Introduction
Colorectal cancer (CRC), a malignant neoplasm
originating from the mucosal lining of the colon or rectum, remains
a significant global health burden. It is often diagnosed in the
first instance at an advanced stage, which complicates both
treatment and prognosis (1,2). Patients typically present with
systemic symptoms such as fatigue, unintended weight loss, anaemia,
abdominal pain and in certain cases, fever, often indicative of
metastatic spread. As a highly heterogeneous disease, CRC
encompasses several subtypes, including mucinous, non-mucinous,
neuroendocrine and undifferentiated forms, each characterised by
distinct molecular and histological features (3). This heterogeneity poses substantial
challenges for clinical management, especially in advanced disease,
where resistance to conventional therapies is common and long-term
survival rates remain low (4).
While early detection through screening and adenoma resection has
contributed to a decline in mortality, CRC incidence and metastatic
potential continue to rise. Globally, >1.85 million new cases
and 850,000 CRC-related mortalities are reported annually (2). Notably, China recorded ~600,000 new
CRC cases and 261,777 CRC-related mortalities in 2019, the highest
national mortality worldwide (5).
The 5-year survival rate for metastatic CRC remains <15%,
despite aggressive treatment (6).
These trends are suggested to be driven by an ageing population,
the widespread adoption of Western dietary habits and rising
obesity, which are well-established risk factors for CRC (7–9).
Collectively, these factors underscore the need for innovative
approaches to CRC prevention, early diagnosis and treatment.
Current treatment strategies for CRC typically
involve a combination of surgery, chemotherapy and radiation
therapy (9). Surgery remains the
primary curative approach for localised CRC, while chemotherapy and
radiotherapy are critical for advanced or metastatic disease.
Targeted therapies, such as monoclonal antibodies against epidermal
growth factor receptor (EGFR) and vascular endothelial growth
factor (VEGF), have improved outcomes in specific patient
subgroups, most notably patients with RAS wild-type tumours for
EGFR-targeted therapy and those receiving combination chemotherapy
for VEGF inhibition. EGFR inhibitors, including cetuximab and
panitumumab, are effective in patients harbouring wild-type RAS
genes by blocking ligand binding and downstream signalling pathways
involved in tumour progression. Similarly, VEGF inhibitors such as
bevacizumab suppress tumour angiogenesis, enhancing the efficacy of
chemotherapy (10). However, the
clinical success of these therapies is often limited by acquired
resistance and immune-related complications (11).
In this context, cancer immunotherapy has emerged as
a transformative modality, offering durable responses in various
malignancies, including CRC (12).
Immunotherapeutic strategies, such as immune checkpoint inhibitors
(ICIs), adoptive cell transfer, cancer vaccines and oncolytic virus
therapy, aim to reactivate antitumour immunity by targeting immune
evasion mechanisms (12). Among
them, ICIs have shown therapeutic potential, especially in tumours
with high microsatellite instability (MSI-H) or mismatch repair
deficiency (dMMR), where they can elicit sustained clinical
responses (13). Nevertheless, the
efficacy of ICIs in the broader CRC population remains limited. A
notable proportion of patients exhibit primary or acquired
resistance to ICIs, and immune-related adverse events, including
dermatological, gastrointestinal, pulmonary and endocrine
toxicities, are frequently observed (12,14).
These challenges highlight the need for reliable immune-related
biomarkers that can guide treatment selection, predict response and
minimise toxicity (12).
Recent studies have implicated dysregulation of the
tumour immune microenvironment as a key mechanism underlying
resistance to immunotherapy (15,16).
Among the emerging regulatory proteins, DDB1- and CUL4-associated
factor 13 (DCAF13) has gained attention due to its multifaceted
roles in oncogenesis and immune modulation. DCAF13 is a conserved
substrate receptor in the CUL4-DDB1 E3 ubiquitin ligase complex,
involved in post-translational modification, RNA metabolism,
protein degradation and cell cycle regulation (17–19).
Functionally, DCAF13 acts as a novel RNA-binding protein, with
pivotal roles in zygotic genome activation and immune homeostasis
(20). DCAF13 deficiency leads to
impaired proliferation in HeLa cells and preimplantation lethality
in murine embryos (20), while
DCAF13 upregulation is associated with a poor prognosis in patients
with breast cancer, where it promotes tumour progression via the
NOTCH4 signalling pathway (21,22).
In addition to breast cancer, DCAF13 has been associated with
tumorigenesis in lung, liver, ovarian and haematological
malignancies (23–26). DCAF13 also modulates immune
responses within the tumour microenvironment (TME); in the TME,
DCAF13 contributes to immune escape and resistance to ICIs, as
demonstrated in lung adenocarcinoma and hepatocellular carcinoma
(26,27). Furthermore, DCAF13 is essential for
T-cell function, and it has been shown that DCAF13 depletion
disrupts T-cell proliferation and homeostasis (28). These findings suggest a dual role
for DCAF13 in promoting tumour progression and mediating immune
evasion, making it a compelling candidate for further exploration
in cancer immunotherapy.
While the oncogenic role of DCAF13 has been
increasingly recognised, its specific function in CRC and
association with immune infiltration remain poorly understood. Sun
et al (29) recently
demonstrated that CRISPR-mediated knockout of DCAF13
significantly impaired CRC cell proliferation. The present study
investigated the biological function of DCAF13 in CRC progression
and immune modulation. Bioinformatics analyses, including ESTIMATE,
CIBERSORT, immune checkpoint profiling, TIDE scoring and GEPIA,
were used to explore the relationship between DCAF13 and the tumour
immune microenvironment. Findings were validated using
transcriptomic data from the GSE40967 dataset and
immunohistochemical (IHC) staining of clinical CRC samples. In
vitro functional assays further confirmed the oncogenic role of
DCAF13. These results highlight DCAF13 as a novel immune-related
biomarker and a promising therapeutic target for improving
immunotherapy efficacy in CRC.
Materials and methods
Data collection and preprocessing
Gene expression profiles and associated clinical
information for patients with CRC in The Cancer Genome Atlas (TCGA)
cohort were obtained from the UCSC Xena database (https://xena.ucsc.edu/, accessed in March 2024). The
data, with 332 primary CRC cases, were screened and samples without
follow-up information, with 0-day survival or representing
duplicate sequencing from the same patient were removed, which
resulted in a final cohort of 282 primary CRC cases for the
training dataset. The gene expression dataset GSE40967 (575 CRC
samples and 10 normal tissue samples) was downloaded from the Gene
Expression Omnibus (GEO) repository (https://ncbi.nlm.nih.gov/geo/) (30); this dataset served as the external
validation set. Samples lacking survival data in the GSE40967
dataset were excluded from further analysis. To minimise technical
variability between datasets, batch effects were corrected using
the R ‘limma’ package (version 3.50.3) (31). Specifically, the ‘removeBatchEffect’
function was applied after merging tumour samples from the
validation cohort in the UCSC Xena data with GSE40967, which were
generated using the same microarray platform [GPL570
(HG-U133_Plus_2); Human Genome U133 Plus 2.0 Array; Affymetrix;
Thermo Fisher Scientific, Inc.]. Subsequently, mRNA expression data
from both datasets were normalised to ensure consistency for
downstream analysis. The demographics and clinicopathological
characteristics of these patients are displayed in Table I.
 | Table I.Clinical features of patients with
CRC in the TCGA (n=332) and GEO (n=585) datasets. |
Table I.
Clinical features of patients with
CRC in the TCGA (n=332) and GEO (n=585) datasets.
| Clinical
feature | TCGA, n (%) | GSE40967, n
(%) |
|---|
| Overall
survival |
|
|
|
Alive | 276 (83.13) | 385 (65.81) |
|
Dead | 56 (16.87) | 195 (33.33) |
| Age, years |
|
|
|
>58 | 238 (71.69) | 441 (75.38) |
|
≤58 | 94 (28.31) | 141 (24.10) |
| Sex |
|
|
|
Male | 180 (54.22) | 322 (55.04) |
|
Female | 152 (45.78) | 263 (44.96) |
| PR status |
|
|
|
Positive | - | 395 (67.52) |
|
Negative | - | 180 (30.77) |
|
Indeterminate | - | 11 (1.89) |
| Stage |
|
|
| I | 106 (31.93) | 38 (6.50) |
| II | 84 (25.30) | 271 (46.32) |
|
III | 92 (27.71) | 210 (35.90) |
| IV | 50 (15.06) | 60 (10.26) |
| T stage |
|
|
| T1 | 18 (5.42) | 12 (2.05) |
| T2 | 56 (16.87) | 49 (8.38) |
| T3 | 228 (68.67) | 379 (64.79) |
| T4 | 28 (8.43) | 119 (20.34) |
| N stage |
|
|
| N0 | 168 (50.60) | 314 (53.68) |
| N1 | 90 (27.11) | 137 (23.42) |
| N2 | 68 (20.48) | 100 (17.09) |
| M stage |
|
|
| M0 | 252 (75.90) | 499 (85.30) |
| M1 | 48 (14.49) | 61 (10.43) |
Pan-cancer function and expression
analyses of DCAF13
Pan-cancer functional analyses of DCAF13 were
performed using the Cancer Single-Cell State TCGA (CancerSEA;
http://biocc.hrbmu.edu.cn/CancerSEA/)
database (32). Expression levels
of DCAF13 across different types of cancer were explored using the
Tumor Immune Estimation Resource (TIMER;
cistrome.shinyapps.io/timer/) (33), which was used to compare DCAF13
expression levels between cancer tissues and matched normal
samples.
For prognostic analysis, patients in the data from
TCGA (https://www.cancer.gov/ccg/research/genome-sequencing/tcga)
were stratified into high- and low-expression groups based on the
median expression levels of DCAF13, which served as the optimal
cut-off value. The median DCAF13 expression was 11.98325, with
patients exhibiting expression levels ≥11.98325 classified into the
high-expression group, and those <11.98325 into the
low-expression group. The association between DCAF13 expression and
overall survival (OS) was evaluated using Kaplan-Meier (KM)
survival analysis via the Kaplan-Meier Plotter online tool
(http://gepia.cancer-pku.cn/), using the
CRC mRNA dataset. Patients were stratified into high- and
low-expression groups based on the median expression of DCAF13, and
OS was analysed using the log-rank test. The findings were
validated using the GSE40967 dataset by performing the same
survival analysis on this external cohort, with patients stratified
into high- and low-expression groups based on the median DCAF13
expression level within the GSE40967 cohort.
Assessment of immune cell
infiltration, immune checkpoints and response to immunotherapy
To assess immune cell infiltration and immune
checkpoint activity, several computational tools and bioinformatics
algorithms were used. The ESTIMATE algorithm (https://bioinformatics.mdanderson.org/estimate/) was
used to evaluate the stromal (StromalScore, SS), immune
(ImmuneScore, IS), and overall ESTIMATE scores for each tumour
sample in the data from TCGA (34).
These scores were derived from gene expression data using the
ESTIMATE R package (version 1.0.13). The relationships between
DCAF13 expression and the SS, IS, and ESTIMATE scores were further
analysed to gain insights into its role in immune regulation within
the TME.
For further assessment of immune infiltration, the
CIBERSORT software (https://sangerbox.com; accessed in March 2024) was
used to estimate the relative proportions of 22 immune cell types
in each tumour sample. This was achieved through the analysis of a
normalised gene expression matrix (35). Differences in immune infiltration
between the high- and low-DCAF13 expression groups were compared
using the Wilcoxon rank-sum test (two-sided).
To explore the role of DCAF13 in regulating cancer
immunity, its correlation with various immune checkpoints,
including B and T lymphocyte attenuator (BTLA),
lymphocyte-activating 3 (LAG-3), signal regulatory protein α
(SIRPα) and V-domain Ig suppressor of T-cell activation (VISTA) was
visualised using Gene Expression Profiling Interactive Analysis
(GEPIA; http://gepia.cancer-pku.cn/)
(36). Additionally, the
correlation between DCAF13 expression and 122 immune
checkpoint-related genes (ICPs) was analysed using Pearson's
correlation analysis (37).
To predict the response to immunotherapy, Tumor
Immune Dysfunction and Exclusion (TIDE; https://tide.dfci.harvard.edu/; accessed in March
2024) analysis was applied to the high- and low-DCAF13 expression
groups (38). In the present study,
the TIDE score, dysfunction score, exclusion score and MSI score
between the high- and low-DCAF13 expression groups were calculated
to evaluate their potential responsiveness to immunotherapy.
Patient specimens and ethics
statement
In the present study, a total of 234
paraffin-embedded CRC tissue samples and their matched adjacent
non-tumorous tissues, collected ≥5 cm away from the tumour margin,
were obtained from patients who underwent surgical resection at the
Department of Pathology, Affiliated Hospital of Inner Mongolia
University (Hohhot, China) between January 2013 and June 2015. All
samples were histologically confirmed as CRC and were obtained
prior to any neoadjuvant therapy. Among the 234 patients, 146 were
male and 88 were female. The age distribution showed 83 patients
were aged ≤60 years and 151 patients were aged >60 years. The
cohort included tumours originating from both the colon and rectum,
including the ascending (n=76), transverse (n=31),
descending/sigmoid (n=68) colon and rectum (n=59). Tumour histology
was classified according to WHO criteria, with the majority being
tubular or papillary adenocarcinomas (n=208), and the rest
categorised as other types (n=26) (39). Tumour differentiation included
well/moderately differentiated (n=141) and poorly differentiated
(n=93) tumours. Staging was based on the AJCC 8th edition TNM
classification, comprising stage I (n=26), stage II (n=62) and
stage III/IV (n=146) cases (40).
Additional T and N staging details were also recorded. The present
study was approved by the Ethical Review Committee of Inner
Mongolia Medical University (approval no. 2022513411; Hohhot,
China) and was performed in accordance with the Declaration of
Helsinki. Informed consent was written from all patients prior to
inclusion in the study. Detailed clinicopathological
characteristics are summarised in Table II.
| Table II.Association of DCAF13 expression with
clinical characteristics and biological markers of patients with
CRC. |
Table II.
Association of DCAF13 expression with
clinical characteristics and biological markers of patients with
CRC.
| Characteristic | Patients, n | High-DCAF13
expression (n=137), n (%) | Low- or no-DCAF13
expression (n=97), n (%) | χ2 | P-value |
|---|
| Age, years |
|
|
| 0.994 | 0.319 |
|
≤60 | 83 | 45 (54.2) | 38 (45.8) |
|
|
|
>60 | 151 | 92 (60.9) | 59 (39.1) |
|
|
| Sex |
|
|
| 0.164 | 0.685 |
|
Male | 146 | 84 (57.5) | 62 (42.5) |
|
|
|
Female | 88 | 53 (60.2) | 35 (39.8) |
|
|
| Tumour size,
cm |
|
|
| 2.012 | 0.156 |
| ≤5 | 182 | 111 (61.0) | 71 (39.0) |
|
|
|
>5 | 52 | 26 (50.0) | 26 (50.0) |
|
|
| Histological
classification |
|
|
| 0.563 | 0.453 |
| Tubular
+ papillary | 208 | 120 (57.7) | 88 (42.3) |
|
|
|
Other | 26 | 17 (65.4) | 9 (34.6) |
|
|
| Location |
|
|
| 6.712 | 0.082 |
|
Ascending colon | 76 | 44 (57.9) | 32 (42.1) |
|
|
|
Transverse colon | 31 | 18 (58.1) | 13 (41.9) |
|
|
|
Descending + sigmoid
colon | 68 | 33 (48.5) | 35 (51.5) |
|
|
|
Rectum | 59 | 42 (71.2) | 17 (28.8) |
|
|
|
Differentiation |
|
|
| 4.080 | 0.043 |
| Well +
moderate | 141 | 90 (63.8) | 51 (36.2) |
|
|
|
Poor | 93 | 47 (50.5) | 46 (49.5) |
|
|
| T stage |
|
|
| 4.158 | 0.125 |
| T1 | 54 | 28 (51.9) | 26 (48.1) |
|
|
| T2 | 100 | 55 (55.0) | 45 (45.0) |
|
|
| T3 +
T4 | 80 | 54 (67.5) | 26 (32.5) |
|
|
| N stage |
|
|
| 2.099 | 0.350 |
| N0 | 141 | 84 (59.6) | 57 (40.4) |
|
|
| N1 | 63 | 39 (61.9) | 24 (38.1) |
|
|
| N2 | 30 | 14 (46.7) | 16 (53.3) |
|
|
| TNM stage |
|
|
| 8.310 | 0.016 |
| I | 26 | 12 (46.2) | 14 (53.8) |
|
|
| II | 62 | 29 (46.8) | 33 (53.2) |
|
|
| III +
IV | 146 | 96 (65.8) | 50 (34.2) |
|
|
Immunohistochemical (IHC)
staining
All tissue samples were fixed in 10%
neutral-buffered formalin (no. G2161; Beijing Solarbio Science
& Technology Co., Ltd.) at room temperature for 24 h, routinely
dehydrated, embedded in paraffin and stored under standard
conditions. No tissues were frozen after paraffin embedding. Prior
to use, the paraffin sections of the 234 CRC samples and their
adjacent tissues were prepared at a thickness of 4 µm,
deparaffinised in xylene, rehydrated through a graded ethanol
series and washed with PBS (0.01 M, pH 7.0) (41). Endogenous peroxidase activity was
quenched by incubation with 3% hydrogen peroxide (cat. no. H792073;
Shanghai Macklin Biochemical Co., Ltd.) at room temperature for 10
min. Antigen retrieval was performed by boiling the slides in
citrate buffer (0.01 M, pH 6.0) at 95–100°C for 15 min, followed by
natural cooling to room temperature. For blocking of non-specific
binding, the sections were incubated with 5% goat serum (cat. no.
SL039; Beijing Solarbio Science & Technology Co., Ltd.) at 37°C
for 30 min. The sections were then incubated at 4°C overnight with
a rabbit anti-DCAF13 antibody (1:500; cat. no. bs-62879R; BIOSS).
After washing with PBS, the sections were incubated with a goat
anti-rabbit HRP secondary antibody (1:2,000; cat. no. ZDR-5306;
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.) at 37°C for 1 h.
For visualisation, the sections were incubated with DAB chromogen
(cat. no. DA1010, Beijing Solarbio Science & Technology Co.,
Ltd.) at room temperature for 5 min, followed by counterstaining
with haematoxylin solution at room temperature for 10 sec.
Immunostained sections were independently evaluated by two
experienced pathologists under blinded conditions using a light
microscope (Olympus BX53; Olympus Corporation). The staining
intensity and percentage of DCAF13-positive cells were
quantitatively analysed using ImageJ software (version 1.53t;
National Institutes of Health).
Cell culture
FHC, Caco2, SW480, HCT116, Lovo and RKO cells were
used in the present study and cultured in a humidified atmosphere
at 37°C and 5% CO2. FHC cells, a normal intestinal cell
line, were purchased from Zhejiang Meisen Cell Technology Co., Ltd.
(cat. no. CTCC-001-0208) and cultured in their respective
commercial medium (cat. no. CTCC-002-047; Zhejiang Meisen Cell
Technology Co., Ltd). Caco2 cells were cultured in MEM medium (cat.
no. PM150410; Procell Life Science & Technology Co., Ltd.)
supplemented with 20% foetal bovine serum (FBS; cat. no. C04001;
Shanghai VivaCell Biosciences, Ltd.) and 100 IU/ml
penicillin/streptomycin (P/S; cat. no. 15070063; Thermo Fisher
Scientific, Inc.). SW480, Lovo and RKO cells were cultured in DMEM
medium (cat. no. PM150210; Procell Life Science & Technology
Co., Ltd.) with 10% FBS and 100 IU/ml P/S. HCT116 cells were
maintained in IMEM medium (cat. no. C12440500BT; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS and 100 IU/ml
P/S.
Western blotting
To identify the CRC cells with the highest
expression levels of DCFA13, protein lysates from CRC cells were
extracted using RIPA lysis buffer (cat. no. DE101; TransGen Biotech
Co., Ltd.) and quantified using a bicinchoninic acid assay kit
(cat. no. PC0020; Beijing Solarbio Science & Technology Co.,
Ltd.) (42,43). Proteins (30 µg per lane) were
resolved by 10% SDS-PAGE (cat. no. P1200; Beijing Solarbio Science
& Technology Co., Ltd.) and transferred to PVDF membranes (cat.
no. PVH00010; Merck KGaA) using 1× Tris-glycine buffer (cat. no.
G2145; Wuhan Servicebio Technology Co., Ltd.). After blocking with
10% non-fat milk at room temperature for 1 h, the membranes were
incubated at 4°C overnight with a rabbit anti-DCAF13 antibody
(1:2,000; cat. no. bs-62879R; BIOSS), a rabbit anti-γ-H2AX antibody
(1:2,000; cat. no. ab81299; Abcam), a rabbit anti-ATM antibody
(1:2,000; cat. no. bsm-52360R; BIOSS), a rabbit anti-E-cadherin
antibody (1:2,000; cat. no. AF0131; Affinity Biosciences), a rabbit
anti-N-cadherin antibody (1:2,000; cat. no. AF6710; Affinity
Biosciences), a rabbit anti-Vimentin antibody (1:2,000; cat. no.
60330-1-Ig; Proteintech Group, Inc.), a rabbit anti-PCNA antibody
(1:2,000; cat. no. 10205-2-AP; Proteintech Group, Inc.) or a mouse
anti-actin antibody (1:2,000; cat. no. CW0096M; Jiangsu CoWin
Biotech Co., Ltd.). After three washes with Tris-HCl with 0.1%
Tween-20, the membranes were incubated with goat anti-rabbit IgG
H&L HPR antibodies (1:2,000; cat. no. S0001; Affinity
Biosciences) or HRP-conjugated Affinipure goat anti-mouse IgG
(1:2,000; cat. no. SA00001-1; Proteintech Group, Inc.) at room
temperature for 1 h. Immunoblots were visualised using enhanced
chemiluminescence (cat. no. W1001; Promega Corporation) using a
ChampChemi system (Shanghai Champ Biomedical Technology Co., Ltd.).
The densitometry analysis of the protein bands was performed using
ImageJ software (version 1.53t; National Institutes of Health).
Lentivirus packaging and
infection
Lentiviral vectors expressing either a short hairpin
RNA (shRNA) targeting the DCAF13 coding region
(5′-CGAATCTTTCCTGTAGACAAA-3′) or a non-targeting control shRNA
sequence (5′-TTCTCCGAACGTGTCACGT-3′), which does not match any
known human gene, were supplied by Shanghai GeneChem Co., Ltd. The
lentiviral plasmid containing the shRNA was packaged using a
second-generation system, with 293T cells (American Type Culture
Collection) used as the interim cell line. For transfection, 10 µg
lentiviral plasmid was mixed with 10 µg lentivirus packaging
plasmid (pMD2.G) and 10 µg packaging plasmid (psPAX2) in a 1:1:1
ratio. The mixture was transfected into 293T cells using
Lipofectamine 3000 (cat. no. L3000008; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Transfection was
carried out at 37°C for 6 h, and the medium was replaced with fresh
growth medium after 6 h. Lentiviral particles were collected 48 h
post-transfection, and the multiplicity of infection was adjusted
to 5 for infection of RKO cells with high baseline expression of
DCAF13. The cells were transduced in the presence of polybrene (8
µg/ml; cat. no. H9268; Sigma-Aldrich; Merck KGaA) for 12 h.
Following transduction, cells were selected with puromycin (1
µg/ml; cat. no. 314Y0415; Beijing Solarbio Science & Technology
Co., Ltd.) for 14 days to establish stable cell lines. Stable
DCAF13 knockdown (shDCAF13) and the non-targeting control (shNC)
cell lines were validated using RT-qPCR and western blotting.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
RNA extraction and qPCR were performed to assess the
efficiency of DCAF13 knockdown. Total RNA was extracted from RKO
cells in both experimental groups using TRIzol® reagent
(cat. no. 79306; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Reverse transcription of 1 µg of RNA was
performed using a PrimeScript™ RT Reagent Kit (cat. no. RR047A;
Takara Biotechnology Co., Ltd.), according to the manufacturer's
protocol, to synthesise cDNA. For amplification, specific forward
and reverse primers for DCAF13 were designed using
PrimerBank (version 2.1; http://pga.mgh.harvard.edu/primerbank/) and
synthesised by Sango Biotech Co., Ltd. qPCR was performed using the
SYBR® Premix Ex Taq™ Kit (cat. no. RR820A; Takara
Biotechnology Co., Ltd.) in a 20-µl reaction volume, using a Roche
LightCycler 480 II System (Roche Diagnostics). The following
thermocycling conditions were used for qPCR: Initial denaturation
at 94°C for 2 min; 40 cycles of denaturation at 95°C for 5 sec;
annealing at 60°C for 15 sec; and extension at 72°C for 10 sec.
Fluorescence signals were detected during the annealing-extension
phase, and GAPDH was used as the internal reference for
normalisation. The relative expression levels of DCAF13 in
each group were calculated using the 2−ΔΔCq method
(44). The following primer pairs
were used for qPCR: DCAF13 forward (F),
5′-ACTGCACAGCTAAAGAACCG-3′ and reverse (R),
5′-TCCCAGACTACTTCCAGTCAC-3′; and GAPDH F,
5′-TGAACGGGAAGCTCACTG-3′ and R, 5′-GCTTCACCACCTTCTTGATG-3′.
In vitro functional assays of DCAF13
in RKO cells
Proliferation assays
RKO cells (1×104 cells per well) from
both groups were seeded in a 96-well plate and imaged at 8, 16 and
24 h using an IncuCyte system (Essen Bioscience) to track
proliferation. The growth curves of each group were then analysed.
In addition, the expression levels of PCNA in RKO cells of both
groups were detected by western blotting (42,45).
Wound healing assay
shNC and shDCAF13 RKO cells (2×103 cells)
were seeded into 6-well plates and cultured at 37°C in DMEM medium
supplemented with 10% FBS and 100 IU/ml P/S overnight. A wound was
created by scratching the monolayer with a 200 µl pipette tip, and
the cells were cultured in DMEM medium without FBS or P/S for an
additional 24 h. The degree of wound closure was observed and
microscopically (Eclipse Ts2 inverted phase contrast microscope;
Nikon Corporation) imaged with the cell migration rate calculated
using the following formula: Migration rate (%)=[(initial wound
width-wound width at 24 h)/initial wound width] × 100% (42,45).
Transwell assay
shNC and shDCAF13 RKO cells (2×103 cells)
were seeded into the upper chamber of a Transwell migration assay
plate (cat. no. CLS3422; Corning, Inc.) without Matrigel
precoating. The medium in the upper chamber was MEM medium (cat.
no. PM150410; Procell system) without serum. In the lower chamber,
DMEM medium (cat. no. PM150210; Procell system) was supplemented
with 20% FBS to attract migrating cells. After 24 h incubation at
37°C, non-migrating cells on the upper side of the membrane were
removed with a cotton swab, and the migrating cells on the lower
membrane were fixed in 4% methanol (cat. no. P1110; Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 15 min and stained with 0.1% crystal violet (cat. no. C0121;
Beyotime Biotechnology) at room temperature for 15 min. Cells were
examined using an inverted phase contrast microscope (Eclipse Ts2;
Nikon Corporation) at 400× magnification. A total of four fields of
view per chamber were randomly selected for quantification
(42,45).
Clonogenicity assay
shNC and shDCAF13 RKO cells (1×103 cells)
were plated into 6-well plates and cultured for 7 days at 37°C in
DMEM medium supplemented with 10% FBS and 100 IU/ml P/S. The media
was changed every 2 days. After 7 days incubation, cells were fixed
with 4% paraformaldehyde (cat. no. P1110; Beijing Solarbio Science
& Technology Co., Ltd.) at room temperature for 15 min,
colonies were stained with 0.1% crystal violet at room temperature
for 15 min. Colonies, defined as clusters of >50 cells, were
examined using an inverted phase contrast microscope (Eclipse Ts2;
Nikon Corporation) and counted using the Image J software (version
1.53t; National Institutes of Health) (42,45).
Adhesion assay
shNC and shDCAF13 RKO cells (5×103 cells)
were plated in 10 µg/ml fibronectin-coated (precoated at 37°C for 1
h) 96-well plates. After 30 min plating at 37°C, the wells were
washed with PBS to remove non-adherent cells. Adherent cells were
fixed with 100% methanol at room temperature for 15 min and stained
with 0.1% crystal violet at room temperature for 15 min. Adherent
cells were examined using an inverted phase contrast microscope
(Eclipse Ts2; Nikon Corporation) and counted using the Image J
software (version 1.53t) (42,45).
Cell cycle analysis
shNC and shDCAF13 RKO cells were fixed overnight in
70% ethanol at 20°C. Following the manufacturer's protocol, cells
were then stained with the cell cycle analysis working solution
(cat. no. CA1510; Beijing Solarbio Science & Technology Co.,
Ltd.) at room temperature for 1 h. The staining solution contained
propidium iodide (PI; cat. no. C0121; Beyotime Biotechnology) for
DNA content analysis, and RNase A (cat. no. C1608; Beyotime
Biotechnology) was used to degrade RNA prior to PI staining.
Intracellular permeabilization was not required in this assay, as
the fixation process with ethanol preserved the cell membranes.
Cell cycle analysis was performed using flow cytometry with a
FACScan flow cytometer (BD Biosciences) equipped with a 488 nm
laser for excitation. PI fluorescence was detected at 620 nm (FL3
channel). Data were analysed using FlowJo software (version 10.8.1;
BD Biosciences) (42,45).
Transcriptomic analysis and
validation
For transcriptomic analysis, total RNA was extracted
from RKO cells using TRIzol® reagent (cat. no. 79306;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions (46). RNA quality and
integrity were assessed using an Agilent 2100 Bioanalyzer (Agilent
Technologies, Inc.), and only samples with an RNA integrity number
(RIN) ≥7.0 were used for subsequent library construction. mRNA was
enriched using Oligo(dT) beads, fragmented into short fragments,
and reverse-transcribed into cDNA using the NEBNext®
Ultra™ RNA Library Prep Kit for Illumina® (cat. no.
7530; New England BioLabs, Inc.). The resulting double-stranded
cDNA fragments were end-repaired and ligated to Illumina sequencing
adapters, followed by purification using AMPure XP Beads (1.0×;
Beckman Coulter, Inc.) and PCR amplification. The final libraries
were quantified using a Qubit™ 4 Fluorometer (Thermo Fisher
Scientific, Inc.), and molar concentrations were calculated.
Libraries were diluted to a final loading concentration of 8 pM
prior to sequencing. Sequencing was performed on an Illumina
NovaSeq 6000 platform (Illumina, Inc.) using paired-end sequencing
with a read length of 150 bp (PE150). Raw sequencing data were
pre-processed using fastp (version 0.18.0; http://github.com/OpenGene/fastp) to remove adapter
sequences and low-quality reads. Clean reads were aligned to the
human reference genome (GRCh38) using HISAT2 (version 2.4;
http://daehwankimlab.github.io/hisat2/). Transcript
abundance was quantified using RSEM (version 1.3.3; http://deweylab.github.io/RSEM/). Differentially
expressed genes (DEGs) were identified using DESeq2 (version
1.40.2; http://bioconductor.org/packages/DESeq2/), with a
false discovery rate (FDR)-adjusted P value <0.05 and an
absolute log2 fold change ≥1 considered statistically
significant. Functional enrichment analyses were performed using
DAVID Bioinformatics Resources (version 6.8; http://david.ncifcrf.gov/). In addition, gene set
enrichment analysis (GSEA) was conducted using GSEA software
(version 4.3.2; Broad Institute; http://www.gsea-msigdb.org/gsea/) to identify
significantly enriched biological processes and signalling pathways
associated with DCAF13 regulation.
Statistical analysis
Statistical analyses were performed using SPSS
(version 19.0; IBM Corp.) and R (version 4.3.1; Posit Software,
PBC). Pearson's χ2 test was used to examine the
associations between categorical variables, including DCAF13
expression status (high vs. low) and clinicopathological
parameters. Pearson's correlation coefficient (r) was used to
assess correlations between continuous variables, including DCAF13
expression levels and immune-related gene expression or immune
infiltration scores. Differences between two independent groups
were analysed using the Wilcoxon rank-sum test (Mann-Whitney U
test). Kaplan-Meier survival curves were generated and compared
using the log-rank test. Variables with prognostic significance in
univariate analysis were further analysed using multivariate Cox
proportional hazards regression. A two-sided P<0.05 was
considered to indicate a statistically significant difference.
Results
Pan-cancer analysis of DCAF13
expression levels and prognostic performance in CRC
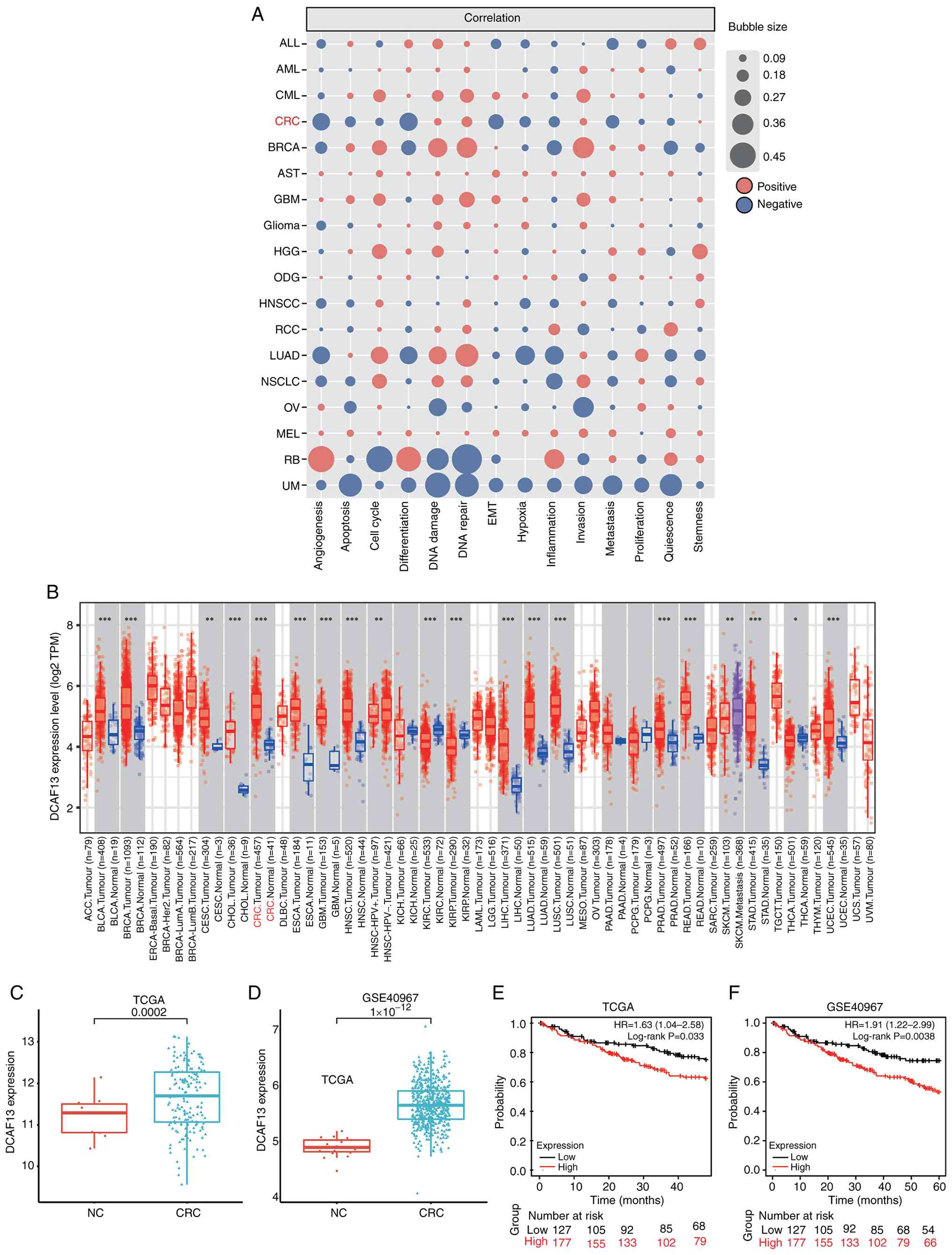
DCAF13 expression was negatively correlated with
angiogenesis (r=−0.29; P<0.05) and differentiation (r=−0.30;
P<0.05) signatures in CRC samples (Fig. 1A). TIMER analysis indicated that
DCAF13 expression levels were significantly elevated in CRC tissues
compared with that of normal colorectal samples. Additionally,
compared with adjacent normal tissues, DCAF13 mRNA expression
levels were increased in 18 other types of cancer (Fig. 1B).
 | Figure 1.DCAF13 is upregulated in pan-cancer
dataset and involved in multiple biological processes. (A)
Functional states of DCAF13 and association with 15 different
cancer types, as analysed using the CancerSEA database. (B)
Expression levels of DCAF13 across various cancer types, based on
data from the TCGA database and analysed using the Tumour Immune
Estimation Resource tool. (C) Differential expression of DCAF13
between CRC and NC tissues in the TCGA cohort. (D) Differential
expression of DCAF13 between CRC and NC tissues in the GSE40967
validation dataset. (E) KM survival analysis comparing high- and
low-expression groups of DCAF13 in the TCGA cohort. (F) KM survival
analysis comparing high- and low-expression groups of DCAF13 in the
GSE40967 validation dataset. *P<0.05, **P<0.01 and
***P<0.001. For box plots, the centre line indicates the median,
the box represents the interquartile range and the whiskers
represent data dispersion. CRC, colorectal cancer; DACF13, DDB1 And
CUL4 Associated Factor 13; TCGA, The Cancer Genome Atlas; KM,
Kaplan-Meier; HR, hazard ratio; NC, normal control. |
DCAF13 was found to be significantly upregulated in
CRC tumour tissue compared with that in the paired adjacent normal
tissue, in both the TCGA (P<0.005; Fig. 1C) and GSE40967 validation datasets
(P<0.005; Fig. 1D). KM analysis
of TCGA data showed that patients with CRC with high expression
levels of DCAF13 had a shorter OS [hazard ratio (HR)=1.63;
P<0.05; Fig. 1E] compared with
that of the low expression group. Similarly, KM analysis of the
GSE40967 validation dataset confirmed that patients with CRC with
high expression levels of DCAF13 had a shorter OS compared with
that of the low expression group (HR=1.91; P<0.005; Fig. 1F).
Immune infiltration, immune
checkpoints and response to immunotherapy
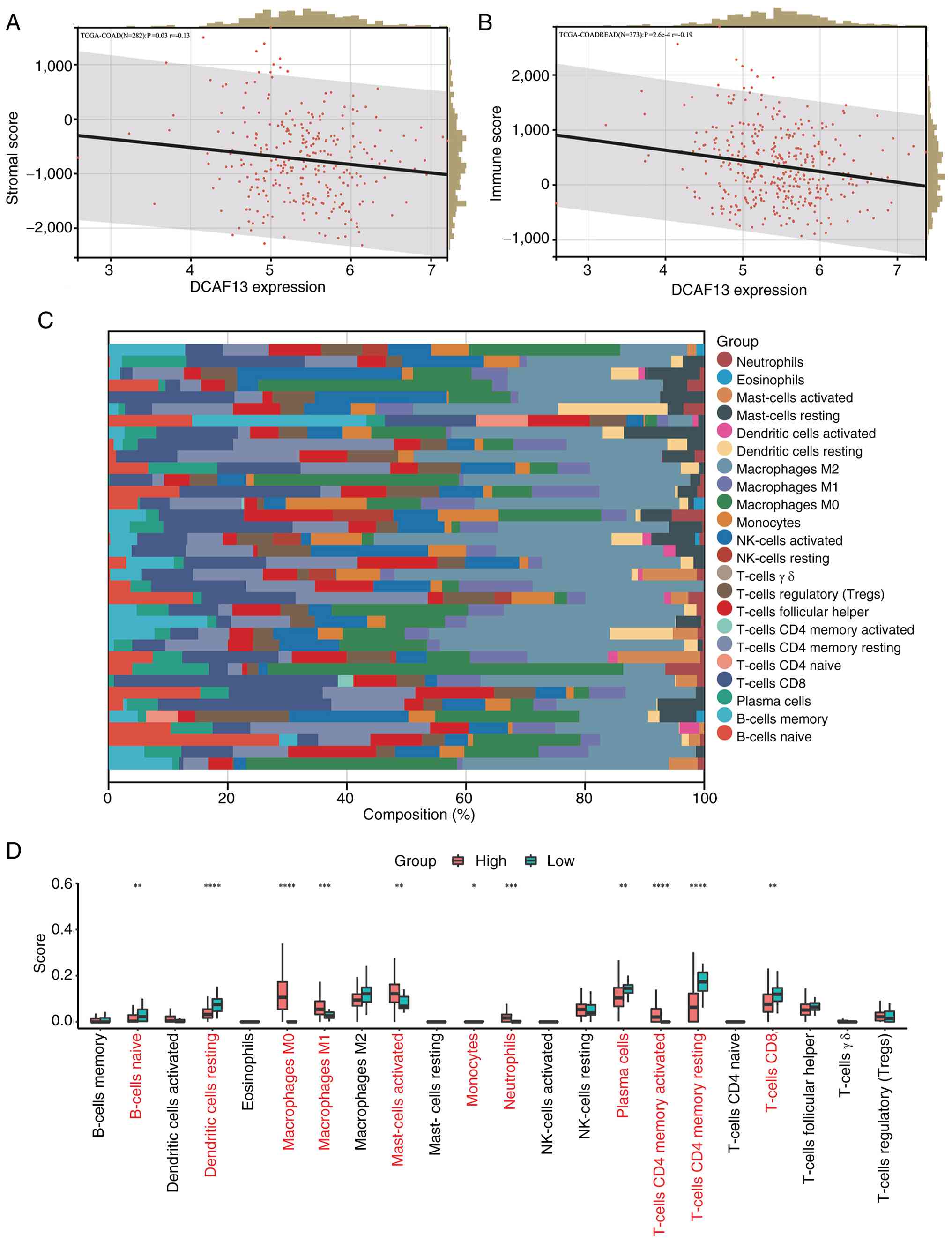
DCAF13 expression exhibited a significant negative
association with both SS (P<0.05; Fig. 2A) and IS (P<0.005; Fig. 2B) in TCGA-COAD cohort after
excluding samples with missing transcriptomic or ESTIMATE score
data. Immune infiltration analysis using the CIBERSORT algorithm
demonstrated the proportions of the 22 immune cell types in the TME
of CRC in TCGA cohort (Fig. 2C).
Comparison of immune cell infiltration between the high- and
low-DCAF13 expression groups showed that macrophages (M0 and M1),
activated mast cells, neutrophils and activated CD4+ T
cells were significantly more prevalent in the high-DCAF13
expression group. Conversely, the infiltration levels of naive B
cells, resting dendritic cells, monocytes, plasma cells, resting
CD4+ T cells and CD8+ T cells were
significantly lower in the high-DCAF13 expression group (Fig. 2D).
Correlation between DCAF13 and immune
checkpoints
Using the GEPIA database, the correlation between
DCAF13 and various immune checkpoints was evaluated. The results
indicated a potential association between DCAF13 and immune
checkpoints, including BTLA (P<0.05; Fig. 3A), LAG-3 (P<0.05; Fig. 3B), SIRPα (P<0.05; Fig. 3C) and VISTA (P<0.05; Fig. 3D). This association with immune
checkpoints led to further investigation into the relationship
between DCAF13 and immunomodulators. ICP analysis demonstrated that
DCAF13 expression was significantly correlated with a large
proportion of immunomodulators in CRC (Fig. 3E).
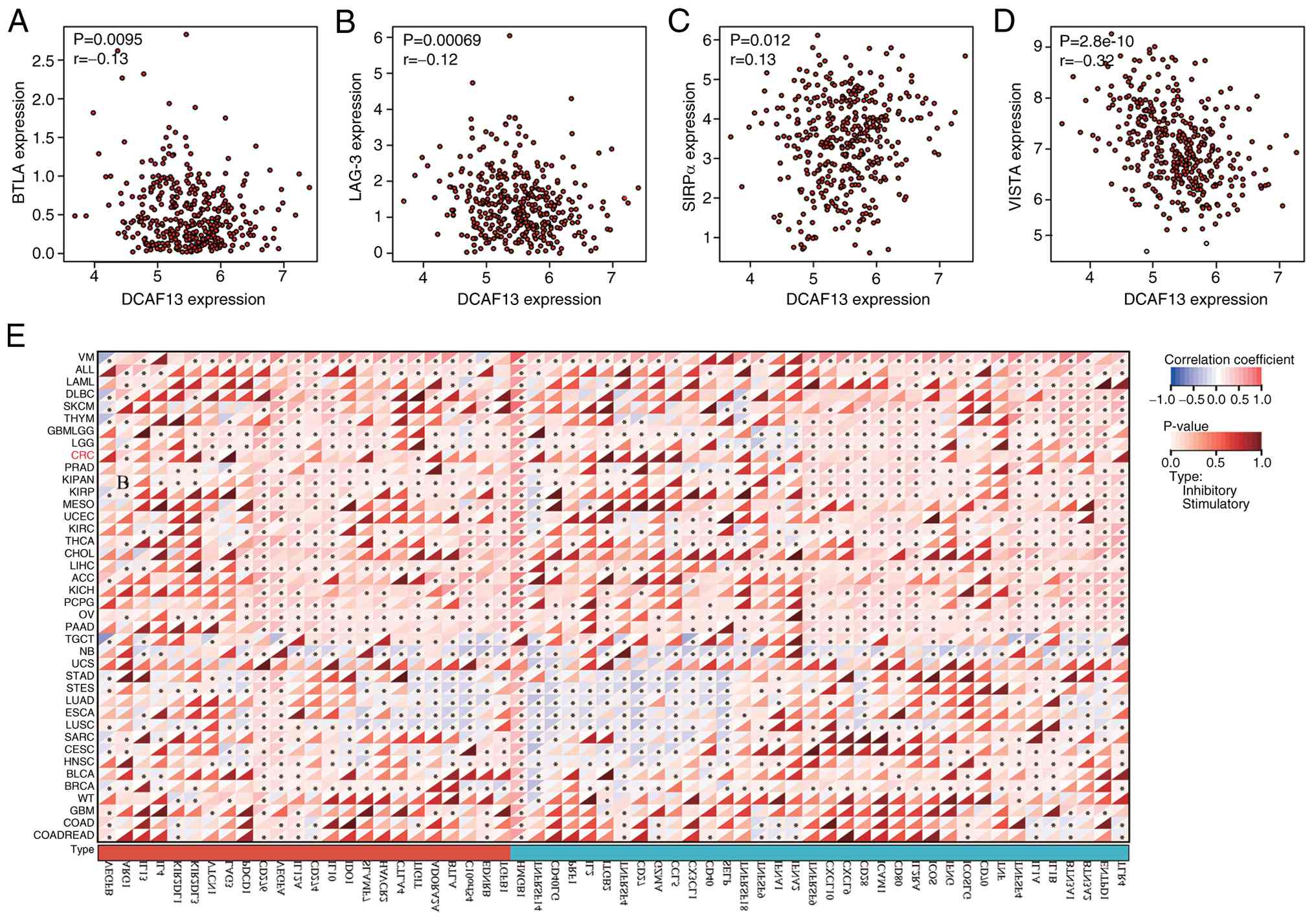 | Figure 3.Correlation between DCAF13 and immune
checkpoints and immune checkpoint gene analysis. (A) Correlation
between DCAF13 expression and BTLA. (B) Correlation between DCAF13
expression and LAG-3. (C) Correlation between DCAF13 expression and
SIRPα. (D) Correlation between DCAF13 expression and VISTA. (E)
Correlation analysis between different types of cancer, including
CRC, and 122 immunomodulators, including chemokines, receptors, MHC
molecules and immunostimulators. *P<0.05. DACF13, DDB1- and
CUL4-associated factor 13; BTLA, B and T lymphocyte attenuator;
LAG-3, lymphocyte-activating 3; SIRPα, signal regulatory protein α;
VISTA, V-domain Ig suppressor of T-cell activation. |
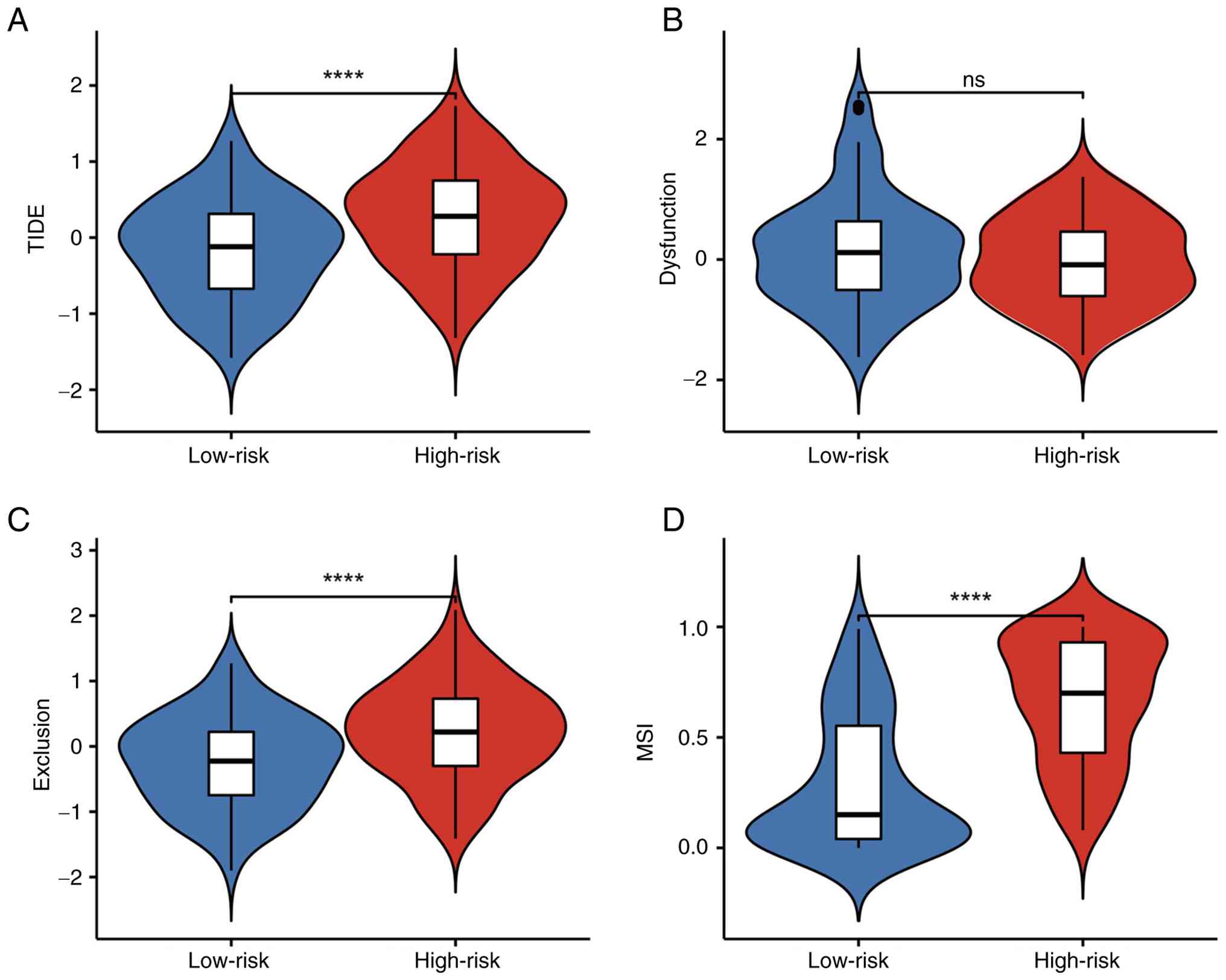
TIDE score and response to
immunotherapy
To predict the potential response of patients with
CRC to immunotherapy, the TIDE algorithm was applied to compare
predicted immunotherapy responses between patients stratified into
high- and low-DCAF13 expression groups. The analysis demonstrated
significantly higher TIDE scores in the high-risk group, suggesting
that patients with high DCAF13 expression may have a worse response
to immunotherapy compared with those in the low-risk group
(P<0.05; Fig. 4A). Notably,
there was no significant difference in T-cell dysfunction scores
between the high- and low-risk groups; however, T-cell exclusion
scores were significantly increased in the high-risk group, which
indicated a greater potential for immune evasion in high-risk
patients (Fig. 4B and C).
Additionally, the high-risk group exhibited significantly increased
MSI scores, which suggested that the high-risk CRC group may have a
higher prognostic potential and poor immunotherapy response
(Fig. 4D).
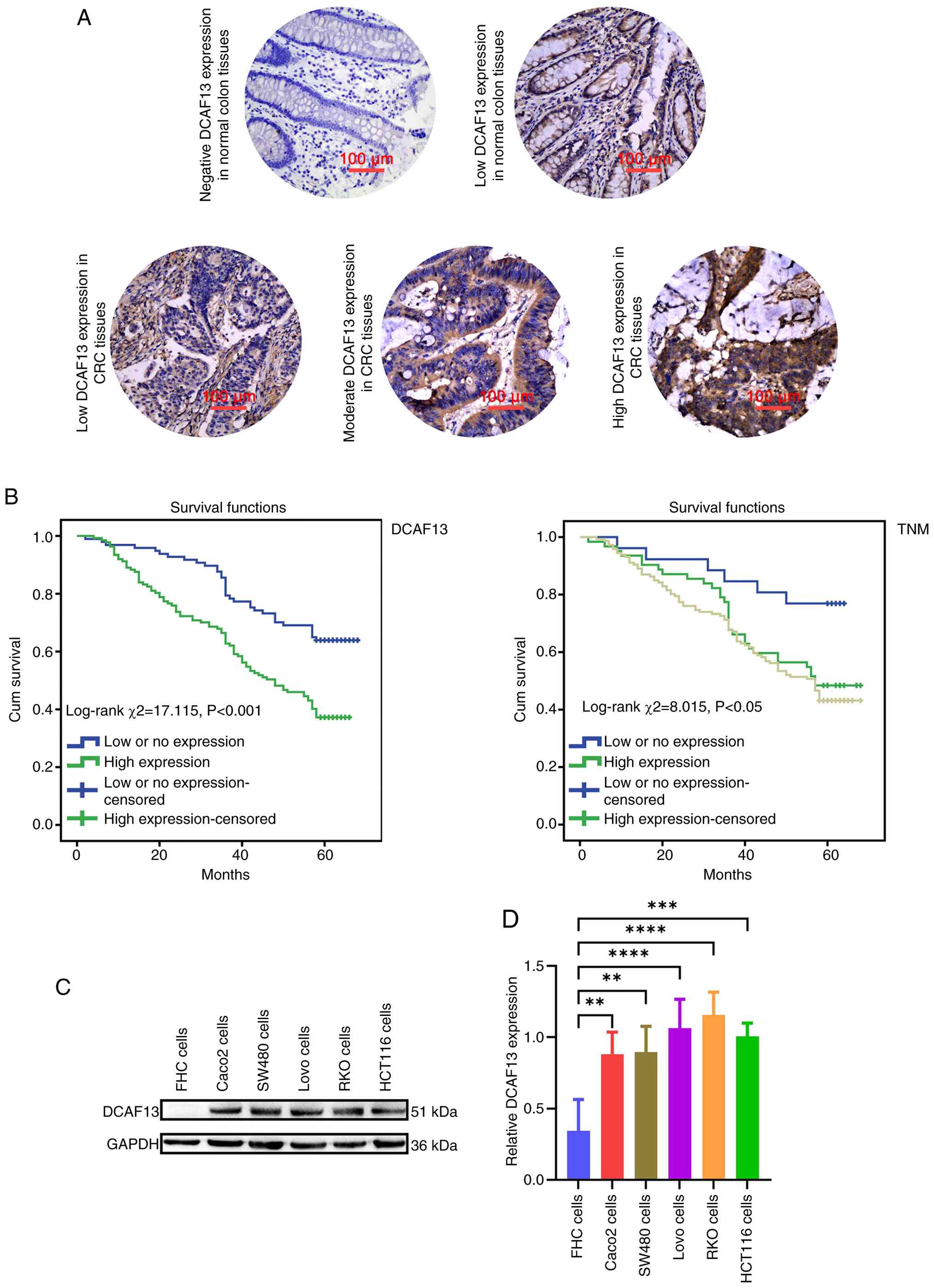
Upregulated expression of DCAF13 is
predictive of a poor prognosis in patients with CRC
DCAF13 expression was predominantly localised to the
cytoplasm in CRC tissues, whereas no notably high positive staining
was observed in normal colon tissue (Fig. 5A). Quantitative analysis using the
χ2 test demonstrated that high DCAF13 expression was
detected in 58.55% (137/234) of CRC tissues, significantly higher
compared with the 41.45% (97/234) of non-cancerous tissues
(P<0.05; Table II). Further
investigation into the association between DCAF13 expression and
the clinicopathological features of patients with CRC is presented
in Table II. Elevated DCAF13
expression was significantly associated with poorer differentiation
and advanced TNM stage in patients with CRC. Specifically, patients
with well or moderately differentiated tumours exhibited a higher
proportion of high DCAF13 expression (63.8%) compared with those
with poorly differentiated tumours (50.5%; P<0.05; Table II), indicating a negative
association between DCAF13 expression and tumour differentiation.
In terms of TNM stage, high DCAF13 expression was more frequent in
advanced stages (65.8% in stage III + IV vs. 46.2% in stage I;
P<0.05; Table II), suggesting a
positive association between DCAF13 expression and disease
progression. However, no significant associations were observed
between DCAF13 expression and clinicopathological factors such as
age, sex, tumour location or tumour size.
To evaluate the clinical relevance of DCAF13
expression, univariable analyses were performed to assess its
relationship with various clinicopathological factors (Table III). High DCAF13 expression
(HR=2.230; P<0.001) and advanced TNM stage (HR=1.464; P<0.05)
were significantly associated with a worse OS in patients with CRC.
Multivariable Cox proportional hazards regression analysis
confirmed that high DCAF13 expression (HR=2.148; P<0.001) and
TNM stage (HR=1.398; P<0.05) were independent prognostic factors
for poor 5-year OS (Table III).
KM survival analysis demonstrated that patients with CRC with high
DCAF13 expression had a significantly shorter OS compared with
those with low or no DCAF13 expression (Fig. 5B). Furthermore, survival analysis
according to TNM stage revealed a clear prognostic stratification,
with stage I patients showing the most favourable survival, stage
II patients exhibiting intermediate outcomes and stage III/IV
patients having the worst prognosis, and the differences among TNM
stages were statistically significant (Fig. 5B). Additionally, western blotting
analysis demonstrated that, compared with normal colon cells (FHC
cells), DCAF13 expression was significantly elevated in CRC cell
lines, particularly in RKO cells (Fig.
5C and D).
| Table III.Univariate and multivariate analysis
of prognostic factors in CRC for 5-year overall survival. |
Table III.
Univariate and multivariate analysis
of prognostic factors in CRC for 5-year overall survival.
|
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|
|---|
| Parameter | Variable | HR | P-value | 95% CI | HR | P-value | 95% CI |
|---|
| Expression of
DCAF13 | High-expression vs.
low- or no-expression | 2.230 |
<0.001a | 1.504–3.305 | 2.148 | <0.001 | 1.447–3.189 |
| Age, years | ≤60 vs. >60 | 0.962 | 0.840 | 0.663–1.396 |
|
|
|
| Sex | Male vs.
female | 0.749 | 0.134 | 0.512–1.094 |
|
|
|
| Tumour size,
cm | ≤5 vs. >5 | 0.936 | 0.768 | 0.603–1.453 |
|
|
|
| Histological
classification | Tubular + papillary
vs. other | 1.106 | 0.723 | 0.633–1.931 |
|
|
|
| Location | Ascending colon vs.
transverse colon vs. descending colon vs. sigmoid colon vs.
rectum | 0.913 | 0.240 | 0.785–1.062 |
|
|
|
|
Differentiation | Well + moderate vs.
poor | 0.899 | 0.562 | 0.626–1.289 |
|
|
|
| T stage | T1 vs. T2 vs. T3 +
T4 | 1.214 | 0.105 | 0.960–1.535 |
|
|
|
| N stage | N0 vs. N1 vs.
N2 | 1.202 | 0.128 | 0.949–1.523 |
|
|
|
| TNM stage | I vs. II vs. III +
IV | 1.464 | 0.010a | 1.096–1.955 | 1.398 | 0.023 | 1.047–1.867 |
Effect of DCAF13 knockdown on CRC
cells
To confirm the pro-tumorigenic role of DCAF13 on CRC
progression, lentivirus-mediated shRNA was used to knock down
DCAF13 expression in RKO cells. RT-qPCR results confirmed that
DCAF13 expression levels in the shDCAF13 were significantly reduced
compared with control cells (Fig.
6A). As confirmed by western blotting analysis, DCAF13
expression levels also showed a similar reduction, validating the
knockdown efficacy (Fig. 6B and C).
Cell proliferation assays revealed that DCAF13 knockdown
significantly impaired the proliferative potential of RKO cells
(Fig. 6D). In parallel, the
expression of the cell proliferation marker PCNA was significantly
decreased in shDCAF13 cells, further confirming the reduction in
proliferation (Fig. 6E and F).
Migration assays, performed using scratch and Transwell assays,
demonstrated that DCAF13 knockdown significantly abrogated the
ability of RKO cells to migrate compared with that of the shNC
group (Fig. 6G-J). Furthermore, the
colony formation and adhesion assays showed significant reductions
in the number of colonies and adherent cells following DCAF13
knockdown compared with that of the control group (Fig. 6K-N). Cell cycle distribution
analysis revealed that DCAF13 knockdown induced a G1
phase arrest, indicating a disruption in cell cycle progression
(Fig. 6O and P). Corresponding
western blotting analysis showed that, compared with shNC cells,
knockdown of DCAF13 resulted in increased expression of the
epithelial marker E-cadherin and decreased expression of the
mesenchymal markers N-cadherin and vimentin in CRC cells (Fig. 6Q and R), further supporting the
involvement of DCAF13 in regulating EMT progression in CRC
cells.
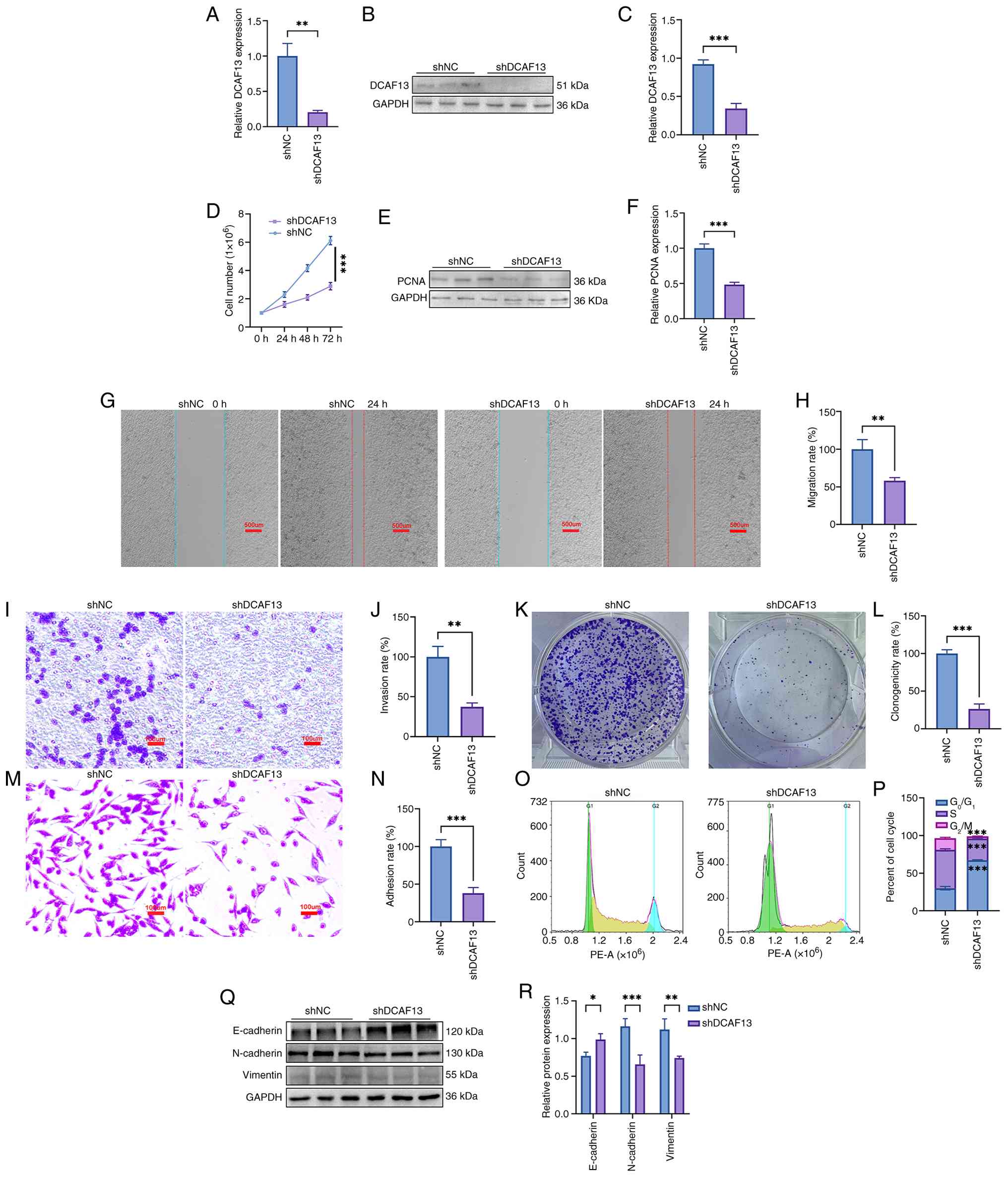 | Figure 6.Effects of DCAF13 knockdown on
inhibiting the malignant behaviour of RKO cells. (A) Detection of
DCAF13 knockdown efficiency using quantitative reverse
transcription-quantitative PCR. (B) Detection of DCAF13 knockdown
efficiency using western blot. (C) Quantitative DCAF13 expression
levels. (D) Cell growth curve. (E) Representative PCNA expression
results. (F) Quantitative PCNA expression levels. (G)
Representative result of cell migration. Scale bar, 500 µm (H)
Quantitative result of cell migration. (I) Representative result of
cell migration by Transwell assay. Scale bar, 100 µm. (J)
Quantitative result of the cell migration assay by Transwell assay.
(K) Representative result of the clonogenicity assay. (L)
Quantitative result of clonogenicity assay. (M) Representative
result of cell adhesion. Scale bar, 100 µm. (N) Quantitative result
of the cell adhesion assay. (O) Cell cycle distribution. (P)
Percentage of cells in each cell cycle phase. (Q) Representative
expression result of EMT-related proteins. (R) Relative expression
levels of ETM-related proteins. *P<0.05, **P<0.01,
***P<0.001. CRC, colorectal cancer; DACF13, DDB1- and
CUL4-associated factor 13; PCNA, proliferating cell nuclear
antigen; sh, short hairpin; EMT, epithelial-to-mesenchymal
transition. |
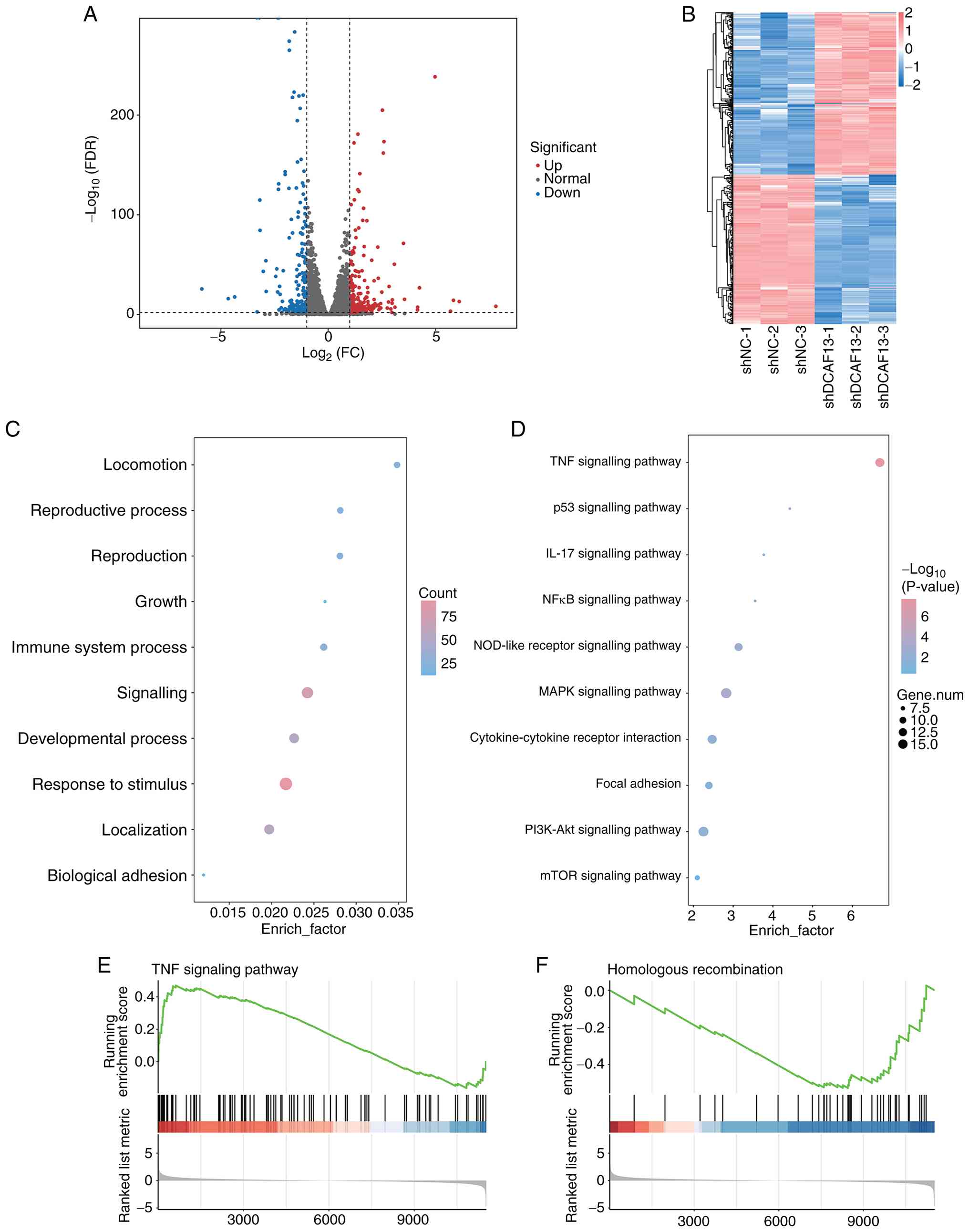
Effect of DCAF13 knockdown on
alterations in the transcriptome of RKO cells
To further uncover the pro-tumourigenic effect of
DCAF13 on CRC progression, the transcriptome characteristics of
DCAF13 knockdown RKO cells were analysed. The results confirmed
that a total of 12,652 transcripts were mined in the RKO cells
between the shNC and shDCAF13 groups. The volcano plot identified
423 DEGs between the shNC and shDCAF13 groups; this comprised 219
upregulated DEGs and 204 downregulated DEGs in the shDCAF13 group
when compared with the shNC group (Fig.
7A). A distinct hierarchical clustering of genes in the
shDCAF13 RKO cells compared with the shNC group was demonstrated in
the heat map (Fig. 7B).
Conventionally, the functional GO enrichment analyses were
performed to determine the overall functional implications of these
identified DEGs. In terms of biological processes (Fig. 7C), the identified DEGs were
predominantly involved in the subcategories of ‘locomotion’,
‘growth’ and ‘biological adhesion’. KEGG pathway enrichment
analysis indicated significant involvement of multiple oncogenic
and immune-related pathways, such as ‘TNF signalling pathway’,
‘MAPK signalling pathway’, ‘cytokine-cytokine receptor interaction’
and ‘NOD-like receptor signalling pathway’, which highlighted the
potential role of DCAF13 in modulating tumour biology (Fig. 7D). GSEA results further demonstrated
the TNF signalling pathway and homologous recombination as central
regulatory pathways that potentially mediate the pro-tumourigenic
effects of DCAF13 in CRC progression (Fig. 7E and F), which was further validated
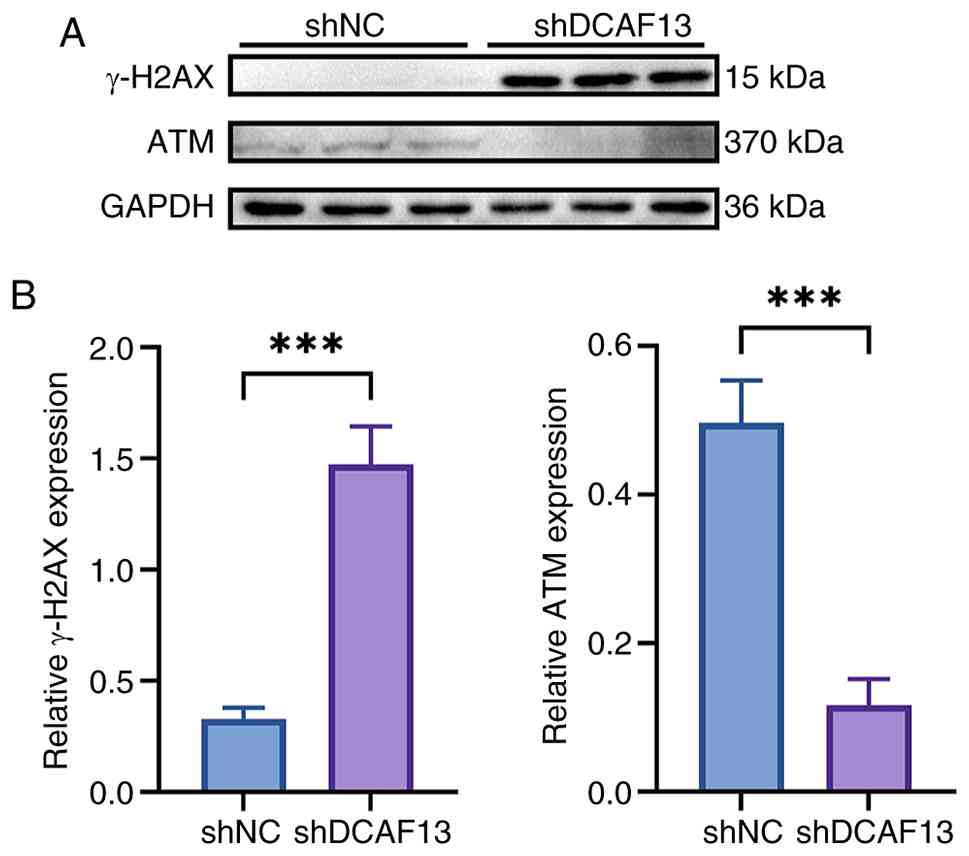
by the increased expression levels of γ-H2AX and decreased ATM
expression levels in the shDCAF13 group when compared with that of
the shNC group (Fig. 8). These
results suggest an association between DCAF13 depletion and
elevated DNA damage signalling.
Discussion
CRC remains a leading cause of cancer-related
mortality globally. Advanced stages of CRC are often characterised
by metastasis, drug resistance and immune evasion, which severely
complicate treatment and contribute to a poor prognosis (47). While traditional therapies,
including chemotherapy and surgery, have led to improvements in
survival rates, metastatic CRC still poses a major challenge
(2). The prognosis for patients
with metastatic CRC remains poor, with a median 5-year survival of
12.5% in the USA (48). Recent
advances in immunotherapy offer hope, particularly for patients
with tumours that are resistant to conventional treatments. The
emergence of ICIs, such as pembrolizumab and nivolumab, has marked
a breakthrough in CRC treatment (48,49).
Approvals from the Food and Drug Administration of ICIs that target
the programmed cell death 1 PD-1/PD-L1 and cytotoxic T lymphocyte
antigen 4 (CTLA4), have shown significant survival benefits in
patients with dMMR or MSI-H CRC, which are characterised by high
mutational burdens (50,51). By contrast, patients with proficient
mismatch repair (pMMR) tumours, including those with microsatellite
stability (MSS) or low-level microsatellite instability (MSI-L),
typically exhibit low levels of tumour-infiltrating lymphocytes and
show only modest responses to immune checkpoint inhibitors
(52,53).
To identify novel biomarkers and therapeutic targets
that may enhance the efficacy of immunotherapy, particularly in
pMMR/MSS CRC, DCAF13, a substrate receptor within the CUL4-DDB1 E3
ubiquitin ligase complex, was assessed in the present study. This
protein complex regulates protein turnover through the
ubiquitin-proteasome system and is known to influence processes
such as DNA damage repair, cell cycle progression and immune
regulation. Recent studies have implicated DCAF13 in tumorigenesis,
metastasis and chemoresistance across several malignancies,
including breast and ovarian cancer (18,25).
These findings prompted further investigation of the expression
patterns, clinical relevance and immunological implications of
DCAF13 in CRC.
The TME serves a central role in immune evasion in
CRC through mechanisms such as the upregulation of immune
checkpoints and the presence of immunosuppressive cells (47,54).
CRC tumours enriched with tumour-infiltrating lymphocytes and an
inflamed TME tend to exhibit improved responses to ICIs (55). Considering these challenges,
combination therapies that pair ICIs with chemotherapy, targeted
therapies or immune modulators are being actively explored.
Personalised immunotherapy, guided by molecular profiles and
predictive biomarkers such as PD-L1 expression, MSI status and
tumour mutational burden (TMB) holds notable potential (56–58).
Despite these advances, the clinical challenges remain,
particularly the heterogeneous nature of CRC. In addition, current
ICIs are largely ineffective in patients with pMMR and MSS or MSI-L
tumours (48). Low TMB and a lack
of immune cell infiltration in these tumours are considered key
mechanisms of immune resistance (48). Ongoing research into combination
strategies, TME modulation and the identification of novel
immunotherapy biomarkers will be critical for optimising
immunotherapeutic outcomes in CRC.
DCAFs regulates the ubiquitin-proteasome system
(UPS) to maintain cellular homeostasis (59). The UPS governs the degradation of
proteins involved in critical cellular functions, including DNA
repair, transcription, cell cycle regulation and apoptosis. DCAFs
are essential for substrate recognition and specificity within the
UPS, making them integral to maintaining cellular functions that,
when disrupted, may contribute to cancer progression (60). An increasing body of research into
DCAF proteins has revealed their involvement in tumorigenesis,
metastasis, immune evasion and chemoresistance, highlighting their
potential as therapeutic targets (61,62).
DCAF1 (also known as VprBP) and DCAF2 (also known as CDT2), for
example, are key regulators of DNA damage repair and cell cycle
progression, processes that are key to cancer development (63,64).
Additionally, DCAF proteins, particularly DCAF13, have been
implicated in chemotherapy resistance through their role in the
CUL4-DDB1 E3 ubiquitin ligase complex, which targets key regulatory
proteins for polyubiquitination and proteasomal degradation.
Although direct modulation of classical drug-metabolising enzymes
by DCAF13 has not been demonstrated, recent studies have shown that
DCAF13 negatively regulates the stability of tumour suppressors
such as PTEN and epigenetic enzymes such as SUV39H1. Furthermore,
in breast cancer, DCAF13 has been identified as an RNA-binding
protein that promotes the degradation of DTX3 mRNA (21,23,27).
Notably, doxorubicin treatment was found to upregulate DCAF13
expression levels, thereby enhancing cancer cell migration, and
EMT, suggesting a potential mechanism by which chemotherapy may
inadvertently promote metastasis in drug-resistant tumours
(65). These findings have prompted
growing interest in targeting DCAF13 to overcome resistance to
therapy.
A previous study has further emphasised the role of
DCAF proteins in CRC, particularly DCAF4L2, which is upregulated in
CRC cell lines and patient tissues (66). Upregulated expression of DCAF4L2 in
CRC is associated with enhanced cell migration and metastatic
potential, suggesting its involvement in tumour initiation and
progression. By modulating the CUL4A-DDB1 complex, DCAF proteins
regulate critical cellular processes such as cell cycle control and
DNA repair, reinforcing their involvement in CRC pathogenesis. In
addition, targeting the DCAF-CUL4A-DDB1 interaction with
small-molecule inhibitors such as NSC1892 has shown promise in
preclinical models, suppressing CRC cell proliferation and
migration (67). Furthermore, the
integration of DCAF-related pathways with immune checkpoint
regulation and TME modulation highlights novel possibilities for
enhancing immunotherapy efficacy (68). As such, DCAF proteins represent a
promising molecular target in CRC, and ongoing research into their
mechanistic roles and therapeutic potential is crucial. Their
modulation could offer novel avenues for precision medicine in CRC,
improving treatment strategies and patient outcomes.
In the present study, it was shown that DCAF13
expression was upregulated in various solid tumours, including CRC.
Specifically, CRC tissues exhibited higher levels of DCAF13
compared with that of adjacent normal tissues. This finding is
consistent with previous studies, such as that of Ieranò et
al (69), who reported elevated
DCAF13 protein expression levels in CRC tissues relative to
adjacent normal samples. The results of the present study, further
confirmed by the results of analysis of data obtained from TCGA and
the GSE40967 cohorts, as well as by the IHC staining of clinical
CRC samples, corroborate these findings. Furthermore, KM survival
analysis demonstrated that higher expression of DCAF13 was
associated with a shorter OS in patients with CRC, suggesting that
DCAF13 may serve as a prognostic biomarker. External validation
with independent cohorts and IHC staining experiments of the
clinical samples confirmed the robustness and reliability of this
marker, further supporting its potential as a therapeutic
target.
According to a previous study, high DCAF13
expression is associated with an immunosuppressive TME and
resistance to immunotherapy (27).
Correspondingly, in the present study, analysis of the immune
microenvironment demonstrated the correlation between DCAF13
expression and TIICs, as well as immune checkpoint molecules. As
TIICs are known to serve significant prognostic and therapeutic
roles in CRC and other types of cancer (70,71),
this finding is particularly relevant. The present study also
demonstrated that high DCAF13 expression was associated with
increased responsiveness to immune checkpoint blockade therapy, as
indicated by immune checkpoint analysis and TIDE scores. TIDE is a
computational framework designed to model tumour immune evasion
mechanisms and to predict patient response to ICIs, such as
anti-programmed cell death 1 (PD-1)/programmed cell death 1 ligand
(PD-L1) therapies. Specifically, TIDE evaluates two major
mechanisms of immune escape. The first is T cell dysfunction, which
occurs in tumours with high levels of cytotoxic T lymphocyte (CTL)
infiltration. In such cases, although CTLs are present in the TME,
their antitumour activity is impaired due to exhaustion or other
suppressive mechanisms. The second mechanism is T cell exclusion,
where CTLs fail to infiltrate the tumour, often due to barriers in
the TME or immunosuppressive signalling pathways that prevent
effective T cell trafficking. In addition to these two components,
TIDE also incorporates an MSI score, which reflects the degree of
dMMR in tumour cells (72,73). High MSI levels are typically
associated with increased neoantigen load and an improved response
to ICIs. These findings suggest that DCAF13 may serve as a valuable
predictor of immunotherapy efficacy in CRC. Nevertheless, further
mechanistic studies are required to clarify how DCAF13 modulates
TIICs and immune checkpoint interactions.
The analysis performed in the present study also
identified significant differences in SSs among patients with CRC,
in agreement with prior research highlighting the contribution of
stromal components to tumour progression, metastasis and resistance
to therapy (74,75). A significant difference in the ISs
among patients with CRC was also observed, which is likely
attributed to variations in the proportions of immune cell
populations. These results suggested that DCAF13 expression may
influence CRC progression and metastasis by modulating the immune
microenvironment. Additionally, DCAF13 expression was positively
correlated with immune cell populations such as M0 and M1
macrophages, activated mast cells, neutrophils and CD4+
memory T cells in CRC, further highlighting the potential
application of targeting DCAF13 for improving the efficacy of
immunotherapy in patients with CRC.
Consistent with the results of Shan et al
(18), the present study
demonstrated that the knockdown of DCAF13 significantly suppressed
proliferation, colony formation, migration and adhesion in CRC
cells, as well as disturbing cell cycle progression, EMT and
transcriptome characteristics of CRC cells. These phenotypic
changes further supported the results of CancerSEA analysis, which
indicated the pro-tumourigenic role of DCAF13 in promoting the
proliferation, migration, clonogenicity, adhesion, metastasis and
EMT progression of CRC cells.
Mechanistically, the KEGG pathway and GSEA results
in the present study underscored the pivotal role of homologous
recombination and the ‘TNF signalling pathway’ as key regulatory
mechanisms mediating the pro-tumourigenic effects of DCAF13 in CRC
progression. Homologous recombination, a highly regulated DNA
repair mechanism essential for maintaining genomic integrity in
response to DNA double-stranded breaks (DSBs) or lesions, protects
cells from exogenous and endogenous DNA damage (76). The homologous recombination
deficiency (HRD) and homologous recombination repair pathway gene
mutations, comprising at least 10 key genes (ATM, BARD1, BRCA1,
BRCA2, BRIP1, CHEK2, NBS1(NBN), PALB2, RAD51C and
RAD51D), are frequently associated with inherited
susceptibility to several types of cancer, including breast,
ovarian, prostate and pancreatic cancer (77,78).
As a well-recognised characteristic of tumours, HRD induces the
activation of error-prone DNA damage repair mechanisms, which,
while attempting to repair DNA, often result in the accumulation of
DSBs, promoting genomic instability and eventual cell death
(79). Additionally, HRD also
enhances cellular sensitivity to DNA-targeted therapies, including
platinum-based chemotherapy, DSB-inducing agents and poly
(ADP-ribose) polymerase (PARP) inhibitors (80,81).
Notably, accumulating evidence has revealed that tumours with HRD
often exhibit a high mutational load, resulting in increased levels
of TMB, tumour neoantigen burden and neoantigen expression, which
may increase tumour-cell recognition by T cells, thus facilitating
an effective lymphoid immune response and potentially improving
prognosis and survival outcomes (82,83).
Notably, Liu et al (84)
demonstrated that HRD suppressed TNF-α expression via the JNK/c-Jun
signalling pathway in triple-negative breast cancer cells and
patient tissues, thereby negatively regulating PD-L1 expression.
This suggests a role for HRD in modulating immune responses,
further supporting the potential of HRD testing for cancer
diagnosis and treatment via immune regulation.
In the present study, knockdown of DCAF13
impaired the homologous recombination process, as indicated by
changes in the expression levels of key homologous recombination
markers, including ATM and γ-H2AX, in RKO cells. These findings
align with previous research showing that DCAF8L2 overexpression
disrupts homologous recombination in breast cancer cells by
promoting the ubiquitination and degradation of BARD1, thereby
increasing sensitivity to DNA damage and facilitating cancer
progression (85). Given the
critical role of DCAF13 in homologous recombination, it could be
suggested that its dysregulation may affect tumour sensitivity to
DNA-damaging agents, such as platinum-based chemotherapeutics and
PARP inhibitors. Notably, multi-omics approaches, particularly
those integrating genomics and pharmacogenomics, have demonstrated
strong predictive power in assessing drug responses and identifying
synergistic targets (86,87). Future studies should incorporate
these approaches to comprehensively evaluate the correlation
between DCAF13 expression and sensitivity to DNA-damaging agents.
Furthermore, considering the established interplay between DNA
repair defects and immunogenicity, combining DCAF13 inhibition with
immune checkpoint blockade (such as anti-PD-1/PD-L1 or CTLA-4
therapies) may yield synergistic effects. This combination strategy
is especially promising for immunologically ‘cold’ CRC tumours,
which are often refractory to conventional ICIs (88). These insights support the rationale
for exploring DCAF13 as a dual-function biomarker and therapeutic
target in precision oncology (27,69).
Several limitations of the present study should be
acknowledged. Notably, the absence of single-cell RNA sequencing
(scRNA-seq) and spatial transcriptomics data restricts the
resolution of the analysis regarding the heterogeneity and spatial
architecture of the tumour immune microenvironment. These
cutting-edge multi-omics techniques, especially when combined with
transcriptomic, proteomic and metabolomic layers, are increasingly
regarded as essential tools for elucidating tumour heterogeneity,
identifying drug resistance pathways and optimising immunotherapy
strategies (86,87). By integrating scRNA-seq and spatial
transcriptomic technologies, future research will more accurately
map the immune cell subtypes and their spatial distributions in
tumours with high DCAF13 expression, thereby providing direct
evidence for the immunomodulatory role of DCAF13 and facilitating
the development of precision immunotherapies.
In conclusion, the present study demonstrated that
DCAF13 is aberrantly upregulated in CRC and is closely associated
with CRC progression. Through comprehensive cellular analyses, it
was demonstrated that DCAF13 served a pivotal role in regulating
key cellular processes such as proliferation, migration,
metastasis, EMT and homologous recombination in CRC. These findings
provide novel insights into the pro-tumourigenic functions of
DCAF13, underscoring its potential as a critical regulator in CRC
pathophysiology. However, further investigations are required to
fully elucidate the precise molecular mechanisms through which
DCAF13 modulates homologous recombination and its impact on
therapeutic responses. Specifically, understanding how DCAF13
influences DNA repair pathways and immune regulation could offer
novel avenues for improving the effectiveness of DNA-targeted
therapies and ICIs in CRC treatment in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Inner Mongolia (grant no. 2025MS08125), the National
Natural Science Foundation of China (grant nos. 82060567 and
82360551), Outstanding Young Talents Cultivation Program of
Grassland Elite in Inner Mongolia (grant no. Q202286), Zhiyuan
Talents Cultivation Program of Mongolia Medical University (grant
no. ZY20242129), Inner Mongolia Medical University Affiliated
Hospital Talent Training Project-Sailing Series, the Health Science
and Technology Project of Chu Zhou (grant no. 2022003) and the
Public Hospital Scientific Research Joint Fund Project (grant no.
2025NMWJKJXM1167).
Availability of data and materials
The data generated in the present study may be
found in the Genome Sequence Archive in National Genomics Data
Center, China National Center for Bioinformation/Beijing Institute
of Genomics, Chinese Academy of Sciences database under accession
number GSA-Human: HRA012999, which is associated with BioProject
PRJCA045205 or at the following URLs: https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA045205
and https://ngdc.cncb.ac.cn/gsa-human/browse/HRA012999.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
WZ, LS and GL performed study conceptualization.
WZ, RZ, MJ, SL, FL, QJ and GL developed the methodology. WZ, RZ, MJ
and SL performed data validation. WZ, MJ and FL performed
bioinformatics data processing and analysis. RZ, MJ and SL
conducted the cell biology and molecular biology experiments. QJ
was responsible for clinical sample collection, immunohistochemical
staining, and patient prognostic analysis. WZ, RZ, MJ and GL
performed formal data analysis. WZ, LS and GL acquired funding. LS
and GL provided resources and supervision. GL wrote the manuscript.
WZ and GL confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Review Committee of Inner Mongolia Medical University (approval no.
2022513411; Hohhot, China) and was performed in accordance with the
Declaration of Helsinki. Informed consent was written from all
patients prior to inclusion in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eng C, Yoshino T, Ruíz-García E, Mostafa
N, Cann CG, O'Brian B, Benny A, Perez RO and Cremolini C:
Colorectal cancer. Lancet. 404:294–310. 2024. View Article : Google Scholar
|
|
2
|
Biller LH and Schrag D: Diagnosis and
treatment of metastatic colorectal cancer: A Review. JAMA.
325:669–685. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Uneyama M, Chambers JK, Nakashima K,
Uchida K and Nakayama H: Histological classification and
immunohistochemical study of feline colorectal epithelial tumors.
Vet Pathol. 58:305–314. 2021. View Article : Google Scholar
|
|
4
|
Marusyk A, Janiszewska M and Polyak K:
Intratumor heterogeneity: The Rosetta Stone of therapy resistance.
Cancer Cell. 37:471–484. 2020. View Article : Google Scholar
|
|
5
|
GBD 2019 Colorectal Cancer Collaborators,
. Global, regional, and national burden of colorectal cancer and
its risk factors, 1990–2019: A systematic analysis for the global
burden of disease study 2019. Lancet Gastroenterol Hepatol.
7:627–647. 2022. View Article : Google Scholar
|
|
6
|
Zygulska AL and Pierzchalski P: Novel
diagnostic biomarkers in colorectal cancer. Int J Mol Sci.
23:8522022. View Article : Google Scholar
|
|
7
|
Gerstberger S, Jiang Q and Ganesh K:
Metastasis. Cell. 186:1564–1579. 2023. View Article : Google Scholar
|
|
8
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar
|
|
9
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar
|
|
10
|
Van Cutsem E, Cervantes A, Adam R, Sobrero
A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson
A, Bodoky G, et al: ESMO consensus guidelines for the management of
patients with metastatic colorectal cancer. Ann Oncol.
27:1386–1422. 2016. View Article : Google Scholar
|
|
11
|
Rui R, Zhou L and He S: Cancer
immunotherapies: advances and bottlenecks. Front Immunol.
14:12124762023. View Article : Google Scholar
|
|
12
|
Naimi A, Mohammed RN, Raji A, Chupradit S,
Yumashev AV, Suksatan W, Shalaby MN, Thangavelu L, Kamrava S,
Shomali N, et al: Tumor immunotherapies by immune checkpoint
inhibitors (ICIs); the pros and cons. Cell Commun Signal.
20:442022. View Article : Google Scholar
|
|
13
|
Le DT, Durham JN, Smith KN, Wang H,
Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et
al: Mismatch repair deficiency predicts response of solid tumors to
PD-1 blockade. Science. 357:409–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin Q, Wu L, Han L, Zheng X, Tong R, Li L,
Bai L and Bian Y: Immune-related adverse events of immune
checkpoint inhibitors: A review. Front Immunol. 14:11679752023.
View Article : Google Scholar
|
|
15
|
Xiao C, Xiong W, Xu Y, Zou J, Zeng Y, Liu
J, Peng Y, Hu C and Wu F: Immunometabolism: A new dimension in
immunotherapy resistance. Front Med. 17:585–616. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kawashima S and Togashi Y: Resistance to
immune checkpoint inhibitors and the tumor microenvironment. Exp
Dermatol. 32:240–249. 2023. View Article : Google Scholar
|
|
17
|
Lee J and Zhou P: DCAFs, the missing link
of the CUL4-DDB1 ubiquitin ligase. Mol Cell. 26:775–780. 2007.
View Article : Google Scholar
|
|
18
|
Shan BQ, Wang XM, Zheng L, Han Y, Gao J,
Lv MD, Zhang Y, Liu YX, Zhang H, Chen HS, et al: DCAF13 promotes
breast cancer cell proliferation by ubiquitin inhibiting PERP
expression. Cancer Sci. 113:1587–1600. 2022. View Article : Google Scholar
|
|
19
|
Yan X, Rong M, Zhou Q and Zhang C: DCAF13
is essential for the pathogenesis of preeclampsia through its
involvement in endometrial decidualization. Mol Cell Endocrinol.
556:1117412022. View Article : Google Scholar
|
|
20
|
Zhang YL, Zhao LW, Zhang J, Le R, Ji SY,
Chen C, Gao Y, Li D, Gao S and Fan HY: DCAF13 promotes pluripotency
by negatively regulating SUV39H1 stability during early embryonic
development. EMBO J. 37:e989812018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Li H, Mao A, Lu J, Liu W, Qie J and
Pan G: DCAF13 promotes triple-negative breast cancer metastasis by
mediating DTX3 mRNA degradation. Cell Cycle. 19:3622–3631. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang K, Li L, Fu L, Yuan Y, Dai H, Zhu T,
Zhou Y and Yuan F: Integrated bioinformatics analysis the function
of RNA binding proteins (RBPs) and their prognostic value in breast
cancer. Front Pharmacol. 10:1402019. View Article : Google Scholar
|
|
23
|
Wei S, Xing J, Chen J, Chen L, Lv J, Chen
X, Li T, Yu T, Wang H, Wang K and Yu W: DCAF13 inhibits the p53
signaling pathway by promoting p53 ubiquitination modification in
lung adenocarcinoma. J Exp Clin Cancer Res. 43:32024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang WJ, Hu CL, Guo BL, Liang XP, Wang CY
and Yang T: STAT5B suppresses ferroptosis by promoting DCAF13
transcription to regulate p53/xCT pathway to promote mantle cell
lymphoma progression. Biologics. 18:181–193. 2024.PubMed/NCBI
|
|
25
|
Tang ZY, Wang XM, Xu CW, Sun QQ, Hua YX,
Zhou QY, Hu HY, Liu SB, Guo YJ, Ao L, et al: DCAF13 promotes
ovarian cancer progression by activating FRAS1-mediated FAK
signaling pathway. Cell Mol Life Sci. 81:4212024. View Article : Google Scholar
|
|
26
|
Liu ZY, Li YH, Zhang QK, Li BW and Xin L:
Development and validation of a ubiquitin-proteasome system gene
signature for prognostic prediction and immune microenvironment
evaluation in hepatocellular carcinoma. J Cancer Res Clin Oncol.
149:13363–13382. 2023. View Article : Google Scholar
|
|
27
|
Wei S, Lu K, Xing J and Yu W: A
multidimensional pan-cancer analysis of DCAF13 and its
protumorigenic effect in lung adenocarcinoma. FASEB J.
37:e228492023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou L, Wang S, Hu W, Liu X, Xu L, Tong B,
Zhang T, Xue Z, Guo Y, Zhao J, et al: T cell proliferation requires
ribosomal maturation in nucleolar condensates dependent on DCAF13.
J Cell Biol. 222:e2022010962023. View Article : Google Scholar
|
|
29
|
Sun Y, Baechler SA, Zhang X, Kumar S,
Factor VM, Arakawa Y, Chau CH, Okamoto K, Parikh A, Walker B, et
al: Targeting neddylation sensitizes colorectal cancer to
topoisomerase I inhibitors by inactivating the DCAF13-CRL4
ubiquitin ligase complex. Nat Commun. 14:37622023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marisa L, de Reyniès A, Duval A, Selves J,
Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D,
Ayadi M, et al: Gene expression classification of colon cancer into
molecular subtypes: Characterization, validation, and prognostic
value. PLoS Med. 10:e10014532013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yuan H, Yan M, Zhang G, Liu W, Deng C,
Liao G, Xu L, Luo T, Yan H, Long Z, et al: CancerSEA: A cancer
single-cell state atlas. Nucleic Acids Res. 47((D1)): D900–D908.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li D, Zhao W, Zhang X, Lv H, Li C and Sun
L: NEFM DNA methylation correlates with immune infiltration and
survival in breast cancer. Clin Epigenetics. 13:1122021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiang S, Li J, Shen J, Zhao Y, Wu X, Li M,
Yang X, Kaboli PJ, Du F, Zheng Y, et al: Identification of
prognostic genes in the tumor microenvironment of hepatocellular
carcinoma. Front Immunol. 12:6538362021. View Article : Google Scholar
|
|
35
|
Yang S, Wen L, Chai X, Song Y, Chen X,
Chen ZF, Li R, Dong C, Qi Z and Cai Z: The protective effects of
taurine and fish oil supplementation on PM2.5-induced heart
dysfunction among aged mice: A random double-blind study. Sci Total
Environ. 851((Pt 1)): 1579662022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45((W1)):
W98–W102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang W, Xie M, Huang Q, Liu H, Liu J, Sun
X and Li C: STEAP3 is a potential preliminary prognostic biomarker
of glioblastoma. Sci Rep. 14:309942024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xia Y, Zhang R, Wang M, Li J, Dong J, He
K, Guo T, Ju X, Ru J, Zhang S and Sun Y: Development and validation
of a necroptosis-related gene prognostic score to predict prognosis
and efficiency of immunotherapy in gastric cancer. Front Immunol.
13:9773382022. View Article : Google Scholar
|
|
39
|
Nagtegaal ID, Odze RD, Klimstra D, Paradis
V, Rugge M, Schirmacher P, Washington KM, Carneiro F and Cree IA;
WHO Classification of Tumours Editorial Board, : The 2019 WHO
classification of tumours of the digestive system. Histopathology.
76:182–188. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weiser MR: AJCC 8th Edition: Colorectal
cancer. Ann Surg Oncol. 25:1454–1455. 2018. View Article : Google Scholar
|
|
41
|
Jin Q, Liu G, Bao L, Ma Y, Qi H, Yun Z,
Dai Y and Zhang S: High Spy1 expression predicts poor prognosis in
colorectal cancer. Cancer Manag Res. 10:2757–2765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang M, Lan S, Song M, Zhang R, Zhang W,
Sun X and Liu G: Synthesis of zinc oxide-doped carbon dots for
treatment of triple-negative breast cancer. Int J Nanomed.
19:13949–13971. 2024. View Article : Google Scholar
|
|
43
|
Zhang R, Lan S, Jia M, Liu F, Wang M, Jin
Q, Su L and Liu G: Theranostic applications of taurine-derived
carbon dots in colorectal cancer: Ferroptosis induction and
multifaceted antitumor mechanisms. Int J Nanomedicine.
20:7613–7635. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu G, Li S, Yuan H, Hao M, Wurihan Yun Z,
Zhao J, Ma Y and Dai Y: Effect of sodium alginate on mouse ovary
vitrification. Theriogenology. 113:78–84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang M, Lan S, Zhang W, Jin Q, Du H, Sun
X, He L, Meng X, Su L and Liu G: Anti-Cancer potency of
copper-doped carbon quantum dots against breast cancer progression.
Int J Nanomed. 19:1985–2004. 2024. View Article : Google Scholar
|
|
46
|
Zhang W, Sun J, Liu F, Li S, Wang X, Su L
and Liu G: Alleviative effect of lactoferrin interventions against
the hepatotoxicity induced by titanium dioxide nanoparticles. Biol
Trace Elem Res. 202:624–642. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shin AE, Giancotti FG and Rustgi AK:
Metastatic colorectal cancer: Mechanisms and emerging therapeutics.
Trends Pharmacol Sci. 44:222–236. 2023. View Article : Google Scholar
|
|
48
|
Ganesh K, Stadler ZK, Cercek A, Mendelsohn
RB, Shia J, Segal NH and Diaz LA Jr: Immunotherapy in colorectal
cancer: Rationale, challenges and potential. Nat Rev Gastroenterol
Hepatol. 16:361–375. 2019. View Article : Google Scholar
|
|
49
|
Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao
X and Lu Y: Immunotherapy in colorectal cancer: Current
achievements and future perspective. Int J Biol Sci. 17:3837–3849.
2021. View Article : Google Scholar
|
|
50
|
Kasi PM, Budde G, Krainock M, Aushev VN,
Koyen Malashevich A, Malhotra M, Olshan P, Billings PR and Aleshin
A: Circulating tumor DNA (ctDNA) serial analysis during progression
on PD-1 blockade and later CTLA-4 rescue in patients with mismatch
repair deficient metastatic colorectal cancer. J Immunother Cancer.
10:e0033122022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Salewski I, Kuntoff S, Kuemmel A,
Feldtmann R, Felix SB, Henze L, Junghanss C and Maletzki C:
Combined vaccine-immune-checkpoint inhibition constitutes a
promising strategy for treatment of dMMR tumors. Cancer Immunol
Immunother. 70:3405–3419. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang W, Mei Z, Chen Y, Jiang J, Qu Y,
Saifuding K, Zhou N, Bulibu G, Tang Y, Zhai X and Jiang Z: Immune
checkpoint inhibitors for patients with mismatch repair deficient
or microsatellite instability-high advanced cancers: A
meta-analysis of phase I–III clinical trials. Int J Surg.
111:1357–1372. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kazama K, Otake J, Satoyoshi T, Shiozawa
M, Sugano N, Sato S, Atsumi Y, Kano K, Murakawa M, Maezawa Y, et
al: Distribution of regulatory T-cells and other phenotypes of
T-cells in tumors and regional lymph nodes of colorectal cancer
patients. In Vivo. 34:849–856. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bai Z, Zhou Y, Ye Z, Xiong J, Lan H and
Wang F: Tumor-Infiltrating lymphocytes in colorectal cancer: the
fundamental indication and application on immunotherapy. Front
Immunol. 12:8089642021. View Article : Google Scholar
|
|
56
|
Yamaguchi H, Hsu JM, Sun L, Wang SC and
Hung MC: Advances and prospects of biomarkers for immune checkpoint
inhibitors. Cell Rep Med. 5:1016212024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang C, Wang HN and Wang L: Biomarkers for
predicting the efficacy of immune checkpoint inhibitors. J Cancer.
13:481–495. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mc Neil V and Lee SW: Advancing cancer
treatment: A review of immune checkpoint inhibitors and combination
strategies. Cancers (Basel). 17:14082025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stier A, Gilberto S, Mohamed WI, Royall
LN, Helenius J, Mikicic I, Sajic T, Beli P, Müller DJ, Jessberger S
and Peter M: The CUL4B-based E3 ubiquitin ligase regulates mitosis
and brain development by recruiting phospho-specific DCAFs. EMBO J.
42:e1128472023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jang SM, Redon CE and Aladjem MI:
Switching DCAFs: Beyond substrate receptors. BioEssays.
43:e21000572021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhou Z, Song X, Wavelet CM and Wan Y:
Cullin 4-DCAF proteins in tumorigenesis. Adv Exp Med Biol.
1217:241–259. 2020. View Article : Google Scholar
|
|
62
|
Miao Q, Kadam VD, Mukherjee A, Tan Z and
Teng M: Unlocking DCAFs To catalyze degrader development: An arena
for innovative approaches. J Med Chem. 66:13369–13383. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pan WW, Zhou JJ, Yu C, Xu Y, Guo LJ, Zhang
HY, Zhou D, Song FZ and Fan HY: Ubiquitin E3 ligase
CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian
surface epithelial cancer. J Biol Chem. 288:29680–29691. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ghate NB, Kim S, Mehmood R, Shin Y, Kim K
and An W: VprBP/DCAF1 regulates p53 function and stability through
site-specific phosphorylation. Oncogene. 42:1405–1416. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sun Z, Zhou D, Yang J and Zhang D:
Doxorubicin promotes breast cancer cell migration and invasion via
DCAF13. FEBS Open Bio. 12:221–230. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang H, Chen Y, Han J, Meng Q, Xi Q, Wu G
and Zhang B: DCAF4L2 promotes colorectal cancer invasion and
metastasis via mediating degradation of NFκb negative regulator
PPM1B. Am J Transl Res. 8:405–418. 2016.PubMed/NCBI
|
|
67
|
Yang C, Wu J, He H and Liu H: Small
molecule NSC1892 targets the CUL4A/4B-DDB1 interactions and causes
impairment of CRL4DCAF4 E3 ligases to inhibit colorectal
cancer cell growth. Int J Biol Sci. 16:1059–1070. 2020. View Article : Google Scholar
|
|
68
|
Kim KT, Lee MH, Shin SJ, Cho I, Kuk JC,
Yun J and Choi YY: Decorin as a key marker of desmoplastic
cancer-associated fibroblasts mediating first-line immune
checkpoint blockade resistance in metastatic gastric cancer.
Gastric Cancer. 28:12–26. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ieranò C, Righelli D, D'Alterio C,
Napolitano M, Portella L, Rea G, Auletta F, Santagata S, Trotta AM,
Guardascione G, et al: In PD-1+ human colon cancer cells NIVOLUMAB
promotes survival and could protect tumor cells from conventional
therapies. J Immunother Cancer. 10:e0040322022. View Article : Google Scholar
|
|
70
|
Ye L, Zhang T, Kang Z, Guo G, Sun Y, Lin
K, Huang Q, Shi X, Ni Z, Ding N, et al: Tumor-infiltrating immune
cells act as a marker for prognosis in colorectal cancer. Front
Immunol. 10:23682019. View Article : Google Scholar
|
|
71
|
Chen Y, Zhao B and Wang X: Tumor
infiltrating immune cells (TIICs) as a biomarker for prognosis
benefits in patients with osteosarcoma. BMC Cancer. 20:10222020.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Qin Y, Liu Y, Xiang X, Long X, Chen Z,
Huang X, Yang J and Li W: Cuproptosis correlates with
immunosuppressive tumor microenvironment based on pan-cancer
multiomics and single-cell sequencing analysis. Mol Cancer.
22:592023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zheng K, Hai Y, Chen H, Zhang Y, Hu X and
Ni K: Tumor immune dysfunction and exclusion subtypes in bladder
cancer and pan-cancer: A novel molecular subtyping strategy and
immunotherapeutic prediction model. J Transl Med. 22:3652024.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hu Y, Zheng M, Wang S, Gao L, Gou R, Liu
O, Dong H, Li X and Lin B: Identification of a five-gene signature
of the RGS gene family with prognostic value in ovarian cancer.
Genomics. 113:2134–2144. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang Z, Wei X, Pan Y, Xu J, Si Y, Min Z
and Yu B: A new risk factor indicator for papillary thyroid cancer
based on immune infiltration. Cell Death Dis. 12:512021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yamamoto H and Hirasawa A: Homologous
recombination deficiencies and hereditary tumors. Int J Mol Sci.
23:3482021. View Article : Google Scholar
|
|
77
|
Villacampa G, Llop-Guevara A, Filmann N,
Herencia-Ropero A, Fasching PA, Karn T, Marmé F, Klare P, Müller V,
Stefek A, et al: RAD51 testing in patients with early HER2-negative
breast cancer and homologous recombination deficiency: A Post-Hoc
analysis of the GeparOLA trial. Clin Cancer Res. 31:808–814. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tao S, Pu Y, Yang EJ, Ren G, Shi C, Chen
LJ, Chen L and Shim JS: Inhibition of GSK3β is synthetic lethal
with FHIT loss in lung cancer by blocking homologous recombination
repair. Exp Mol Med. 57:167–183. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wagener-Ryczek S, Merkelbach-Bruse S and
Siemanowski J: Biomarkers for homologous recombination deficiency
in cancer. J Pers Med. 11:6122021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
van der Wiel AMA, Schuitmaker L, Cong Y,
Theys J, Van Hoeck A, Vens C, Lambin P, Yaromina A and Dubois LJ:
Homologous recombination deficiency scar: Mutations and
beyond-implications for precision oncology. Cancers (Basel).
14:41572022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hong CS, Ho W, Zhang C, Yang C, Elder JB
and Zhuang Z: LB100, a small molecule inhibitor of PP2A with potent
chemo- and radio-sensitizing potential. Cancer Biol Ther.
16:821–833. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhou X, Ying H, Sun Y, Zhang W, Luo P, Zhu
S and Zhang J: Homologous recombination deficiency (HRD) is
associated with better prognosis and possibly causes a non-inflamed
tumour microenvironment in nasopharyngeal carcinoma. J Pathol Clin
Res. 10:e123912024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hong L, Li J and Shao W: A risk model
developed based on homologous recombination deficiency genes for
evaluating the drug sensitivity and prognostic prediction of lung
adenocarcinoma. Curr Med Chem. 32:6847–6866. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu C, Qian X, Yu C, Xia X, Li J, Li Y,
Xie Y, Gao G, Song Y, Zhang M, et al: Inhibition of ATM promotes
PD-L1 expression by activating JNK/c-Jun/TNF-α signaling axis in
triple-negative breast cancer. Cancer Lett. 586:2166422024.
View Article : Google Scholar
|
|
85
|
Deng J, Zhang T, Liu F, Han Q, Li Q, Guo
X, Ma Y, Li L and Shao G: CRL4-DCAF8L2 E3 ligase promotes
ubiquitination and degradation of BARD1. Biochem Biophys Res
Commun. 611:107–113. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang Z, Zhao Y and Zhang L: Emerging
trends and hot topics in the application of multi-omics in drug
discovery: A bibliometric and visualized study. Current
Pharmaceutical Analysis. 21:20–32. 2024. View Article : Google Scholar
|
|
87
|
Chen J, Lin A and Luo P: Advancing
pharmaceutical research: A comprehensive review of cutting-edge
tools and technologies. Current Pharmaceutical Analysis. 21:1–19.
2024. View Article : Google Scholar
|
|
88
|
Liu JL, Yang M, Bai JG, Liu Z and Wang XS:
‘Cold’ colorectal cancer faces a bottleneck in immunotherapy. World
J Gastrointest Oncol. 15:240–250. 2023. View Article : Google Scholar
|