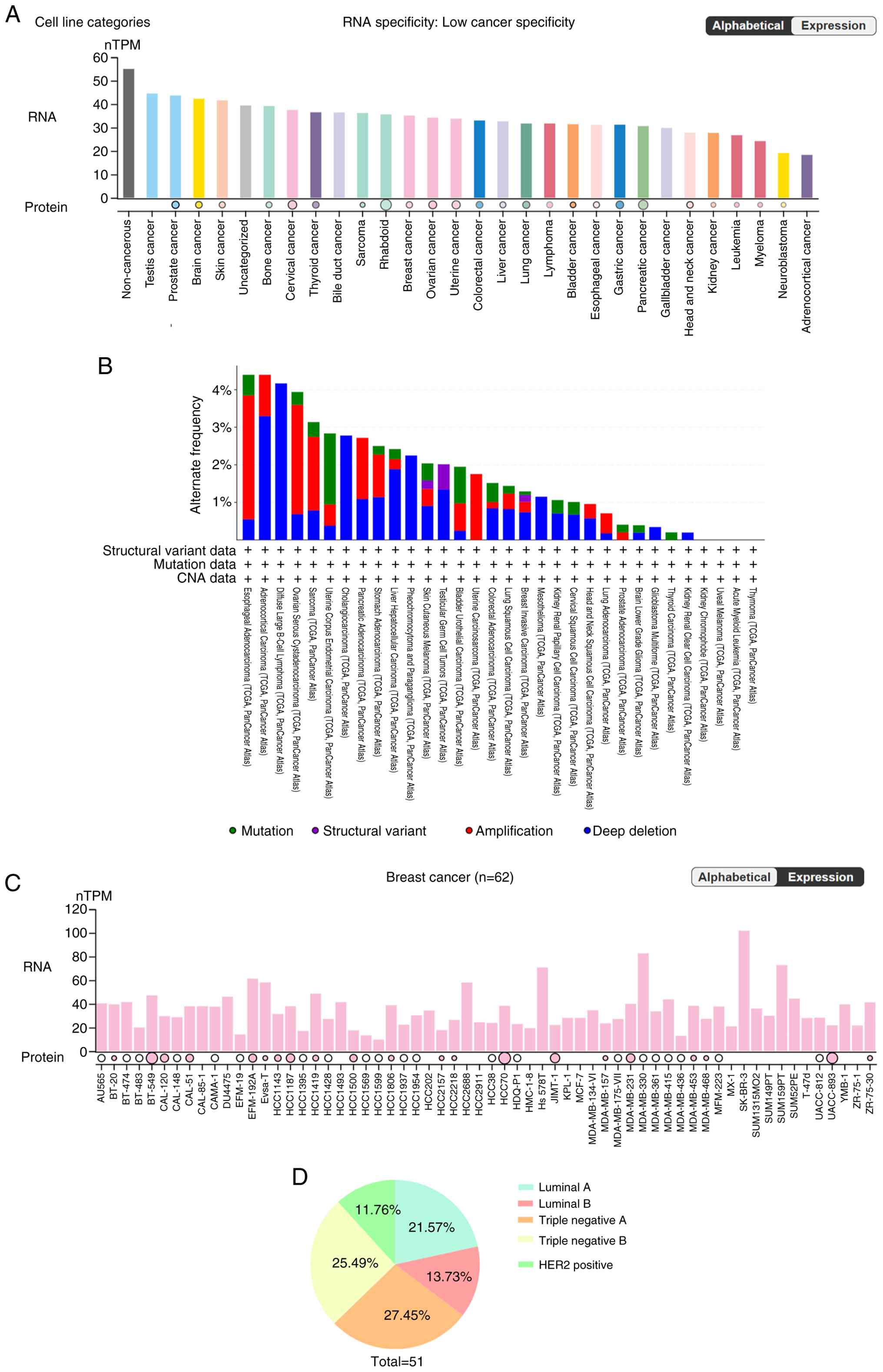
Introduction
Protein prenylation is an essential
post-translational modification that regulates the function of
specific proteins involved in eukaryotic cell biology, particularly
those implicated in tumor initiation and progression. This
modification primarily occurs on CAAX motifs and involves three key
steps: First, a 15-carbon (farnesyl) or 20-carbon (geranyl)
isoprenoid lipid is covalently attached to a cysteine residue by
protein farnesyltransferase (FTase) or protein
geranlygeranyltransferase I (GGTase I), for instance: The HRAS can
only be FTase catalytic, but KRAS and NRAS can be catalyzed by
FTase and GGTase I (1). Second,
the-AAX tripeptide is cleaved by Ras-converting enzyme 1 (RCE1), a
protease that catalyzes endoproteolytic hydrolysis (2,3).
Finally, isoprenylcysteine carboxymethyltransferase (ICMT)
catalyzes the methylation of the cysteine-terminal α-carboxylic
acid, completing the modification (4), as indicated in Fig. 1. The processing of CAAX proteins has
attracted substantial attention due to the pivotal role of mutant
Ras proteins in cancer development. Despite the potential of FTase
inhibitors in cancer therapy, their efficacy is limited by the
compensatory activation of GGTase I when FTase is inhibited,
highlighting the need for alternative therapeutic strategies
(5,6). ICMT, however, uniquely modifies CAAX
proteins, making it a more promising target. In RCE1-deficient
fibroblasts, Ras mislocalization and markedly reduced cell growth
are observed, and RCE1-deficient mice typically die in late
gestation or shortly after birth (7). By contrast, ICMT-deficient mice
exhibit more pronounced phenotypic defects, including severe growth
retardation during embryonic development and lethality by embryonic
day 10.5–11.5 (8). These findings
underscore the irreplaceable role of ICMT in cellular homeostasis.
As such, targeting ICMT may offer a novel approach for cancer
treatment by disrupting the modification and localization of CAAX
proteins, such as Ras, thus inhibiting tumor cell proliferation and
migration. This emerging strategy presents new avenues for
therapeutic intervention in cancer.
 | Figure 1.Three steps of CAAX protein
prenylation. After the RAS gene is transcribed and translated on
the ribosome, it is anchored to the endoplasmic reticulum membrane
for further modification. First, a 15-carbon (farnesyl) or
20-carbon (geranyl) isoprenoid lipid is covalently attached to a
cysteine residue by protein FTase or GGTase I. Subsequently, the
-AAX tripeptide is cleaved by RCE1, a protease that catalyzes
endoproteolytic hydrolysis. Finally, ICMT catalyzes the methylation
of the cysteine-terminal α-carboxylic acid, completing the
modification. FTase, farnesyltransferase; GGTase I,
geranlygeranyltransferase I; RCE1, Ras-converting enzyme 1; ICMT,
isoprenylcysteine carboxymethyltransferase. |
To systematically summarize the ICMT-related
findings, the PubMed (https://pubmed.ncbi.nlm.nih.gov/) and X-MOL
(https://www.x-mol.com/) databases were searched
without a lower date limit through December 2025, sorting results
‘most recent first’, and Boolean logic coupled with MeSH terms was
used to link ‘prenylcysteine carboxyl methyltransferase’ to breast,
pancreatic, gastric, hepatocellular and epithelial ovarian cancers
as well as non-tumor diseases, RAS family, Ras homolog gene family,
member A (RhoA), Rab GTPases, apoptosis, autophagy, cell cycle,
migration, invasion, breast-cancer stemness and DNA-damage repair,
retaining only full-length, data-complete original studies while
excluding abstracts, conference papers, letters, duplicates and
articles loosely connected to ICMT regulation and disease.
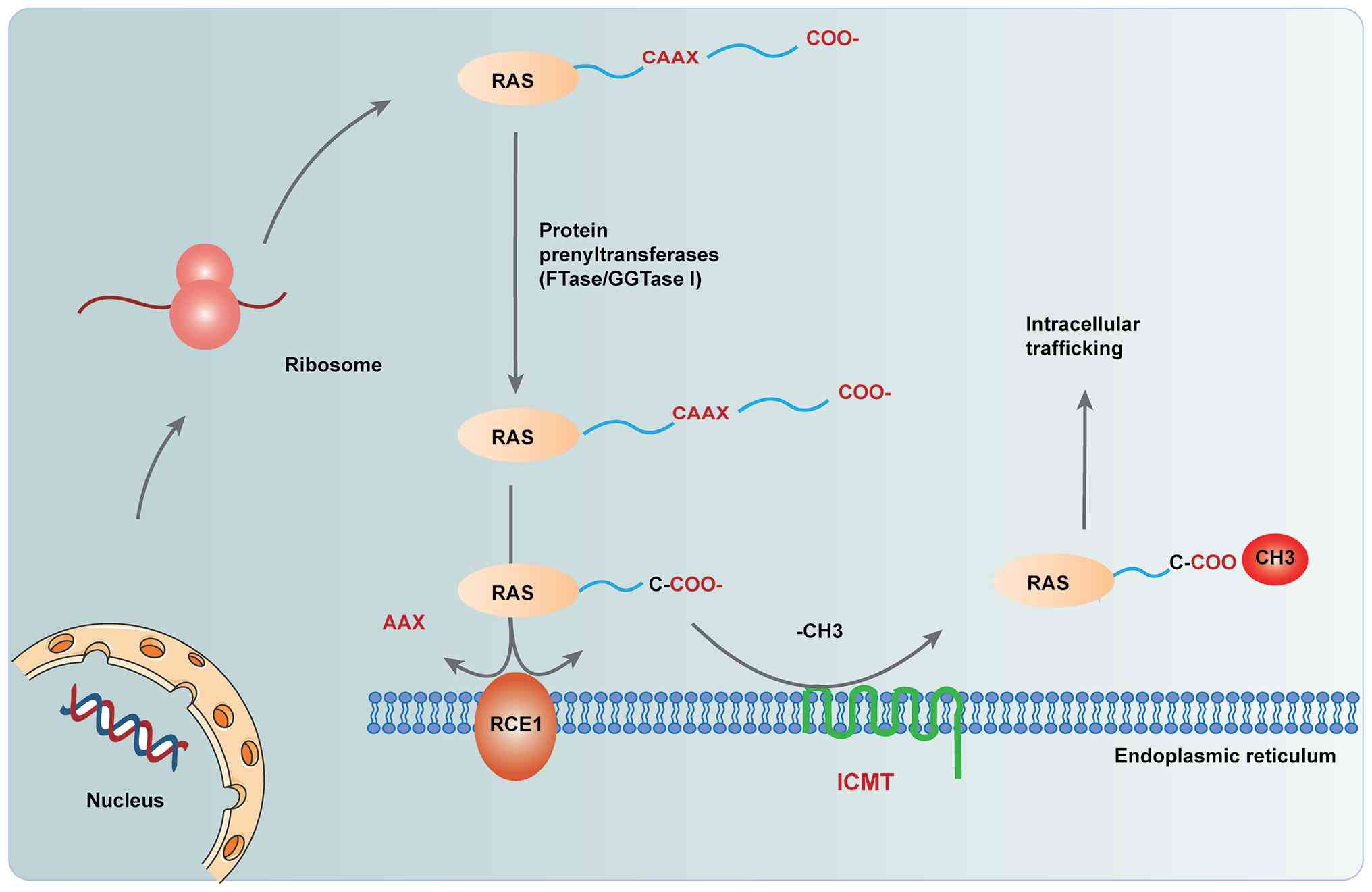
Fig. 2
systematically illustrates the research milestones of ICMT. The
journey began in 1978 with the discovery of the farnesyl-like
isoprene moiety in fungal rhodotorucine A, providing the first
evidence of cysteine-linked thioether bonding (9). By 1988, the confirmation of human RAS
protein farnesylation established a critical link between
post-translational modification and oncogenesis (10,11).
The 1990s saw the elucidation of the ‘CAAX’ modification
cascade-catalyzed by FTase/GGTase I, RCE1 and ICMT, positioning
ICMT as the final essential step (2,12–15).
Functional breakthroughs occurred in 2004 and 2010, demonstrating
that ICMT deficiency or inhibition suppresses K-Ras/B-Raf-mediated
transformation and triggers autophagy-dependent apoptosis (16,17).
More recent advances in 2015 and 2021 expanded its role to
mitochondrial metabolic reprogramming and DNA damage repair
(18,19). Recently, findings published in 2024
reveal that ICMT promotes invasive pseudopodia and metastasis,
underscoring its multifaceted regulatory roles in tumor progression
(20). Building upon these
evolutionary milestones, this review offers a holistic perspective
on the biochemical intricacies of ICMT-driven modifications and
their complex regulatory networks. We seek to delineate how recent
mechanistic breakthroughs can be leveraged to develop ICMT-targeted
interventions, thereby offering a forward-looking perspective on
its therapeutic viability in oncology.
 | Figure 2.Timeline of ICMT research. In 1978,
chemical analyses from a study on fungal mating factor
rhodotorucine A revealed a farnesyl-like isoprene linked to the Cys
residue of the peptide via a thioether bond (9); in 1988, human RAS proteins were found
to be indeed farnesylated and this modification was required for
oncogenic RAS-transformed cells (10,11);
in the 1990s, the three-step modification process of CAAX protein
was reported to be catalyzed by FTase or GGTase I, RCE1 and ICMT,
respectively (2,12–15);
in 2004, inactivation of ICMT was found to inhibit the
transformation of oncogenic K-Ras and B-Raf (16); in 2010, inhibition of ICMT was
indicated to induce autophagy-dependent apoptosis and impair tumor
growth (17); in 2015, ICMT was
reported to regulate mitochondrial respiration and cancer cell
metabolism (18); in 2021,
inhibition of ICMT was indicated to impair DNA damage repair
(19); in 2024, ICMT was found to
promote the formation of invasive pseudopods and metastasis of
cancer cells (20). FTase,
farnesyltransferase; GGTase I, geranlygeranyltransferase I; RCE1,
Ras-converting enzyme 1; ICMT, isoprenylcysteine
carboxymethyltransferase. |
Structure and function of ICMT
The direct homologue of ICMT in Saccharomyces
cerevisiae, STE14p, was the first enzyme of this class to be
cloned and sequenced, establishing it as a founding member of the
eukaryotic protein methyltransferase family (21). Subsequently, a human homologue of
STE14 was identified, sharing significant sequence similarity with
its yeast counterpart (21). The
human and mouse genes were subsequently designated ICMT2 and ICMT,
respectively. ICMT is composed of eight transmembrane α-helices
(M1-M8), predominantly located within the endoplasmic reticulum
membrane. Notably, the M1-M3 region is unique to animal ICMT, where
it interacts closely with M4 and the M4-M5 junction (15,21).
These regions are stabilized within the transmembrane domain
through interactions between M1, M2 and M3, as well as between the
M6-M7 and M5-M6 junctions. The largest cytoplasmic region of ICMT
contains the AdoHcy binding site, which is defined by an extension
of M8 and the M6-M7 linkage (22).
Genetic deletion of M1 and M2 leads to inactivation of human ICMT
(23).
Protein isoprenylation is a critical
post-translational modification that enables proteins bearing-CAAX
or -CXC motifs to acquire their full biological activity after
transcription and translation (24). ICMT serves as the terminal enzyme in
this process, catalyzing the methylation of isoprenylated proteins,
with major substrates including Ras proteins, various Rho GTPases
and the γ-subunit of G proteins (1). In summary, ICMT, as a key
isoprenyl-cysteine carboxymethyltransferase, plays a vital role
within the eukaryotic protein methyltransferase family. Its unique
transmembrane structure and functional domains facilitate its
stable residence in the endoplasmic reticulum membrane, where it
catalyzes the isoprenylation modification of numerous critical
proteins, thereby influencing their biological functions. In this
review, the comprehensive role of ICMT in regulating different
diseases and their progression will be discussed, particularly for
cancer.
ICMT in cancer therapy
Mutations in Ras genes are among the most common
genetic alterations in human cancers, occurring in ~30% of cancer
cases (25). Protein isoprenylation
is a crucial post-translational modification that allows proteins
with -CAAX or -CXC motifs to achieve their full biological
function. ICMT catalyzes the final step in this process,
methylating isoprenylated proteins. However, current targeted
therapies aimed at disrupting the isoprenylation process face
significant challenges.
Although inhibitors of farnesyltransferase can block
the isoprenylation of certain Ras proteins (e.g., KRAS and NRAS),
these proteins can still be modified through the alternative
geranylgeranylation pathway, resulting in drug resistance (6). While concurrent inhibition of FTase
and GGTase I has been shown to block the isoprenylation of
oncogenic Ki-Ras4B in animal models, the dose required to achieve
effective anti-tumor efficacy causes rapid animal mortality
(26). Furthermore, inhibition of
RCE1 in the second step of isoprenylation has shown variable
inhibitory effects in different tissues (27,28).
This variability can lead to severe side effects, including
disruption of normal visual and cardiac cell functions, and may
even promote the development of acute myeloid leukemia (1,29).
Given these limitations, targeting the methylation
step catalyzed by ICMT-the final step in the isoprenylation
pathway-may represent a more effective and specific approach for
cancer therapy. By focusing on the methylation of isoprenylated
proteins, such as Ras, it may be possible to reduce resistance and
minimize off-target effects, offering a promising therapeutic
strategy (30).
The expression of ICMT in various cancers has
garnered significant attention due to the broad spectrum of ICMT
substrates. However, significantly higher expression levels were
observed in testicular and prostate cancers, while the lowest
expression was noted in adrenal cancer. This pattern suggests that
while ICMT impacts a wide range of cancer cells, its expression
varies considerably across different cancers (20,31).
Notably, the variability in ICMT expression within the same cancer
type implies that its function may not be determined solely by its
abundance in the cell. Instead, it is likely influenced by the role
of ICMT's substrates and their ability to regulate relevant
cellular processes, as indicated in Fig. 3. The following content will mainly
focus on ICMT-regulated cancer development, according to different
cancer types, especially pancreatic cancer and breast cancer. Also,
some critical substrates of ICMT will be discussed in this
review.
Role of ICMT in pancreatic cancer
ICMT regulates the initiation of
pancreatic cancer
Human pancreatic ductal adenocarcinoma (PDA) is
characterized by oncogenic KRAS mutations, which occur in >90%
of PDAC cases (32). Activating
mutations in the KRAS proto-oncogene have been identified in
patients with pancreatic intraepithelial neoplasia (PanIN),
indicating the significant role of KRAS mutations in the
pathogenesis of PDA (33). Court
et al (34) developed a
Pdx1-Cre, LSL-KrasG12D mouse model with ICMT knockout at the
embryonic stage and demonstrated that the progression from PanIN to
PDA was significantly accelerated in ICMT-deficient pancreata.
Similarly, in a Notch1-deficient mouse model, the effects of Notch1
and ICMT deficiency were comparable, suggesting that both may
influence pancreatic cancer development through a shared pathway,
potentially due to the dependence of the Notch1 signaling pathway
on ICMT activity. By contrast, in a mouse model without
Kras-induced carcinogenesis, ICMT deficiency alone did not result
in tumor formation, indicating that ICMT deficiency alone is
insufficient to drive pancreatic tumorigenesis (34). On the other hand, in a pancreatic
cancer cell model, the ICMT inhibitor compound 8.12 not only
induced cell cycle arrest and inhibited anchorage-independent
growth in HepG2 and PC3 cells, but also promoted autophagy by
dose-dependently increasing light chain 3 (LC3)-II levels in both
cell types (35). In summary, while
ICMT inhibition accelerates tumor progression in animal models, it
shows promising therapeutic effects in cellular models,
highlighting the differential impact of ICMT inhibition depending
on the model used.
ICMT regulates apoptosis and autophagy
in pancreatic cancer
Autophagy is a highly conserved intracellular
degradation process that maintains cellular homeostasis by
degrading damaged, misfolded or aged proteins and organelles via
the lysosomal system (36–38). While apoptosis, a programmed cell
death process, is activated by death signals and can be mediated
through extrinsic receptor-dependent pathways or intrinsic
mitochondrial pathways (39,40).
Mitochondrial respiration plays a pivotal role in tumor cell
survival (41). Since Warburg et
al (42) first established the
link between mitochondria and cancer via the Warburg effect in the
1920s, subsequent investigations have consistently demonstrated
that targeted inhibition of mitochondrial respiration can potently
suppress tumor progression and reverse therapeutic resistance
(43,44). ICMT was also recently reported to
play a critical role in regulating cell survival and death through
its influence on apoptosis and autophagy. In sensitive pancreatic
cancer cells, inhibition of ICMT led to mitochondrial respiratory
defects and cellular energy depletion, resulting in significant
upregulation of p21. However, cells resistant to ICMT inhibition
did not show mitochondrial dysfunction or alterations in p21
signaling, and instead, apoptosis was induced through p21-activated
BCL2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3)
expression, rather than autophagy (45). Treatment of HepG2 cells with an ICMT
inhibitor caused a marked increase in LC3-II protein levels, with
LC3-II translocation to autophagosomes, which is a widely used
indicator of autophagy induction. These findings suggest that ICMT
inhibition triggers autophagy in pancreatic cancer cells by
increasing LC3-II levels (17).
Thus, inhibition of ICMT appears to have a negative effect on tumor
cell survival, inducing autophagy and potentially contributing to
therapeutic efficacy.
ICMT regulates the cell cycle in
pancreatic cancer
The cell cycle is a tightly regulated process
through which cells divide, ensuring accurate replication and
distribution of genetic material. Dysregulation of the cell cycle
can lead to uncontrolled cell proliferation, a hallmark of cancer
(46,47). ICMT directly influences cell cycle
progression by regulating the expression and activity of cell
cycle-related proteins (45).
Inhibition of ICMT in pancreatic cancer cells led to cell cycle
arrest and apoptosis via the p21 and p21-regulated BNIP3 pathway
(45). Further experiments showed
that treatment of HepG2 cells with ICMT inhibitors resulted in a
significant increase in the proportion of cells in the G1 phase,
along with enhanced cell death, especially with prolonged
treatment. Notably, the activity of caspase 3 was significantly
increased in the cell lysates, and there was a marked elevation in
p27 protein levels (17). These
results collectively confirm that ICMT inhibition effectively
induces cell cycle arrest and promotes apoptosis in HepG2 cells.
Therefore, in pancreatic cancer cells, ICMT inhibition not only
induces cell cycle arrest but also triggers apoptosis through
multiple pathways, providing a solid foundation for understanding
the role of ICMT in cell cycle regulation and highlighting it as a
potential therapeutic target for pancreatic cancer.
Role of ICMT in breast cancer
ICMT regulates breast cancer
tumorigenesis
ICMT plays a crucial regulatory role in the
initiation and transformation of breast cancer. Lau et al
(48) investigated the effect of
ICMT on KRAS mutant (G12V) cells by constructing an HME1-shp53 cell
model. Their results showed that deletion of ICMT inhibited colony
formation, while restoration of ICMT expression re-established this
ability, indicating that ICMT is critical for KRAS-driven cancer
progression (48). In
KRAS-transfected fibroblasts, the absence of carboxymethylation led
to rapid turnover of RhoA and upregulation of the cyclin-dependent
kinase inhibitor p21Cip1. Of note, deletion of the p21Cip1 gene
abolished the effects of ICMT inactivation on cell growth and
KRAS-induced soft agar colony formation, suggesting that ICMT
influences cell growth and KRAS transformation through interaction
with p21Cip1 (16). In summary,
ICMT is essential for the initiation and maintenance of KRAS-driven
cancers, and its loss significantly impairs cell transformation and
colony formation.
ICMT regulates autophagy and apoptosis
in breast cancer
Studies have reported that inhibiting ICMT in breast
cancer can induce autophagy and apoptosis, like the phenotypes
observed in pancreatic cancer. Autophagy and apoptosis often work
synergistically to induce cancer cell death, particularly during
chemotherapy or radiotherapy in breast cancers, and typically occur
in cells with intact apoptotic pathways (49,50).
Inhibition of ICMT impairs the function of mitochondrial complexes
I, II and III in cancer cells, leading to decreased ATP production
and depletion of tricarboxylic acid cycle metabolites. This
disruption inhibits cell growth and metabolism, triggering a
significant autophagic response (18). Treatment with the ICMT inhibitor
Cysmethynil led to a significant increase in intracellular LC3-II
levels and the formation of autophagic lysosomes, indicating that
ICMT inhibition induces autophagic cell death (51). These findings suggest that ICMT
affects both autophagy and apoptosis, underlining its potential as
a therapeutic target for cancer treatment by modulating
mitochondrial function and cellular metabolism.
ICMT regulates breast cancer migration
and invasion
Cancer metastasis involves several steps: In
situ tumor growth, angiogenesis, epithelial-mesenchymal
transition (EMT), invasion, endocytosis, circulation, exocytosis,
dormancy and metastatic growth (52). In the highly metastatic human breast
cancer cell line MDA-MB-231, inhibition of ICMT significantly
reduced cell adhesion and spreading ability. Further experiments
showed that ICMT inhibition also impaired cell migration,
suggesting that ICMT plays a key role in promoting the migratory
and invasive abilities of breast cancer cells (53). Integrin β3 is strongly associated
with invasiveness and cancer lethality in various tissues (54,55).
In the breast cancer cell line MCF-7, forced expression of integrin
β3 led to a more polarized cellular morphology, increased levels of
phosphorylated focal adhesion kinase and enhanced actin stress
fiber formation. However, these effects were completely reversed
upon ICMT inhibition, indicating that the function of integrin β3
in promoting cancer cell polarity and migration is dependent on
ICMT activity (56). These findings
underscore the importance of ICMT in regulating the migration and
invasion of breast cancer cells, highlighting it as a potential
therapeutic target in breast cancer metastasis.
ICMT regulates breast cancer stem
cells (CSCs)
CSCs are a subpopulation of tumor cells with stem
cell-like properties. They have a significantly higher
proliferative capacity compared to other cancer cells within the
same tumor and play an essential role in tumor initiation,
progression and recurrence (57–59).
CSCs are characterized by high tumorigenicity, drug resistance,
unlimited self-renewal capacity and dysregulated cell cycles,
making them a major contributor to cancer lethality (60). CSCs are prevalent in all types of
cancer, with metastatic breast cancer being particularly enriched
in CSCs (58). In the developing
organism, activation of the Yes-associated protein
(YAP)/transcriptional co-activator with PDZ-binding motif (TAZ)
pathway imparts stem cell-like properties to cells, triggering
uncontrolled proliferation and tumor formation (61,62).
Positive interactions between KRAS and YAP/TAZ have been observed
in several cancer types (63,64).
For instance, inhibition of ICMT reduced the self-renewal capacity
of KRAS-driven breast cancer cells, with silencing ICMT leading to
a significant reduction in TAZ protein levels and loss of
self-renewal potential. This effect could be reversed by
overexpressing mutant KRAS, demonstrating that ICMT modifies KRAS
regulation of TAZ stability and function (65). These findings highlight the crucial
role of ICMT in regulating the stemness of cancer cells and its
interaction with KRAS and YAP/TAZ, providing a new therapeutic
perspective for targeting CSCs.
ICMT regulates the cell cycle in
breast cancer
ICMT has been reported to regulate cell cycle
progression in various cancer cell types, including breast cancer.
Treatment of cancer cells with ICMT inhibitors has been shown to
increase the proportion of cells in G1 phase, with elevated p27
levels and reduced cyclin D1 levels, suggesting cell cycle arrest
at G1 (51). Further investigation
revealed that loss of ICMT function results in cell cycle arrest at
the G2/M phase, as evidenced by increased levels of phosphorylated
CDC2 (Thr15) and cyclin B1, confirming that ICMT-deficient cells
are arrested at G2/M (19).
Additionally, ICMT deletion in cancer cells decreased cyclin D1
levels and increased p27 levels, leading to G0/G1 phase arrest
(66). These results suggest that
ICMT regulates cell cycle progression, and its loss leads to
abnormal cell cycle arrest, which could be exploited for
therapeutic purposes in breast cancer.
ICMT regulates DNA damage repair in
breast cancer
DNA integrity is essential for normal cellular
function and stability, and DNA damage, whether caused by
environmental factors or endogenous toxic agents such as free
radicals, can compromise genome stability and contribute to cancer
development (67,68). DNA repair mechanisms are complex
processes that ensure the repair of DNA damage to maintain genomic
stability. These mechanisms include base excision repair,
nucleotide excision repair, mismatch repair and double-strand break
repair (DSBR), with DSBR further subdivided into non-homologous end
joining and homologous recombination (69,70).
In MDA-MB-231 breast cancer cells, deletion of ICMT resulted in
massive apoptosis and a significant increase in p-γH2AX and cleaved
caspase 7 levels, indicating that ICMT deletion induced DNA damage
and cell death. p-γH2AX persisted in ICMT-deficient cells following
DNA damage, whereas it rapidly decreased in control cells, further
confirming that ICMT loss impairs DNA damage repair (19). These findings provide new insights
into the role of ICMT in DNA repair and its critical function in
maintaining genomic stability in breast cancer cells.
Regulation of ICMT in other cancers
Gastric cancer
ICMT is not only strongly associated with breast
cancer but also plays a role in various other cancers. Although the
incidence and mortality rates of gastric cancer have declined over
the past 50 years, it remains the fifth most diagnosed cancer
worldwide and the third leading cause of cancer-related deaths
globally (71,72). In 2022, Ma et al (73) demonstrated that licoricidin
downregulated ICMT expression and inhibited gastric cancer growth.
Notably, active Ras GTP levels were significantly reduced in tumors
treated with glycyrrhizin in a dose-dependent manner compared to
controls. Furthermore, the inactivation of Ras effectively blocked
Raf and Erk phosphorylation in the glycyrrhizin-treated group,
preventing the activation of downstream signaling pathways
essential for cancer progression. These results highlight the
potential anticancer activity of glycyrrhizin through the ICMT/Ras
pathway, suggesting that ICMT inhibition may be a valuable
therapeutic strategy for gastric cancer treatment (73). Thus, by inhibiting ICMT,
glycyrrhizin shows promising synergistic effects in gastric cancer
therapy.
Hepatocellular carcinoma (HCC)
HCC is the fourth most common cancer globally and
the third leading cause of cancer-related deaths in China (74). In 2019, Xu et al (75) reported that ICMT protein levels were
significantly higher in ~70% of HCC tissues and cell lines compared
to their normal counterparts. ICMT was shown to suppress the growth
and migration of Adriamycin-resistant HCC cells while inducing
apoptosis. Conversely, overexpression of ICMT promoted the growth
and migration of normal hepatocytes. Inhibition of ICMT suppressed
Ras/Raf/Mek/Erk signaling and EMT in HCC cells, suggesting that
ICMT plays an essential role in HCC biology (75). Therefore, targeting ICMT may help
suppress the development and progression of HCC.
Epithelial ovarian cancer
Epithelial ovarian cancer is the leading cause of
gynecological cancer-related deaths among women worldwide (76). In 2018, Liu et al (77) found that upregulation of ICMT
expression in malignant ovarian tissues compared to normal ovarian
tissues is a common phenomenon in ovarian epithelial cancer.
Additionally, SKOV3 and TOV-112D cells, which exhibit lower ICMT
expression, were significantly more sensitive to paclitaxel and
cisplatin treatment than PA1, Caov3, and SW626 cells, which express
higher levels of ICMT (77). These
findings suggest that ICMT contributes to the efficacy of
chemotherapy drugs, enhancing the therapeutic effects of standard
treatments.
Molecular mechanisms of ICMT regulation in
cancers
ICMT is a key protein methyltransferase that
modulates numerous substrates through diverse regulatory
mechanisms. The following discusses the major molecular mechanisms
by which ICMT exerts its effects, particularly focusing on Ras and
its regulation in cancer. In this chapter, the major pathways of
mutant Ras and related RAS inhibitors are discussed, and they are
also presented in Fig. 4 and
Table I.
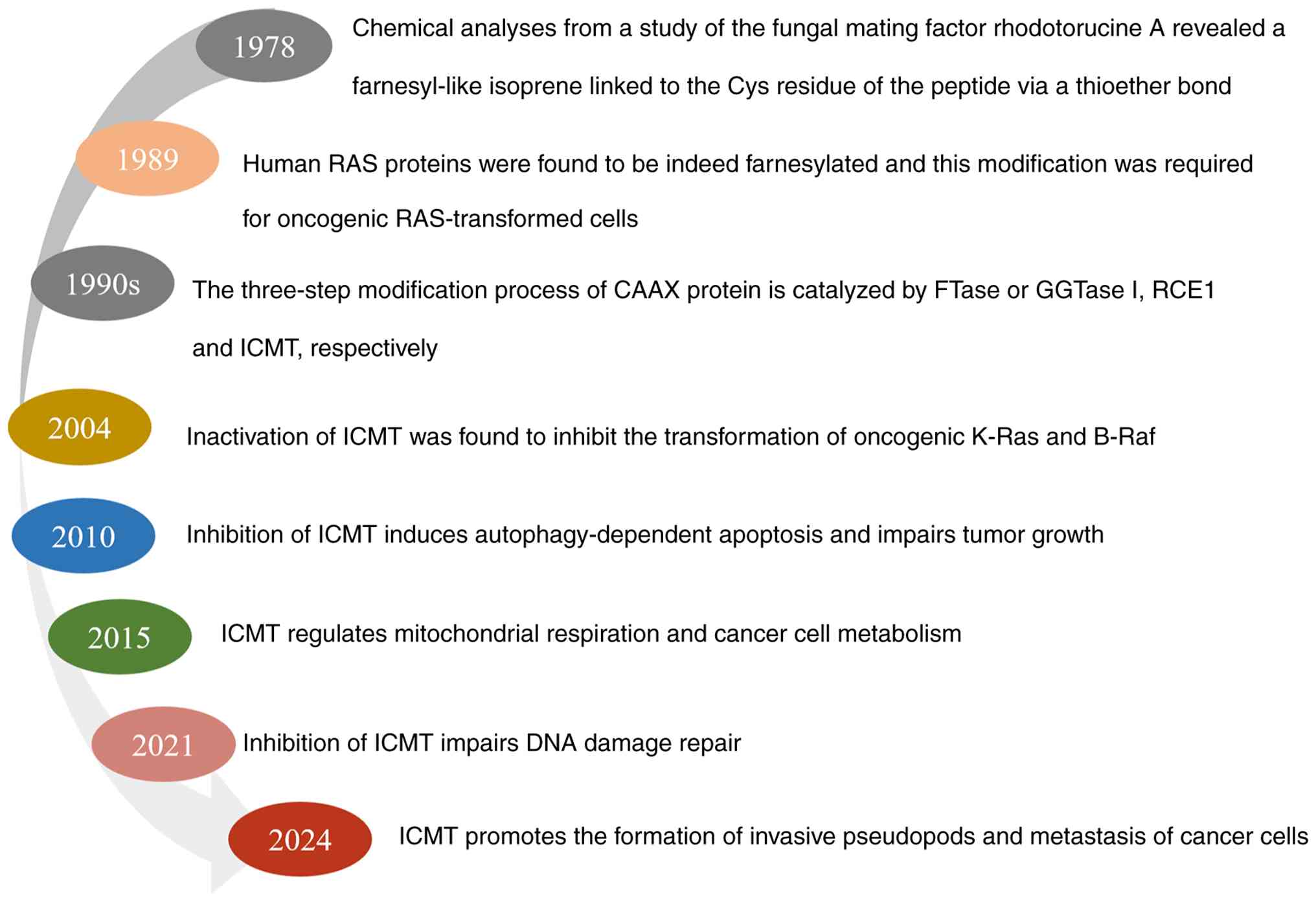
 | Figure 4.Summary of critical inhibitors
targeting Ras according to its molecular mechanism. Ras serves as a
crucial signaling node that coordinates multiple cellular processes
through three major effector pathways. The RAS-MAPK pathway,
activated by receptor tyrosine kinases (RTKs), transduces signals
through the RAF-MEK-ERK cascade to regulate cell proliferation and
differentiation. Simultaneously, the PI3K-AKT pathway promotes cell
survival and growth by converting PIP2 to PIP3 through PI3K, which
subsequently activates AKT to inhibit apoptotic signals.
Additionally, the RALGDS pathway modulates cytoskeletal dynamics
and vesicular trafficking, thereby influencing cell migration and
secretory functions. RTKs, receptor tyrosine kinases; MAPK,
mitogen-activated protein kinase; RAF, rapidly accelerated
fibrosarcoma kinase; MEK, MAPK kinase; ERK, extracellular
signal-regulated kinase; PI3K, phosphatidylinositol 3-kinase; PIP2,
phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol
3,4,5-trisphosphate; RALGDS, ral guanine nucleotide dissociation
stimulator. |
 | Table I.Summary of classification, mechanism
of action and study phase of inhibitors targeting the Ras signaling
pathway. |
Table I.
Summary of classification, mechanism
of action and study phase of inhibitors targeting the Ras signaling
pathway.
| Ras-related
pathway | Inhibitor
class | Drug name | Mechanism of
action | Study phase |
|---|
| Ras membrane
localization | Inhibitors of
membrane association | GGTI-298 | Disruption of Ras
membrane localization | Preclinical |
|
|
| GGTI-2154 | Disruption of Ras
membrane localization | Preclinical |
|
|
| UCM-1336 | Disruption of Ras
membrane localization | Preclinical |
| Direct Ras | Direct | ARS-853 | Targeting of mutant
Ras proteins | Preclinical |
| inhibition | inhibitors | AMG510 | Targeting of mutant
Ras proteins | Preclinical |
|
|
| MRTX849 | Targeting of mutant
Ras proteins | Preclinical |
|
|
| SCH-54292 | Targeting of mutant
Ras proteins | Preclinical |
|
|
| RMC6236 | Targeting of mutant
Ras proteins | Preclinical |
| RAS-MAPK effector
pathway | Inhibitors of
effector signaling-MEK inhibitor | GDC-0623 | Inhibiting of MEK
in the RAF-MEK-ERK cascade | Preclinical |
|
| Inhibitors of
effector signaling-Raf inhibitor | Dabrafenib | Inhibiting of Raf
in the RAF-MEK-ERK cascade | Clinical |
| PI3K-AKT effector
pathway | Inhibitors of
effector | INCB 0504655 | Inhibiting of PI3K
to block PIP2 to PIP3 conversion | Clinical |
|
| signaling-PI3K
inhibitor | GDC 0084 | Inhibiting of PI3K
to block PIP2 to PIP3 conversion | Clinical |
| RALGDS effector
pathway | Inhibitors of
effector | RBC8 | Inhibiting of Ral
in the RALGDS pathway | Preclinical |
|
| Signaling-Ral
inhibitor |
|
|
|
| Metabolic
intervention | Inhibitors of
metabolism | GLS | Targeting of
metabolic vulnerabilities | Preclinical |
|
|
| Phenformin | Exploitation of
metabolic vulnerabilities (biguanide class) | Preclinical |
|
|
| Metformin | Exploitation of
metabolic vulnerabilities (biguanide class) | Clinical |
| Synthetic
lethal | Inhibitors of | Trametinib | Targeting of
synthetic lethal interactions | Clinical |
| interactions | synthetic lethal
interactors | PD032590 | Targeting of
synthetic lethal interactions | Clinical |
| Autophagy
modulation | Inhibitors of
autophagy | Hydroxychloro
quine | Modulation of
autophagy | Clinical |
|
|
| Flunarizine | Modulation of
autophagy | Clinical |
Ras
Ras proteins are small GTP-binding proteins that
belong to the Ras superfamily. Active Ras proteins have significant
roles in cell growth, differentiation, cytoskeletal dynamics,
protein transport and secretion (78). They are localized to the plasma
membrane and regulate various cellular functions (79). Ras proteins undergo
post-translational modifications at their C-terminal CAAX motifs,
including farnesylation, proteolytic cleavage and
carboxymethylation, which facilitate their membrane affinity
(31). Ras proteins are crucial
components of the MAPK signaling pathway; when activated, Ras
transduces signals via RAF kinase, MEK kinase and MAPK (e.g., ERK),
regulating cell proliferation (80). ICMT inhibition prevents Ras
translocation to the plasma membrane, thereby inhibiting Ras
activity, which has therapeutic potential for Ras-driven
tumorigenesis (81). ICMT regulates
the interleukin-1 receptor-associated kinase
(IRAK)-mitogen-activated protein kinase kinase
(MAPKK)-MAPK-activator protein 1 (AP-1) pathway involved in
inflammation in a Ras-dependent manner (82). Furthermore, ICMT interacts with
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4),
influencing the RAS/AKT pathway and cell migration (83). An ICMT inhibitor, UCM-1336, induces
Ras mislocalization, reduces GTP-loaded Ras activity, inhibits the
PI3K/AKT and MEK/ERK pathways, and promotes autophagy and apoptosis
(84). In glioblastoma cells, ICMT
inhibition reduced Ras-GTP levels and decreased the phosphorylation
of Raf, ERK, p90RSK, Akt and mTOR, suggesting that the Ras/Raf/Mek
and Ras/PI3K/Akt/mTOR pathways are inhibited by ICMT inhibition
(85). These findings suggest that
ICMT drives tumor development by interacting with Ras, and its
inhibition disrupts Ras localization and downstream signaling,
offering therapeutic implications for Ras-associated cancers.
Rho
The Rho subfamily, a member of the Ras superfamily,
plays a critical role in regulating cell signaling pathways
involved in cell proliferation, differentiation, migration and
morphology (86). Rho proteins are
processed through the prenylation modification, and their activity
impacts various cellular processes. A previous study by Bergo et
al (16) showed that ICMT
inactivation inhibited cell growth and K-Ras-induced oncogenic
transformation in both soft agar assays and nude mouse models. This
was accompanied by a significant increase in the Ras/Erk1/2
dependence of p21(Cip1), possibly due to reduced levels of RhoA.
Papaharalambus et al (87)
discovered that ICMT inhibition affects the interaction between
Rac1 and its inhibitor, Rho guanine nucleotide dissociation
inhibitor (RhoGDI). In ICMT-deficient cells, lower levels of Rho
proteins were observed (88). ICMT
inhibition in MDA-MB-231 cells significantly impaired thrombin- and
EGF-induced activation of Rho and Rac, and increased RhoGDI binding
to RhoA and Rac1. Overexpression of RhoGTPases partially rescued
the migratory impairment caused by ICMT inhibition, likely due to
the involvement of other Rho proteins such as cell division cycle
42 (CDC42) (53). These findings
indicate that ICMT inhibition disrupts RhoA and Rac activation,
impairing cell morphology, adhesion and migration, and that these
effects can be partially rescued by exogenous expression of RhoA or
Rac1. ICMT, therefore, plays a crucial role in RhoA and Rac
activation, affecting cellular processes critical to cancer
metastasis.
Rab
Rab GTPases represent the largest subfamily of small
GTPases within the Ras family, with nearly 70 members (89). Activated Rab proteins regulate the
trafficking of specific proteins to cellular compartments and
organelles, playing a central role in membrane trafficking. ICMT
deficiency causes mislocalization of GFP-RAB7 and GFP-RAB8 from the
inner membrane to the cytoplasmic lysate, enhancing their binding
to RABGDI and reducing their GTP-bound state. This results in
defective Notch1 processing and mislocalization (90). RAB4A, involved in integrin β3
recycling, is crucial for cell migration in response to
pro-migratory stimuli like platelet-derived growth factor (PDGF)
(91,92). Knockdown of ICMT in MDA-MB-231 and
HT-1080 cells reduced the basal and PDGF-stimulated localization of
integrin β3 and RAB4 at the cell membrane, suggesting that ICMT
regulates the function of RAB4A in integrin β3 recycling and cell
migration (56). In conclusion,
ICMT affects the localization and activity of Rab proteins,
influencing the Notch signaling pathway and cell migration, with
significant implications for cancer metastasis.
ICMT's regulation in other diseases
In addition to its significant role in cancer, ICMT
has been implicated in a range of other diseases. For example, ICMT
inhibitors such as cysmethynil and
3-methoxy-N-[2-2,2,6,6-tetramethyl-4-phenyltetrahydropyran-4-yl)
ethyl] aniline (MTPA) have been shown to attenuate the
lipopolysaccharide-induced ICMT/Ras/AP-1 signaling pathway, thereby
inhibiting inflammatory responses (93). Marín-Ramos et al (84) reported a potent ICMT inhibitor,
compound 3, which significantly impaired membrane binding of four
Ras isoforms, leading to reduced Ras activity and inhibition of
downstream Ras signaling pathways, ultimately improving survival in
an in vivo model of acute myeloid leukemia. Similarly,
compound C75, another potent ICMT inhibitor, delayed the
progression of acute myeloid leukemia, blocked the methylation of
progerin, disrupted its interaction with AKT and enhanced AKT
signaling (94). In
Hutchinson-Gilford progeria syndrome (HGPS) cells and
Zmpste24-deficient mouse fibroblasts, compound C75 stimulated
growth and development by targeting progerin mislocalization and
activating AKT signaling (95).
Furthermore, ICMT inhibitor UCM-13207 significantly improved the
proliferative capacity of HGPS fibroblasts, reduced progerin
localization to the nuclear membrane, decreased DNA damage,
increased cell viability, and notably improved body weight and
lifespan while reducing tissue senescence in mice (96). In diabetes, knockdown of ICMT
attenuated glucose-induced insulin secretion and Rac1 activation,
as well as reactive oxygen species (ROS) production, suggesting
that ICMT regulates glucose-induced insulin secretion, Rac1
activation and ROS production through its methylation of Rac1
(97). These findings highlight the
broad therapeutic potential of ICMT inhibition, not only in cancer
but also in a variety of other diseases.
Discussion
Enzymes that post-translationally modify the CAAX
motif of Ras proteins have long been considered promising targets
for anti-Ras drug discovery. After several preclinical studies
demonstrated that ICMT is essential for KRAS-mediated cellular
transformation and in vivo tumorigenesis (16,81),
and with evidence suggesting that pharmacological inhibition of
ICMT blocks Ras-dependent transformation (30,94),
ICMT has emerged as a valuable drug target. For example, in
pancreatic cancer, ICMT inhibition induces mitochondrial
dysfunction, leading to cell death (45). Additionally, ICMT deficiency results
in cell-cycle arrest, further driving cell death (17). However, in the Pdx1-Cre,
LSL-KrasG12D mouse model of pancreatic cancer, ICMT knockout
unexpectedly accelerated tumor progression. This may be due to the
involvement of multiple ICMT substrates in the pancreas, with
differential expression of these substrates potentially explaining
why ICMT may act as a tumor suppressor in certain tissues (e.g.,
pancreas) but not others (e.g., bone marrow). Accumulating evidence
indicates that depletion of Ras family members in mice leads to
profound developmental abnormalities, frequently resulting in
embryonic or neonatal lethality. These phenotypes include pulmonary
immaturity, as well as cardiac and placental defects (98–100).
Within the Rho GTPase family, CDC42 is essential for meiosis and
the establishment of cell polarity, whereas Rac1 is required for
gastrulation and epiblast survival; loss of either gene results in
early embryonic lethality (101–103). Similarly, Rab family GTPases play
indispensable roles during development. Genetic ablation of Rab
proteins in mice commonly causes early embryonic lethality-for
example, Rab1a deletion induces developmental arrest. Notably,
distinct subtype-specific effects have been observed among Rab5
isoforms: Homozygous deletion of Rab5a or Rab5b is compatible with
viability, whereas loss of Rab5c is lethal, and Rab5c heterozygous
mice exhibit hematopoietic and metabolic abnormalities (104,105). Furthermore, the acceleration of
PanINs in the absence of ICMT could be due to enhanced stromal
responses, including increased inflammatory cell infiltration and
fibrosis in ICMT-deficient pancreata expressing KRASG12D (81). Collectively, these essential
developmental functions underscore that ICMT-catalyzed prenylation
is indispensable for the survival and homeostasis of multiple
tissues; consequently, its early global loss in the Pdx1-Cre;
LSL-KrasG12D model is likely to unleash context-specific stress and
compensatory hyperplasia that accelerate PanIN
formation-highlighting the need to weigh developmental toxicity
against tumor-suppressive potential when timing ICMT inhibition in
pancreatic cancer therapy. Besides, the roles of ICMT substrates
other than Ras should be carefully considered in future therapeutic
strategies for pancreatic cancer. Furthermore, the bulk of current
work relies on fully transformed, established tumor lines,
indirectly suggesting that ICMT is actionable only as a therapeutic
target, not as a candidate for cancer prevention. In conclusion,
ICMT has considerable potential for the development of novel
therapeutic approaches for pancreatic cancer, and targeting ICMT or
its substrates offers promising avenues for treatment.
The research on ICMT extends beyond pancreatic
cancer, with significant implications for other cancers, such as
breast cancer. ICMT has been shown to influence the transformation
of cells with Ras mutations (48)
and plays a crucial role in regulating cancer cell invasion and
metastasis. By targeting RhoA, one of ICMT's substrates, cancer
cell migration and metastasis can be modulated (53,56).
Furthermore, ICMT inhibition leads to mitochondrial dysfunction,
which triggers cellular autophagy and apoptosis (18). ICMT also impacts cell cycle
regulation and DNA damage repair (19,56).
In breast cancer, combining ICMT inhibition with existing therapies
such as radiotherapy, chemotherapy or targeted therapies (e.g.,
carboplatin) could enhance the anti-tumor effect and improve
prognosis.
ICMT's substrates are diverse, and its regulatory
mechanisms extend to Ras, RhoA, and Rab proteins. For instance,
ICMT interacts with PFKFB4 to influence the RAS/AKT pathway and
cell migration (83), and
inhibition of ICMT impairs thrombin-mediated RhoA and Rac
activation, leading to disrupted cell migration (53). ICMT also regulates the
mislocalization of Rab proteins, such as Rab7 and Rab8, which
impacts the Notch signaling pathway and cellular processes like
migration (90). Future studies
should explore additional signaling pathways and mechanisms of
action related to ICMT to fully understand its therapeutic
potential.
Although the observed phenotypes are largely driven
by canonical ICMT substrates, synergistic contributions from
additional, less-characterized substrates cannot be excluded.
Hypomethylated CDC42 disrupts cytoskeletal dynamics in MDA-MB-231
cells and precipitates mitotic errors (53); RAB-focused experiments in
KRAS-mutant breast-cancer models reveal that ICMT inhibition
simultaneously cripples RAS-dependent metastatic signaling
(56); yet even KRAS-wild-type
lines accumulate γH2AX-positive DNA damage after ICMT knockdown,
pointing to RAS-independent routes to genomic instability (19). To determine whether these outcomes
reflect single- or multi-substrate perturbation, future work should
couple single-cell multi-omics with a stepwise HMEC transformation
cell model in which individual prenylated proteins (KRAS, RhoA,
RAB4, etc.) can be toggled on or off while ICMT is acutely
inhibited. Such precision-controlled systems will clarify which
combinations of hypomethylated substrates must be co-targeted to
maximize therapeutic efficacy.
Beyond pancreatic and breast cancer, ICMT has
promising applications in the treatment of other cancers. For
example, licoricidin has shown promising anti-gastric cancer
activity through the ICMT/Ras pathway (73), while in HCC, ICMT inhibition
impaired cell growth and migration, and induced apoptosis (75). KRAS also requires palmitoylation to
reinforce its membrane anchoring function, so future therapeutic
strategies could consider combination therapies targeting this
modification (106). ICMT also has
important roles in non-cancer diseases, including inflammation,
acute myeloid leukemia and premature aging (93–95).
At present, the small-molecule ICMT inhibitor cysmethynil has low
aqueous solubility and remains in the pre-clinical stage;
therefore, no clinical data are available to support a therapeutic
window. However, published studies allow us to speculate: In a
Hutchinson-Gilford progeria mouse model (95,96,107),
ICMT knockdown produced no overt toxicity, yet targeting the second
step of prenylation (e.g., RCE1) sensitizes retinal neurons
(28,108), so ICMT inhibition may have
potential for clinical treatment with low toxicity, but might
elicit similar side effects. Overall, ICMT inhibition demonstrates
significant therapeutic potential in both cancer and other
diseases, and its targeted inhibition is expected to contribute to
advancements in human healthcare in the future.
Besides its therapeutic potential as a single-agent
target, ICMT holds considerable promise in combination treatment
strategies. The immediate argument is a mechanistic fit with
poly(ADP-ribose) polymerase (PARP) inhibitors: By silencing
multiple DNA-repair circuits, ICMT inhibition imposes a ‘BRCAness’
state on otherwise BRCA-proficient breast cancers, converting them
into PARP-inhibitor-responsive tumors (19,109,110). This synthetic-lethal logic can be
extended further. So far, current research cannot exclude the
possibility that concomitant blockade of the YAP arm of the Hippo
pathway may elicit synthetic-lethal interactions in specific
breast-CSC contexts; although, in one KRAS-mutant pancreatic and
one KRAS-mutant breast cancer line, the Hippo-YAP axis did not
emerge as the decisive downstream effector of the ICMT-KRAS module,
broadening the cell-line panel or interrogating non-KRAS-mutant
tumors could still uncover settings in which YAP co-inhibition is
genuinely synthetically lethal (65,111–113) Likewise, the line-to-line
variability in ICMT-mediated cell-cycle arrest positions ICMT
inhibitors as natural partners for cyclin-dependent kinases or
other cell-cycle checkpoints blockers (17,19,35,45,51).
Mechanistically, ICMT suppression appears to kill by
stripping cancer cells of their redundant repair and survival
modules, such as ICMT deletion causing ‘BRCAness’ (19). However, the metabolic adaptations
that might eventually restore viability, and thus drive acquired
resistance, remain largely uncharted. Building clinically relevant,
drug-resistant models to map these escape routes should be a
high-priority follow-up for ICMT inhibitor clinical study. For the
biomarker development field, a validated predictor of
ICMT-inhibitor benefit is still limited. ICMT transcript or protein
levels display no systematic elevation in tumors vs. matched normal
tissues and vary widely across cancer types, depriving clinicians
of a straightforward expression-based biomarker. Attention
therefore centers on tumors whose oncogenic circuitry is already
wired through ICMT substrates-RAS-mutant cancers, lesions with
hyper-active RAS signaling and metastases addicted to Rho-family
GTPases-rather than on ICMT abundance per se. Future
ICMT-directed trials must therefore embed parallel
biomarker-discovery arms that link pharmacodynamic readouts to
multi-omic signatures, ensuring that patients are selected for the
dependency state created by the enzyme rather than for the enzyme
itself (30,109,114).
Acknowledgements
The authors would like to thank Dr Gang Xiang
(School of Pharmacy and Bioengineering, Chongqing University of
Technology, Chongqing, China) for providing the website resources
for this article and Professor Tingxiu Xiang (Chongqing Key
Laboratory for the Mechanism and Intervention of Cancer Metastasis,
Chongqing University Cancer Hospital, Chongqing University,
Chongqing, China), who gave suggestions for the content.
Funding
This work was supported by Science and Technology Research
Program of Chongqing Municipal Education Commission (grant no.
KJQN202501152) and the Scientific Research Foundation of Chongqing
University of Technology (to JT).
Availability of data and materials
Not applicable.
Authors' contributions
JH contributed to the drafting of the manuscript and
literature search. YX contributed to drafting the manuscript and
provided some of the images included. JT proposed ideas, provided
the framework of the manuscript and made revisions. All authors
have read and approved the final manuscript. Data authentication is
not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FTase
|
farnesyltransferase
|
|
GGTase I
|
geranly-geranyltransferase I
|
|
RCE1
|
Ras-converting enzyme 1
|
|
ICMT
|
isoprenylcysteine
carboxymethyltransferase
|
|
RhoA
|
Ras homolog family member A
|
|
PDA
|
pancreatic ductal adenocarcinoma
|
|
PanIN
|
pancreatic intraepithelial
neoplasia
|
|
BNIP3
|
BCL2/adenovirus E1B 19 kDa-interacting
protein 3
|
|
EMT
|
epithelial-mesenchymal transition
|
|
CSCs
|
cancer stem cells
|
|
TAZ
|
transcriptional co-activator with
PDZ-binding motif
|
|
IRAK
|
interleukin-1 receptor-associated
kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MAPKK
|
mitogen-activated protein kinase
kinase
|
|
AP-1
|
activator protein 1
|
|
PFKFB4
|
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4
|
|
CDC42
|
cell division cycle 42
|
|
HGPS
|
Hutchinson-Gilford progeria
syndrome
|
|
RTKs
|
receptor tyrosine kinases
|
References
|
1
|
Wang M and Casey PJ: Protein prenylation:
Unique fats make their mark on biology. Nat Rev Mol Cell Biol.
17:110–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boyartchuk VL, Ashby MN and Rine J:
Modulation of Ras and a-factor function by carboxyl-terminal
proteolysis. Science. 275:1796–1800. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Freije JM, Blay P, Pendás AM, Cadiñanos J,
Crespo P and López-Otín C: Identification and chromosomal location
of two human genes encoding enzymes potentially involved in
proteolytic maturation of farnesylated proteins. Genomics.
58:270–280. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gutierrez L, Magee AI, Marshall CJ and
Hancock JF: Post-translational processing of p21ras is two-step and
involves carboxyl-methylation and carboxy-terminal proteolysis.
EMBO J. 8:1093–1098. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
James G, Goldstein JL and Brown MS:
Resistance of K-RasBV12 proteins to farnesyltransferase inhibitors
in Rat1 cells. Proc Natl Acad Sci USA. 93:4454–4458. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Whyte DB, Kirschmeier P, Hockenberry TN,
Nunez-Oliva I, James L, Catino JJ, Bishop WR and Pai JK: K- and
N-Ras are geranylgeranylated in cells treated with farnesyl protein
transferase inhibitors. J Biol Chem. 272:14459–14464. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bergo MO, Ambroziak P, Gregory C, George
A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A and Young SG:
Absence of the CAAX endoprotease Rce1: Effects on cell growth and
transformation. Mol Cell Biol. 22:171–181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bergo MO, Leung GK, Ambroziak P, Otto JC,
Casey PJ, Gomes AQ, Seabra MC and Young SG: Isoprenylcysteine
carboxyl methyltransferase deficiency in mice. J Biol Chem.
276:5841–5845. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kamiya Y, Sakurai A, Tamura S and
Takahashi N: Structure of rhodotorucine A, a novel lipopeptide,
inducing mating tube formation in Rhodosporidium toruloides.
Biochem Biophys Res Commun. 83:1077–1083. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Casey PJ, Solski PA, Der CJ and Buss JE:
p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci
USA. 86:8323–8327. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clarke S, Vogel JP, Deschenes RJ and Stock
J: Posttranslational modification of the Ha-ras oncogene protein:
Evidence for a third class of protein carboxyl methyltransferases.
Proc Natl Acad Sci USA. 85:4643–4647. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reiss Y, Goldstein JL, Seabra MC, Casey PJ
and Brown MS: Inhibition of purified p21ras farnesyl: Protein
transferase by Cys-AAX tetrapeptides. Cell. 62:81–88. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Seabra MC, Reiss Y, Casey PJ, Brown MS and
Goldstein JL: Protein farnesyltransferase and
geranylgeranyltransferase share a common alpha subunit. Cell.
65:429–434. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Otto JC, Kim E, Young SG and Casey PJ:
Cloning and characterization of a mammalian prenyl protein-specific
protease. J Biol Chem. 274:8379–8382. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dai Q, Choy E, Chiu V, Romano J, Slivka
SR, Steitz SA, Michaelis S and Philips MR: Mammalian prenylcysteine
carboxyl methyltransferase is in the endoplasmic reticulum. J Biol
Chem. 273:15030–15034. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bergo MO, Gavino BJ, Hong C, Beigneux AP,
McMahon M, Casey PJ and Young SG: Inactivation of Icmt inhibits
transformation by oncogenic K-Ras and B-Raf. J Clin Invest.
113:539–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang M, Hossain MS, Tan W, Coolman B, Zhou
J, Liu S and Casey PJ: Inhibition of isoprenylcysteine
carboxylmethyltransferase induces autophagic-dependent apoptosis
and impairs tumor growth. Oncogene. 29:4959–4970. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teh JT, Zhu WL, Ilkayeva OR, Li Y, Gooding
J, Casey PJ, Summers SA, Newgard CB and Wang M: Isoprenylcysteine
carboxylmethyltransferase regulates mitochondrial respiration and
cancer cell metabolism. Oncogene. 34:3296–3304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang J, Casey PJ and Wang M: Suppression
of isoprenylcysteine carboxylmethyltransferase compromises DNA
damage repair. Life Sci Alliance. 4:e2021011442021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Borini Etichetti C, Arel Zalazar E, Di
Benedetto C, Cocordano N, Valente S, Bicciato S, Menacho-Márquez M,
Larocca MC and Girardini J: Isoprenylcysteine carboxyl
methyltransferase (ICMT) promotes invadopodia formation and
metastasis in cancer cells. Biochimie. 222:28–36. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bergo MO, Leung GK, Ambroziak P, Otto JC,
Casey PJ and Young SG: Targeted inactivation of the
isoprenylcysteine carboxyl methyltransferase gene causes
mislocalization of K-Ras in mammalian cells. J Biol Chem.
275:17605–17610. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diver MM, Pedi L, Koide A, Koide S and
Long SB: Atomic structure of the eukaryotic intramembrane RAS
methyltransferase ICMT. Nature. 553:526–529. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Diver MM and Long SB: Mutational analysis
of the integral membrane methyltransferase isoprenylcysteine
carboxyl methyltransferase (ICMT) reveals potential substrate
binding sites. J Biol Chem. 289:26007–26020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang FL and Casey PJ: Protein
prenylation: Molecular mechanisms and functional consequences. Annu
Rev Biochem. 65:241–269. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cox AD, Der CJ and Philips MR: Targeting
RAS membrane association: Back to the future for Anti-RAS drug
discovery? Clin Cancer Res. 21:1819–1827. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
deSolms SJ, Ciccarone TM, MacTough SC,
Shaw AW, Buser CA, Ellis-Hutchings M, Fernandes C, Hamilton KA,
Huber HE, Kohl NE, et al: Dual protein
farnesyltransferase-geranylgeranyltransferase-I inhibitors as
potential cancer chemotherapeutic agents. J Med Chem. 46:2973–2984.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma C, Yang Y, Xu L, Tu W, Chen F and Wang
J: Rce1 suppresses invasion and metastasis of hepatocellular
carcinoma via epithelial-mesenchymal transition induced by the
TGF-β1/H-Ras signaling pathway. J Cell Physiol. 235:2506–2520.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aiyagari AL, Taylor BR, Aurora V, Young SG
and Shannon KM: Hematologic effects of inactivating the Ras
processing enzyme Rce1. Blood. 101:2250–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karlsson C, Akula MK, Staffas A, Cisowski
J, Sayin VI, Ibrahim MX, Lindahl P and Bergo MO: Knockout of the
RAS endoprotease RCE1 accelerates myeloid leukemia by
downregulating GADD45b. Leukemia. 35:606–609. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Winter-Vann AM, Baron RA, Wong W, dela
Cruz J, York JD, Gooden DM, Bergo MO, Young SG, Toone EJ and Casey
PJ: A small-molecule inhibitor of isoprenylcysteine carboxyl
methyltransferase with antitumor activity in cancer cells. Proc
Natl Acad Sci USA. 102:4336–4341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wright LP and Philips MR: Thematic review
series: Lipid posttranslational modifications. CAAX modification
and membrane targeting of Ras. J Lipid Res. 47:883–891. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kurniansyah N, Goodman MO, Khan AT, Wang
J, Feofanova E, Bis JC, Wiggins KL, Huffman JE, Kelly T, Elfassy T,
et al: Evaluating the use of blood pressure polygenic risk scores
across race/ethnic background groups. Nat Commun. 14:32022023.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Almoguera C, Shibata D, Forrester K,
Martin J, Arnheim N and Perucho M: Most human carcinomas of the
exocrine pancreas contain mutant c-K-ras genes. Cell. 53:549–554.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Court H, Amoyel M, Hackman M, Lee KE, Xu
R, Miller G, Bar-Sagi D, Bach EA, Bergö MO and Philips MR:
Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates
KRAS-driven pancreatic neoplasia via Notch suppression. J Clin
Invest. 123:4681–4694. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lau HY, Ramanujulu PM, Guo D, Yang T,
Wirawan M, Casey PJ, Go ML and Wang M: An improved
isoprenylcysteine carboxylmethyltransferase inhibitor induces
cancer cell death and attenuates tumor growth in vivo. Cancer Biol
Ther. 15:1280–1291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yamamoto H, Zhang S and Mizushima N:
Autophagy genes in biology and disease. Nat Rev Genet. 24:382–400.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gómez-Virgilio L, Silva-Lucero MD,
Flores-Morelos DS, Gallardo-Nieto J, Lopez-Toledo G,
Abarca-Fernandez AM, Zacapala-Gómez AE, Luna-Muñoz J, Montiel-Sosa
F, Soto-Rojas LO, et al: Autophagy: A key regulator of homeostasis
and disease: An overview of molecular mechanisms and modulators.
Cells. 11:22622022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nagata S and Golstein P: The fas death
factor. Science. 267:1449–1456. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang J, Zhang A, Wu M and Qin S:
Manipulating mitochondrial electron flow: A novel approach to
enhance tumor immunogenicity. Mol Biomed. 5:102024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang X, Xu Z, Wang J, Wu C, Zhang L, Qian
C, Luo Y, Gu Y, Wong WT and Xiang D: A Mitochondria-targeted
biomimetic nanomedicine capable of reversing drug resistance in
colorectal cancer through mitochondrial dysfunction. Adv Sci
(Weinh). 12:e24106302025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Feng J, Lian Z, Xia X, Lu Y, Hu K, Zhang
Y, Liu Y, Hu L, Yuan K, Sun Z, et al: Targeting metabolic
vulnerability in mitochondria conquers MEK inhibitor resistance in
KRAS-mutant lung cancer. Acta Pharm Sin B. 13:1145–1163. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Manu KA, Chai TF, Teh JT, Zhu WL, Casey PJ
and Wang M: Inhibition of isoprenylcysteine
carboxylmethyltransferase induces Cell-Cycle Arrest and Apoptosis
through p21 and p21-Regulated BNIP3 induction in pancreatic cancer.
Mol Cancer Ther. 16:914–923. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cavalu S, Abdelhamid AM, Saber S, Elmorsy
EA, Hamad RS, Abdel-Reheim MA, Yahya G and Salama MM: Cell cycle
machinery in oncology: A comprehensive review of therapeutic
targets. FASEB J. 38:e237342024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lau HY, Tang J, Casey PJ and Wang M:
Isoprenylcysteine carboxylmethyltransferase is critical for
malignant transformation and tumor maintenance by all RAS isoforms.
Oncogene. 36:3934–3942. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gewirtz DA, Hilliker ML and Wilson EN:
Promotion of autophagy as a mechanism for radiation sensitization
of breast tumor cells. Radiother Oncol. 92:323–328. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Seyrek K, Wohlfromm F, Espe J and Lavrik
IN: The cross-talk of autophagy and apoptosis in breast carcinoma:
Implications for novel therapies? Biochem J. 479:1581–1608. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang M, Tan W, Zhou J, Leow J, Go M, Lee
HS and Casey PJ: A small molecule inhibitor of isoprenylcysteine
carboxymethyltransferase induces autophagic cell death in PC3
prostate cancer cells. J Biol Chem. 283:18678–18684. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ziemys A, Yokoi K, Kai M, Liu YT, Kojic M,
Simic V, Milosevic M, Holder A and Ferrari M: Progression-dependent
transport heterogeneity of breast cancer liver metastases as a
factor in therapeutic resistance. J Control Release. 291:99–105.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cushman I and Casey PJ: Role of
isoprenylcysteine carboxylmethyltransferase-catalyzed methylation
in Rho function and migration. J Biol Chem. 284:27964–27973. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wen S, Hou Y, Fu L, Xi L, Yang D, Zhao M,
Qin Y, Sun K, Teng Y and Liu M: Cancer-associated fibroblast
(CAF)-derived IL32 promotes breast cancer cell invasion and
metastasis via integrin β3-p38 MAPK signalling. Cancer Lett.
442:320–332. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wei L, Zhou Q, Tian H, Su Y, Fu GH and Sun
T: Integrin β3 promotes cardiomyocyte proliferation and attenuates
hypoxia-induced apoptosis via regulating the PTEN/Akt/mTOR and
ERK1/2 pathways. Int J Biol Sci. 16:644–654. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Do MT, Chai TF, Casey PJ and Wang M:
Isoprenylcysteine carboxylmethyltransferase function is essential
for RAB4A-mediated integrin β3 recycling, cell migration and cancer
metastasis. Oncogene. 36:5757–5767. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells-perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zanconato F, Cordenonsi M and Piccolo S:
YAP/TAZ at the roots of cancer. Cancer Cell. 29:783–803. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cordenonsi M, Zanconato F, Azzolin L,
Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR,
Poletti A, et al: The Hippo transducer TAZ confers cancer stem
cell-related traits on breast cancer cells. Cell. 147:759–772.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shao DD, Xue W, Krall EB, Bhutkar A,
Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et
al: KRAS and YAP1 converge to regulate EMT and tumor survival.
Cell. 158:171–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang W, Nandakumar N, Shi Y, Manzano M,
Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, et
al: Downstream of mutant KRAS, the transcription regulator YAP is
essential for neoplastic progression to pancreatic ductal
adenocarcinoma. Sci Signal. 7:ra422014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chai TF, Manu KA, Casey PJ and Wang M:
Isoprenylcysteine carboxylmethyltransferase is required for the
impact of mutant KRAS on TAZ protein level and cancer cell
self-renewal. Oncogene. 39:5373–5389. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang S, Wang W, Zhang S, Yang F, Qiu J,
Guo Q, Zheng J and Chen Z: Isoprenylcysteine carboxyl
methyltransferase promotes the progression of tongue squamous cell
carcinoma via the K-Ras and RhoA signaling pathways. Arch Oral
Biol. 134:1053202022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin L, Cheng X and Yin D: Aberrant DNA
methylation in esophageal squamous cell carcinoma: Biological and
clinical implications. Front Oncol. 10:5498502020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Klintman J, Appleby N, Stamatopoulos B,
Ridout K, Eyre TA, Robbe P, Pascua LL, Knight SJL, Dreau H, Cabes
M, et al: Genomic and transcriptomic correlates of Richter
transformation in chronic lymphocytic leukemia. Blood.
137:2800–2816. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Richardson C, Moynahan ME and Jasin M:
Homologous recombination between heterologs during repair of a
double-strand break. Suppression of translocations in normal cells.
Ann N Y Acad Sci. 886:183–186. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Iliakis G, Wang Y, Guan J and Wang H: DNA
damage checkpoint control in cells exposed to ionizing radiation.
Oncogene. 22:5834–5847. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI
|
|
72
|
Ajani JA, D'Amico TA, Bentrem DJ, Chao J,
Cooke D, Corvera C, Das P, Enzinger PC, Enzler T, Fanta P, et al:
Gastric cancer, version 2.2022, NCCN clinical practice guidelines
in oncology. J Natl Compr Canc Netw. 20:167–192. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ma H, Wu F, Bai Y, Wang T, Ma S, Guo L,
Liu G, Leng G, Kong Y and Zhang Y: Licoricidin combats gastric
cancer by targeting the ICMT/Ras pathway in vitro and in vivo.
Front Pharmacol. 13:9728252022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
75
|
Xu J, Zhu Y, Wang F, Zhou Y, Xia G and Xu
W: ICMT contributes to hepatocellular carcinoma growth, survival,
migration and chemoresistance via multiple oncogenic pathways.
Biochem Biophys Res Commun. 518:584–589. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015.PubMed/NCBI
|
|
77
|
Liu Q, Chen J, Fu B, Dai J, Zhao Y and Lai
L: Isoprenylcysteine carboxylmethyltransferase regulates ovarian
cancer cell response to chemotherapy and Ras activation. Biochem
Biophys Res Commun. 501:556–562. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bourne HR, Sanders DA and McCormick F: The
GTPase superfamily: A conserved switch for diverse cell functions.
Nature. 348:125–132. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Takashima A and Faller DV: Targeting the
RAS oncogene. Expert Opin Ther Targets. 17:507–531. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wahlstrom AM, Cutts BA, Liu M, Lindskog A,
Karlsson C, Sjogren AK, Andersson KM, Young SG and Bergo MO:
Inactivating Icmt ameliorates K-RAS-induced myeloproliferative
disease. Blood. 112:1357–1365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yang WS, Kim HG, Kim E, Han SY, Aziz N, Yi
YS, Kim S, Lee Y, Yoo BC, Han JW, et al: Isoprenylcysteine carboxyl
methyltransferase and its substrate ras are critical players
regulating TLR-Mediated inflammatory responses. Cells. 9:12162020.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Sittewelle M, Kappès V, Zhou C, Lécuyer D
and Monsoro-Burq AH: PFKFB4 interacts with ICMT and activates
RAS/AKT signaling-dependent cell migration in melanoma. Life Sci
Alliance. 5:e2022013772022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Marín-Ramos NI, Balabasquer M,
Ortega-Nogales FJ, Torrecillas IR, Gil-Ordóñez A, Marcos-Ramiro B,
Aguilar-Garrido P, Cushman I, Romero A, Medrano FJ, et al: A potent
isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor
improves survival in Ras-Driven acute myeloid leukemia. J Med Chem.
62:6035–6046. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wan W, Xiao W, Pan W, Chen L, Liu Z and Xu
J: Isoprenylcysteine carboxyl methyltransferase is critical for
glioblastoma growth and survival by activating Ras/Raf/Mek/Erk.
Cancer Chemother Pharmacol. 89:401–411. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Colicelli J: Human RAS superfamily
proteins and related GTPases. Sci STKE. 2004:Re132004. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Papaharalambus C, Sajjad W, Syed A, Zhang
C, Bergo MO, Alexander RW and Ahmad M: Tumor necrosis factor alpha
stimulation of Rac1 activity. Role of isoprenylcysteine
carboxylmethyltransferase. J Biol Chem. 280:18790–18796. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cushman I and Casey PJ: RHO methylation
matters: A role for isoprenylcysteine carboxylmethyltransferase in
cell migration and adhesion. Cell Adh Migr. 5:11–15. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Schwartz SL, Cao C, Pylypenko O, Rak A and
Wandinger-Ness A: Rab GTPases at a glance. J Cell Sci.
120:3905–3910. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Court H, Ahearn IM, Amoyel M, Bach EA and
Philips MR: Regulation of NOTCH signaling by RAB7 and RAB8 requires
carboxyl methylation by ICMT. J Cell Biol. 216:4165–4182. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Roberts M, Barry S, Woods A, van der
Sluijs P and Norman J: PDGF-regulated rab4-dependent recycling of
alphavbeta3 integrin from early endosomes is necessary for cell
adhesion and spreading. Curr Biol. 11:1392–1402. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
White DP, Caswell PT and Norman JC: Alpha
v beta3 and alpha5beta1 integrin recycling pathways dictate
downstream Rho kinase signaling to regulate persistent cell
migration. J Cell Biol. 177:515–525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yang WS, Kim HG, Lee Y, Yoon K, Kim S, Kim
JH and Cho JY: Isoprenylcysteine carboxyl methyltransferase
inhibitors exerts anti-inflammatory activity. Biochem Pharmacol.
182:1142192020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Judd WR, Slattum PM, Hoang KC, Bhoite L,
Valppu L, Alberts G, Brown B, Roth B, Ostanin K, Huang L, et al:
Discovery and SAR of methylated tetrahydropyranyl derivatives as
inhibitors of isoprenylcysteine carboxyl methyltransferase (ICMT).
J Med Chem. 54:5031–5047. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen X, Yao H, Kashif M, Revêchon G,
Eriksson M, Hu J, Wang T, Liu Y, Tüksammel E, Strömblad S, et al: A
small-molecule ICMT inhibitor delays senescence of
Hutchinson-Gilford progeria syndrome cells. Elife. 10:e632842021.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Marcos-Ramiro B, Gil-Ordóñez A,
Marín-Ramos NI, Ortega-Nogales FJ, Balabasquer M, Gonzalo P,
Khiar-Fernández N, Rolas L, Barkaway A, Nourshargh S, et al:
Isoprenylcysteine Carboxylmethyltransferase-Based Therapy for
Hutchinson-Gilford Progeria Syndrome. ACS Cent Sci. 7:1300–1310.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Jayaram B, Syed I, Singh A, Subasinghe W,
Kyathanahalli CN and Kowluru A: Isoprenylcysteine carboxyl
methyltransferase facilitates glucose-induced Rac1 activation, ROS
generation and insulin secretion in INS 832/13 β-cells. Islets.
3:48–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Koera K, Nakamura K, Nakao K, Miyoshi J,
Toyoshima K, Hatta T, Otani H, Aiba A and Katsuki M: K-ras is
essential for the development of the mouse embryo. Oncogene.
15:1151–1159. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Nakamura K, Ichise H, Nakao K, Hatta T,
Otani H, Sakagami H, Kondo H and Katsuki M: Partial functional
overlap of the three ras genes in mouse embryonic development.
Oncogene. 27:2961–2968. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Fuentes-Mateos R, Jimeno D, Gómez C,
Calzada N, Fernández-Medarde A and Santos E: Concomitant deletion
of HRAS and NRAS leads to pulmonary immaturity, respiratory failure
and neonatal death in mice. Cell Death Dis. 10:8382019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Cui XS, Li XY and Kim NH: Cdc42 is
implicated in polarity during meiotic resumption and blastocyst
formation in the mouse. Mol Reprod Dev. 74:785–794. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sugihara K, Nakatsuji N, Nakamura K, Nakao
K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, et
al: Rac1 is required for the formation of three germ layers during
gastrulation. Oncogene. 17:3427–3433. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
He X, Liu J, Qi Y, Brakebusch C,
Chrostek-Grashoff A, Edgar D, Yurchenco PD, Corbett SA, Lowry SF,
Graham AM, et al: Rac1 is essential for basement membrane-dependent
epiblast survival. Mol Cell Biol. 30:3569–3581. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Dickinson ME, Flenniken AM, Ji X, Teboul
L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu
H, et al: High-throughput discovery of novel developmental
phenotypes. Nature. 537:508–514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wu Y, Yang D and Chen GY: Targeted
disruption of Rab1a causes early embryonic lethality. Int J Mol
Med. 49:462022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Dekker FJ, Rocks O, Vartak N, Menninger S,
Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M,
Schölermann B, et al: Small-molecule inhibition of APT1 affects Ras
localization and signaling. Nat Chem Biol. 6:449–456. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ibrahim MX, Sayin VI, Akula M, Liu M, Fong
LG, Young SG and Bergo MO: Targeting isoprenylcysteine methylation
ameliorates disease in a mouse model of progeria. Science.
340:1330–1333. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bergo MO, Lieu HD, Gavino BJ, Ambroziak P,
Otto JC, Casey PJ, Walker QM and Young SG: On the physiological
importance of endoproteolysis of CAAX proteins: Heart-specific RCE1
knockout mice develop a lethal cardiomyopathy. J Biol Chem.
279:4729–4736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang MT, Holderfield M, Galeas J,
Delrosario R, To MD, Balmain A and McCormick F: K-Ras promotes
tumorigenicity through suppression of Non-canonical wnt signaling.
Cell. 163:1237–1251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Sinnett-Smith J, Anwar T, Reed EF, Teper
Y, Eibl G and Rozengurt E: Opposite effects of src family kinases
on YAP and ERK activation in pancreatic cancer cells: Implications
for targeted therapy. Mol Cancer Ther. 21:1652–1662. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wang H, Tao Y, Han J, Shen J, Mu H, Wang
Z, Wang J, Jin X, Zhang Q, Yang Y, et al: Disrupting YAP1-mediated
glutamine metabolism induces synthetic lethality alongside ODC1
inhibition in osteosarcoma. Cell Oncol (Dordr). 47:1845–1861. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ren F, Yi Y, Lu T, Liu X, Cui G, Huang S,
Parada LF and Chen J: Synthetic lethality through Gsk3β inhibition
in glioma stem cells via the WNT-WWC1-YAP axis. Oncogene.
44:2427–2439. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Vasan N, Baselga J and Hyman DM: A view on
drug resistance in cancer. Nature. 575:299–309. 2019. View Article : Google Scholar : PubMed/NCBI
|