Introduction
Cancer is characterized by the uncontrolled growth
of somatic cells driven by mutations in proto-oncogenes and tumor
suppressor genes (1), leading to
the invasion of surrounding tissues and the progressive spread to
secondary organs (2). According to
the World Health Organization, in 2020 cancer was responsible for
nearly 10 million deaths globally, making it one of the leading
causes of mortality (2). Tumor
growth and progression is accompanied by systemic effects,
including cachexia, that cannot be explained solely by the effects
of tumor cells on their immediate tumor microenvironment. In
particular, cachexia, often associated with late stages of cancer
as well as other chronic inflammatory diseases, markedly
contributes to increased mortality, reduced quality of life and
physical dysfunction of affected individuals (3).
Cachexia is defined as a multifactorial catabolic
syndrome characterized by the loss of skeletal muscle mass, reduced
appetite and persistent fatigue. Its prevalence ranges from 35–90%
in patients with advanced stages of cancer, being particularly
prevalent in patients with malignant neoplasms that arise in the
digestive system (4,5). Indeed, loss of skeletal muscle mass,
or muscle wasting, is a central feature of the cachectic syndrome
(6). Among primary mechanisms
underlying muscle wasting is the upregulation of catabolic pathways
that create an energy imbalance favoring the degradation of
myofibrillar proteins. This metabolic imbalance is the result of
paracrine signals in the form of soluble proinflammatory cytokines
and/or extracellular vesicles (EVs) released from tumor cells or
stroma cells in response to tumor cells (7).
EVs are membrane-bound particles produced and
released by most cells. It is currently considered that EVs
represent a complex mechanism of intercellular communication that
involves the simultaneous transfer of multiple biomolecules (such
as cargoes) that may regulate various processes in target cells
(8). In particular, EVs released by
tumor cells can function as carriers of biomolecules destined to
several different tissues, including skeletal muscles, likely
contributing to the muscle wasting observed in patients with cancer
(9).
In addition to a vast and functionally diverse array
of proteins, EVs also contain micro RNAs (miRNAs), a group of small
non-coding RNAs that participate in post-transcriptional mechanisms
of control of gene expression by binding to the 3′UTR of the coding
mRNAs, leading to silencing the respective target genes. While
several miRNAs carried by EVs released by cancer cells can regulate
the expression of tumor suppressor genes and proto-oncogenes,
thereby controlling tumor development and progression (10), they may also regulate inflammatory
processes, protein degradation and the inhibition of myogenesis in
muscle cells. So far, however, there is limited evidence indicating
that these miRNA-dependent mechanisms are important contributors to
cancer-related cachexia (11).
The present review summarized the available
information linking cancer-derived extracellular vesicles (EVs) to
inflammation and proteolytic mechanisms of muscle wasting in
patients with cachexia.
Cancer-associated cachexia
Cancer-associated cachexia is a systemic condition
characterized by a negative energy balance due to increased energy
expenditure and reduced food intake (anorexia), resulting in loss
of adipose tissue followed by loss of muscle mass, ultimately
leading to dysfunction of multiple organs (12).
Tumor cells are characterized by a deregulated and
inefficient metabolism, which results in a high demand for glucose
and an increase in the production of lactate, even in the context
of normal concentrations of oxygen (Warburg effect) (13). Due to metabolic inefficiency and
overactivation of the Cori cycle, catabolic pathways are activated
in the adipose tissue and skeletal muscle cells in order to
mobilize glucose precursors for hepatic gluconeogenesis needed to
sustain the energy requirements of tumor cells (13). This systemic metabolic reprogramming
leads to a higher resting energy expenditure and a decrease in
muscle strength and function, ultimately leading to a poor quality
of life and reduced survival rates of patients with cancer
(14).
Cachexia is observed in >50% of patients with
cancer and is responsible for nearly 20% of cancer-related deaths.
The incidence of cachexia varies depending on the type and stage of
the tumor, with rates >80% in advanced gastric and pancreatic
cancers (15). Overall, the
cachectic syndrome is clinically classified into three stages: i)
Pre-cachexia, where patients lose <5% of body weight and begin
experiencing early anorexia-related symptoms; ii) cachexia, where
patients lose >5% of body weight over a 6 months-period; this
stage is often accompanied by increased release of pro-inflammatory
factors by cancer cells, leading to organ dysfunction; and iii)
refractory cachexia, where patients have a survival prognosis of
<3 months. This latter stage is characterized by an exacerbated
catabolism, neural disorders and dysfunction of the immune system,
all factors that contribute to the rapid progression of cachexia
and multi-organ functional decline (16).
It is worth mentioning that, in addition to factors
released by tumor cells (or by stromal cells in response to tumor
cells), most antitumoral treatments are known to cause adverse
effects, including nausea, vomiting, dysphagia and mood disorders,
that likely contribute to the development of anorexia and thus can
exacerbate cachexia (17). However,
as aforementioned, the increase in energy expenditure observed in
cachectic patients is mainly attributed to the effects of systemic
inflammation and the energy demands of tumor cells. Thus,
inflammatory mediators and the increase in energy requirements of
tumors cells leads to the degradation of muscle proteins through
activation of the ubiquitin-proteasome system. Released amino acids
serve in turn as glucose precursors for the hepatic synthesis of
glucose, a process known as gluconeogenesis. In addition, muscle
protein synthesis is reduced due to inhibition of the PI3K-AKT-mTOR
pathway, which further contributes to muscle wasting in patients
with cachexia (18).
Role of proinflammatory cytokines in
cancer-associated cachexia
Inflammation is a known enabling feature of cancer
(19). Several aspects of chronic
inflammation may contribute to tumorigenesis, including the release
of local and systemic proinflammatory and mitogenic mediators, as
well as the increased accumulation of reactive oxygen species
(ROS), with the consequent oxidative stress and genotoxic damage
(19). In addition to promoting
tumor initiation, features of chronic inflammation are also present
during tumor progression. Thus, the tumor microenvironment often
encompasses a heterogeneous population of cells in addition to
cancer cells. These ‘stromal cells’ may include cellular components
of the immune system capable of releasing pro-inflammatory
mediators such as cytokines that favor tumor progression (20).
Cellular components of the immune system, both
innate and adaptive, contribute to inflammation through the release
of soluble mediators, namely cytokines, chemokines and other
molecules (21). Cytokines may aid
cancer cells through the promotion of angiogenesis and by
increasing glucose availability within the tumor microenvironment,
both processes being crucial for cancer progression (22). Proinflammatory cytokines and
chemokines, such as IL-6, TNF-α, IL-8 and IL-1, can promote
cachexia and rapid skeletal muscle functional deterioration
(7). Indeed, the functional link
between soluble proinflammatory mediators and muscle wasting is at
the core of the most accepted mechanism through which chronic
inflammation induces cachexia (23). Notably, it has been shown that
increased expression and release of IL-5 by tumor cells leads to an
increase in the serum levels of IL-6 and IL-8, which is accompanied
by a reduction in the expression of the secreted glycoprotein
Folistatin-like protein-1 (FSTl-1) in muscle cells; reduced
expression of FSTI-1 leads, in turn, to decreased vascularization
and hindered muscle regeneration in patients with cancer with
cachexia (24). Other studies have
shown that IL-6 causes atrophy of C2C12-derived myotubes (25–27).
Not surprisingly, IL-6 produced by pancreatic adenocarcinoma cells
contributes to systemic inflammation and chronic lipolysis in the
adipose tissue, resulting in increased fatty acid accumulation
around muscle cells, myosteatosis and lipotoxicity-driven cachexia
(25). IL-6 is further linked to
neuromuscular junction dysfunction by increasing Noggin expression
in muscle cells (26). Noggin, a
signaling protein that regulates tissue development and
differentiation, inhibits the BMP-SMAD1/5/814 pathway in skeletal
muscles, which is crucial for muscle growth and maintenance
(26). Inhibition of this pathway
during cancer-associated cachexia therefore may also promote
neuromuscular junction denervation (27).
Local release of proinflammatory cytokines at the
tumor microenvironment can also contribute to the generation of
neuroinflammation and lead to changes in the
hypothalamus-pituitary-adrenal (HPA) axis (28). This latter physiological axis, a key
regulator of the stress response, includes corticotropin-releasing
hormone, corticotropin, and the cortisol-glucocorticoid receptor
interaction (29). Dysfunction of
the HPA axis has been linked to the adverse effects of
proinflammatory cytokines, potentially leading to systemic
dysregulation of energy metabolism and increased fatigue in
patients with cancer (28). For
example, IL-1β has been shown to upregulate leukemia inhibitory
factor mRNA in the central nervous system, which may in turn
contribute to HPA axis dysfunction, becoming apparent as appetite
loss, reduced muscle mass and increased release of amino acids from
muscle fibers, in the context of the increased energy demands of
tumor cells (30,31). In addition to these systemic,
HPA-mediated effects, cytokines may also affect more directly
muscle contractile abilities and strength. For example, IL-1α and
IL-1β are implicated in the decrease of myotube width and reduced
amounts of sarcomeric actin, through the activation of catabolic
pathways that lead to an upregulation of muscle atrophy F-box
(Atrogin1/MAFbx), as well as muscle RING finger 1 (MuRF1) mRNA
levels, which are components of the ubiquitin-proteasome
proteolytic system that promote muscle protein loss (32).
TNF-α, a key proinflammatory cytokine linked to
cachexia, can stimulate the ubiquitin-proteasome system in muscle
cells, resulting in muscle mass loss and altered metabolism in
patients with cancer (33). Indeed,
TNF-α has been used as a serum biomarker of muscle wasting in
individuals with colorectal cancer (34). In addition, a positive correlation
between TNF-α and the upregulation of high-mobility group protein
B1 (HMGB1) in the serum has been reported (35). HMGB1, a non-histone nuclear protein
that is released by cancer cells or cells subjected to stressful
stimuli, stimulates the ATP-dependent ubiquitin-proteasome pathway
via activation of the NF-κB pathway, leading to autophagy in
skeletal muscle cells, the mobilization of amino acids from muscle
cells and their release into the blood stream to finally be used
for gluconeogenesis in the liver (34,35).
TNF-α also modulates the production and function of various
non-coding RNAs in muscle cells, including long non-coding RNAs
(lncRNAs) that are linked to morphological alterations in C2C12
cells (36). Powrózek et al
(36) reported an inverse
correlation between TNF-α levels and the expression of the lncRNAs
Gm4117 and Ccdc41os1, implying that increased TNF-α levels lead to
the suppression of these lncRNAs and the upregulation of another
lncRNA, 5830418P13Rik. According to this study, the functional
interaction between Gm4117 and Npm1, the gene encoding the
chromatin modifier protein Nucleophosphin, interferes with a
phosphoprotein involved in the control of muscle differentiation, a
process mediated by several transcription factors, including c-Myc,
NF-κB, Yin Yang 1 and interferon regulatory factor 1 (IRF-1)
(36). Thus, a TNF-α-mediated
reduction in Gm4117 levels contributes to muscle breakdown.
Conversely, targeting of miRNA-125 by 5830418P13Rik positively
regulates proteolysis by activating MAPK and forkhead box O (FOXO)
signaling (36).
IL-8, a chemokine-type inflammatory cytokine, plays
a critical role in local and systemic inflammation by promoting
neutrophil chemotaxis (37).
Similarly, activation of macrophages, induced by IL-6 secretion,
further intensifies inflammatory responses mediated by IL-8
(37). In patients with cancer and
cachexia, elevated IL-8 levels are associated with a poorer
prognosis, typically indicating reduced survival (38). High IL-8 levels have also been
linked to muscle atrophy in cachectic patients (37). IL-8 exerts its biological effects
through its binding to the G protein-coupled receptor CXCR2
(38). Activation of CXCR2
activates the ERK signaling, which has been shown to markedly
reduce the diameter of C2C12 myotubes treated with IL-8-containing
conditioned media derived from human pancreatic cancer cells
(38).
Recent studies have identified other members of the
interleukin family linked to cachexia, such as IL-17A, which is
primarily released by CD8+ T cells and plays a critical role in
inducing inflammation (39,40). By synergizing with TNF-α, IL-17A
also promotes the progression of pancreatic ductal adenocarcinoma
(41). Conversely, head and neck
cancers with low IL-17A expression levels tend to be associated
with more advanced cancer stages. Therefore, IL-17A may have a dual
function being both pro- and anti-tumoral, depending of the type of
malignancy (42). Notably, this
suggests a mechanism involving the regulation of T and B cells that
infiltrate the tumor (41). In
terms of muscle wasting, a study by Ying et al (42) found that lung patients with cancer
with reduced muscle mass exhibited elevated serum levels of
C-reactive protein and IL-17A. Additionally, lung carcinoma cells
have been shown to induce muscle wasting in part through the
release of increased levels of IL-17A, leading to the
phosphorylation and activation of the JAK2/STAT3 signaling pathway
in muscle cells. Despite the identification of several inflammatory
factors associated with cancer cachexia, further research is needed
to pinpoint the specific interleukins involved in the etiology of
cancer-associated cachexia (Fig.
1). An improved understanding of the pivotal role of
proinflammatory cytokines, as well as of the intracellular pathways
they activate in muscle cells, will set the stage for a deeper
examination of the mechanisms governing myofibrillar protein
degradation in cachectic patients.
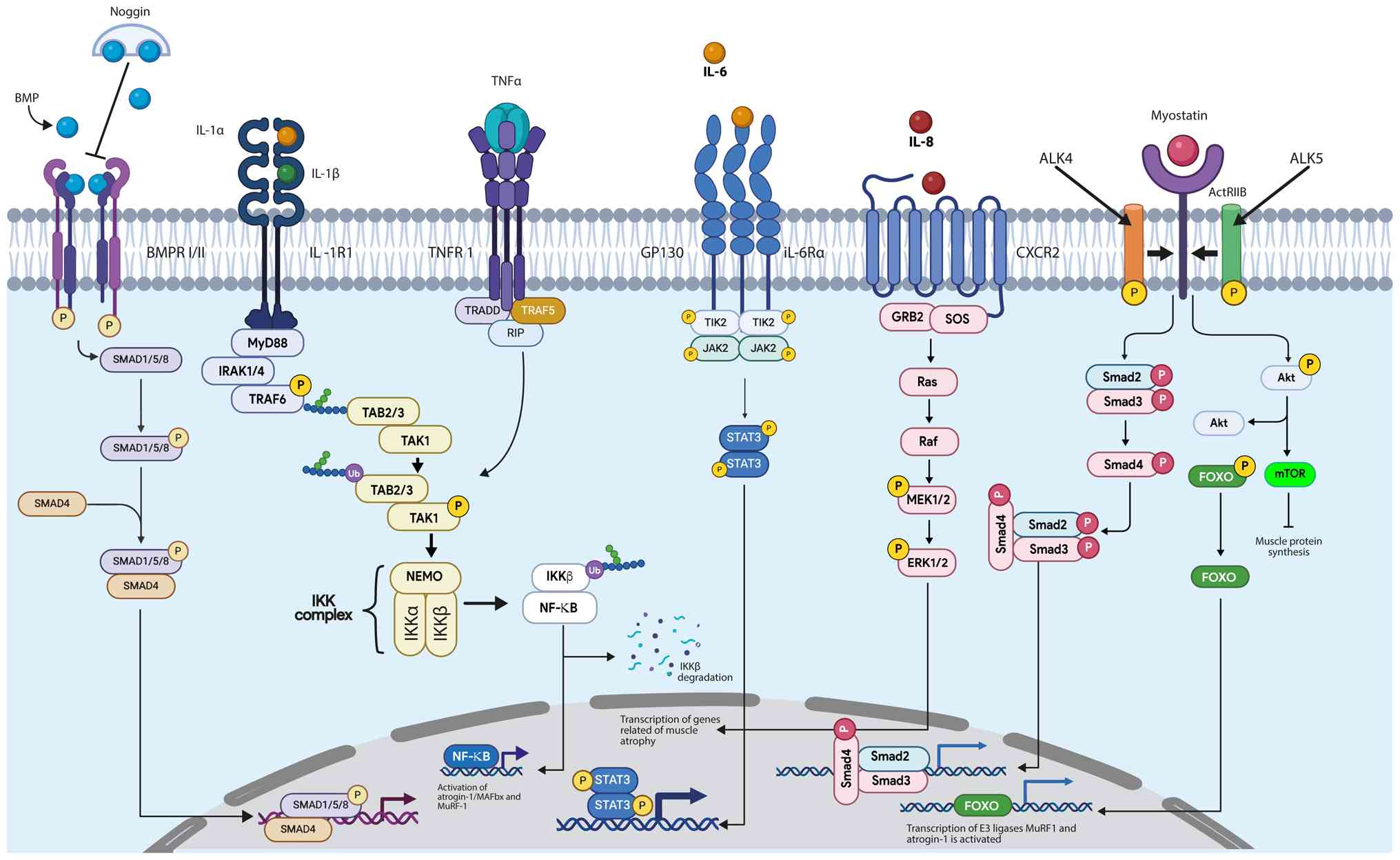 | Figure 1.Main cell signaling pathways involved
in myofibrillar protein degradation in cancer-associated cachexia.
BMP, bone morphogenetic protein; BMPR I/II, bone morphogenetic
protein receptor type I/type II; SMAD1/5/8, mothers against
decapentaplegic homologs 1, 5 and 8; SMAD4, mothers against
decapentaplegic homolog 4; IL-1R1, interleukin-1 receptor type 1;
MyD88, myeloid differentiation primary response protein 88;
IRAK1/4, IL-1R-associated kinase 1/4; TRAF6, TNF
receptor-associated factor 6; IKK, IκB kinase; IκB, inhibitor of
κB; NF-κB, nuclear factor κ-light-chain-enhancer of activated B
cells; TNF-α, tumor necrosis factor α; TNFR1, TNF receptor 1;
TRADD, TNF receptor-associated death domain; RIP,
receptor-interacting protein; TRAF5, TNF receptor-associated factor
5; TAB2/3, TAK1-binding protein 2/3; TAK1, transforming growth
factor-β activated kinase 1; NEMO, NF-κB essential modulator;
GP130, glycoprotein 130; JAK1/2, janus kinase 1/2; STAT3, signal
transducer and activator of transcription 3; CXCR2, C-X-C chemokine
receptor 2; GRB2, growth factor receptor-bound protein 2; SOS, son
of sevenless; Ras, rat sarcoma viral oncogene homolog; Raf, rapidly
accelerated fibrosarcoma kinase; MEK1/2, mitogen-activated protein
kinase kinase 1/2; ERK1/2, extracellular signal-regulated kinase
1/2; ActRIIB, activin receptor type IIB; ALK4/ALK5, activin
receptor-like kinase 4/5; Smad2/Smad3, mothers against
decapentaplegic homologs 2/3; FOXO, forkhead box o; Akt, protein
kinase B; mTOR, mechanistic target of rapamycin; MuRF1, muscle ring
finger 1; Atrogin-1 (MAFbx), muscle atrophy F-box protein. |
Myofibrillar protein degradation in
cancer-associated cachexia
It has now become clear that the metabolic and
morphologic alterations in muscle fibers leading to muscle mass
loss in cancer-associated cachexia is the result of an upregulation
of the ubiquitin-proteasome system and/or the activation of the
autophagy-lysosomal process (43).
In a study by Zhang et al (44), changes in myocyte morphology,
including misalignment of Z-lines and sarcomeres, were noticed in
cachectic patients with gastric cancer. Additionally, these
findings were accompanied by a reduction in the cross-sectional
area of muscle fibers along with an increased expression of LC3B,
an autophagy marker, and MuRF1 (45). MuRF1, a muscle-specific RING finger
protein 1, acts as an E3 ubiquitin ligase in skeletal muscle
catabolism (46). It binds directly
to myofibrillar proteins, thereby initiating the proteolytic
process that eventually leads to muscle wasting or atrophy
(46). According to this model,
increased proteolysis is mediated by cytokines and proinflammatory
factors released by neoplastic cells, particularly TNF-α. Upon
TNF-α binding to its receptor the signal transducer tumor necrosis
factor receptor adaptor protein α (TRAF6) is activated (47), which phosphorylates the IKK kinase
complex, leading to activation of NF-κB. This, in turn, activates
the transcription of MuRF-1, which promotes the ubiquitination of
sarcomere thick filaments (Myosin), ultimately resulting in
proteasome-mediated myofibrillar protein degradation (47).
As aforementioned, autophagy is also involved in the
pathogenesis of muscle wasting. In this multistep process, a
double-membrane structure called a phagophore is first formed
(48). The phagophore sequesters
cellular components or protein complexes to be degraded; the fully
formed cargo-containing doble-membrane vesicle, known as the
autophagosome, then fuses with the lysosome to become an
autolysosome, the site in which the degradation of the sequestered
cargoes occurs (49). Upregulation
of LC3B, a protein essential for autophagosome formation, has been
reported in the quadriceps femoris muscle of pre-cachectic and
cachectic individuals with lung cancer (49). Similarly, elevated LC3B-II/I ratios
(an indication of an increased autophagy flux) have been documented
in the vastus lateralis muscle of patients with esophageal cancer
(49). Such elevated levels of LC3B
in muscle tissue are indicative of autophagosome accumulation,
disrupted trafficking and cytoskeletal organization in myofibrils,
all of which contribute to loss of muscle mass (49).
A study by Johns et al (50) set out to determine the structure and
morphological composition of muscle fibers in cachectic patients,
as well as various signaling pathways involved in
cancer-associated-cachexia. The authors observed that an increase
in the levels of SMAD3 protein was associated with >5% body
weight loss in patients with pancreatic cancer (51). SMAD proteins constitute a family of
intracellular proteins that act as signal transducers and
transcriptional regulators downstream activated TGF-β receptors,
thus transmitting signals involved in cell growth and
differentiation (51).
Specifically, it has been shown that SMAD3, in complex with SMAD2,
is part of a signaling cascade that contributes to muscle wasting
following the binding of myostatin to phosphorylated activin
receptor type IIB, a serine/threonine kinase TGF-β receptor that is
fundamental in muscle development, metabolism and other
physiological processes in humans (52). Activation of this cascade inhibits
the AKT/mTOR signaling pathway, which when active promotes muscle
hypertrophy and protein synthesis (53). Additionally, SMAD3 activates the
transcription factor FOXO3a, which regulates the autophagy pathway
during muscle atrophy through an upregulation of LC3 (53). SMAD3 also increases the
transcription of MuRF-1 and Atrogin-1, leading to increased protein
degradation (53).
Regarding mechanistic target of rapamycin complex 1
(mTORC1), some discrepancy exists on its role in muscle wasting in
the context of cancer-associated cachexia. Although mTORC1
inhibition does not directly lead to muscle atrophy, an
upregulation of the process of mitophagy has been reported
(54). Following the injection of
C26 colon cancer cells in conditional knockout mice that are
deficient in Raptor (a key protein of the mTOR complex 1, mTORC1)
specifically in skeletal muscle cells, a significant increase in
LC3 lipidation was observed in the gastrocnemius muscle compared to
non-inoculated mice after colchicine treatment. Accordingly,
AKT-mTORC1 activation alone in muscle fibers was sufficient to
reduce myofibrillar protein ubiquitination and autophagy in the
context of cachexia (55). These
findings suggested the participation of mTORC1 as a integrating
node in multiple signaling pathways that regulate muscle wasting in
cachectic patients with cancer.
Furthermore, in vivo results using B16F10
mouse melanoma cells have demonstrated a markedly higher activation
of the Ubiquitin-proteasome proteolytic system in the extensor
digitorum longus muscle of tumor-bearing animals when compared to
controls (56). This study also
revealed a correlation between increased expression of MuRF-1 and
reduced weight and muscle strength in tumor-bearing mice with
cachexia. Reinforcing this association, inhibition of MuRF-1 led to
a reduction in muscle mass loss (56). These and other findings highlight
MuRF-1 as a key player in muscle wasting observed in tumor-bearing
animals.
Another signaling pathway involved in muscle
catabolism during cachexia is the JAK/STAT signaling pathway.
Increased activation levels of STAT3, myostatin, as well as
Atrogin-1 and MuRF1 ligases, have been reported in skeletal muscles
of cachectic patients with liver cancer (57). In addition, the interaction between
Heat Shock Protein 90 (HSP90) and STAT3 seems to be enhanced in
muscles of cachectic individuals. HSP90, a chaperone protein that
facilitates the proper folding of other proteins, also facilitates
protein degradation and may regulate STAT3 activation during
cancer-associated cachexia (58).
STAT3 alone is sufficient to induce muscle atrophy both in
vivo and in vitro models, resulting in a 30% reduction
in the diameter of C2C12 myofibers (59). Conversely, STAT3 inhibition prevents
IL-6-mediated muscle atrophy in vitro and attenuates
cachexia in patients with colorectal or lung cancers (59,60).
These findings suggest that JAK/STAT activation, driven by
chronically elevated IL-6 levels, promotes skeletal muscle
deterioration by activating autophagy and subsequently increasing
myofibrillar protein degradation.
The effect of EVs derived from tumors on
muscle cells in cachectic syndrome
EVs are lipid bilayer-encased structures that carry
complex cargoes that include proteins, lipids, and nucleic acids
(61). EVs are released from a
variety of cells and act on distant target cells, facilitating
paracrine or endocrine intercellular communication (61). EVs can be classified into different
sub-types according to latest guidelines of ‘Minimal information
for studies of extracellular vesicles’ (62). Small extracellular vesicles,
typically with a diameters of <200 nm, are generated within the
endosomal compartment region referred to as the multivesicular body
(MVB), which must fuse with the plasma membrane in order to be
released (61). Microvesicles, on
the other hand, range in size from 50–1,000 nm and are shed
directly from the plasma membrane through exocytosis (62). Finally, apoptotic bodies are even
larger vesicles (1–5 µm) produced as a consequence of the
fragmentation of apoptotic cells (62). The term ‘exosome’, although widely
used, refers specifically to an EV of ~100 nm in diameter, formed
as an intraluminal vesicle within an MVB and released outside the
cell through the process of exocytosis. Essentially, an exosome is
a specific type of EV, but not all EVs can be classified as
exosomes (63).
It has been well established that, depending on the
type of cargo, EVs released by cancer cells can regulate their
growth, survival and dissemination to distant sites (64). For example, tumor-derived EVs can
transport metalloproteinases that facilitate invasion of cancer
cells through remodeling of the extracellular matrix (65). This process is accompanied by
epithelial-mesenchymal transition, which enhances migration and
invasiveness of cancer cells (66).
In addition, EVs can provide spatial information for the formation
of new blood vessels in tumor niches (67). Notably, some tumor-derived EVs
display in their surface the protein programmed death ligand 1
(PD-L1), which facilitates the development of an immunosuppressive
tumor microenvironment, thereby allowing the tumor cells to escape
the detection of the adaptive immune system (68). These findings highlight the crucial
role of EVs in shaping the tumor microenvironment and in
establishing a selection mechanism that ultimately increases
intratumoral heterogeneity. Overall, tumor-derived EVs are emerging
as important mediators of paracrine, autocrine and endocrine
communication by virtue of their capacity to transport various
cargoes that affect the behavior of target cells (64). This communication promotes
interactions between cancer cells and non-cancerous cells,
explaining, at least in part, how tumor cells exert distant effects
on other tissues (69), including
muscle cells in the context of cancer-associated cachexia.
A study shed light on the role of tumor-derived EVs
in driving catabolism in tissues distant from the primary tumor in
the context of cachexia (69). For
example, C2C12 myoblasts exposed to conditioned-medium containing
EVs derived from an AH-130 hepatoma cells exhibited delayed
differentiation, marked by a reduced expression of Myogenin and
Myosin Heavy Chain (MHC), as well as an overall decrease in protein
synthesis compared to controls (70). Additionally, tumor-derived EVs
carrying miR-21a-5p and miR-148a-3p enhance mitophagy in muscle
cells by upregulating Bcl-2 interacting protein 3 (Bnip3)
expression, leading to reduced mRNA levels of PPARGC1A (70), a gene encoding peroxisome
proliferator-activated receptor gamma coactivator 1α (PGC-1α) that
particularly regulates mitochondrial biogenesis (71). This suggests that an increased tumor
burden triggers an energy crisis that prompts a shift from an
oxidative to glycolytic metabolism, contributing to significant
mitochondrial dysfunction and to an increased lactate production in
muscle cells (70). In a study
using a mouse model of esophageal squamous cell carcinoma-induced
cachexia, the authors observed that tumor-derived EVs containing
the beta subunit of prolyl 4-hydroxylase (P4HB), a protein
disulfide isomerase (PDI) that can trigger cell death upon
accumulation in response to misfolded proteins, could activate
caspase-3 and caspase-8 in C2C12 myoblasts, accelerating the
development of muscle wasting (72). Likewise, differentiated C2C12
myotubes treated with P4HB-containing EVs displayed reduced
diameters and a downregulation in the expression of MHC, while the
levels of MURF1, as well as the autophagy marker LC3, were
concomitantly elevated (72).
Finally, inoculating esophageal cancer cells overexpressing P4HB in
mice generated a greater loss of body weight and a reduction in the
cross-sectional area of myofibrils (72). Taken together, these observations
suggest that P4HB may play a crucial role in inducing muscle
wasting and therefore represent a key mediator of cachexia.
However, further research is required to fully understand the role
of P4HB in the proteolysis of myofibrils.
Hu et al (73) reported the effects of
IL-6-containing exosomes released by Lewis lung carcinoma cells on
muscle wasting. Notably, the authors noted that the activation of
the STAT3-dependent proteolytic pathway initiated by ligand binding
to IL6R resulted in atrophy of C2C12-derived myotubes, as well as
adipocyte lipolysis. Similarly, it has been reported that EVs
derived from a Lewis lung carcinoma cells contain the chaperone
proteins HSP70 and HSP90, which, once incorporated into C2C12
myotubes, activate p38MAPK, resulting in an increased expression of
the ubiquitin ligases Atrogin1 and UBR2 (74). Accordingly, the inhibition of HSP70
and HSP90 reduces the catabolic response triggered by Lewis lung
carcinoma cells in C2C12 myotubes, as evidenced by the
downregulation of the ubiquitin ligases Atrogin1/MAFbx and UBR2, as
well as a reduced loss of myofibrillar protein (74). Finally, exosomes derived from colon
adenocarcinoma cells containing Growth Differentiation Factor 15
(GDF-15) have also been reported to contribute to muscle wasting
(75). In a study by Zhang et
al (75), myocytes exposed to
GDF-15-containing exosomes displayed a reduced expression of Bcl-2
and an increased expression of BAX which is associated with
activation of caspase 3 and apoptosis, thereby promoting loss of
muscle mass.
Kuang et al (76) reported that small EVs released from
murine colon carcinoma-26 cells were enriched in miR-183-5p, a
miRNA associated with a reduction in the diameter in C2C12-derived
myotubes. In addition, it was observed that miR-183-5p, through
activation of the Myostatin/Smad3 signaling pathway, upregulates
the expression of MuRF-1 and Atrogin-1, promoting the degradation
of proteins in myotubes. These responses were accompanied by a
significant decrease in basal respiration and ATP production
(76). It has also been observed
that miR-195a-5p- and miR-125b-1-3p-containing EVs derived from
colon adenocarcinoma cells can decrease Bcl-2 expression and induce
apoptosis in muscle cells through caspase 3 activation (77). Another microRNA involved in muscle
wasting is miR-122, found in EVs derived from MDA-MB-231 breast
cancer cells. When added to C2C12-derived myotubes, these EVs
induce a reduction in the expression of the tumor suppressor p53,
Tfam, PGC-1α, SCO2 and MFN2, involved in negative regulation of
mitochondrial function, thus leading to increased production of
ROS, reduced ATP levels, decreased mitochondrial content and
altered energy production (78).
This may suggest that the induction of mitophagy upon inhibition of
p53 might play a role in the disruption of energy homeostasis and
mitochondrial regulation (79). Of
note, while the effects of p53 inhibition in skeletal muscle were
observed under stress conditions, there is no evidence for the
relevance of this pathway in the context of muscle wasting observed
in cancer-associated cachexia. Similarly, it has been shown that
miR-122-containing EVs released by MDA-MB-231 breast cancer cells
leads to a reduction in cytosolic O-GlcNAcylation with an
upregulation of ryanodine receptor 1 (RyR1), a calcium release
channel located in the sarcoplasmic reticulum of skeletal muscles,
and the calcium-activated protease Calpain, ultimately promoting
the calcium-dependent proteolysis of gastrocnemius myofibrillar
proteins in female nonobese diabetic-scid-IL-2γ chain receptor null
mice (80). On the other hand, it
has been observed that myeloma-derived EVs contain receptors for
advanced glycation endpoints (RAGE), which facilitate muscle
wasting of myotubes through the activation of the Toll-like
receptor 4/NF-κB p65 pathway (81).
Taken together, these data seem to indicate that EVs
released by tumor cells may primarily induce mitochondrial
dysfunction and an enhancement of glycolytic metabolism in skeletal
muscle cells, leading to an energy imbalance that activate the
Ubiquitin-Proteasome and/or autophagy, ultimately ending with the
degradation of myofibrillar units that is typical of muscle wasting
(Fig. 2). So far, however, little
is known about signaling pathways that tumor-derived EVs activate
or repressed in target cells (such as skeletal muscle cells).
Similarly, we have a rather incomplete understanding of the cargoes
carried by tumor-derived EVs that render them more or less likely
to induce degradation of muscle components (Table I).
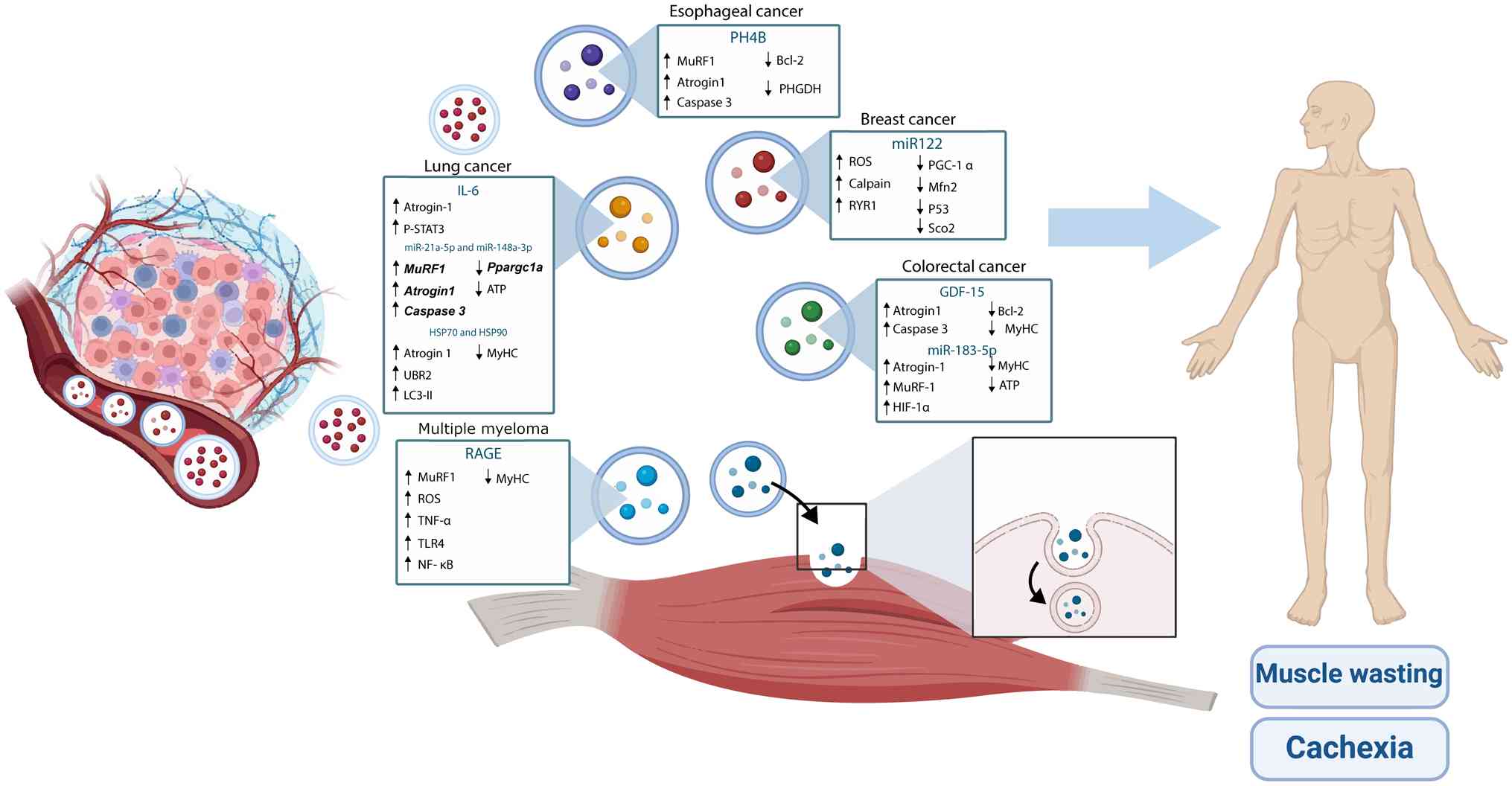 | Figure 2.Characteristics of tumor EVs in
cachectic syndrome. EVs, extracellular vesicles. ATP, adenosine
triphosphate; Atrogin-1 (MAFbx), muscle atrophy f-box protein;
Bcl-2, B-cell lymphoma 2; Caspase 3, cysteine-aspartic protease 3;
GDF-15, growth differentiation factor 15; HIF-1α, hypoxia-inducible
factor 1α; HSP90, heat shock protein 90; IL-6, interleukin-6;
LC3-II, microtubule-associated protein light chain 3-II;
miR-21a-5p, microRNA-21a-5p; Mfn-2, Mitofusin-2; MuRF1, muscle ring
finger protein 1; MyHC, myosin heavy chain; NF-κB, nuclear factor
κ-light-chain-enhancer of activated B cells; PGC-1α, peroxisome
proliferator-activated receptor gamma coactivator 1α; PHGDH,
phosphoglycerate dehydrogenase; PPARGC1A (Ppargc1a), gene encoding
PGC-1α; RAGE, receptor for advanced glycation end products; ROS,
reactive oxygen species; RyR1, ryanodine receptor 1; SOD2,
superoxide dismutase 2; STAT3/p-STAT3, signal transducer and
activator of transcription 3 (phosphorylated); TLR4, toll-like
receptor 4; TNF-α, tumor necrosis factor α; UBR2, ubiquitin protein
ligase E3 component N-recognin 2. |
 | Table I.Studies evaluating the effect of EVs
tumor-derived on skeletal muscle. |
Table I.
Studies evaluating the effect of EVs
tumor-derived on skeletal muscle.
| First author/s,
year | Cells of
origin | Origin of the
EVs | Isolation of
EVs | Characteristic of
EVs | Treatment in in
vitro and in vivo models | Results | (Refs.) |
|---|
| Hu et al,
2019 | •Lewis lung | •Culture
medium |
•Ultracentrifugation | •Diameter: | In vivo
models | In vivo
models | (73) |
|
| carcinoma
cells. | •Human serum |
| 106.4 nm | •Male Lean C57BL
mice | •Body weight
(↓) |
|
|
| •C57BL mice | and murine
serum |
| •CD9 | •6 to 10 weeks
old | •Muscle mass
TA, |
|
|
|
|
|
| •TSG101 | •Subcutaneous
LLC | GN, quadriceps
(↓) |
|
|
|
|
|
| •HSP70 | injection | •Atrogin 1 (↑) |
|
|
|
|
|
| •IL-6 | •Sacrificed 28 days
after | •P-STAT3 (↑) |
|
|
|
|
|
|
| tumor
implantation. | •Myotube diameter
(↓) |
|
|
|
|
|
|
| In vitro
models | In vitro
models |
|
|
|
|
|
|
| •Conditioned
culture | •Atrogin1 (↑) |
|
|
|
|
|
|
| medium for
murine | •P-STAT3/STAT3
(↑) |
|
|
|
|
|
|
| C2C12 myotubes |
|
|
|
|
|
|
|
| •Concentration of 5
to |
|
|
|
|
|
|
|
| 20 µg of EVs |
|
|
|
|
|
|
|
| •EVs labeled with
PKH67 |
|
|
|
|
|
|
|
|
internalization |
|
|
|
|
|
|
|
| •Analysis at 12 h
and 24 h |
|
|
|
|
|
|
|
| of treatment |
|
|
| Gao et al,
2021 | •Squamous cell | •Culture
medium |
•Ultracentrifugation | •Diameter: | In vivo
models | In vivo
models | (72) |
|
| carcinoma of |
| •ExoQuickTC | 30 −150 nm | •Male BALB/c nude
and | •Body weight
(↓) |
|
|
| the esophagus. |
| Exosome
Isolation | •ALIX | C57BL/6J mice | •Muscle mass GA
(↓) |
|
|
| •Human |
| reagent kit | •FLOT-1 | •6 weeks old | •Myod1
(↓) |
|
|
|
|
|
| •TSG101 | •ESCC 100 µl
subcutaneous | •Atrogin1
(↑) |
|
|
|
|
|
| •P4HB | injection. | •MURF1
(↑) |
|
|
|
|
|
|
| •Sacrificed 18 days
after | •MURF1 (↑) |
|
|
|
|
|
|
| tumor
implantation. | •PHGDH (↓) |
|
|
|
|
|
|
| In vitro
models | •Cleaved caspase-3
(↑) |
|
|
|
|
|
|
| •Conditioned
culture | In vitro
models |
|
|
|
|
|
|
| medium for
C2C12 | •MURF1 (↑) |
|
|
|
|
|
|
| murine
myoblasts | •PHGDH (↓) |
|
|
|
|
|
|
| •Concentration of
10 µg | •Bcl-2 (↓) |
|
|
|
|
|
|
| of EVs | •Cleaved caspase-3
(↑) |
|
|
|
|
|
|
| •Analysis after 24
h of | •Bax (↑) |
|
|
|
|
|
|
| treatment |
|
|
|
|
|
|
|
| •EVs marked with
PKH67- |
|
|
|
|
|
|
|
|
Internalization |
|
|
| Miao et
al, | •Murine colon | •Culture
medium | •ExoQuick-TC | •Diameter: | In vivo
models | In vivo
models | (77) |
| 2021 | carcinoma
cells | and mouse
serum | Exosome
Isolation | ~100 nm | •BALB/c and C57BL/6
mice | •Grip strength
(↓) |
|
|
| C26 |
| reagent kit | •miR-195a-5p | •6-8 weeks | •Body weight
(↓) |
|
|
|
|
|
| •miR-125b- | •Subcutaneous
injection | •Bcl-2(↓) |
|
|
|
|
|
| 1-3p | 1×106 of
C26 cells | •Muscle mass TA
(↓) |
|
|
|
|
|
|
| •Intramuscular
injection | In vitro
models |
|
|
|
|
|
|
| of C26 EVs (GN,
TA) | •Myotube diameter
(↓) |
|
|
|
|
|
|
| •Sacrificed 16 and
24 days | •MyHC (↓) |
|
|
|
|
|
|
| after tumor
implantation | •MURF1 (↑) |
|
|
|
|
|
|
| In vitro
models | •Bcl-2(↓) |
|
|
|
|
|
|
| •Conditioned
culture | •Cleaved caspase-3
(↑) |
|
|
|
|
|
|
| medium for
myoblasts |
|
|
|
|
|
|
|
| and murine
myotube |
|
|
|
|
|
|
|
| C2C12 |
|
|
|
|
|
|
|
| •Concentration of
50 µg of |
|
|
|
|
|
|
|
| EVs |
|
|
|
|
|
|
|
| •EVs marked with
PKH67- |
|
|
|
|
|
|
|
|
Internalization |
|
|
|
|
|
|
|
| •Analysis after 48
h of |
|
|
|
|
|
|
|
| treatment |
|
|
| Pin et al,
2022 | •Murine Lewis | •Culture
medium |
•Ultracentrifugation | •Diameter: | In vivo
models | In vivo
models | (70) |
|
| lung carcinoma | and
murine/rats |
| ND | •Male Wistar
rats | •Muscle mass
GN, |
|
|
| cells | plasma |
| •miR-21a-5p | •Subcutaneous
injection | TA (↓) |
|
|
| •Murine colon |
|
| •miR-148a-3p | 5×105 of
C26 cells |
•Becn1/Beclin1 (↑) |
|
|
| carcinoma
cells |
|
|
| •Sacrificed on day
7 and 14 |
•MURF1(↑) |
|
|
| C26 |
|
|
| after tumor
implantation | •Atrogin1
(↑) |
|
|
|
|
|
|
| In vitro
models | In vitro
models |
|
|
|
|
|
|
| •Conditioned
culture |
•MURF1(↑) |
|
|
|
|
|
|
| medium for murine
myo- | •Atrogin1
(↑) |
|
|
|
|
|
|
| tube and myoblast
C2C12 | •Caspase 3 (↑) |
|
|
|
|
|
|
| •Analysis at 2, 4
and 6 days | •Myh7
(↓) |
|
|
|
|
|
|
| of treatment
(myoblast) | •Bnip3
(↑) |
|
|
|
|
|
|
| •Analysis at 24, 48
and 72 h | •Lactate (↑) |
|
|
|
|
|
|
| of treatment
(myotube) | •Ppargc1a
(↓) |
|
|
|
|
|
|
|
| •ATP (↓) |
|
| Yan et al,
2022 | •Triple
negative | •Culture
medium |
•Ultracentrifugation | •Diameter: | In vivo
models | In vivo
models | (80) |
|
| breast cancer |
|
| ND | •15-week-old NSG
mice | •O- |
|
|
| cells |
|
| •CD9 | •Intravenous EV
injection |
GlucosilNAcetilación |
|
|
| •MDA-MB-231 |
|
| •CD63 | •~10 µg of EVs for
5 weeks | RYR1 (↓) |
|
|
| •Human | |
| •CD81 | (2× weeks) | •Muscle mass
GN- |
|
|
|
|
|
| •miR-122 | In vitro
models | TA (↓) |
|
|
|
|
|
|
| •Conditioned
culture | •Muscle
Contrac- |
|
|
|
|
|
|
| medium for murine
C2C12 | tility(↓) |
|
|
|
|
|
|
| myotube. | •Desmin
filament |
|
|
|
|
|
|
| •Concentration of ~
2 µg of | cleavage (↑) |
|
|
|
|
|
|
| EVs | •Vimentin
filament |
|
|
|
|
|
|
|
| cleavage (↑) |
|
|
|
|
|
|
|
| In vitro
models |
|
|
|
|
|
|
|
| •RYR1 (↑) |
|
|
|
|
|
|
|
| •Calpain (↑) |
|
|
|
|
|
|
|
| •Vimentin
filament |
|
|
|
|
|
|
|
| cleavage (↑) |
|
|
|
|
|
|
|
| •Desmin
filament |
|
|
|
|
|
|
|
| cleavage (↑) |
|
| Liu et al,
2022 | •Murine Lewis | •Culture
medium | •ExoQuick-TC | •Diameter: | In vivo
models | In vivo
models | (74) |
|
| lung carcinoma |
| Exosome
Isolation | ND | •8-week-old male
C57BL/6 | •Body weight
(↓) |
|
|
| cells | | reagent kit | •HSP70 | mice | •Muscle mass
TA, |
|
|
|
|
|
| •HSP90 | •Subcutaneous
injection | EDL (↓) |
|
|
|
|
|
|
| 1×106 of
LLC | •Grip strength
(↓) |
|
|
|
|
|
|
| •Sacrificed on day
21 | •MyHC (↓) |
|
|
|
|
|
|
| In vitro
models | •LC3-II (↑) |
|
|
|
|
|
|
| •Conditioned
culture | In vitro
models |
|
|
|
|
|
|
| medium for murine
C2C12 | •Atrogin1/MAFbx
(↑) |
|
|
|
|
|
|
| myotubes. | • p38 MAPK (↑) |
|
|
|
|
|
|
| •48 h of
treatment. | •Atrogin 1 (↑) |
|
|
|
|
|
|
|
| •UBR2 (↑) |
|
|
|
|
|
|
|
| •MyHC (↓) |
|
|
|
|
|
|
|
| •Myotube diameter
(↓) |
|
| Zhang et
al, | •Murine colon | •Culture
medium | •ExoQuick-TC | •Diameter: | In vivo
models | In vivo
models | (75) |
| 2022 | carcinoma
cells |
| Exosome
Isolation | 30 −150 nm | •Male BALB/c
andC57BL/ | • MyHC (↓) |
|
|
| C26 and MC38 |
| reagent kit | •CD9 | 6 J mice | •Myotube diameter
(↓) |
|
|
|
|
|
| •CD63 | •6-8 weeks | •Bcl-2/Bax (↓) |
|
|
|
|
|
| •CD81 | •Subcutaneous
injection of | •Atrogin1 (↑) |
|
|
|
|
|
| •TSG101 | C26 tumor
cells | •Cleaved
caspase-3/ |
|
|
|
|
|
| •GDF-15 | •Sacrificed on day
16 and | caspase-3 (↑) |
|
|
|
|
|
|
| day 22 | In vitro
models |
|
|
|
|
|
|
| In vitro
models | •MyHC (↓) |
|
|
|
|
|
|
| •Conditioned
culture | •Bcl-2/Bax (↓) |
|
|
|
|
|
|
| medium for murine
C2C12 |
•Caspase-3/cleaved |
|
|
|
|
|
|
| myotubes. | caspase-3 (↑) |
|
|
|
|
|
|
| •48 h of
treatment |
|
|
| Kuang et
al, | •Murine colon | •Culture
medium | •ExoQuick-TC | •Diameter: | In vitro
models | In vitro
models | (76) |
| 2022 | cancer C26 and |
| Exosome
Isolation | ND | •Conditioned
culture | •MyHC (↓) |
|
|
| MC38 | | reagent kit | •miR-183-5p | medium for C2C12
murine | •FHL1 (↓) |
|
|
|
|
|
|
| myotubes | •HIF-1α (↑) |
|
|
|
|
|
|
| •Concentration of
50 µg of | •Atrogin-1 (↑) |
|
|
|
|
|
|
| EVs | •MuRF-1 (↑) |
|
|
|
|
|
|
| •From the fourth
day of | •ATP (↓) |
|
|
|
|
|
|
|
differentiation |
|
|
|
|
|
|
|
| •48 h of
treatment |
|
|
| Ruan et
al, | •Triple
negative | •Culture
medium |
•Ultracentrifugation | •Diameter: | In vivo
models | In vivo
models | (78) |
| 2023 | breast cancer |
|
| ND | •8-week-old female
NSG | •p53 (↓) |
|
|
| cell line MDA- |
|
| •miR-122 | mice | •mtDNA (↓) |
|
|
| MB-231 |
|
|
| •Intravenous EVs
injection | •ROS (↑) |
|
|
| •Human |
|
|
| ~10 µg | •mitochondrial |
|
|
|
| |
|
| •For 5 weeks (2 ×
weeks) | contents (↓) |
|
|
|
|
|
|
| In vitro
models | •Pgc-1 α (↓) |
|
|
|
|
|
|
| •Conditioned
culture | •Mfn2 (↓) |
|
|
|
|
|
|
| medium for murine
C2C12 | •Sco2 (↓) |
|
|
|
|
|
|
| myotubes. | •Distance traveled
(↓) |
|
|
|
|
|
|
| •Concentration of 2
µg of | In vitro
models |
|
|
|
|
|
|
| EVs for 105 cells
cultured | •p53 (↓) |
|
|
|
|
|
|
| in 2 ml of
medium. | •ATP (↓) |
|
|
|
|
|
|
| •24 h of
treatment | •ROS (↑) |
|
|
|
|
|
|
|
| •Pgc-1 α (↓) |
|
|
|
|
|
|
|
| •Mfn2 (↓) |
|
|
|
|
|
|
|
| •Sco2 (↓) |
|
| Wu et al,
2024 | •Multiple | •Culture
medium |
•Ultracentrifugation | •Diameter: | In vivo
models | In vivo
models | (81) |
|
| myeloma |
|
| 65-70 nm | •6–8 weeks male
BALB/c | •Body weight
(↓) |
|
|
| •Murine Sp2/0- |
|
| •CD9 | mice | •Muscle mass
TA, |
|
|
| Ag14 cell line | |
| •CD63 | •Subcutaneous
injection of | GA (↓) |
|
|
|
|
|
| •CD81 | Sp2/0-Ag14 tumor
cells | •CSA of muscle |
|
|
|
|
|
| •TSG101 | •Injected into the
TA | fiber (↓) |
|
|
|
|
|
| •RAGE | muscles 400 µg of
EVs | •Grip strength
(↓) |
|
|
|
|
|
|
| Sacrificed on day
22 | •Serum levels ROS
(↑) |
|
|
|
|
|
|
| In vitro
models | •Serum levels |
|
|
|
|
|
|
| •Conditioned
culture | TNF-α (↑) |
|
|
|
|
|
|
| medium for murine
C2C12 | •Serum levels IL-6
(↑) |
|
|
|
|
|
|
| myotubes. | In vitro
models |
|
|
|
|
|
|
| •Concentration 50
µg of | •ROS (↑) |
|
|
|
|
|
|
| EVs | •MuRF-1 (↑) |
|
|
|
|
|
|
| •24 h of
treatment | •MyHC (↓) |
|
|
|
|
|
|
|
| •IL-6 (↑) |
|
|
|
|
|
|
|
| •TNF-α (↑) |
|
|
|
|
|
|
|
| •TLR4 (↑) |
|
|
|
|
|
|
|
| •NF- κB (↑) |
|
Clinical and therapeutic implications of
tumor-derived EVs in cachectic syndrome
Currently, the clinical approach to cachexia
involves a nutritional intervention based on the administration of
food supplements, with enteral or parenteral nutrition depending on
the clinical condition of the patient (82). This nutritional approach can be
complemented with drugs such as Anamorelin, a potent and specific
agonist of the ghrelin receptor that markedly stimulates appetite
in patients with cancer (83).
Nutritional and pharmacological approaches can both be effective in
increasing body weight; however, no improvements in survival have
been reported (82). Notably, Fan
et al (84) showed that CT26
colon cancer cells treated with atractylenolide I, a component of a
Chinese medicinal herb, produced and released fewer EVs, which was
associated with an increase in food intake and an attenuation in
the decrease of body weight in tumor-carrying BALB/c mice. Another
study assessed the effects of the drug Amiloride (an inhibitor of
Na/H and Na/K exchangers) on colon cancer cell lines (CT26) and
primary Lewis lung carcinoma cells. The authors observed a
significant reduction in the release of exosomes and reported
drastic decreases in the plasma density of exosomes in a murine
model (85).
Notably, a nanovesicular drug called Physiactisome
has been recently patented. The drug is made up of vesicles
containing the 60 kDa heat shock protein Hsp60, derived from
HSPD1-overexpressing C2C12 cells (86). Although there is still no evidence
regarding the effectiveness of Physiactisome, it is hypothesized
that it could improve the expression of PGC-1α in cachectic muscle,
increase mitochondrial biogenesis, thus reducing the generation of
ROS and decreasing muscle waste (86). Furthermore, EVs have therapeutic
potential when they contain IL-6 signal transducer decoy receptors,
as they can inhibit STAT3 phosphorylation in skeletal muscle and
reverse myotube reduction in the C2C12 cell line. This may
contribute to reducing the activation of inflammatory cytokine
signaling pathways and decreasing NF-κB translocation in cachectic
syndrome (87). These preliminary
data suggest the possibility of using EVs as therapeutic vehicles
to counteract muscle wasting that occurs in cachexia or,
alternatively, reduce the secretion of EVs released by tumor cells.
Furthermore, nanovesicles can be generated to deliver drugs and/or
proteins that may improve myofibrillar protein synthesis.
Conclusions and future perspectives
Cachexia-associated muscle wasting is driven by
pro-inflammatory and catabolic factors released by the tumor cells
or stromal cells in response to tumor cells. These factors create
an energy imbalance, activating the ubiquitin-proteasome signaling
pathway while, at the same time, suppressing the PI3K/AKT/mTOR
pathway. This cascade exacerbates myofibril proteolysis and
autophagy and reduces protein synthesis. Despite the considerable
amount of information available concerning the mechanisms of muscle
wasting in cancer-associated cachexia, there is still rather
limited evidence available on the role of tumor-derived EVs in
these processes. Skeletal muscle mass markedly affects cancer
patient prognosis, survival rates and treatment effectiveness.
Therefore, investigating the mechanisms through which EVs
contribute to tumor cells-muscle cells crosstalk in different
malignancies is clinically relevant. It is considered that such a
research program will lead to the development of novel therapeutic
strategies aimed to improve the survival of patients with cancer
with muscle wasting. Future research should focus on identifying
systemic molecular markers associated with different stages of the
cachectic syndrome and muscle wasting. Insights gained from such
studies could help identify prognostic factors and enhance early
pre-cachexia interventions to improve cancer patient functionality,
quality of life and to reduce treatment-related side effects.
Acknowledgements
The authors would like to thank Miss Consuelo
Berríos-Contreras of the Corporate Communications Department,
University of Talca (Talca, Chile) for the digitalization of the
figures.
Funding
The present study was supported by the ANID-FONDECYT-Regular
(grant no. 1210644) and Fondo de Investigación Avanzada en Áreas
Prioritarias (grant nos. 15130011 and 1523A0008) (all to AFGQ).
Availability of data and materials
Not applicable.
Authors' contributions
LBC, MMV, NB, CV and RMC conceived the study and
wrote the manuscript. LBC conducted the literature search/selection
and data extraction. AFGQ, SB and FLS revised and edited the
manuscript. All authors read and approved the final manuscript.
Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Williams MJ, Sottoriva A and Graham TA:
Measuring clonal evolution in cancer with genomics. Annu Rev
Genomics Hum Genet. 20:309–329. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Ervik M, Lam F, Colombet M, Mery
L and Piñeros M: Global cancer observatory: Cancer today. Int
Agency Res Cancer; Lyon: 2019
|
|
3
|
Dolly A, Dumas JF and Servais S: Cancer
cachexia and skeletal muscle atrophy in clinical studies: What do
we really know? J Cachexia Sarcopenia Muscle. 11:1413–1428. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun L, Quan XQ and Yu S: An
epidemiological survey of cachexia in advanced cancer patients and
analysis on its diagnostic and treatment status. Nutr Cancer.
67:1056–1062. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vagnildhaug OM, Balstad TR, Almberg SS,
Brunelli C, Knudsen AK and Kaasa S: A cross-sectional study
examining the prevalence of cachexia and areas of unmet need in
patients with cancer. Support Care Cancer. 26:1871–1880.
2018.PubMed/NCBI
|
|
6
|
Fearon K: Definition and classification of
cancer cachexia: An international consensus. Lancet Oncol.
12:489–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schmidt SF, Rohm M, Herzig S and Berriel
Diaz M: Cancer cachexia: More than skeletal muscle wasting. Trends
Cancer. 4:849–860. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pathan M, Fonseka P, Chitti SV, Kang T,
Sanwlani R, Van Deun J, Hendrix A and Mathivanan S: Vesiclepedia
2019: A compendium of RNA, proteins, lipids and metabolites in
extracellular vesicles. Nucleic Acids Res. 47:D516–D519. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang X, Zhao Y and Yan W: The role of
extracellular vesicles in skeletal muscle wasting. J Cachexia
Sarcopenia Muscle. 14:2462–2472. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi Y, Liu Z, Lin Q, Luo Q, Cen Y, Li J,
Fang X and Gong C: MiRNAs and cancer: Key link in diagnosis and
therapy. Genes (Basel). 12:12892021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marinho R, Alcântara PSM, Ottoch JP and
Seelaender M: Role of exosomal microRNAs and myomiRs in the
development of cancer cachexia-associated muscle wasting. Front
Nutr. 4:692018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Argilés JM, Stemmler B and López-Soriano
FJ: Inter-tissue communication in cancer cachexia. Nat Rev
Endocrinol. 15:9–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tisdale MJ: Cachexia in cancer patients.
Nat Rev Cancer. 2:862–871. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohmae N, Yasui-Yamada S, Furumoto T, Wada
K, Hayashi H, Kitao M, Yamanaka A, Kubo M, Matsuoka M, Kamimura S,
et al: Muscle mass, quality, and strength; physical function and
activity; and metabolic status in cachectic patients with head and
neck cancer. Clin Nutr ESPEN. 53:113–119. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stewart GD, Skipworth RJ and Fearon KC:
Cancer cachexia and fatigue. Clin Med (Lond). 6:140–143. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang YF, An ZY, Lin DH and Jin WL:
Targeting cancer cachexia: Molecular mechanisms and clinical study.
MedComm (2020). 3:e1642022. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yeom E and Yu K: Understanding the
molecular basis of anorexia and tissue wasting in cancer cachexia.
Exp Mol Med. 54:426–432. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Law ML: Cancer cachexia: Pathophysiology
and association with cancer-related pain. Front Pain Res.
3:9712952022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishida A and Andoh A: The role of
inflammation in cancer: Mechanisms of tumor initiation,
progression, and metastasis. Cells. 14:4882025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang M, Chen S, He X, Yuan Y and Wei X:
Targeting inflammation as cancer therapy. J Hematol Oncol.
17:132024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khandia R and Munjal A: Interplay between
inflammation and cancer. Adv Protein Chem Struct Biol. 119:199–245.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Briukhovetska D, Dörr J, Endres S, Libby
P, Dinarello CA and Kobold S: Interleukins in cancer: From biology
to therapy. Nat Rev Cancer. 21:481–499. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inácio Pinto N, Carnier J, Oyama LM, Otoch
JP, Alcântara PS, Tokeshi F and Nascimento CM: Cancer as a
proinflammatory environment: Metastasis and cachexia. Mediators
Inflamm. 2015:7910602015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Castro GS, Correia-Lima J, Simoes E,
Orsso CE, Xiao J, Gama LR, Gomes SP, Gonçalves DC, Costa RGF,
Radloff K, et al: Myokines in treatment-naïve patients with
cancer-associated cachexia. Clin Nutr. 40:2443–2455. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rupert JE, Narasimhan A, Jengelley DHA,
Jiang Y, Liu J, Au E, Silverman LM, Sandusky G, Bonetto A, Cao S,
et al: Tumor-derived IL-6 and trans-signaling among tumor, fat, and
muscle mediate pancreatic cancer cachexia. J Exp Med.
218:e201904502021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sartori R, Hagg A, Zampieri S, Armani A,
Winbanks CE, Viana LR, Haidar M, Watt KI, Qian H, Pezzini C, et al:
Perturbed BMP signaling and denervation promote muscle wasting in
cancer cachexia. Sci Transl Med. 13:eaay95922021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sartori R, Schirwis E, Blaauw B,
Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L,
Ferry A, et al: BMP signaling controls muscle mass. Nat Genet.
45:1309–1318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rusin A, Seymour C, Cocchetto A and
Mothersill C: Commonalities in the features of cancer and chronic
fatigue syndrome (CFS): Evidence for stress-induced phenotype
instability. Int J Mol Sci. 23:6912022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smith SM and Vale WW: The role of the
hypothalamic-pituitary-adrenal axis in neuroendocrine responses to
stress. Dialogues Clin Neurosci. 8:383–395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laird BJ, McMillan D, Skipworth RJE,
Fallon MT, Paval DR, McNeish I and Gallagher IJ: The emerging role
of interleukin 1β (IL-1β) in cancer cachexia. Inflammation.
44:1223–1228. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grossberg AJ, Scarlett JM, Zhu X, Bowe DD,
Batra AK, Braun TP and Marks DL: Arcuate nucleus
proopiomelanocortin neurons mediate the acute anorectic actions of
leukemia inhibitory factor via gp130. Endocrinology. 2:606–616.
2010. View Article : Google Scholar
|
|
32
|
Li W, Moylan JS, Chambers MA, Smith J and
Reid MB: Interleukin-1 stimulates catabolism in C2C12 myotubes. Am
J Physiol Cell Physiol. 297:C706–C714. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Patel HJ and Patel BM: TNF-α and cancer
cachexia: Molecular insights and clinical implications. Life Sci.
170:56–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ohmori H, Kawahara I, Mori T, Nukaga S,
Luo Y, Kishi S, Fujiwara-Tani R, Mori S, Goto K, Sasaki T and
Kuniyasu H: Evaluation of parameters for cancer-induced sarcopenia
in patients autopsied after death from colorectal cancer.
Pathobiology. 86:306–314. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li L, Liu H, Tao W, Wen S, Fu X and Yu S:
Pharmacological inhibition of HMGB1 prevents muscle wasting. Front
Pharmacol. 12:7313862021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Powrózek T, Pigoń-Zając D, Mazurek M,
Ochieng Otieno M, Rahnama-Hezavah M and Małecka-Massalska T:
TNF-α-induced myotube atrophy in C2C12 cell line uncovers putative
inflammatory-related lncRNAs mediating muscle wasting. Int J Mol
Sci. 23:38782022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Murai M, Higashiguchi T, Futamura A, Ohara
H, Tsuzuki N, Itani Y, Kaneko T, Chihara T, Shimpo K and Nakayama
N: Interleukin-8 and clinical symptoms can be prognostic indicators
for advanced cancer patients with cachexia. Fujita Med J.
6:117–121. 2020.PubMed/NCBI
|
|
38
|
Callaway CS, Delitto AE, Patel R, Nosacka
RL, D'Lugos AC, Delitto D, Deyhle MR, Trevino JG, Judge SM and
Judge AR: IL-8 released from human pancreatic cancer and
tumor-associated stromal cells signals through a CXCR2-ERK1/2 axis
to induce muscle atrophy. Cancers (Basel). 11:18632019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rex DAB, Dagamajalu S, Gouda MM, Suchitha
GP, Chanderasekaran J, Raju R, Prasad TSK and Bhandary YP: A
comprehensive network map of IL-17A signaling pathway. J Cell
Commun Signal. 17:209–215. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Picard FSR, Lutz V, Brichkina A, Neuhaus
F, Ruckenbrod T, Hupfer A, Raifer H, Klein M, Bopp T, Pfefferle PI,
et al: IL-17A-producing CD8+ T cells promote PDAC via induction of
inflammatory cancer-associated fibroblasts. Gut. 72:1510–1522.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu M, Qian XX, Li G, Cheng Z and Lin Z:
Prognostic biomarker IL17A correlated with immune infiltrates in
head and neck cancer. World J Surg Oncol. 20:2432022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ying L, Yao Y, Lv H, Lu G, Zhang Q, Yang Y
and Zhou J: IL-17A contributes to skeletal muscle atrophy in lung
cancer-induced cachexia via JAK2/STAT3 pathway. Am J Physiol Cell
Physiol. 322:C814–C824. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pang X, Zhang P, Chen X and Liu W:
Ubiquitin-proteasome pathway in skeletal muscle atrophy. Front
Physiol. 14:12895372023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Wang J and Wang X, Gao T, Tian H,
Zhou D, Zhang L, Li G and Wang X: The autophagic-lysosomal and
ubiquitin proteasome systems are simultaneously activated in the
skeletal muscle of gastric cancer patients with cachexia. Am J Clin
Nutr. 111:570–579. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Peris-Moreno D, Cussonneau L, Combaret L,
Polge C and Taillandier D: Ubiquitin ligases at the heart of
skeletal muscle atrophy control. Molecules. 26:4072021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xia Q, Huang X, Huang J, Zheng Y, March
ME, Li J and Wei Y: The role of autophagy in skeletal muscle
diseases. Front Physiol. 12:6389832021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Op den Kamp CM, Langen RC, Snepvangers FJ,
de Theije CC, Schellekens JM, Laugs F, Dingemans AM and Schols AM:
Nuclear transcription factor κB activation and protein turnover
adaptations in skeletal muscle of patients with progressive stages
of lung cancer cachexia. Am J Clin Nutr. 98:738–748. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tardif N, Klaude M, Lundell L, Thorell A
and Rooyackers O: Autophagic-lysosomal pathway is the main
proteolytic system modified in the skeletal muscle of esophageal
cancer patients. Am J Clin Nutr. 98:1485–1492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Aversa Z, Pin F, Lucia S, Penna F, Verzaro
R, Fazi M, Colasante G, Tirone A, Rossi Fanelli F, Ramaccini C, et
al: Autophagy is induced in the skeletal muscle of cachectic cancer
patients. Sci Rep. 6:303402016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Johns N, Hatakeyama S, Stephens NA, Degen
M, Degen S, Frieauff W, Lambert C, Ross JA, Roubenoff R, Glass DJ,
et al: Clinical classification of cancer cachexia: Phenotypic
correlates in human skeletal muscle. PLoS One. 9:e836182014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Itoh Y, Saitoh M and Miyazawa K:
Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim
Biophys Sin (Shanghai). 50:82–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thompson TB, Woodruff TK and Jardetzky TS:
Structures of an ActRIIB:Activin A complex reveal a novel binding
mode for TGF-beta ligand:receptor interactions. EMBO J.
22:1555–1566. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J,
et al: FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab. 6:458–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Matsuyama T, Ishikawa T, Okayama T, Oka K,
Adachi S, Mizushima K, Kimura R, Okajima M, Sakai H, Sakamoto N, et
al: Tumor inoculation site affects the development of cancer
cachexia and muscle wasting. Int J Cancer. 137:2558–2565. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Geremia A, Sartori R, Baraldo M, Nogara L,
Balmaceda V, Dumitras GA, Ciciliot S, Scalabrin M, Nolte H and
Blaauw B: Activation of Akt-mTORC1 signalling reverts
cancer-dependent muscle wasting. J Cachexia Sarcopenia Muscle.
13:648–661. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Adams V, Gußen V, Zozulya S, Cruz A,
Moriscot A, Linke A and Labeit S: Small-molecule chemical knockdown
of MuRF1 in melanoma-bearing mice attenuates tumor
cachexia-associated myopathy. Cells. 9:22722020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Niu M, Song S, Su Z, Wei L, Li L, Pu W,
Zhao C, Ding Y, Wang J, Cao W, et al: Inhibition of heat shock
protein 90 reverses STAT3-mediated muscle wasting in cancer
cachexia mice. Br J Pharmacol. 178:4485–4500. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan
R, Puzis L, Koniaris LG and Zimmers TA: JAK/STAT3 pathway
inhibition blocks skeletal muscle wasting downstream of IL-6 and in
experimental cancer cachexia. Am J Physiol Endocrinol Metab.
303:E410–E421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Miller A, McLeod L, Alhayyani S, Szczepny
A, Watkins DN, Chen W, Enriori P, Ferlin W, Ruwanpura S and Jenkins
BJ: Blockade of the IL-6 trans-signalling/STAT3 axis suppresses
cachexia in Kras-induced lung adenocarcinoma. Oncogene.
36:3059–3066. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Guo D, Wang C, Wang Q, Qiao Z and Tang H:
Pantoprazole blocks the JAK2/STAT3 pathway to alleviate skeletal
muscle wasting in cancer cachexia by inhibiting inflammatory
response. Oncotarget. 8:39640–39648. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jeppesen DK, Zhang Q, Franklin JL and
Coffey RJ: Extracellular vesicles and nanoparticles: Emerging
complexities. Trends Cell Biol. 33:667–681. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Welsh JA, Goberdhan DCI, O'Driscoll L,
Buzas EI, Blenkiron C, Bussolati B, Cai H, Di Vizio D, Driedonks
TAP, Erdbrügger U, et al: Minimal information for studies of
extracellular vesicles (MISEV2023): From basic to advanced
approaches. J Extracell Vesicles. 13:e124042024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Saint-Pol J and Culot M: Minimum
information for studies of extracellular vesicles (MISEV) as a
toolbox for rigorous, reproducible and homogeneous studies on
extracellular vesicles. Toxicol In Vitro. 106:1060492025.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu YJ and Wang C: A review of the
regulatory mechanisms of extracellular vesicle-mediated
intercellular communication. Cell Commun Signal. 21:772023.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lopez K, Lai SWT, Lopez Gonzalez EJ,
Dávila RG and Shuck SC: Extracellular vesicles: A dive into their
role in the tumor microenvironment and cancer progression. Front
Cell Dev Biol. 11:11545762023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hoshino A, Costa-Silva B, Shen TL,
Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di
Giannatale A, Ceder S, et al: Tumour exosome integrins determine
organotropic metastasis. Nature. 527:329–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Costa-Silva B, Aiello NM, Ocean AJ, Singh
S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et
al: Pancreatic cancer exosomes initiate pre-metastatic niche
formation in the liver. Nat Cell Biol. 17:816–826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang S, Yang J and Shen L: Extracellular
vesicle-mediated regulation of tumor angiogenesis: Implications for
anti-angiogenesis therapy. J Cell Mol Med. 25:2776–2785. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chitti SV, Fonseka P and Mathivanan S:
Emerging role of extracellular vesicles in mediating cancer
cachexia. Biochem Soc Trans. 46:1129–1136. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Pin F, Beltrà M, Garcia-Castillo L,
Pardini B, Birolo G, Matullo G, Penna F, Guttridge D and Costelli
P: Extracellular vesicles derived from tumour cells as a trigger of
energy crisis in the skeletal muscle. J Cachexia Sarcopenia Muscle.
13:481–494. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Abu Shelbayeh O, Arroum T, Morris S and
Busch KB: PGC-1α is a master regulator of mitochondrial lifecycle
and ROS stress response. Antioxidants (Basel). 12:10752023.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Gao X, Wang Y, Lu F, Chen X, Yang D, Cao
Y, Zhang W, Chen J, Zheng L, Wang G, et al: Extracellular vesicles
derived from oesophageal cancer containing P4HB promote muscle
wasting via regulating PHGDH/Bcl-2/caspase-3 pathway. J Extracell
Vesicles. 10:e120602021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hu W, Ru Z, Zhou Y, Xiao W, Sun R, Zhang
S, Gao Y, Li X, Zhang X and Yang H: Lung cancer-derived
extracellular vesicles induce myotube atrophy and adipocyte
lipolysis via extracellular IL-6-mediated STAT3 pathway. Biochim
Biophys Acta Mol Cell Biol Lipids. 1864:1091–1102. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu Z, Xiong J, Gao S, Zhu MX, Sun K, Li
M, Zhang G and Li YP: Ameliorating cancer cachexia by inhibiting
cancer cell release of Hsp70 and Hsp90 with omeprazole. J Cachexia
Sarcopenia Muscle. 13:636–647. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang W, Sun W, Gu X, Miao C, Feng L, Shen
Q, Liu X and Zhang X: GDF-15 in tumor-derived exosomes promotes
muscle atrophy via Bcl-2/caspase-3 pathway. Cell Death Discov.
8:1622022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kuang JX, Shen Q, Zhang RQ, Fang QY, Deng
X, Fan M, Cheng CR, Zhang XW and Liu X: Carnosol attenuates atrophy
of C2C12 myotubes induced by tumour-derived exosomal miR-183-5p
through inhibition of Smad3 pathway activation and preservation of
mitochondrial respiration. Basic Clin Pharmacol Toxicol.
131:500–513. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Miao C, Zhang W, Feng L, Gu X, Shen Q, Lu
S, Fan M, Li Y, Guo X, Ma Y, et al: Cancer-derived exosome miRNAs
induce skeletal muscle wasting by Bcl-2-mediated apoptosis in colon
cancer cachexia. Mol Ther Nucleic Acids. 24:923–938. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ruan X, Cao M, Yan W, Jones YZ, Gustafsson
ÅB, Patel HH, Schenk S and Wang SE: Cancer-cell-secreted
extracellular vesicles target p53 to impair mitochondrial function
in muscle. EMBO Rep. 24:e564642023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Tasdemir E, Maiuri MC, Galluzzi L, Vitale
I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C,
Harper F, et al: Regulation of autophagy by cytoplasmic p53. Nat
Cell Biol. 10:676–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yan W, Cao M, Ruan X, Jiang L, Lee S,
Lemanek A, Ghassemian M, Pizzo DP, Wan Y, Qiao Y, et al:
Cancer-cell-secreted miR-122 suppresses O-GlcNAcylation to promote
skeletal muscle proteolysis. Nat Cell Biol. 24:793–804. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wu Y, Yao X, Shi X, Xu Z, Ren J, Shi M, Li
M, Liu J and Du X: Myeloma extracellular vesicle-derived RAGE
increases inflammatory responses and myotube atrophy in multiple
myeloma through activation of the TLR4/NF-κB p65 pathway.
Apoptosis. 29:849–864. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Soria Rivas A, Escobar Álvarez Y, Blasco
Cordellat A, Majem Tarruella M, Molina Mata K, Motilla de la Cámara
M, Del Mar Muñoz Sánchez M, Zafra Poves M, Beato Zambrano C and
Cabezón Gutierrez L: SEOM clinical guidelines for cancer
anorexia-cachexia síndrome (2023). Clin Transl Oncol. 26:2866–2876.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Fujii H, Yamada Y, Iihara H, Kobayashi R
and Suzuki A: Anamorelin in the management of cancer cachexia:
Clinical efficacy, challenges and future directions. Anticancer
Res. 45:865–881. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Fan M, Gu X, Zhang W, Shen Q, Zhang R,
Fang Q, Wang Y, Guo X, Zhang X and Liu X: Atractylenolide I
ameliorates cancer cachexia through inhibiting biogenesis of IL-6
and tumour-derived extracellular vesicles. J Cachexia Sarcopenia
Muscle. 13:2724–2739. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhou L, Zhang T, Shao W, Lu R, Wang L, Liu
H, Jiang B, Li S, Zhuo H, Wang S, et al: Amiloride ameliorates
muscle wasting in cancer cachexia through inhibiting tumor-derived
exosome release. Skelet Muscle. 11:172021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Di Felice V, Barone R, Trovato E, D'Amico
D, Macaluso F, Campanella C, Marino Gammazza A, Muccilli V, Cunsolo
V, Cancemi P, et al: Physiactisome: A new nanovesicle drug
containing heat shock protein 60 for treating muscle wasting and
cachexia. Cells. 11:14062022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Conceição M, Forcina L, Wiklander OPB,
Gupta D, Nordin JZ, Vrellaku B, McClorey G, Mäger I, Gӧrgens A,
Lundin P, et al: Engineered extracellular vesicle decoy
receptor-mediated modulation of the IL6 trans-signalling pathway in
muscle. Biomaterials. 266:1204352021. View Article : Google Scholar : PubMed/NCBI
|














