Introduction
Rapid advancements in cytogenetics and molecular
biology, together with their clinical translation, have advanced
the mechanistic understanding of acute myeloid leukemia (AML),
including its pathogenesis, risk stratification and prognosis.
These advancements have provided critical evidence supporting risk
assessment, minimal residual disease (MRD) monitoring and the
development of precision therapeutic strategies (1). Notably, key genomic alterations such
as chromosomal translocations, copy number alterations (CNAs),
mutations in pan-tumor genes such as TP53, NRAS and
BRAF, and lineage-specific driver mutations such as
nucleophosmin 1 (NPM1) and fms-related receptor tyrosine
kinase 3 (FLT3), are highly prevalent in hematopoietic
malignancies, and have increasingly assumed a central role in
laboratory diagnostics (2,3). Furthermore, technological innovations
integrating conventional chromosome banding, fluorescence in
situ hybridization (FISH) and high-throughput sequencing have
improved the accuracy of risk stratification, supported the
establishment of prognostic models, and facilitated the
implementation of personalized treatment protocols for AML
(4,5). However, this progress has not been
translated into clinical benefits for certain elderly patients with
AML, such as those harboring NPM1 mutations, who continue to
face issues with ambiguous prognostic assessment and suboptimal
risk stratification due to the incomplete integration of genomic
profiling.
NPM1 represents one of the most prevalent
driver mutations in AML and is ubiquitously expressed across
diverse cell types (6).
Accumulating evidence indicates that approximately one-third of
adult patients with AMLharbor NPM1 variants distributed
across all French-American-British (FAB) subtypes (7). Although several studies have suggested
that the prognosis of patients with NPM1-mutated AML,
particularly isolated NPM1-mutated AML, is generally more
favorable compared with that of patients with TP53-mutated
AML, the current diagnostic and therapeutic framework continues to
face substantial challenges in elderly patients, defined as
patients ≥60 years old herein. Elderly patients often present with
multiple comorbidities and age-related organ dysfunction, resulting
in markedly lower treatment tolerance compared with that of their
younger counterparts. More importantly, this population exhibits
substantial prognostic heterogeneity: Even within the
NPM1-mutated subtype, overall survival (OS) may range from
several months to several years (8). A key contributor to this heterogeneity
is suboptimal risk stratification, such as ‘pseudo-isolated’
NPM1 mutations accompanied by undetected CNAs or
copy-neutral loss of heterozygosity (CN-LOH). The latter is a loss
of heterozygosity without changes in copy number, typically
involving duplication of the retained allele. Such risk
misclassification leads to biased risk stratification, ultimately
resulting in the overtreatment of low-risk patients and the
undertreatment of high-risk individuals (9). The core underlying issue is that
current risk stratification systems inadequately account for
genomic integrity in elderly patients: They focus primarily on
single-gene mutations, such as FLT3-internal tandem
duplication (ITD), or basic cytogenetic abnormalities, while
failing to fully integrate key genomic alterations such as CNAs and
CN-LOH.
Clinically actionable CNAs have emerged as pivotal
molecular markers for prognosis and therapeutic decision-making in
AML, and the characterization of their structural variants,
including deletions, duplications and amplifications, has become
central to basic and translational research (10). However, conventional cytogenetic
techniques, including G-banding and FISH, are limited by low
resolution and incomplete genome-wide coverage, leading to missed
detection of relevant genetic information in a subset of AML
patients and hindering accurate diagnosis and risk stratification
(4,11). As a complementary tool, shallow
whole-genome sequencing (sWGS) enables the comprehensive,
high-sensitivity capture of genome-wide CNAs and CN-LOH, with
higher sensitivity for large genomic segments compared with
conventional cytogenetic testing such as G-banding. The sWGS
approach also holds significant promise for the identification of
critical biomarkers to support risk-adapted stratification and the
refinement of prognostic models in AML (12).
Against this background, the present study aims to
address the challenges of prognostic heterogeneity, diagnostic
misclassification and suboptimal treatment decision-making in
elderly patients with NPM1-mutated AML by systematically
analyzing CNAs and CN-LOH in bone marrow specimens from 61 such
patients using sWGS. By integrating these genomic data with
hematological parameters, myeloid gene mutation profiles, such as
those of FLT3, isocitrate dehydrogenase 2 (IDH2) and
NRAS/KRAS, and relevant clinical variables, including age
and FAB subtype, the study seeks to identify potential integrative
genetic and molecular biomarkers. Ultimately, the present study
aims to provide preliminary evidence to optimize risk
stratification and prognostic assessment in this vulnerable patient
population, thereby contributing to improved stratification and
treatment decision-making in elderly patients with
NPM1-mutated AML.
Materials and methods
Patient characteristics
The present single-center retrospective study was
approved by the Clinical Ethics Committee of China-Japan Friendship
Hospital (Beijing, China; approval no. 2023-KY-200). A total of 61
adult patients (27 men and 34 women) with NPM1-mutated AML
were included. All patients in this study were diagnosed at the
China-Japan Friendship Hospital between January 2012 and December
2023, based on the criteria outlined in the World Health
Organization Classification of Tumours: Haematolymphoid Tumours
(5th edition, 2022) (13). The
median age of the cohort was 73 years (range, 60–88 years), with a
median follow-up duration of 14 months from diagnosis to the last
follow-up or death. Patients with incomplete medical records or
concurrent active malignancies were excluded. Baseline demographic
and clinical characteristics of the study cohort are summarized in
Table I. The definitions of key
characteristics are provided as follows: FAB subtype was determined
by morphological review of Wright-Giemsa-stained bone marrow
aspirate smears at diagnosis and was classified by two independent
hematopathologists according to the FAB cooperative group criteria
(7). Previous hematological
disorder was defined as a documented history of myelodysplastic
syndrome (MDS), myeloproliferative neoplasm (MPN), MDS/MPN overlap
syndrome or therapy-related myeloid neoplasm prior to the current
AML diagnosis. Complete remission (CR) and relapse were defined
according to the 2022 ELN recommendations (14). CR required bone marrow blasts
<5%, absence of circulating blasts and extramedullary disease,
absolute neutrophil count >1.0×109/l, platelet count
>100×109/l, and independence from red cell and
platelet transfusions. Relapse was defined as the reappearance of
>5% leukemic blasts in the bone marrow (not attributable to
regeneration) or the development of extramedullary disease after
achieving CR. The 1-year relapse rate was calculated as the
proportion of patients who experienced relapse within 12 months
from the date of CR attainment.
 | Table I.Comparison of clinical manifestations
and laboratory features in elderly patients with
NPM1-mutated acute myeloid leukemia stratified by CNA/CN-LOH
status. |
Table I.
Comparison of clinical manifestations
and laboratory features in elderly patients with
NPM1-mutated acute myeloid leukemia stratified by CNA/CN-LOH
status.
| Characteristic | Overall (n=61) | <3 CNA/CN-LOH
(n=50) | ≥3 CNA/CN-LOH
(n=11) | P-value |
|---|
| Age, years | 72.97 (60–88) | 72.12 (60–88) | 76.82 (65–87) | 0.047 |
| Sex,
male/female | 27/34 | 25/25 | 2/9 | 0.078 |
| White cell counts
at presentation, ×109/l | 61.23 | 58.74 | 72.32 | 0.611 |
|
| (1.23–417.23) | (1.23–417.23) | (1.27–206.45) |
|
| Hemoglobin at
presentation, g/l | 74.47 (35–111) | 73.59 (35–111) | 78.36 (52–104) | 0.415 |
| Platelets at
presentation, ×109/l | 65.65 (8–349) | 69.43 (8–349) | 48.82 (11–101) | 0.315 |
| Bone marrow blasts,
% | 63.42
(20.00–97.00) | 62.33
(20.00–96.50) | 68.37
(20.00–97.00) | 0.507 |
| NPM1
mutation type, n |
|
|
|
|
| Type
A | 47 | 39 | 8 | 1.000 |
| Type
B | 3 | 2 | 1 | 0.455 |
| Type
D | 3 | 3 | 0 | 1.000 |
|
Others | 8 | 6 | 2 | 0.955 |
| FAB subtype, n |
|
|
|
|
| M2 | 13 | 12 | 1 | 0.492 |
| M4 | 5 | 2 | 3 | 0.037 |
| M5 | 31 | 26 | 5 | 0.694 |
|
Others | 12 | 10 | 2 | 1.000 |
| Previous
hematological disorder, n | 15 | 12 | 3 | 0.601 |
| Induction
chemotherapy regimens, n |
|
|
|
|
|
Containing cytarabine | 16 | 13 | 3 | 1.000 |
|
Containing demethylation
agent | 35 | 30 | 5 | 0.585 |
|
Containing venetoclax | 16 | 12 | 4 | 0.642 |
|
Others | 15 | 11 | 4 | 0.539 |
| Complete remission,
n | 23 | 19 | 4 | 1.000 |
| 1-year relapse,
n | 16 | 13 | 3 | 1.000 |
| 2-year overall
survival, n | 11 | 11 | 0 | 0.188 |
DNA extraction, targeted
next-generation sequencing (NGS) and sWGS
Genomic DNA was extracted from bone marrow samples
remaining from routine hematology laboratory tests performed at the
initial diagnostic consultation using the Genomic DNA Extraction
Kit (cat. no. 8.02.0014; Amoy Diagnostics Co., Ltd.;) in strict
accordance with the manufacturer's recommended protocol.
For targeted NGS, a custom-designed gene panel
comprising 36 genes, including those recommended by the European
Leukemia Net (ELN) and other frequently mutated genes in myeloid
malignancies, was used (14). A
list of all 36 target genes is provided in Table SI. Library preparation was carried
out using the KAPA HyperPlus Kit (KAPA Biosystems; Roche
Diagnostics), and sequencing was performed on an Illumina MiSeq
platform (Illumina, Inc.) with a minimum sequencing depth of 2,000
reads per base. Raw sequencing data were processed using the Genome
Analysis Toolkit (version 4.4.0.0; Broad Institute) with the
following steps: Base calling for raw signal-to-base conversion,
read alignment to the reference genome, and variant calling of
single-nucleotide polymorphisms and small insertions/deletions. All
identified variants were categorized following the joint guidelines
of the American College of Medical Genetics and Genomics and the
Association for Molecular Pathology. Only mutations classified as
pathogenic or likely pathogenic with a variant allele frequency ≥2%
were retained for further analysis (15). NPM1 mutations were
categorized into types A, B, D, or others based on the
characteristic 4-bp insertions in exon 12, as previously defined
(6).
For sWGS, ~500 ng high-quality genomic DNA
(OD260/280 ratio, 1.8–2.0) was used for library construction with
the KAPA HyperPrep Kit (KAPA Biosystems; Roche Diagnostics), using
dual-indexed adapters. The final libraries were quantified using a
Qubit fluorometer (Thermo Fisher Scientific, Inc.) and pooled at an
equimolar concentration of 2 nM for sequencing. Sequencing was
conducted on an Illumina NovaSeq platform (Illumina, Inc.) in a
2×150 bp paired-end mode, achieving a mean genomic coverage depth
of 1 read per base. The sWGS data were processed using established
bioinformatic pipelines to identify CNAs and CN-LOH events
(11,16). Following quality control and data
preprocessing, the CNA and CN-LOH datasets were formatted to meet
the input requirements of the R package GenVisR (version 1.34.0;
http://bioconductor.org/packages/GenVisR). To
visualize the genomic alterations, the plotKaryotype function of
GenVisR was used to create karyotype plots of the CNA/CN-LOH
profiles. The CNA/CN-LOH burden was categorized as negative (0
events), low (1 or 2 events) or high (≥3 events) based on the total
number of distinct events per patient.
Chromosome karyotype analysis and FISH
detection
Bone marrow cells were harvested following culture
for 24–48 h in RPMI 1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin, at 37°C in a humidified
atmosphere containing 5% CO2, without stimulation to
preserve the native cytogenetic status of the cells. For
conventional karyotype analysis, G-banded metaphase spreads were
prepared from cultured bone marrow aspirates following unstimulated
culture, and cytogenetic evaluation was carried out using standard
laboratory techniques, which involved G-banding by trypsin
digestion followed by Giemsa staining (GTG-banding) to produce
characteristic light and dark bands for chromosome identification
and analysis. A total of 20 metaphase spreads per sample were
analyzed to ensure sufficient representation of the cell
population, and the karyotypes were documented in strict accordance
with the guidelines outlined in the International System for Human
Cytogenetic Nomenclature (2020) (17).
In addition to karyotype analysis, FISH testing was
performed in a subset of 36 patients to detect specific chromosomal
aberrations. This testing used a FISH probe panel (MetaSystems)
including the following probes: XL 5q31/5q33/5p15 (cat. no.
D-5081-100-TC), XL del(7)(q22q31) (cat. no. D-5068-100-TC), XCE 8
(cat. no. D-0808-050-FI), XL TP53 (cat. no. D-5103-100-OG), XL ETV6
(cat. no. D-5139-100-OG), XL RB1/DLEU/LAMP (cat. no.
D-5070-100-TC), XL 20q12/20qter (cat. no. D-5121-100-OG), XL RUNX1
(cat. no. D-5096-100-OG) and XCE X/Y (cat. no. D-0825-200-OG).
Statistical analysis
Fisher's exact test was used to compare categorical
variables. For continuous variables, normality was first assessed
using the Shapiro-Wilk test. One-way analysis of variance was used
for comparisons among different subgroups if variables were
normally distributed, while the Kruskal-Wallis H test was used if
normality was violated. OS was defined as the time from diagnosis
to death (event) or last follow-up (censored). OS distributions
were estimated by Kaplan-Meier analysis and differences between
survival curves were assessed using the log-rank test. In addition,
hazard ratios (HRs) with 95% confidence intervals (CI) were
calculated to assess survival differences. Multivariate analyses
were performed using binary logistic regression and Cox
proportional hazards models for OS, with a limited backward
elimination procedure used to exclude redundant variables. For
analyses involving multiple comparisons, the Benjamini-Hochberg
(BH) method was used to control the false discovery rate, with a
corrected Q<0.05 considered to indicate statistical
significance. All statistical analyses were performed using SPSS
Statistics version 22.0 (IBM Corp.), and two-sided P<0.05 was
considered to indicate a statistically significant result unless
otherwise specified.
Results
Gene mutation landscape analysis
A total of 61 bone marrow samples from elderly
patients with AML were subjected to NGS using a 36-gene panel.
Genomic profiling identified NPM1 mutation subtypes A, B and
D and other variants in 47 cases (77.05%), 3 cases (4.92%), 3 cases
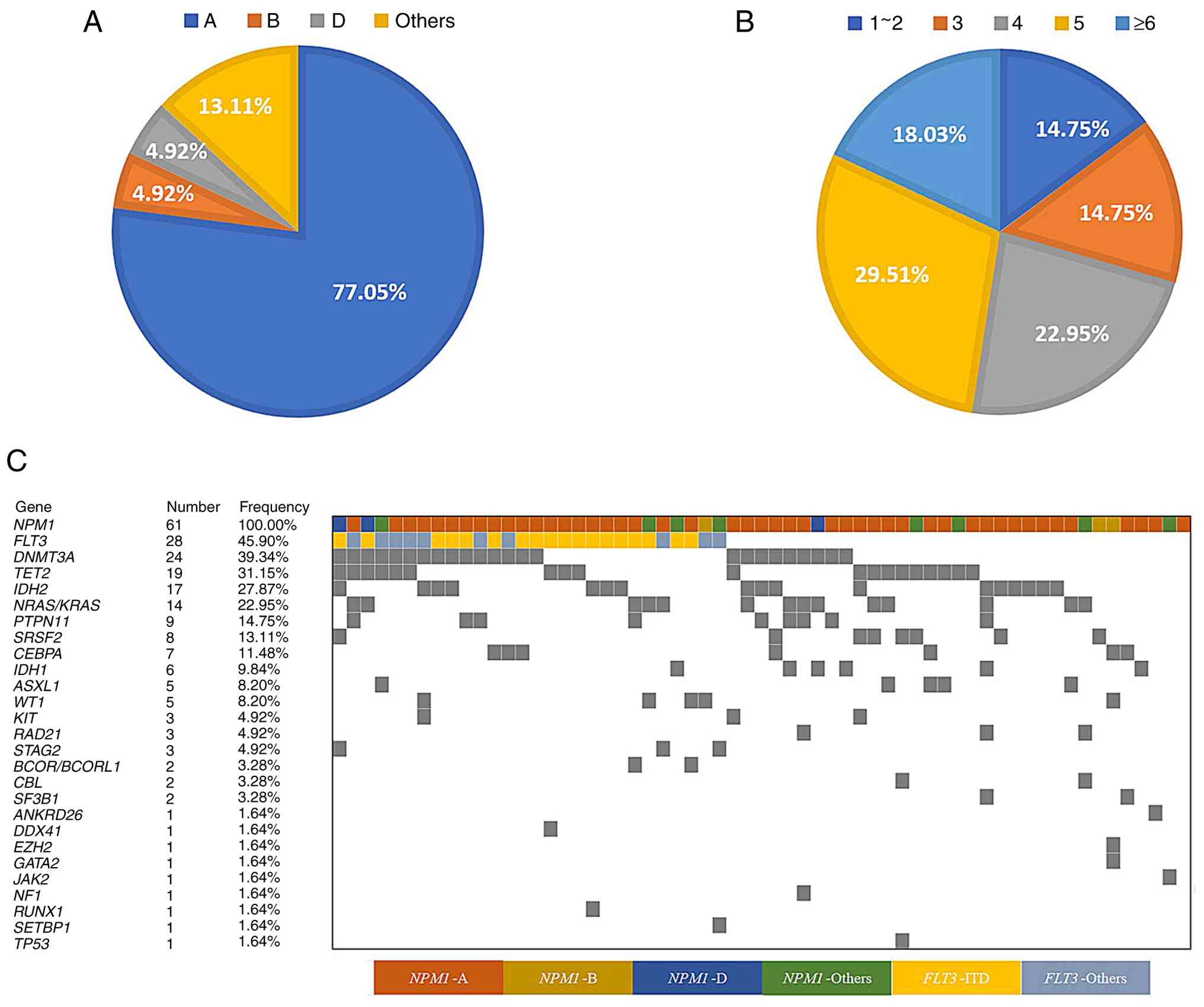
(4.92%) and 8 cases (13.11%), respectively (Fig. 1A). The majority of patients (n=32;
52.46%) exhibited a total mutational burden of 4 or 5 concurrent
somatic gene mutations, with the NPM1 mutation consistently
included in this mutational spectrum. Specifically, 14 cases
(22.95% of the cohort) presented with a total of 4 mutations and 18
cases (29.51%) had a total of 5 mutations (Fig. 1B). The most prevalent co-mutations
were detected in FLT3 (45.90%), DNMT3A (39.34%),
TET2 (31.15%), IDH2 (27.84%) and
NRAS/KRAS (22.95%) (Fig.
1C). Notably, all samples exhibited wild-type status for
BRAF, CSF3R, ETV6, PHF6, PPM1D, U2AF1, and ZRSR2. By
contrast, ANKRD26, DDX41, EZH2, GATA2, JAK2, NF1, RUNX1,
SETBP1 and TP53 were each only mutated in a single
case.
Analysis of CNAs and CN-LOH
Of the 61 patients with NPM1-mutated AML, 31
patients (50.82%) were found to harbor CNAs, while the remaining 30
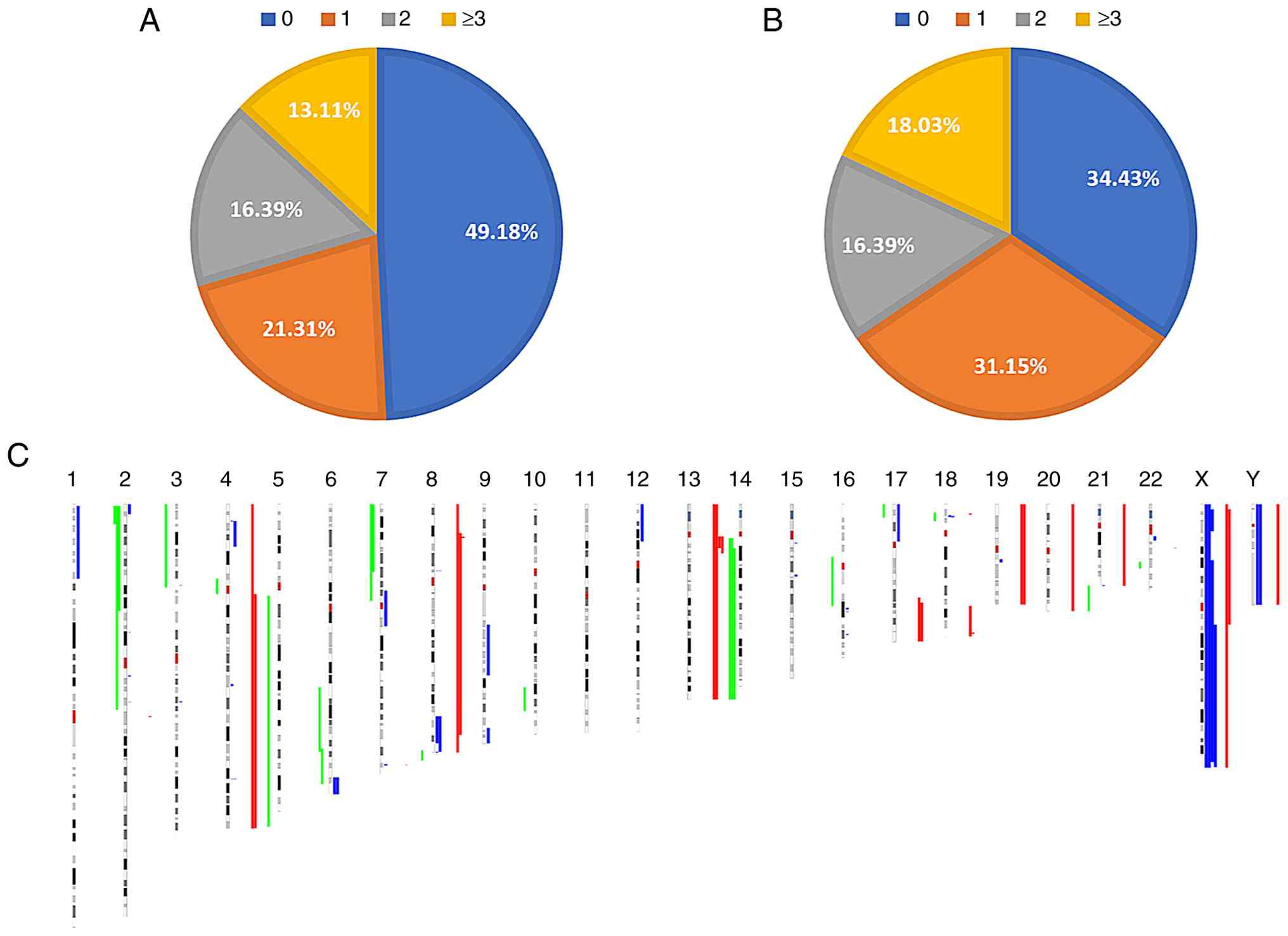
patients (49.18%) had no detectable CNAs (Fig. 2A). The patients harboring CNAs
included 13 patients (21.31%) with one CNA, 10 patients (16.39%)
with 2 CNAs and 8 patients (13.11%) with ≥3 CNAs. In addition, 40
patients (65.57%) were identified to have CNA/CN-LOH events,
whereas the remaining 21 patients (34.43%) had no detectable
CNA/CN-LOH events (Fig. 2B). The
cohort included 19 patients (31.15%) with 1 CNA/CN-LOH event, 10
patients (16.39%) with 2 such events and 11 patients (18.03%) with
≥3 CNA/CN-LOH events. The most frequently observed altered regions
included dup(4)(q11q35) (4 cases), dup(18)(p11.31p11.23) (3 cases)
and del(16)(q21q23) (3 cases); these represent putative recurrent
alterations given the small sample size. Fig. 2C illustrates the genome-wide
distribution of CNAs and CN-LOH across human chromosomes 1–22, X
and Y.
The 61 patients were stratified into two groups
based on the presence or absence of CNA/CN-LOH: The
CNA/CN-LOH-positive group comprising 40 cases and the
CNA/CN-LOH-negative group comprising 21 cases. Additionally, for
further analysis, the patients were categorized by the burden of
CNA/CN-LOH events: The <2 CNA/CN-LOH group, comprising all 21
CNA/CN-LOH-negative cases and the 19 CNA/CN-LOH-positive cases with
1 event (n=40), and the ≥2 CNA/CN-LOH group (n=21). Statistical
analysis revealed no significant intergroup differences in the
distribution of NPM1 mutation subtypes, indicating genetic
homogeneity in this aspect across the cohort. Subsequent
comparative analyses demonstrated no significant differences
between the groups in FAB subtype classification, sex distribution,
age, baseline blood counts (including white blood cell count,
platelet count and hemoglobin levels), CR rates or 2-year OS rates
(Tables SII and SIII). However, these results were limited
by the small single-center cohort and may not be generalizable.
Gene mutation profiling identified recurrent alterations in
FLT3, DNMT3A, TET2, IDH2, and NRAS in both the
CNA/CN-LOH-positive and -negative groups, as well as in the <2
CNA/CN-LOH and ≥2 CNA/CN-LOH subgroups (Tables SIV and SV). However, no statistically significant
differences in mutation frequencies were observed between these
cohorts. To further explore the potential impact of a high
CNA/CN-LOH burden on clinical and genetic characteristics, patients
were stratified into two subgroups based on the number of
CNA/CN-LOH variants: The ≥3 CNA/CN-LOH group (n=11) and the <3
CNA/CN-LOH group (n=50). Analysis demonstrated that patients
harboring ≥3 CNA/CN-LOH events were older (P=0.047) and more
frequently assigned to the M4 subtype according to the FAB
classification (P=0.037; Table I).
However, after adjusting P-values via the BH method, these
differences failed to reach the statistical significance threshold
of Q<0.05 (Q=0.447 and 0.703, respectively), indicating the
absence of a robust association. In addition, no significant
discrepancies were observed between the two subgroups regarding
other hematological parameters, mutation frequencies, clinical
indices or other relevant metrics (Tables I and II).
| Table II.Comparison of genetic alterations in
elderly patients with NPM1-mutated acute myeloid leukemia
stratified by CNA/CN-LOH status. |
Table II.
Comparison of genetic alterations in
elderly patients with NPM1-mutated acute myeloid leukemia
stratified by CNA/CN-LOH status.
| Variant | Whole cohort
(n=61) | <3 CNA/CN-LOH
(n=50) | ≥3 CNA/CN-LOH
(n=11) | P-value |
|---|
| FLT3 | 28 | 22 | 6 | 0.525 |
|
FLT3-ITD | 23 | 19 | 4 | 1.000 |
| DNMT3A | 24 | 20 | 4 | 1.000 |
| IDH2 | 17 | 12 | 5 | 0.287 |
| TET2 | 19 | 16 | 3 | 1.000 |
|
NRAS/KRAS | 14 | 12 | 2 | 0.984 |
| PTPN11 | 9 | 8 | 1 | 0.908 |
| CEBPA | 7 | 7 | 0 | 0.426 |
| IDH1 | 6 | 4 | 2 | 0.294 |
| SRSF2 | 8 | 7 | 1 | 1.000 |
| WT1 | 5 | 4 | 1 | 1.000 |
| ASXL1 | 6 | 5 | 1 | 1.000 |
G-banding and FISH analysis
Chromosomal analysis was performed on all 61 elderly
patients with NPM1-mutated AML using G-banding. Cytogenetic
abnormalities were detected in 8 patients (13.11%), while the
remaining 53 cases exhibited normal karyotypes. Among the 36 cases
who additionally underwent FISH testing, 7 cases (19.44%) showed
abnormalities in the targeted genomic regions, including 6 cases
with CNAs and 1 case with a chromosomal translocation. Integrative
analysis of G-banding and FISH results identified chromosomal
aberrations in 11 patients, while the remaining 50 cases had no
detectable abnormalities. In a subset analysis of the 6
FISH-positive patients with CNA-type abnormalities, the comparison
of FISH findings with sWGS-detected CNAs revealed the following
cytogenetic abnormalities: Loss of chromosome X (−X), loss of
chromosome Y (−Y), gain of chromosome X (+X), trisomy 8 (+8), 17p
deletion (17p-) and concurrent gains of 21q/13q/20q, each occurring
one in 1 patient. Representative FISH images are presented in
Fig. S1. Quantitative evaluation
using Cohen's κ coefficient demonstrated perfect inter-method
agreement for CNA detection (κ=1.0; 95% CI, 1.0–1.0; 100%
concordance).
Survival analysis
To elucidate the impact of mutational burden on
survival outcomes, the 61 elderly patients with NPM1-mutated
AML were stratified into mutational burden subgroups (≥3 vs. <3,
≥4 vs. <4, and ≥5 vs. <5 mutations) and subjected to
univariate survival analysis. No significant differences in
survival were observed between the ≥3 vs. <3 mutation groups
(P=0.166; Fig. 3A) or ≥4 vs. <4
mutation groups (P=0.055; Fig. 3B)
subgroups. However, patients with ≥5 mutations demonstrated a
statistically significant reduction in OS compared with that of
patients with <5 mutations (HR, 2.446; 95% CI, 1.312–4.561;
P=0.002; Fig. 3C). Subgroup
analyses of the frequently mutated genes FLT3, DNMT3A, IDH2,
TET2, NRAS/KRAS and protein tyrosine phosphatase
non-receptor type 11 (PTPN11) revealed that patients
harboring FLT3-ITD mutations exhibited a significantly
shorter OS compared with that of patients without such mutations
(HR, 2.194; 95% CI, 1.192–4.036; P=0.023, Fig. 3D). However, mutations in DNMT3A,
IDH2, TET2, NRAS/KRAS and PTPN11 were not found
to exert a significant impact on survival outcomes
(spendi-replFigs. 3E-I).
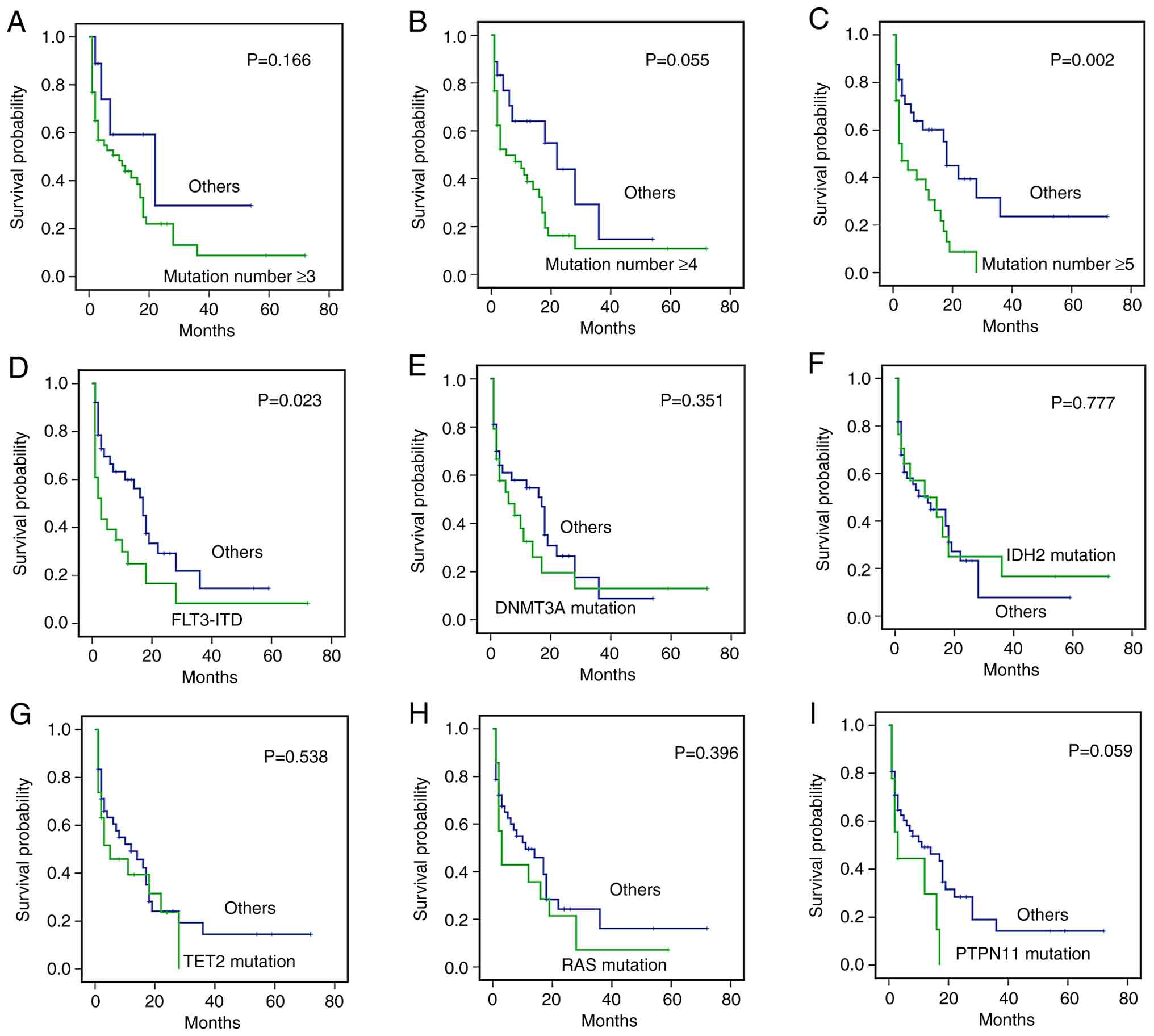 | Figure 3.Kaplan-Meier analysis of overall
survival in 61 patients with acute myeloid leukemia, stratified by
genetic features. (A-C) Survival curves by cumulative mutation
number: (A) ≥3 vs. others, (B) ≥4 vs. others and (C) ≥5 vs. others.
(D-I) Survival by individual genetic mutations: (D) FLT3-ITD
vs. others, (E) DNMT3A mutation vs. others, (F) IDH2
mutation vs. others, (G) TET2 mutation vs. others, (H)
RAS (NRAS/KRAS) mutation vs. others and (I)
PTPN11 mutation vs. others. FLT3-ITD, fms-related
receptor tyrosine kinase-internal tandem duplication;
DNMT3A, DNA methyltransferase 3; IDH2, isocitrate
dehydrogenase 2; TET2, tet methylcytosine dioxygenase 2;
PTPN11, protein tyrosine phosphatase non-receptor type
11. |
The prognostic impact of CNA/CN-LOH on OS was then
investigated. Initial survival analyses focused on the genomic
aberration dup (4)(q11q35), which
was detected in 4 patients. Univariate analysis revealed no
significant difference in OS between patients harboring dup
(4)(q11q35) and those without this
aberration (P=0.397; Fig. 4A). To
determine whether CNA/CN-LOH status influenced clinical outcomes,
patients were stratified into CNA/CN-LOH-positive and -negative
groups, and no significant difference in OS was observed between
these groups (P=0.148; Fig. 4B).
However, subgroup analysis based on the number of CNA/CN-LOH events
revealed a trend toward shorter OS in patients with ≥2 events (HR,
1.375; 95% CI, 0.730–2.590; P=0.082; Fig. 4C) and a statistically significant
reduction in OS among those with ≥3 CNA/CN-LOH events (HR, 2.275;
95% CI, 1.101–4.698; P=0.024; Fig.
4D).
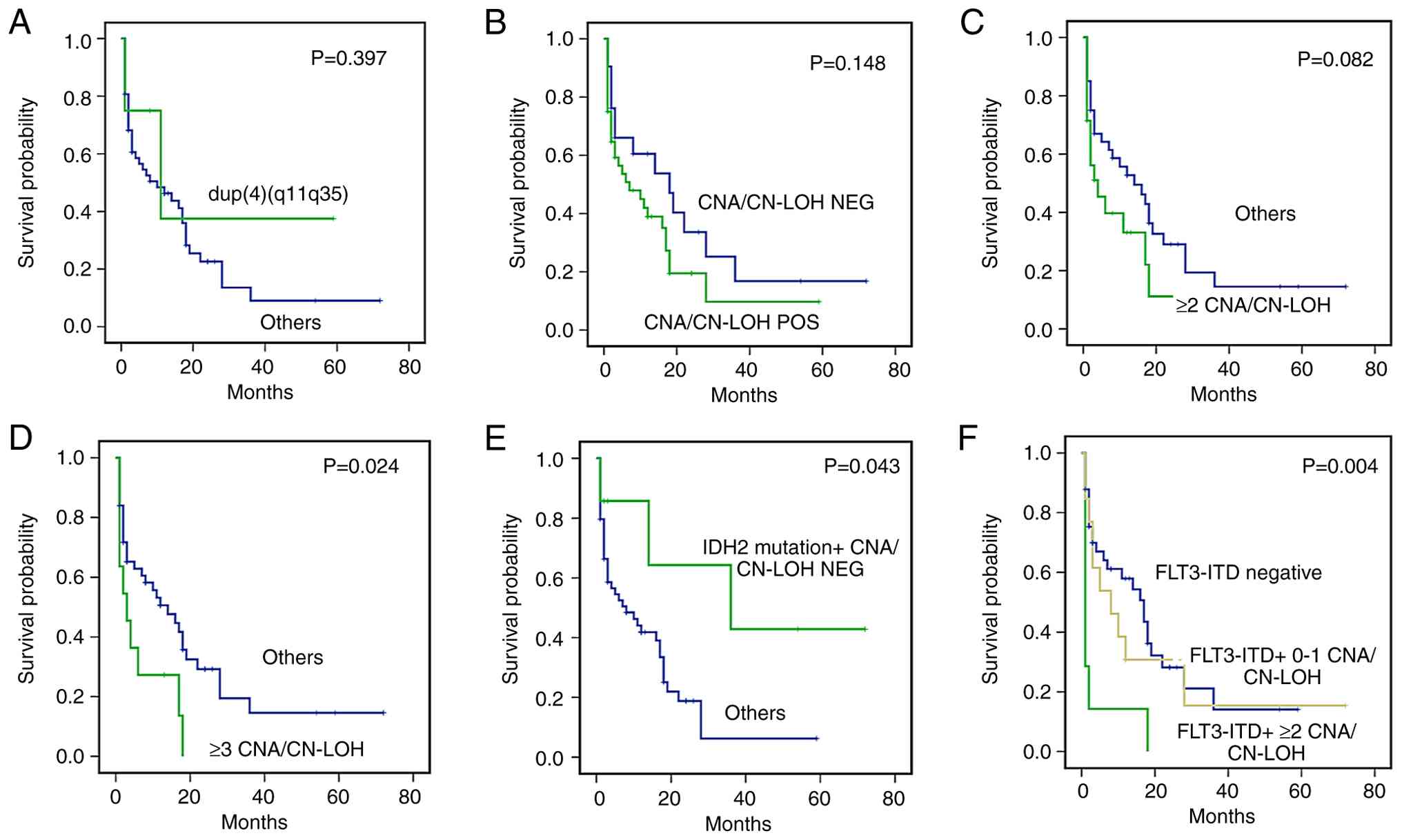 | Figure 4.Kaplan-Meier analysis of overall
survival in 61 patients with acute myeloid leukemia, stratified by
distinct genetic and genomic alteration patterns. Survival analyses
of (A) dup(4)(q11q35) vs. others, (B) CNA/CN-LOH NEG vs. CNA/CN-LOH
POS, (C) ≥2 CNA/CN-LOH events vs. others, (D) ≥3 CNA/CN-LOH events
vs. others, (E) IDH2 mutation + CNA/CN-LOH NEG vs. others
and (F) FLT3-ITD/CNA/CN-LOH combinations, namely
FLT3-ITD negative, FLT3-ITD + 0–1 CNA/CN-LOH events
and FLT3-ITD + ≥2 CNA/CN-LOH events. CNAs, copy number
alteration; CN-LOH, copy-neutral loss of heterozygosity; NEG,
negative; POS, positive; IDH2, isocitrate dehydrogenase 2;
FLT3-ITD, fms-related receptor tyrosine kinase-internal
tandem duplication. |
The integration of gene mutation and CNA/CN-LOH
profiles demonstrated that while an IDH2 mutation alone had
no distinct prognostic significance, patients with a concurrent
IDH2 mutation and no CNA/CN-LOH had a statistically
significant improvement in OS compared with other patients (HR,
0.349; 95% CI, 0.103–1.185; P=0.043; Fig. 4E). Furthermore, patients with
concurrent FLT3-ITD mutations and ≥2 CNA/CN-LOH events
displayed poorer OS outcomes than those who were FLT3-ITD
negative or had FLT3-ITD mutations and 0–1 CNA/CN-LOH events
(HR, 4.528; 95% CI, 1.956–10.478; P=0.004; Fig. 4F).
A preliminary multivariate Cox regression analysis
was performed incorporating the key molecular features that had
been identified. The analysis revealed that high mutational burden
(≥5 mutations; HR, 2.545, P=0.007) was independently associated
with an increased risk of unfavorable OS (95% CI, 1.458–4.441).
Similarly, the presence of ≥3 CNA/CN-LOH (HR, 1.978, P=0.018)
conferred an elevated risk of adverse OS (95% CI: 1.012–3.867) in
elderly patients with NPM1-mutated AML (Table III). Exploratory analyses further
suggested potential prognostic trends, albeit without reaching
statistical significance. Specifically, FLT3-ITD combined
with ≥2 CNA/CN-LOH events exhibited a trend toward an increased
risk of adverse outcomes (HR, 2.399; 95% CI, 0.770–7.479; P=0.136).
By contrast, the co-occurrence of IDH2 mutation in the
absence of CNA/CN-LOH was associated with a trend toward a reduced
risk of adverse outcomes (HR, 0.443; 95% CI, 0.200–0.977; P=0.222).
These associations did not achieve the statistical significance
threshold.
| Table III.Multivariate analysis of prognostic
variables for overall survival in 61 elderly patients with
NPM1-mutated acute myeloid leukemia. |
Table III.
Multivariate analysis of prognostic
variables for overall survival in 61 elderly patients with
NPM1-mutated acute myeloid leukemia.
| Variables | P-value | Hazard ratio (95%
confidence interval) |
|---|
| ≥5
co-mutations | 0.007 | 2.545
(1.458–4.441) |
| FLT3-ITD +
≥2 CNA/CN-LOH | 0.136 | 2.399
(0.770–7.479) |
| IDH2 mut +
CNA/CN-LOH NEG | 0.222 | 0.443
(0.201–0.977) |
| ≥3 CNA/CN-LOH | 0.018 | 1.978
(1.012–3.867) |
Discussion
With rapid advancements in genetic and molecular
biology detection techniques, particularly high-throughput
sequencing, substantial progress has been achieved in the precise
diagnostic classification, prognostic stratification and MRD
monitoring of AML (5,18,19).
Concurrently, the development and clinical application of novel
therapeutic agents have significantly improved the survival
outcomes of patients with AML (20,21).
However, a substantial proportion of patients continue to be
misclassified with respect to diagnostic subtype and prognostic
risk. This prevents the matching of individuals with optimal
treatment regimens, leading to suboptimal clinical outcomes,
particularly among elderly patients. Although some patients with
NPM1 mutations can achieve a favorable prognosis, a subset
of patients, particularly older individuals, experience poor
outcomes. Therefore, the present study analyzed gene mutations,
CNAs and CN-LOH in 61 elderly patients with NPM1-mutated
AML, with the aim of preliminarily identifying genomic features
that may further refine risk stratification and inform management
strategies within existing precision medicine frameworks.
Targeted NGS was performed on 61 bone marrow samples
obtained from elderly patients with NPM1-mutated AML using a
36-gene panel designed for myeloid malignancy profiling. Consistent
with previous reports (22,23), NPM1 mutation subtype A
predominated, accounting for 77.05% of cases, affirming its status
as the most common NPM1 subtype in AML cohorts. Notably,
52.46% of patients exhibited a total of 4 or 5 gene mutations,
including the pathogenic NPM1 mutation, with FLT3,
DNMT3A, TET2, IDH2, and NRAS/KRAS being the most
frequently co-occurring genetic alterations. These findings are
consistent with an independent study that has identified these
genes as core components of the mutational landscape in
NPM1-mutated AML (24). The
high prevalence of such co-mutations underscores the genetic
complexity of AML, where the accumulation of these mutations drives
leukemogenesis and disease progression (25).
With respect to TP53 mutations, a
well-established high-risk genetic aberration in AML, only a single
patient in the present study cohort carried a TP53 mutation,
which is consistent with previous studies reporting low TP53
mutation frequencies in NPM1-mutated AML (26). Furthermore, within this elderly
NPM1-mutated AML cohort, the mutation frequencies of genes
such as WT1, RUNX1 and ASXL1 were markedly lower than
those reported in patients with conventional
(non-NPM1-mutated) AML, and the incidence of fusion genes
was also relatively low (1,2,19).
A primary objective of the present study was to
delineate the genomic landscape of elderly patients with
NPM1-mutated AML, with particular emphasis on CNA, CN-LOH
and their clinical implications, an area that remains relatively
unexplored in elderly-specific cohorts (27). The data demonstrated a high
CNA/CN-LOH rate (65.57%) in this population. Notably, recurrent
amplifications, such as dup(4q11q35), were identified and are
hypothesized to activate oncogenes or disrupt tumor suppressor
pathways in AML, although the specific driver genes within these
loci remain to be characterized. By contrast, chromosomal
translocations constitute a defining cytogenetic feature of
conventional AML (5). In elderly
patients with AML, while the overall incidence of chromosomal
translocations is relatively low, specific chromosomal
abnormalities, including 5q deletion (−5/5q-), 7q deletion
(−7/7q-), as well as trisomy 8 (+8), are disproportionately
represented (4,5). These findings provide a preliminary
rationale for the systematic exploration of these recurrent
amplification hotspots, with the aim of identifying uncharacterized
specific drivers as druggable targets within these genomic
regions.
In the present study, elderly patients with
NPM1-mutated AML harboring ≥5 concomitant mutations
exhibited a significantly shorter OS, supporting the hypothesis
that high mutational burden reflects underlying genomic instability
and clonal heterogeneity (28).
This observation provides preliminary evidence for the clinical
relevance of mutation burden in elderly patients with NPM1
mutations, a population that is often underrepresented in
large-scale genomic studies of AML. Importantly, in routine
clinical practice, a subset of elderly patients with
NPM1-mutated AML, even those lacking ELN 2022-defined
adverse-risk features (29),
display distinct genomic profiles and experience suboptimal
outcomes, underscoring the necessity for further investigation.
Using sWGS for the detection of CNAs and CN-LOH, it
was further identified that the presence of ≥3 CNA/CN-LOH events
may serve as an independent predictor of poor survival outcomes.
This is a tentative but potentially novel finding, as the present
study is among the first to use sWGS to quantify CNA/CN-LOH burden
as a prognostic marker in elderly patients with NPM1-mutated
AML. By contrast, previous studies in this field have primarily
focused on individual chromosomal aberrations, such as del(5q) and
monosomy 7, rather than using CNA/CN-LOH burden as a composite
prognostic indicator (27,30).
In subtype-specific analyses, consistent with
previous studies, isolated IDH2 mutations did not
demonstrate intrinsic prognostic value (22,31).
However, by integrating mutational profiles with CNA/CN-LOH data,
the present study observed that the co-occurrence of IDH2
mutations and absence of CNA/CN-LOH events was associated with
significantly improved clinical outcomes. Although multivariate Cox
regression analysis did not confirm independent prognostic value,
likely due to the small sample size and limited statistical power,
these preliminary findings suggest that the combined assessment of
IDH2 mutation and CNA/CN-LOH status may help to tentatively
identify subgroups with favorable prognosis among patients with
NPM1-mutated AML. Future large-scale, multicenter studies
are warranted to validate the generalizability of this combined
molecular marker, including its prognostic implications in
conventional AML subtypes.
With respect to FLT3-ITD mutations, the 2022
ELN guidelines classify patients with NPM1-mutated AML
harboring FLT3-ITD as intermediate risk (29); however, some such patients still
experience very poor outcomes. In the present study, it was found
that among FLT3-ITD-positive, NPM1-mutated cases,
those with ≥2 CNA/CN-LOH events exhibited the worst survival
outcomes, underscoring the interplay between mutational status and
genomic instability as a potential prognostic axis. Although this
association did not reach statistical significance in the
multivariate analysis, these observations tentatively suggest a
mechanistic basis for the pronounced prognostic heterogeneity
within this vulnerable subgroup and highlight the need to refine
risk stratification beyond conventional AML classification
systems.
Notably, in the exploratory analysis of associations
between high CNA/CN-LOH burden (≥3 events) and clinical
characteristics, the present study initially identified
associations with advanced patient age and FAB subtype M4. However,
these associations did not retain statistical significance
following BH correction, underscoring that cautious interpretation
is necessary, likely reflecting the impact of multiple testing
adjustment and the limited sample size of the high-burden subgroup
(n=11). These limitations highlight the necessity of validating
these preliminary findings in larger, independent cohorts to
clarify their potential clinical implications.
To define high-risk thresholds for genomic burden,
the CNA/CN-LOH cut-off was first determined. A threshold of ≥3
events was statistically derived by rounding the sum of the cohort
mean (1.3770) plus 1 standard deviation (1.5723), yielding a value
of 2.9494. This threshold included 11 patients (18.03%), which
provides sufficient discriminatory power for survival analysis
while preserving the ability to distinguish a clinically relevant
high-risk subgroup. A higher threshold (≥4 events) reduced the
subgroup to 5 patients (8.20%), compromising statistical
reliability. Kaplan-Meier analysis showed that the separation of OS
was optimal with ≥3 events, whereas a lower threshold exhibited a
weaker association. For mutation burden, the high-risk threshold of
≥5 mutations was selected based on the distribution within the
61-patient cohort (mean, 4.34; median, 4.0), to effectively
identify patients with above-average mutational burden. This
cut-off included 29 patients (47.54%), ensuring balanced group
sizes and adequate analytical power. Importantly, the ≥5 mutation
threshold was associated with shorter OS, supporting its utility
for risk stratification, while lower thresholds lacked robust
prognostic relevance.
Regarding the clinical implications of integrated
genomic stratification, the insufficiently defined prognosis of
NPM1-mutated AML in current clinical practice underscores
the urgent requirement for precise risk stratification. Integrating
CNA/CN-LOH burden (≥3 events) with mutational load (≥5 mutations)
may help to refine existing risk stratification systems and offer
translatable clinical value by providing a more personalized basis
for tailoring therapeutic intensity and disease surveillance
strategies. Specifically, patients with low genomic burden (<3
CNA/CN-LOH events and <5 mutations) may benefit from
de-escalated therapy, potentially minimizing treatment-related
toxicity while maintaining therapeutic efficacy. By contrast,
patients with high genomic burden (≥3 CNA/CN-LOH events and/or≥5
mutations), which is associated with shorter OS, may require more
aggressive upfront treatment regimens. Regarding surveillance,
patients with a high burden may benefit from more frequent
monitoring to enable the early detection of relapse, whereas those
with a low burden could be surveilled less frequently. Beyond
overall genomic burden, subtype-specific analyses suggest the
potential value of integrating CNA/CN-LOH with driver mutations.
Among FLT3-ITD-positive patients, those with ≥2 CNA/CN-LOH
events exhibited the poorest OS, indicating that combinatorial
therapies including FLT3 inhibitors may be explored, while
accounting for the potential contribution of high CNA/CN-LOH burden
to treatment resistance. Conversely, in patients with IDH2
mutations, those without CNA/CN-LOH exhibited a longer OS,
suggesting that CNA/CN-LOH status may serve as a stratification
marker for the efficacy of IDH2 inhibitors and provide guidance for
the design of future clinical trials.
The present study has several inherent limitations
that warrant careful consideration when interpreting the results.
As a single-center retrospective analysis with a modest cohort
size, the generalizability of the findings may be limited. The
small sample size was particularly pronounced in clinically
relevant subgroups, including patients with ≥3 CNA/CN-LOH events
(n=11) and those harboring FLT3-ITD alongside ≥2 CNA/CN-LOH
events (n=10). This may compromise the robustness of inter-subgroup
comparisons and locus-specific CNA/CN-LOH analyses. In addition,
the relatively low number of OS events may introduce variability in
the prognostic parameters, including HRs and 95% CIs, generated by
the preliminary Cox regression model. This also restricts the
ability of the model to adjust for potential confounding variables
or to detect subtle interactions among covariates. Therefore, the
observed independent prognostic value of high mutational burden and
elevated CNA/CN-LOH burden necessitates external validation in
larger, multi-institutional cohorts. Furthermore, the clinical
applicability of these findings is currently constrained by the
lack of associative analyses linking genomic aberrations to
responses to targeted therapies.
To address these limitations, subsequent studies
should prioritize the following: i) Validation of prognostic
thresholds for mutational and CNA/CN-LOH burdens in large,
prospectively curated cohorts; and ii) integrative,
multidimensional analyses of co-occurring gene mutations and
CNA/CN-LOH events, and their association with therapeutic outcomes.
Such efforts will be critical for refining risk stratification
models and informing personalized treatment strategies for older
patients with NPM1-mutated AML. In alignment with these
objectives, the current research team is actively conducting a
multicenter study to systematically compare CNA/CN-LOH profiles
across AML molecular subtypes, including NPM1-mutated AML
and conventional subtypes, such as TP53-, RAS- and
ASXL1-mutated AML. The study is structured around two
pivotal analytical axes: i) Quantification of CNA/CN-LOH burden at
the patient level to define subtype-specific distribution patterns,
and ii) genome-wide mapping of recurrent events, such as
dup(4q11q35) enrichment in NPM1-mutated AML. By constructing
a provisional subtype-resolved CNA/CN-LOH landscape and associating
these genomic features with clinical outcomes, the study aims to
clarify inter-subtype heterogeneity in burden distributions,
identify subtype-discriminatory recurrent events, and elucidate the
mechanistic interplay between CNA/CN-LOH and driver mutations.
In conclusion, the present study preliminarily
delineates the genomic landscape of NPM1-mutated AML in
elderly patients, with a focus on co-mutational patterns, CNAs and
CN-LOH, which are genomic features that have been relatively
unexplored in previous studies. It identifies high mutational
burden (≥5 mutations) and ≥3 CNA/CN-LOH events as potential
indicators of a poor prognosis, tentatively supporting the
established concept that genomic instability and clonal complexity
are associated with adverse outcomes in this subgroup. In addition,
the present study suggests that the combination of IDH2
mutation with the absence of CNA/CN-LOH may be associated with
favorable outcomes, while FLT3-ITD positivity combined with
≥2 CNA/CN-LOH events is associated with poor survival. These
findings may partially explain the prognostic heterogeneity
observed within intermediate- and favorable-risk categories defined
by the ELN 2022 guidelines, highlighting the potential value of
integrating multidimensional genomic profiles, rather than
single-gene mutations alone, to optimize risk stratification beyond
existing guidelines. However, it should be emphasized that the
generalizability of the genomic thresholds proposed in the present
study requires validation in large-scale multicenter studies, and
may ultimately provide a preliminary framework for advancing the
personalized diagnosis, treatment selection and precise management
of elderly patients with NPM1-mutated AML.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science
Foundation of China (grant no. 8130045) and the Clinical Service
Expenses for High-level Hospitals of CJFH (grant no.
2023-NHLHCRF-PY-05).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The raw sequencing data
have been deposited in the China National Genomics Data Center
(NGDC) under the BioProject accession number PRJCA053981
(https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA053981).
The sample-level data can be accessed via GSA accession number
HRA015549 (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA015549).
Authors' contributions
ZL, LG and CZ conceived and designed the study, and
drafted the manuscript. ZL, CZ and MG contributed to the collection
of clinical data and patient management. LG, ZC, CL and SW
performed the laboratory work. ZL, LG and CZ confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
China-Japan Friendship Hospital (approval no. 2023-KY-200) and
conducted in strict adherence to the principles outlined in the
Declaration of Helsinki. Prior to the initiation of the study,
written informed consent was obtained from all enrolled patients or
their legal representatives in accordance with ethical
requirements.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AML
|
acute myeloid leukemia
|
|
BM
|
bone marrow
|
|
CNA
|
copy number alteration
|
|
CN-LOH
|
copy-neutral loss of
heterozygosity
|
|
ELN
|
European Leukemia Net
|
|
FAB
|
French-American-British
|
|
FISH
|
fluorescence in situ
hybridization
|
|
FLT3-ITD
|
FLT3 internal tandem
duplication
|
|
HR
|
hazard ratio
|
|
MDS
|
myelodysplastic syndrome
|
|
MPN
|
myeloproliferative neoplasm
|
|
MRD
|
minimal residual disease
|
|
NGS
|
next-generation sequencing
|
|
OS
|
overall survival
|
|
sWGS
|
shallow whole-genome sequencing
|
References
|
1
|
Shimony S, Stahl M and Stone RM: Acute
myeloid leukemia: 2025 update on diagnosis, risk-stratification,
and management. Am J Hematol. 100:860–891. 2025. View Article : Google Scholar
|
|
2
|
Kayser S and Levis MJ: The clinical impact
of the molecular landscape of acute myeloid leukemia.
Haematologica. 108:308–320. 2023. View Article : Google Scholar
|
|
3
|
Wysota M, Konopleva M and Mitchell S:
Novel therapeutic targets in acute myeloid leukemia (AML). Curr
Oncol Rep. 26:409–420. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bidet A, Quessada J, Cuccuini W, Decamp M,
Lafage-Pochitaloff M, Luquet I, Lefebvre C and Tueur G; Groupe
Francophone de Cytogénétique Hématologique (GFCH), : Cytogenetics
in the management of acute myeloid leukemia and
histiocytic/dendritic cell neoplasms: Guidelines from the Groupe
Francophone de Cytogénétique Hématologique (GFCH). Curr Res Transl
Med. 71:1034212023.PubMed/NCBI
|
|
5
|
Snaith O, Poveda-Rogers C, Laczko D, Yang
G and Morrissette JJD: Cytogenetics and genomics of acute myeloid
leukemia. Best Pract Res Clin Haematol. 37:1015332024. View Article : Google Scholar
|
|
6
|
Falini B, Brunetti L, Sportoletti P and
Martelli MP: NPM1-mutated acute myeloid leukemia: From bench to
bedside. Blood. 136:1707–1721. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hindley A, Catherwood MA, McMullin MF and
Mills KI: Significance of NPM1 gene mutations in AML. Int J Mol
Sci. 22:100402021. View Article : Google Scholar
|
|
8
|
Patel SS: NPM1-mutated acute myeloid
leukemia: Recent developments and open questions. Pathobiology.
91:18–29. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen EC, Shimony S, Luskin MR and Stone
RM: Biology and management of acute myeloid leukemia with mutated
NPM1. Am J Hematol. 100:652–665. 2025. View Article : Google Scholar
|
|
10
|
Giles Doran C and Pennington SR: Copy
number alteration signatures as biomarkers in cancer: A review.
Biomark Med. 16:371–386. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lyu X, Li T, Zhu D, Cheng Y, Chen Y, He X,
Li Z, Li S, Wu W, Geng S, et al: Whole-genome sequencing as an
alternative to analyze copy number abnormalities in acute myeloid
leukemia and myelodysplastic syndrome. Leuk Lymphoma. 63:2301–2310.
2022. View Article : Google Scholar
|
|
12
|
Mareschal S, Palau A, Lindberg J, Ruminy
P, Nilsson C, Bengtzén S, Engvall M, Eriksson A, Neddermeyer A,
Marchand V, et al: Challenging conventional karyotyping by
next-generation karyotyping in 281 intensively treated patients
with AML. Blood Adv. 5:1003–1016. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cree IA: The WHO classification of
haematolymphoid tumours. Leukemia. 36:1701–1702. 2022. View Article : Google Scholar
|
|
14
|
Rausch C, Rothenberg-Thurley M, Dufour A,
Schneider S, Gittinger H, Sauerland C, Görlich D, Krug U, Berdel
WE, Woermann BJ, et al: Validation and refinement of the 2022
European LeukemiaNet genetic risk stratification of acute myeloid
leukemia. Leukemia. 37:1234–1244. 2023. View Article : Google Scholar
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jamieson A, Sobral de Barros J, Cochrane
DR, Douglas JM, Shankar S, Lynch BJ, Leung S, Martin S, Senz J, Lum
A, et al: Targeted and shallow whole-genome sequencing identifies
therapeutic opportunities in p53abn endometrial cancers. Clin
Cancer Res. 30:2461–2474. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McGowan-Jordan J, Hastings RJ and Moore S:
An International System for Human Cytogenomic Nomenclature (2020)
Reprint of 'Cytogenetic and Genome Research 2020, Vol. 160, No.
7–8S. Karger; Basel, Switzerland: pp. 37–106. 2020
|
|
18
|
Mrózek K: Molecular cytogenetics in acute
myeloid leukemia in adult patients: Practical implications. Pol
Arch Intern Med. 132:163002022.
|
|
19
|
Döhner H, Wei AH, Appelbaum FR, Craddock
C, DiNardo CD, Dombret H, Ebert BL, Fenaux P, Godley LA, Hasserjian
RP, et al: Diagnosis and management of AML in adults: 2022
recommendations from an international expert panel on behalf of the
ELN. Blood. 140:1345–1377. 2022. View Article : Google Scholar
|
|
20
|
Falini B: NPM1-mutated acute myeloid
leukemia: New pathogenetic and therapeutic insights and open
questions. Am J Hematol. 98:1452–1464. 2023. View Article : Google Scholar
|
|
21
|
Bhansali RS, Pratz KW and Lai C: Recent
advances in targeted therapies in acute myeloid leukemia. J Hematol
Oncol. 16:292023. View Article : Google Scholar
|
|
22
|
Poonsombudlert K, Yodsuwan R, Mott S,
Crawford K, Hornberg S, Snow AN, Sutamtewagul G,
Magalhaes-Silverman M and Dhakal P: Effect of NPM1 mutation subtype
and co-mutation patterns on the outcomes of acute myeloid leukemia.
Eur J Haematol. 115:29–35. 2025. View Article : Google Scholar
|
|
23
|
Ishikawa Y, Ushijima Y and Kiyoi H: Recent
advances in AML with mutated NPM1. Int J Hematol. 120:556–565.
2024. View Article : Google Scholar
|
|
24
|
Othman J, Potter N, Ivey A, Tazi Y,
Papaemmanuil E, Jovanovic J, Freeman SD, Gilkes A, Gale R,
Rapoz-D'Silva T, et al: Molecular, clinical, and therapeutic
determinants of outcome in NPM1-mutated AML. Blood. 144:714–728.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kishtagari A, Levine RL and Viny AD:
Driver mutations in acute myeloid leukemia. Curr Opin Hematol.
27:49–57. 2020. View Article : Google Scholar
|
|
26
|
Shahzad M, Amin MK, Daver NG, Shah MV,
Hiwase D, Arber DA, Kharfan-Dabaja MA and Badar T: What have we
learned about TP53-mutated acute myeloid leukemia? Blood Cancer J.
14:2022024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu X, Bryke C, Sukhanova M, Huxley E, Dash
DP, Dixon-Mciver A, Fang M, Griepp PT, Hodge JC, Iqbal A, et al:
Assessing copy number abnormalities and copy-neutral
loss-of-heterozygosity across the genome as best practice in
diagnostic evaluation of acute myeloid leukemia: An evidence-based
review from the cancer genomics consortium (CGC) myeloid neoplasms
working group. Cancer Genet. 228–229. 218–235. 2018.
|
|
28
|
Papaemmanuil E, Gerstung M, Bullinger L,
Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F,
Bolli N, et al: Genomic classification and prognosis in acute
myeloid leukemia. N Engl J Med. 374:2209–2221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Salman H: Comparative analysis of AML
classification systems: Evaluating the WHO, ICC, and ELN frameworks
and their distinctions. Cancers (Basel). 16:29152024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pitel BA, Sharma N, Zepeda-Mendoza C,
Smadbeck JB, Pearce KE, Cook JM, Vasmatzis G, Sachs Z,
Kanagal-Shamanna R, Viswanatha D, et al: Myeloid malignancies with
5q and 7q deletions are associated with extreme genomic complexity,
biallelic TP53 variants, and very poor prognosis. Blood Cancer J.
11:182021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Babakhanlou R, DiNardo C and Borthakur G:
IDH2 mutations in acute myeloid leukemia. Leuk Lymphoma.
64:1733–1741. 2023. View Article : Google Scholar
|