitory effect on STOML2 (73). Concurrently, long non-coding RNAs (lncRNAs), such as LINC00662 and HOTAIR, or circular RNAs (circRNAs), including circ_0000517, which are highly expressed in HCC, can function as competitive endogenous RNAs (ceRNAs), acting as ‘molecular sponges’. LINC00662 can sequester miR-101, reducing its binding opportunity with STOML2 mRNA, thereby relieving the miRNA-mediated translational repression of STOML2 and further promoting its upregulation (74). It has also been demonstrated that this lncRNA/circRNA/miRNA/STOML2 ceRNA regulatory network in HCC can enhance activation of the PI3K/AKT pathway by upregulating STOML2, ultimately promoting malignant biological behaviors, such as tumor cell proliferation and invasion (75).
Post-transcriptional and translational regulation
Post-transcriptional and translational regulation may influence STOML2 mRNA stability and translation efficiency. The competitive regulatory mechanisms involving monocyte chemotactic protein-induced protein 2 and insulin-like growth factor 2 mRNA-binding protein 1 observed in breast cancer angiogenesis may similarly apply to HCC (76). With regard to mRNA stability, RNA-binding proteins can regulate STOML2 mRNA half-life by recognizing specific sequences, including AU-rich elements in the 3′-UTR (77). Oncoproteins abnormally upregulated in HCC may bind to STOML2 mRNA, inhibiting its degradation by nucleases and notably prolonging mRNA survival. By contrast, downregulation of tumor suppressor RNA-binding proteins may weaken this inhibitory effect on mRNA stability, indirectly promoting STOML2 mRNA accumulation (78). Furthermore, in terms of translation efficiency, STOML2 mRNA translation may be regulated by certain intracellular signaling pathways. The mTOR pathway, frequently activated in HCC, can promote translation initiation by phosphorylating translation initiation factors, such as eukaryotic translation initiation factor 4E (EIF4) binding protein 1, thereby releasing its inhibition on EIF4 and facilitating ribosomal binding to STOML2 mRNA to accelerate translation initiation (79,80). Furthermore, the 5′-UTR structure of STOML2 mRNA may influence translation recognition and binding efficiency, thereby regulating STOML2 protein synthesis. These potential post-transcriptional and translational regulatory mechanisms may synergize with transcriptional control to contribute to maintaining high STOML2 expression levels in HCC, which would provide a sufficient protein basis for its activation of the PI3K/AKT pathway (81).
Regulation of protein stability
Accumulation of the STOML2 protein in HCC not only depends on transcriptional and translational upregulation, but is also closely associated with enhanced protein stability, a process primarily regulated by the intricate network of post-translational modifications. The ubiquitin-proteasome system, as a core pathway in protein degradation, serves a pivotal role in regulating STOML2 stability (82). Under normal conditions, STOML2 is recognized by specific E3 ubiquitin ligases and undergoes ubiquitination, leading to degradation by the proteasome (83). However, in HCC, impaired function of these E3 ubiquitin ligases, due to downregulation (as observed for regulators such as MDM2 or FBXW7 in certain HCC contexts), genetic mutations or blockade by oncoprotein binding, markedly reduces STOML2 ubiquitination. Concurrently, upregulated deubiquitinating enzymes in HCC; for example, ubiquitin-specific peptidases such as USP7 or USP22, which are often upregulated in HCC and known to stabilize various oncoproteins, may specifically remove ubiquitin chains from STOML2 molecules, further inhibiting its degradation (84). This dual mechanism collectively prolongs the STOML2 protein half-life and enhances its stability, ultimately leading to notable intracellular accumulation. Additionally, other post-translational modifications may indirectly influence its stability. Kinase-mediated phosphorylation of STOML2, potentially by kinases such as AKT, protein kinase C or ERK, which are frequently hyperactivated in HCC and known to phosphorylate scaffold or membrane-associated proteins, can alter its spatial conformation, making it harder for E3 ligases to recognize or promote its binding to chaperone molecules, including heat shock proteins, to form stable complexes (19). Acetylation modifications may enhance structural stability through similar mechanisms (85). Furthermore, these modifications may synergistically regulate the subcellular localization of STOML2, maintaining its enrichment within lipid rafts to ensure effective activation of the PI3K/AKT pathway, thereby amplifying its oncogenic functions (6).
Therapeutic strategies and prospects for liver cancer targeting the STOML2/PI3K/AKT axis
Direct strategies targeting STOML2
Among direct therapeutic interventions targeting STOML2, gene silencing technology currently represents the primary approach (86). RNA interference employs small interfering RNA (siRNA) or short hairpin RNA designed to target STOML2 mRNA. Delivered into HCC cells through viral (adenovirus) or non-viral vectors, these agents specifically degrade the target mRNA and knock down STOML2 expression (87). Both in vitro and in vivo studies have demonstrated that this intervention can effectively suppress HCC cell proliferation, invasion and migration, while inducing apoptosis and enhancing sensitivity to chemotherapeutic agents (88–90).
The development of small-molecule inhibitors faces notable challenges. Since STOML2 primarily functions as a scaffold protein through protein-protein interactions and lacks the typical active pockets found in enzymes, directly designing small molecules that can block its interactions is difficult (91). This underscores the importance of resolving the precise structural details of the interaction interfaces of STOML2 with key partners. Only by elucidating the molecular basis of these interactions can techniques, such as virtual screening, structure-based drug design or fragment screening, be employed to identify small-molecule compounds or peptide mimetics capable of competitively binding to the interaction surfaces. Such compounds would disrupt the binding of STOML2 to downstream molecules and block its function (92). Despite current technical challenges, the field retains potential for developments with ongoing advancements in structural biology techniques.
Targeting the downstream PI3K/AKT pathway
PI3K inhibitors
Inhibitors include pan-PI3K inhibitors, subtype-selective PI3K inhibitors and dual PI3K/mTOR inhibitors (93). However, monotherapy yields limited efficacy due to compensatory pathway activation, such as upregulation of the MAPK/ERK pathway, feedback enhancement of RTK signaling, or mTORC2-mediated re-activation of AKT, and frequently induces adverse reactions, including hyperglycemia, rash and hepatotoxicity. Therefore, combination with other therapies is necessary to enhance outcome (94).
AKT inhibitors
AKT inhibitors can be categorized into the following two types: i) Allosteric inhibitors, such as MK-2206, which block AKT activation by binding to its pleckstrin homology domain; and ii) ATP-competitive inhibitors, such as ipatasertib and capivasertib (95). Preclinical studies have indicated that MK-2206 can markedly inhibit tumor growth in HCC mouse models (96,97), with its efficacy potentially being associated with STOML2 upregulation. STOML2-activated AKT enhances drug sensitivity. However, the majority of AKT inhibitors remain in the early clinical trial phases (98).
mTOR inhibitors
mTORC1 inhibitors, such as rapamycin and its derivative everolimus, have been approved for the second-line treatment of advanced HCC. In a preclinical study it has been suggested that dual mTORC1/mTORC2 inhibitors demonstrate more potent antitumor activity than single-pathway inhibitors, by further blocking downstream AKT signaling (99). Further related clinical trials are currently underway (100,101).
Other approaches
Given the complexity of the PI3K/AKT pathway and tumor heterogeneity, combination therapy has emerged as a key strategy to enhance treatment efficacy. Combining EGFR inhibitors (erlotinib), MEK inhibitors (trametinib) or anti-angiogenic agents (sorafenib and lenvatinib) works to synergistically block numerous oncogenic pathways to enhance outcomes. This combinatorial approach of co-targeting parallel pathways demonstrates broader therapeutic potential beyond specific cancer types. For example, the conceptual rationale, that vertical or horizontal pathway blockade can overcome resistance, is supported by evidence in other malignancies. In glioblastoma, combining EGFR inhibitors with PI3K inhibitors can simultaneously suppress both the EGFR/ERK and PI3K/AKT/mTOR pathways, markedly inhibiting tumor growth and prolonging survival (102). This provides a proof-of-concept that informs analogous combination strategies in HCC. Pathway inhibitors may also be combined with immune checkpoint inhibitors. Activation of the PI3K/AKT pathway induces tumor immune escape, with preclinical studies in cancers such as osteosarcoma (103) and glioma (104) demonstrating that PI3Kβ inhibitors can enhance the efficacy of anti-programmed cell death protein 1 antibodies (103,104).
Targeting upstream regulatory factors
Intervening in the upstream regulatory factors of STOML2 to indirectly reduce its expression, offers an alternative approach for targeted therapy. This primarily involves modulating non-coding RNA regulatory networks to restore the expression of tumor-suppressing miRNAs (105). In HCC, downregulated tumor-suppressing miRNAs, such as miR-101 and miR-139-5p, enhance STOML2 expression. Increasing tumor-suppressing miRNA levels (through mimics) can reinstate inhibition of STOML2 mRNA and reduce STOML2 expression. Current challenges for this strategy include ensuring the stability and delivery efficiency of miRNA mimics in vivo to guarantee consistent function within HCC tissues (106). Inhibiting oncogenic lncRNAs or circRNAs represents another key pathway. LINC00662 and circ_0000517, which are upregulated in HCC, can act as ‘molecular sponges’, trapping large quantities of tumor-suppressing miRNAs and thereby elevating STOML2 expression (107). For such regulatory factors, specific antisense oligonucleotides or siRNAs can be designed to degrade target lncRNAs/circRNAs or inhibit their function, thereby reducing their adsorption effect. This allows miRNAs to re-target STOML2 and suppress its expression. However, this approach faces particular uncertainties, such as ensuring sufficient in vivo delivery to tumor tissue, avoiding off-target effects and overcoming the potential instability of these oligonucleotide agents (108). Other notable challenges include the risk of unintended immune stimulation, achieving tissue-specific targeting, and the unknown long-term safety and feasibility of large-scale production for clinical use (109).
In comparison with previous reviews on STOML2 or the PI3K/AKT pathway in cancer (54,75), the present review provides a dedicated and mechanistic summary focusing specifically on HCC. Not only was the established evidence that STOML2 drives HCC malignancy through PI3K/AKT activation outlined, and its potential association with other pathways (such as MAPK and Wnt pathways) indicated by enrichment analyses noted, but the present review also critically delineated the boundaries between validated mechanisms and speculative extrapolations from other cancer types (such as angiogenesis and PTEN regulation). By integrating clinical data, upstream regulatory networks and therapeutic targeting strategies, the present review presents a comprehensive framework that highlights STOML2 as a central node and pinpoints precise knowledge gaps, thereby offering a clearer direction for future translational research in HCC.
Conclusions and outlook
Overall, the STOML2/PI3K/AKT signaling axis represents a key mechanism driving malignant progression in HCC. Further elucidating the molecular mechanisms of this axis and developing effective, precision-targeted therapeutic strategies represent promise for enhancing HCC diagnosis, prognosis assessment and treatment efficacy, thereby offering patients with liver cancer greater survival prospects.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
ML was responsible for literature retrieval, performed the bioinformatics analyses and generated the figures, drafted manuscript sections on stomatin-like protein 2 expression and downstream mechanisms, and revised the manuscript. JC conceived the review framework, designed and supervised the bioinformatics analyses, interpreted the results and approved the final version of the manuscript. Both authors read and approved the final manuscript. ML and JC confirm the authenticity of all the raw data.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Use of artificial intelligence tools
During the preparation of this work, artificial intelligence tools were used to improve the readability and language of the manuscript or to generate images, and subsequently, the authors revised and edited the content produced by the artificial intelligence tools as necessary, taking full responsibility for the ultimate content of the present manuscript.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
STOML2
|
stomatin-like protein 2
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Mak LY, Liu K, Chirapongsathorn S, Yew KC, Tamaki N, Rajaram RB, Panlilio MT, Lui R, Lee HW, Lai JC, et al: Liver diseases and hepatocellular carcinoma in the Asia-Pacific region: Burden, trends, challenges and future directions. Nat Rev Gastroenterol Hepatol. 21:834–851. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen J, He F, Peng H and Guo J: The underlying mechanism and targeted therapy strategy of miRNAs cross-regulating EMT process through multiple signaling pathways in hepatocellular carcinoma. Front Mol Biosci. 11:13783862024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jin X, Dong H, Wang J, Ou G, Lai X, Tian X, Wang L, Zhuang H, Li T and Xiang K: HBx facilitates drug resistance in hepatocellular carcinoma via CD133-regulated self-renewal of liver cancer stem cells. J Clin Transl Hepatol. 13:15–24. 2025.PubMed/NCBI
|
|
4
|
Mitsopoulos P, Lapohos O, Weraarpachai W, Antonicka H, Chang YH and Madrenas J: Stomatin-like protein 2 deficiency results in impaired mitochondrial translation. PLoS One. 12:e01799672017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li P, Fan Z, Huang Y, Luo L and Wu X: Mitochondrial dynamics at the intersection of macrophage polarization and metabolism. Front Immunol. 16:15208142025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma W, Chen Y, Xiong W, Li W, Xu Z, Wang Y, Wei Z, Mou T, Wu Z, Cheng M, et al: STOML2 interacts with PHB through activating MAPK signaling pathway to promote colorectal Cancer proliferation. J Exp Clin Cancer Res. 40:3592021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qin C, Wang Y, Zhao B, Li Z, Li T, Yang X, Zhao Y and Wang W: STOML2 restricts mitophagy and increases chemosensitivity in pancreatic cancer through stabilizing PARL-induced PINK1 degradation. Cell Death Dis. 14:1912023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun F, Ding W, He JH, Wang XJ, Ma ZB and Li YF: Stomatin-like protein 2 is overexpressed in epithelial ovarian cancer and predicts poor patient survival. BMC Cancer. 15:7462015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao W, Zhang B, Li J, Liu Y, Liu Z and Sun B: SLP-2 overexpression could serve as a prognostic factor in node positive and HER2 negative breast cancer. Pathology. 43:713–718. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Team RC, . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2023
|
|
11
|
Therneau TM: A package for survival analysis in R. CRAN2023.
|
|
12
|
Zhao R, Ge Y, Gong Y, Li B, Xiao B and Zuo S: NAP1L5 targeting combined with MYH9 inhibit HCC progression through PI3K/AKT/mTOR signaling pathway. Aging (Albany NY). 14:9000–9019. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yin J, Zhang S, Yang C, Wang Y, Shi B, Zheng Q, Zeng N and Huang H: Mechanotransduction in skin wound healing and scar formation: Potential therapeutic targets for controlling hypertrophic scarring. Front Immunol. 13:10284102022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hussain MS, Moglad E, Afzal M, Gupta G, Almalki WH, Kazmi I, Alzarea SI, Kukreti N, Gupta S, Kumar D, et al: Non-coding RNA mediated regulation of PI3K/Akt pathway in hepatocellular carcinoma: Therapeutic perspectives. Pathol Res Pract. 258:1553032024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Subramanian AT, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P and Mesirov JP: Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27:1739–1740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li X, Zheng Y, Yu K, Hou S, Cui H, Yin R, Zhou Y, Sun Q, Zhang J and Huang C: Stomatin-like protein 2 promotes cell proliferation and survival under 5-Fluorouracil stress in hepatocellular carcinoma. Mol Biol Rep. 51:2282024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu W, Li W, Geng Q, Wang X, Sun W, Jiang H and Pu X: Silence of stomatin-like protein 2 represses migration and invasion ability of human liver cancer cells via inhibiting the nuclear factor kappa B (NF-κB) pathway. Med Sci Monit. 24:7625–7632. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng Y, Huang C, Lu L, Yu K, Zhao J, Chen M, Liu L, Sun Q, Lin Z, Zheng J, et al: STOML2 potentiates metastasis of hepatocellular carcinoma by promoting PINK1-mediated mitophagy and regulates sensitivity to lenvatinib. J Hematol Oncol. 14:162021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo H, Liang S, Wang Y, Zhou S, Yin D, Zhang S, Wang J, Wu D, Ma K, Liu Y, et al: Cytochrome B5 type A alleviates HCC metastasis via regulating STOML2 related autophagy and promoting sensitivity to ruxolitinib. Cell Death Dis. 13:6232022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu H, Zhang H, Han S, Chen J and Xie Y: STOML2 interacts with PHB to activate the MEK/ERK signaling pathway and mediates autophagy-related proteins in the progression of hepatocellular carcinoma. Int J Mol Med. 57:382026.PubMed/NCBI
|
|
22
|
Feng N, Zhang R, Wen X, Wang W, Zhang N, Zheng J, Zhang L and Liu N: RABIF promotes hepatocellular carcinoma progression through regulation of mitophagy and glycolysis. Commun Biol. 7:13332024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Q, Lan L and Zeng J and Zeng J: The effect of microvascular invasion on hepatocellular carcinoma with portal vein tumor thrombus after hepatectomy: A retrospective study. Cancer Control. 31:107327482412652572024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Song X, Li C and Tian Y: Expression and clinical significance of SLP-2 in ovarian tumors. Oncol Lett. 17:4626–4632. 2019.PubMed/NCBI
|
|
25
|
Blanche PD, Dartigues JF and Jacqmin-Gadda H: Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored survival times. Statistics in Medicine. 5381–5397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fox M, Mott HR and Owen D: Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem Soc Trans. 48:1397–1417. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghalamkari S, Alavi S, Mianesaz H, Khosravian F, Bahreini A and Salehi M: A novel carcinogenic PI3Kα mutation suggesting the role of helical domain in transmitting nSH2 regulatory signals to kinase domain. Life Sci. 269:1187592021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li X, Lau AYT, Ng ASN, Aldehaiman A, Zhou Y, Ng PKS, Arold ST and Cheung LWT: Cancer-associated mutations in the p85α N-terminal SH2 domain activate a spectrum of receptor tyrosine kinases. Proc Natl Acad Sci USA. 118:e21017511182021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shaw AL and Burke JE: Molecular insight on the role of the phosphoinositide PIP3 in regulating the protein kinases Akt, PDK1, and BTK. Biochem Soc Trans. 53:737–749. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chowdhury SM, Zhu X, Aloor JJ, Azzam KM, Gabor KA, Ge W, Addo KA, Tomer KB, Parks JS and Fessler MB: Proteomic analysis of ABCA1-null macrophages reveals a role for stomatin-like protein-2 in raft composition and toll-like receptor signaling. Mol Cell Proteomics. 14:1859–1870. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gong H, Chen S, Liu S, Hu Q, Li Y, Li Y, Li G, Huang K, Li R and Fang L: Overexpressing lipid raft protein STOML2 modulates the tumor microenvironment via NF-κB signaling in colorectal cancer. Cell Mol Life Sci. 81:392024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Delicato A, Montuori E, Angrisano T, Pollice A and Calabrò V: YB-1 oncoprotein controls PI3K/Akt pathway by reducing pten protein level. Genes (Basel). 12:15512021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uzunhisarcıklı E, Bozkurt NM and Sağlam A: ROCK inhibition suppresses glioblastoma via a PTEN-associated reduction in PI3K/AKT signaling. Med Oncol. 42:3722025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen J, Zheng W, Li Q, Xu R, Bai T and Pan C: Disruption of lipid raft reverses drug resistance in colorectal cancer cells through the phosphatase and tensin homolog/phosphoinositide 3-kinase/protein kinase B pathway and P-glycoprotein. Neoplasma. 71:571–580. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qu H, Gao-Wa H, Hou Y, Ren M, Li J, Jing B and Du Y: TRIM37 interacts with PTEN to promote the growth of human T-cell acute lymphocytic leukemia cells through regulating PI3K/AKT pathway. Front Oncol. 12:10167252022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Keller CR, Martinez SR, Keltz A, Chen M and Li W: Lactate oxidase disrupts lactate-activated RAS and PI3K oncogenic signaling. Cancers (Basel). 16:28172024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sang BT, Wang CD, Liu X, Guo JQ, Lai JY and Wu XM: PDGF-BB/PDGFRβ induces tumour angiogenesis via enhancing PKM2 mediated by the PI3K/AKT pathway in Wilms' tumour. Med Oncol. 40:2402023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu Y, Jiang H, Xu Y, Chen G, Fan R, Zhou Y, Liu Y, Yao Y, Liu R, Chen W, et al: Stomatin-like protein 2 deficiency exacerbates adverse cardiac remodeling. Cell Death Discov. 9:632023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hong M, Zhu H, Liu W, Zhang P, Yu S, Gao Q, Shen G, Li B and Wang G: Scoparone suppresses proliferation and cell cycle of hepatocellular carcinoma cells via inhibiting AKT/GSK-3β/cyclin D1 signaling pathway. Transl Cancer Res. 14:1638–1650. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Y, Liu X, Wang H, Liu S, Hu N and Li X: Akt regulated phosphorylation of GSK-3β/Cyclin D1, p21 and p27 contributes to cell proliferation through cell cycle progression from G1 to S/G2M phase in low-dose arsenite exposed HaCat cells. Front Pharmacol. 10:11762019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Park S, Kang M, Kim S, An HT, Gettemans J and Ko J: α-Actinin-4 promotes the progression of prostate cancer through the Akt/GSK-3β/β-catenin signaling pathway. Front Cell Dev Biol. 8:5885442020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang F, Chen E, Yang Y, Han F, Han S, Wu G, Zhang M, Zhang J, Han J, Su L and Hu D: The Akt/FoxO/p27Kip1 axis contributes to the anti-proliferation of pentoxifylline in hypertrophic scars. J Cell Mol Med. 23:6164–6172. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shah MA, Kang JB, Kim MO and Koh PO: Chlorogenic acid alleviates the reduction of Akt and Bad phosphorylation and of phospho-Bad and 14-3-3 binding in an animal model of stroke. J Vet Sci. 23:e842022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shultz JC, Goehe RW, Wijesinghe DS, Murudkar C, Hawkins AJ, Shay JW, Minna JD and Chalfant CE: Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 70:9185–9196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu C, Xing G, Wu C, Zhu J, Wei M, Liu D, Ge Y, Chen Y, Lei T and Yang Y: Inhibition of expression of the S100A8 gene encoding the S100 calcium-binding protein a8 promotes apoptosis by suppressing the phosphorylation of protein kinase B (Akt) in endometrial carcinoma and HEC-1A cells. Med Sci Monit. 24:1836–1846. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chibaya L, Karim B, Zhang H and Jones SN: Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc Natl Acad Sci USA. 118:e20031931182021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Allgayer H, Mahapatra S, Mishra B, Swain B, Saha S, Khanra S, Kumari K, Panda VK, Malhotra D, Patil NS, et al: Epithelial-to-mesenchymal transition (EMT) and cancer metastasis: The status quo of methods and experimental models 2025. Mol Cancer. 24:1672025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Feng K, Di Y, Han M, Yan W and Wang Y: SORBS1 inhibits epithelial to mesenchymal transition (EMT) of breast cancer cells by regulating PI3K/AKT signaling and macrophage phenotypic polarization. Aging (Albany NY). 16:4789–4810. 2024.PubMed/NCBI
|
|
49
|
Li Y, Zhang T, Qin S, Wang R, Li Y, Zhou Z, Chen Y, Wu Q and Su F: Effects of UPF1 expression on EMT process by targeting E-cadherin, N-cadherin, Vimentin and Twist in a hepatocellular carcinoma cell line. Mol Med Rep. 19:2137–2143. 2019.PubMed/NCBI
|
|
50
|
Wang X, Xiao Y, Dong Y, Wang Z, Yi J, Wang J, Wang X, Zhou H, Zhang L and Shi Y: A20 interacts with mTORC2 to inhibit the mTORC2/Akt/Rac1 signaling axis in hepatocellular carcinoma cells. Cancer Gene Ther. 30:424–436. 2023.PubMed/NCBI
|
|
51
|
Novoseletskaya E, Grigorieva O, Nimiritsky P, Basalova N, Eremichev R, Milovskaya I, Kulebyakin K, Kulebyakina M, Rodionov S, Omelyanenko N and Efimenko A: Mesenchymal stromal cell-produced components of extracellular matrix potentiate multipotent stem cell response to differentiation stimuli. Front Cell Dev Biol. 8:5553782020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang QZ, Guo YD, Li HM, Wang RZ, Guo SG and Du YF: Protection against cerebral infarction by Withaferin A involves inhibition of neuronal apoptosis, activation of PI3K/Akt signaling pathway, and reduced intimal hyperplasia via inhibition of VSMC migration and matrix metalloproteinases. Adv Med Sci. 62:186–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Delgado-Bellido D, Oliver FJ, Padilla MVV, Lobo-Selma L, Chacón-Barrado A, Díaz-Martin J and de Álava E: VE-cadherin in cancer-associated angiogenesis: A deceptive strategy of blood vessel formation. Int J Mol Sci. 24:93432023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin Y, Jiang Y, Xian H, Cai X and Wang T: Expression and correlation of the Pi3k/Akt pathway and VEGF in oral submucous fibrosis. Cell Prolif. 56:e134912023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Amir MS, Chiba N, Seong CH, Kusuyama J, Eiraku N, Ohnishi T, Nakamura N and Matsuguchi T: HIF-1α plays an essential role in BMP9-mediated osteoblast differentiation through the induction of a glycolytic enzyme, PDK1. J Cell Physiol. 237:2183–2197. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang J, Yang L, Liang F, Chen Y and Yang G: Integrin alpha × stimulates cancer angiogenesis through PI3K/Akt signaling-mediated VEGFR2/VEGF-A overexpression in blood vessel endothelial cells. J Cell Biochem. 120:1807–1818. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao C, Zhang L, Sorensen MD, Reifenberger G, Kristensen BW, McIntyre TM and Lin F: D-2-hydroxyglutarate regulates human brain vascular endothelial cell proliferation and barrier function. J Neuropathol Exp Neurol. 82:921–933. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fendt SM: 100 years of the Warburg effect: A cancer metabolism endeavor. Cell. 187:3824–3828. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Reinfeld BI, Rathmell WK, Kim TK and Rathmell JC: The therapeutic implications of immunosuppressive tumor aerobic glycolysis. Cell Mol Immunol. 19:46–58. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tian LY, Smit DJ and Jücker M: The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism. Int J Mol Sci. 24:26522023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fontana F, Giannitti G, Marchesi S and Limonta P: The PI3K/Akt pathway and glucose metabolism: A dangerous liaison in cancer. Int J Biol Sci. 20:3113–3125. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Atkins RJ, Dimou J, Paradiso L, Morokoff AP, Kaye AH, Drummond KJ and Hovens CM: Regulation of glycogen synthase kinase-3 beta (GSK-3β) by the Akt pathway in gliomas. J Clin Neurosci. 19:1558–1563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Guo J, Jiang X, Lian J, Li H, Zhang F, Xie J, Deng J, Hou X, Du Z and Hao E: Evaluation of the effect of GSK-3β on liver cancer based on the PI3K/AKT pathway. Front Cell Dev Biol. 12:14314232024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Paskeh MDA, Ghadyani F, Hashemi M, Abbaspour A, Zabolian A, Javanshir S, Razzazan M, Mirzaei S, Entezari M, Goharrizi MAS, et al: Biological impact and therapeutic perspective of targeting PI3K/Akt signaling in hepatocellular carcinoma: Promises and challenges. Pharmacol Res. 187:1065532023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang L, Gao A and Peng K: PIMREG modulation of PI3K/Akt pathway enhances sorafenib resistance in Huh7 cells. Arab J Gastroenterol. 26:241–249. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yu L, Xu H, Zhang S, Chen J and Yu Z: SDC1 promotes cisplatin resistance in hepatic carcinoma cells via PI3K-AKT pathway. Human Cell. 33:721–729. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yin R, Tao Y, Han J, Zhang J, Yu K, Zheng Y, Li X and Huang C: STOML2 inhibits sorafenib-induced ferroptosis in hepatocellular carcinoma via p-AKT signaling pathway. Am J Cancer Res. 15:1614–1628. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pu X, Dong C, Zhu W, Li W and Jiang H: Silencing stomatin-like protein 2 attenuates tumor progression and inflammatory response through repressing CD14 in liver cancer. Onco Targets Ther. 12:7361–7373. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhao Z, Bai M, Wang S, Zhang Y, Liu S, Bao M, Lu S, Cao D, Shen S, Xie L, et al: The inwardly rectifying potassium channel KCNJ12 regulates the stemness of hepatocellular carcinoma cells through the Wnt/β-catenin pathway. J Mol Cell Biol. 2:mjaf0482025. View Article : Google Scholar
|
|
70
|
Zhou C, Li Y, Wang G, Niu W, Zhang J, Wang G, Zhao Q and Fan L: Enhanced SLP-2 promotes invasion and metastasis by regulating Wnt/β-catenin signal pathway in colorectal cancer and predicts poor prognosis. Pathol Res Pract. 215:57–67. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bam S, Buchanan E, Mahony C and O'Ryan C: DNA methylation of PGC-1α is associated with elevated mtDNA copy number and altered urinary metabolites in autism spectrum disorder. Front Cell Dev Biol. 9:6964282021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ryu HY: Histone modification pathways suppressing cryptic transcription. Epigenomes. 8:422024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gupta M, Chandan K and Sarwat M: Role of microRNA and long non-coding RNA in hepatocellular carcinoma. Curr Pharm Design. 26:415–428. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Du K, Bai X, Chen L, Shi Y, Wang HD, Cai MC, Sun WQ, Wang J, Chen SY, Jia XB and Lai SJ: Integrated analysis of microRNAs, circular RNAs, long non-coding RNAs, and mRNAs revealed competing endogenous RNA networks involved in brown adipose tissue whitening in rabbits. BMC Genomics. 23:7792022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang X, Zang W, Xuan X, Wang Z, Liu Z, Wang J, Cui J and Zhao G: miRNA-1207-5p is associated with cancer progression by targeting stomatin-like protein 2 in esophageal carcinoma. Int J Oncol. 46:2163–2171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lu W, Li H, Liu X, Li A and Xiu R: The RNA-binding proteins MCPIP2 and IGF2BP1 competitively modulate breast tumor angiogenesis by antagonizing VEGFA mRNA stability and expression. FASEB J. 39:e705942025. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ma X, Liu S, Fan B, Jin D, Miao L, Liu L, Du S and Lin J: Enhancing mRNA translation efficiency by introducing sequence optimized AU-rich elements in 3′ UTR via HuR anchorage. Mol Ther Nucleic Acids. 36:1024852025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ren X, Yang W, Yan X and Zhang H: Exploring RNA binding proteins in hepatocellular carcinoma: Insights into mechanisms and therapeutic potential. J Exp Clin Cancer Res. 44:1302025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Golob-Schwarzl N, Krassnig S, Toeglhofer AM, Park YN, Gogg-Kamerer M, Vierlinger K, Schröder F, Rhee H, Schicho R, Fickert P and Haybaeck J: New liver cancer biomarkers: PI3K/AKT/mTOR pathway members and eukaryotic translation initiation factors. Eur J Cancer. 83:56–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gufler S, Seeboeck R, Schatz C and Haybaeck J: The translational bridge between inflammation and hepatocarcinogenesis. Cells. 11:5332022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Murgia M, Tan J, Geyer PE, Doll S, Mann M and Klopstock T: Proteomics of Cytochrome C oxidase-negative versus -positive muscle fiber sections in mitochondrial myopathy. Cell Rep. 29:3825–3834. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mookherjee D, Das S, Mukherjee R, Bera M, Jana SC, Chakrabarti S and Chakrabarti O: RETREG1/FAM134B mediated autophagosomal degradation of AMFR/GP78 and OPA1-a dual organellar turnover mechanism. Autophagy. 17:1729–1752. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zeng P, Mao J, Guo J, Yang X, Shi Y, Wang X, Song J, Zhou J, Hou L and Liu J: The E3 ubiquitin ligase STUB1 inhibits Senecavirus A replication by mediating VP1 ubiquitination and proteasomal degradation. J Virol. 99:e01152252025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tian Z, Xu C, He W, Lin Z, Zhang W, Tao K, Ding R, Zhang X and Dou K: The deubiquitinating enzyme USP19 facilitates hepatocellular carcinoma progression through stabilizing YAP. Cancer Lett. 577:2164392023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Serricchio M and Bütikofer P: A conserved mitochondrial chaperone-protease complex involved in protein homeostasis. Front Mol Biosci. 8:7670882021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kumari S, Sankhwar P, Tripathi R, Kawale AK, Gupta S and Jha RK: Stomatin-like protein-2 promotes aggregation, colonization and migration of endometriotic cells. Reprod Sci. 30:1854–1866. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Chuang CC, Lan YH, Lu YJ, Weng YL and Chen JP: Targeted delivery of irinotecan and SLP2 shRNA with GRP-conjugated magnetic graphene oxide for glioblastoma treatment. Biomater Sci. 10:3201–3222. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lu YJ, Lan YH, Chuang CC, Lu WT, Chan LY, Hsu PW and Chen JP: Injectable thermo-sensitive chitosan hydrogel containing CPT-11-loaded EGFR-targeted graphene oxide and SLP2 shRNA for localized drug/gene delivery in glioblastoma therapy. Int J Mol Sci. 21:71112020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hu G, Zhang J, Xu F, Deng H, Zhang W, Kang S and Liang W: Stomatin-like protein 2 inhibits cisplatin-induced apoptosis through MEK/ERK signaling and the mitochondrial apoptosis pathway in cervical cancer cells. Cancer Sci. 109:1357–1368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cheng X, Deng X, Zeng H, Zhou T, Li D and Zheng WV: Silencing of TMED5 inhibits proliferation, migration and invasion, and enhances apoptosis of hepatocellular carcinoma cells. Adv Clin Exp Med. 32:677–688. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Liu Q, Li A, Wang L, He W, Zhao L, Wu C, Lu S, Ye X, Zhao H, Shen X, et al: Stomatin-like protein 2 promotes tumor cell survival by activating the JAK2-STAT3-PIM1 pathway, suggesting a novel therapy in CRC. Mol Ther Oncolytics. 17:169–179. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zanon A, Guida M, Lavdas AA, Corti C, Rueda MP, Negro A, Pramstaller PP, Domingues FS, Hicks AA and Pichler I: Intracellular delivery of Parkin-RING0-based fragments corrects Parkin-induced mitochondrial dysfunction through interaction with SLP-2. J Transl Med. 22:592024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Xu H, Chen K, Shang R and Chen X, Zhang Y, Song X, Evert M, Zhong S, Li B, Calvisi DF and Chen X: Alpelisib combination treatment as novel targeted therapy against hepatocellular carcinoma. Cell Death Dis. 12:9202021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ye L, Mayerle J, Ziesch A, Reiter FP, Gerbes AL and Toni END: The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 5:862019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Crabb SJ, Griffiths G, Marwood E, Dunkley D, Downs N, Martin K, Light M, Northey J, Wilding S, Whitehead A, et al: Pan-AKT inhibitor capivasertib with docetaxel and prednisolone in metastatic castration-resistant prostate cancer: A randomized, placebo-controlled phase II trial (ProCAID). J Clin Oncol. 39:190–201. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mroweh M, Roth G, Decaens T, Marche PN, Lerat H and Jílková ZM: Targeting Akt in hepatocellular carcinoma and its tumor microenvironment. Int J Mol Sci. 22:17942021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Liu G, Shi A, Wang N, Li M, He X, Yin C, Tu Q, Shen X, Tao Y, Wang Q and Yin H: Polyphenolic Proanthocyanidin-B2 suppresses proliferation of liver cancer cells and hepatocellular carcinogenesis through directly binding and inhibiting AKT activity. Redox Biol. 37:1017012020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pervanidis KA, D'Angelo GD, Weisner J, Brandherm S and Rauh D: Akt inhibitor advancements: From capivasertib approval to covalent-allosteric promises. J Med Chem. 67:6052–6063. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yu K, Shi C, Toral-Barza L, Lucas J, Shor B, Kim JE, Zhang WG, Mahoney R, Gaydos C, Tardio L, et al: Beyond rapalog therapy: Preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res. 70:621–631. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hernández-Prat A, Rodriguez-Vida A, Juanpere-Rodero N, Arpi O, Menéndez S, Soria-Jiménez L, Martínez A, Iarchouk N, Rojo F, Albanell J, et al: Novel Oral mTORC1/2 inhibitor TAK-228 has synergistic antitumor effects when combined with paclitaxel or PI3Kα inhibitor TAK-117 in preclinical bladder cancer models. Mol Cancer Res. 17:1931–1944. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hu YT, Shu ZY, Jiang JH, Xie QF and Zheng SS: Torin2 overcomes sorafenib resistance via suppressing mTORC2-AKT-BAD pathway in hepatocellular carcinoma cells. Hepatobiliary Pancreat Dis Int. 19:547–554. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Guo T, Wu C, Zhang J, Yu J, Li G, Jiang H, Zhang X, Yu R and Liu X: Dual blockade of EGFR and PI3K signaling pathways offers a therapeutic strategy for glioblastoma. Cell Commun Signal. 21:3632023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang Y, Liu Y, Ma Y, Xu Y, Wang G and Han X: CD93 aggravates cell proliferation, angiogenesis and immune escape in osteosarcoma through triggering the PI3K/AKT pathway. J Orthop Sci. 30:685–691. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Sun X, Chen Y, Tao X, Zhang W, Wang X, Wang X, Ruan Z and Chen Z: INPP4B inhibits glioma cell proliferation and immune escape via inhibition of the PI3K/AKT signaling pathway. Front Oncol. 12:9835372022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Li B, Feng C, Zhang W, Sun S, Yue D, Zhang X and Yang X: Comprehensive non-coding RNA analysis reveals specific lncRNA/circRNA-miRNA-mRNA regulatory networks in the cotton response to drought stress. Int J Biol Macromol. 253:1265582023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Tang W and Ma X: Identification of causal plasma proteins in hepatocellular carcinoma via two-sample mendelian randomization and integrative transcriptomic-proteomic analysis. Cancer Res Commun. 5:433–443. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Orang FN and Shadbad MA: CircRNA and lncRNA-associated competing endogenous RNA networks in medulloblastoma: A scoping review. Cancer Cell Int. 24:2482024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yum J, Aulia F, Kamiya K, Hori M, Qiao N, Kim BS, Naito M, Ogura S, Nagata T, Yokota T, et al: Hydrophobicity tuning of cationic polyaspartamide derivatives for enhanced antisense oligonucleotide delivery. Bioconjug Chem. 35:125–131. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Liu XM, Zhang Z, Zhong J, Li N, Wang T, Wang L and Zhang Q: Long non-coding RNA MALAT1 modulates myocardial ischemia-reperfusion injury through the PI3K/Akt/eNOS pathway by sponging miRNA-133a-3p to target IGF1R expression. Eur J Pharmacol. 916:1747192022. View Article : Google Scholar : PubMed/NCBI
|















![Paired analysis of STOML2 expression
in HCC and matched adjacent non-cancerous tissues. STOML2
expression levels [log2(TPM+1)] are shown in paired
tumor and adjacent non-tumor tissue samples from the same patients
with HCC. Data were obtained from The Cancer Genome Atlas-Liver
Hepatocellular Carcinoma cohort. Statistical significance was
determined by paired Student's t-test (***P<0.001). STOML2,
stomatin-like protein 2; HCC, hepatocellular carcinoma; TPM,
transcripts per million.](../../../article_images/ol/31/4/ol-31-04-15488-g01.jpg)