Introduction
Melanoma, a highly aggressive cutaneous malignancy,
has a rising global incidence (1,2).
Current treatment encompasses surgery, radiation therapy,
chemotherapy, immunotherapy and targeted therapy (3). While surgical resection remains the
cornerstone for early-stage disease, targeted therapy and
immunotherapy have emerged as essential modalities for advanced or
metastatic melanoma (4). In recent
years, advances in immunotherapy and targeted therapy have brought
new hope to patients with melanoma (5). In particular, targeted therapy plays a
pivotal role in contemporary melanoma management (6).
BRAF mutations are among the most frequent genetic
alterations in melanoma, occurring in ~40-60% of patients with
cutaneous melanoma, with the BRAFV600E variant predominating
(7). The BRAF gene encodes a
serine/threonine protein kinase that plays a crucial role in
cellular signaling. The BRAFV600E point mutation results in the
substitution of valine for glutamate at position 600, leading to
constitutive activation of signaling pathways such as MAPK and
thereby driving aberrant cell growth and proliferation (8). Targeted agents, including BRAF
inhibitors (e.g., vemurafenib, dabrafenib) and MAPK kinase (MEK)
inhibitors (e.g., trametinib), have demonstrated considerable
clinical efficacy against this alteration (9). Nevertheless, despite substantial
advances in targeted therapies for BRAF-mutant melanoma, the
emergence of drug resistance remains a major therapeutic challenge,
as tumor cells evade treatment through multiple mechanisms,
including acquired mutations, activation of alternative pathways
and modifications of the tumor microenvironment (TME) (10). This review aims to systematically
delineate the role of metabolic reprogramming in drug resistance
and identify research gaps in the combined targeting of metabolic
pathways.
Targeted therapy for BRAF-mutant melanoma:
Current status and evolving mechanisms of drug resistance
Current evidence indicates that ~50% of patients
with melanoma harbor activating BRAF mutations. As a central
component of the MAPK cascade, the B-RAF kinase functions as a
critical node in the RAS/RAF/MEK/ERK signaling axis, primarily
regulating cellular proliferation and survival (9). Oncogenic BRAF mutations lead to
constitutive MAPK pathway activation (11), which drives uncontrolled melanoma
cell proliferation, evasion of cell death and malignant
transformation (12). Given its
pivotal role in melanoma initiation, progression, metastasis and
therapeutic resistance (13),
targeting the MAPK pathway has emerged as a cornerstone of
treatment for BRAF-mutant melanoma (14).
Targeted therapies that block the MAPK signaling
pathway primarily include BRAF inhibitors (BRAFi) and MEKi
(15). BRAFi selectively suppress
the mutated BRAF protein, thereby blocking downstream signal
transduction and impeding tumor growth (16), whereas MEKi directly inhibit MEK
activity to achieve a similar pathway blockade (17). Current clinical practice relies on
BRAFi monotherapy or BRAFi/MEKi combination regimens, which have
demonstrated substantial efficacy, significantly improving
objective response rates and overall survival (18). Notably, the Food and Drug
Administration approved vemurafenib and dabrafenib over a decade
ago for advanced BRAF-mutant melanoma, with both agents achieving
high response rates and even complete tumor regression in a subset
of patients (12).
Although drugs targeting the MAPK pathway have made
significant progress in melanoma treatment, resistance to targeted
therapy remains an obstacle to curative treatment for melanoma. The
expansion of research on immune-checkpoint inhibitor-related
mechanisms in melanoma underscores the growing clinical importance
of understanding immune-metabolic interactions (19). The combination of BRAFi and MEKi as
a first-line treatment option for locally advanced or metastatic
melanoma with BRAF mutations has demonstrated excellent response
rates. However, these responses are not durable and nearly all
patients develop resistance (1,2).
Statistical analysis revealed that in BRAF-mutated melanoma, the
median time to clinically detectable acquired resistance to
BRAFi/MEKi was 9–11 months (20).
Metabolic reprogramming as a driver of
targeted therapy resistance in BRAF-mutant melanoma
Melanoma metabolic reprogramming is intricately
linked to targeted therapy resistance (21). As a hallmark of cancer, this process
involves the rewiring of cellular metabolic pathways to meet
bioenergetic and biosynthetic demands, supporting rapid
proliferation and survival (22,23).
In melanoma, an aggressive malignancy with high metabolic
plasticity, such reprogramming fuels tumor progression and enables
adaptation to therapeutic stress (24). Critically, metabolic adaptation not
only drives melanoma growth and metastasis but also underlies the
development of treatment resistance (21,25).
Studies indicate that targeted inhibition of
BRAF-mutant melanoma cells induces profound metabolic shifts,
including enhanced oxidative phosphorylation (OXPHOS), increased
glutamine flux through the tricarboxylic acid (TCA) cycle, and
altered fatty acid (FA) synthesis and catabolism (9). BRAFi and MEKi therapies trigger
metabolic reprogramming in BRAF-mutant melanoma cells, thereby
conferring resistance (26).
Consequently, targeting these adaptive metabolic pathways offers a
promising strategy to overcome resistance and suppress tumor
progression (21).
In this section, the context of targeted therapy
resistance was first outlined, key metabolic adaptations and their
mechanistic roles in promoting resistance were then described in
detail, and finally, emerging therapeutic targets and translational
implications were discussed. Elucidating these metabolic mechanisms
not only reveals actionable vulnerabilities but also informs the
identification of predictive biomarkers for personalized treatment.
Furthermore, growing evidence underscores the interplay between
metabolic rewiring and immune dysregulation, highlighting the need
to integrate metabolic and immune profiling in future research to
develop more effective combinatorial strategies (19).
Lactate accumulation
Lactate accumulation in melanoma cells is closely
associated with monocarboxylate transporters (MCTs), a class of
membrane proteins responsible for transporting monocarboxylic acids
such as lactate and pyruvate (27).
MAPK pathway inhibition has been shown to downregulate the
expression and activity of specific MCT isoforms, particularly MCT1
and MCT4, thereby impairing lactate efflux and promoting
intracellular lactate accumulation (28).
Research into the resistance mechanisms of BRAFV600E
mutant melanoma to BRAFi and MEKi revealed that in
BRAFi/MEKi-resistant melanoma, lactate accumulation drives the
lactylation of lysine specific demethylase 1 (LSD1), thereby
promoting its interaction with FOS like 1, AP-1 transcription
factor subunit (FosL1) and preventing LSD1 degradation by the E3
ligase tripartite motif containing 21. Lactylated LSD1 and FosL1
jointly regulate gene transcription, inhibiting ferroptosis by
interfering with iron uptake mediated by the transferrin receptor
(TFR) (29).
Ferroptosis is characterized by iron overload-driven
mitochondrial dysfunction, excessive reactive oxygen species (ROS)
generation and subsequent peroxidation of polyunsaturated FAs in
cellular membranes, ultimately leading to oxidative cell damage
(30). Tumor cells can negatively
regulate ferroptosis via lactylated LSD1. Inhibition of ferroptosis
markedly enhances the oxidative stress resistance of melanoma
cells, enabling them to adapt and survive under the pressure of
targeted therapy, thereby diminishing the therapeutic efficacy of
targeted agents against melanoma (31) (Fig.
1).
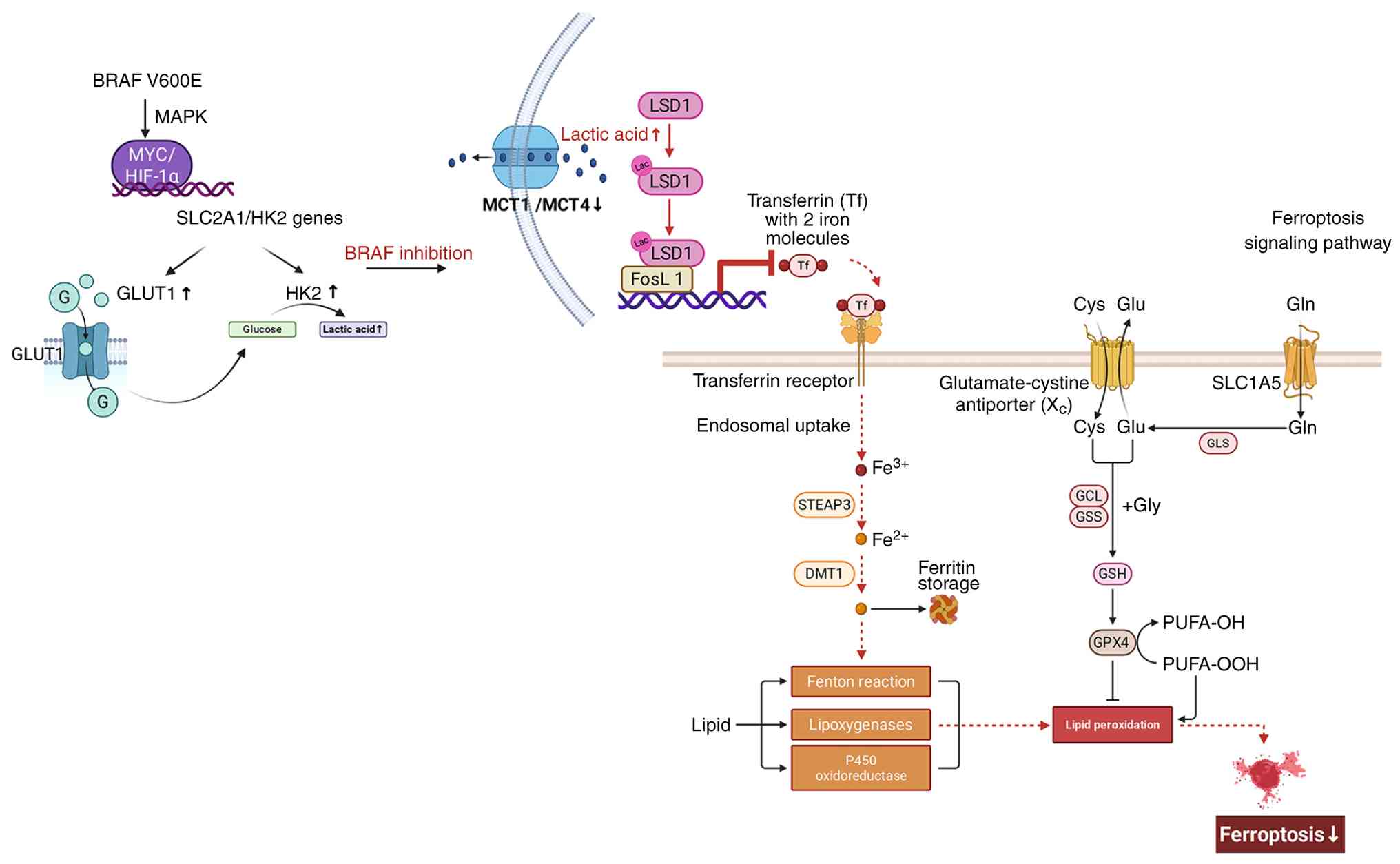 | Figure 1.Lactic acid accumulation induces
targeted therapy resistance by inhibiting ferroptosis. After the
MAPK signaling pathway is inhibited, suppression of MCTs expression
and activity impairs intracellular lactate efflux, leading to
lactate accumulation. This accumulated lactate drives the
lactylation of LSD1. The lactylated LSD1, in conjunction with
FosL1, regulates gene transcription, which interferes with
TFR-mediated iron uptake to inhibit ferroptosis. This inhibition of
ferroptosis significantly enhances the resistance of melanoma cells
to oxidative stress. MAPK, mitogen-activated protein kinase; MCTs,
monocarboxylate transporters; LSD1, lysine-specific demethylase 1;
FosL1, Fos-like antigen 1; TFR, transferrin receptor. |
Increased mitochondrial OXPHOS
In melanoma, although BRAF mutations initially
suppress OXPHOS (11), resistance
to targeted therapy is consistently associated with OXPHOS
upregulation in both experimental models and clinical specimens of
BRAF-mutant melanoma (9,32). This metabolic shift enhances tumor
invasion and metastatic potential, thereby driving resistance to
MAPK pathway inhibitors (11).
Furthermore, the phenotypic plasticity accompanying OXPHOS
activation may complicate this metabolic adaptation and contribute
to acquired resistance against BRAFi and MEKi (e.g., trametinib,
cobimetinib, binimetinib) (33)
(Fig. 2).
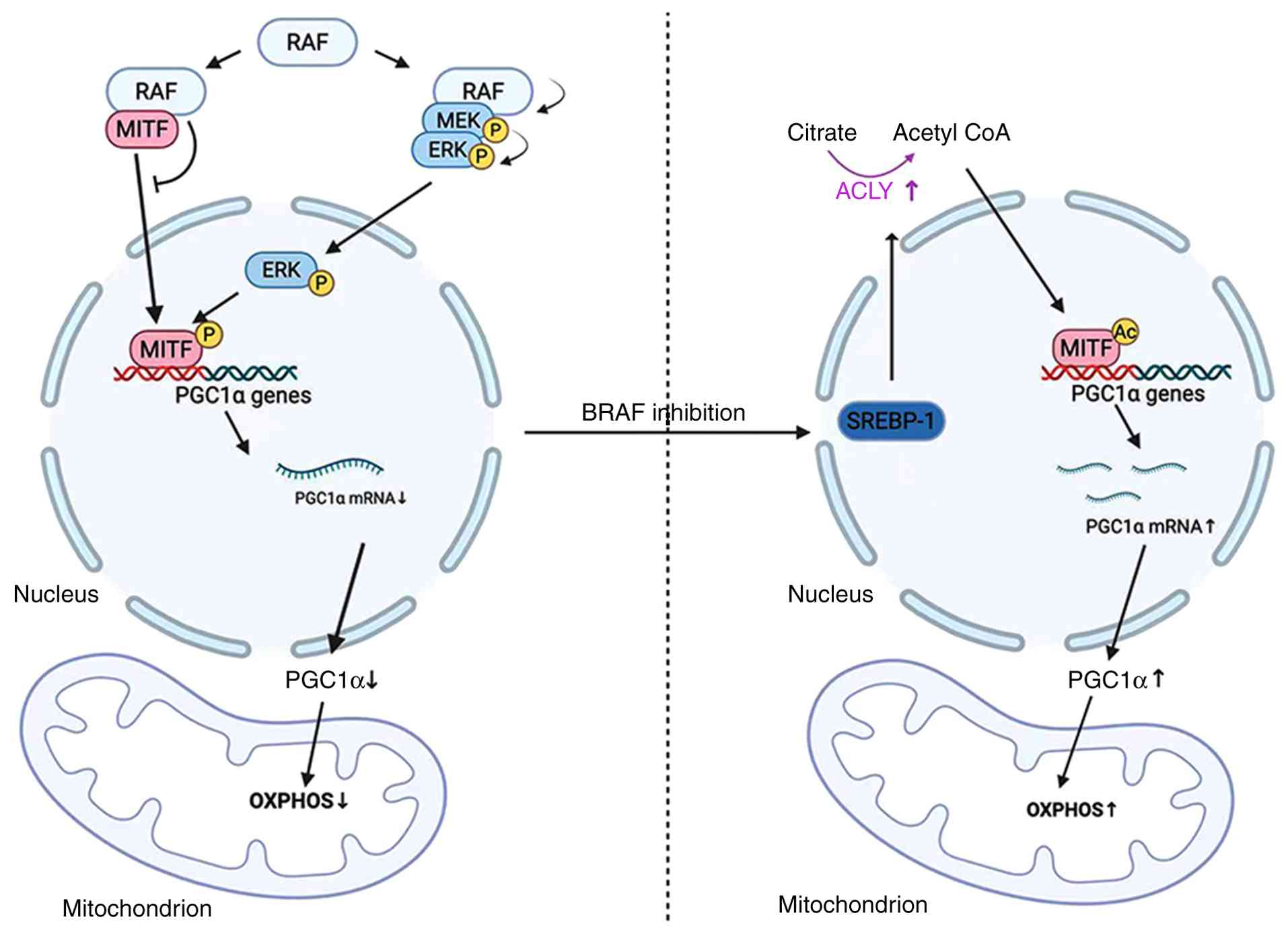 | Figure 2.OXPHOS enhancement leads to targeted
therapy resistance by boosting metabolism. Following MAPK pathway
inhibition, SREBP-1-mediated transcriptional activation increases
ACLY expression, which elevates acetyl-CoA levels. This leads to
enhanced EP300 activity and subsequent activation of the MITF-PGC1α
axis, ultimately resulting in enhanced OXPHOS and augmented tumor
invasion and metastatic capacity. MAPK, mitogen-activated protein
kinase; SREBP-1, sterol regulatory element-binding protein 1; ACLY,
ATP-citrate lyase; EP300, E1A binding protein p300; MITF,
melanocyte-inducing transcription factor; PGC1α, peroxisome
proliferator-activated receptor gamma coactivator 1-alpha; OXPHOS,
oxidative phosphorylation. |
OXPHOS-driven bioenergetic
adaptation
The BRAFV600E mutation leads to
microphthalmia-associated transcription factor (MITF) inactivation
via ERK1/2-mediated phosphorylation or direct RAF binding, which in
turn suppresses the transcription of peroxisome
proliferator-activated receptor gamma Coactivator 1α (PGC1α), a
master regulator of mitochondrial biogenesis and function (34). Consequently, under constitutive MAPK
signaling, reduced MITF and PGC1α levels attenuate oxidative
metabolism. Conversely, BRAF/MAPK pathway inhibition restores MITF
expression and activity, thereby upregulating PGC1α and its
downstream targets (35).
Following MAPK pathway inhibition by BRAF-targeted
therapeutics, ATP citrate lyase (ACLY) expression is enhanced via
sterol regulatory element binding transcription factor 1
transcriptional activation. ACLY enhances histone acetyltransferase
p300 (EP300) activity by supplying sufficient acetyl-CoA. This
increases histone acetylation at MITF binding sites, thereby
promoting transcription via the MITF-PGC1α axis. The MITF-PGC1α
axis activation activates OXPHOS. Expression of MITF and PGC1α
correlates with OXPHOS genes, and PGC1α drives increased glutamine
and serine metabolism dependency, thereby enhancing OXPHOS
(36). Melanoma cells can enhance
energy production and biosynthetic capacity by boosting OXPHOS,
leading to bioenergetic adaptation that sustains their survival and
proliferation (37). Furthermore,
MITF, a transcription factor specific to the typical melanocyte
lineage, is induced and promotes downstream PGC1α-dependent OXPHOS.
This pathway also represents a classic signaling cascade involved
in adaptive resistance to MAPK inhibitor-targeted therapies in
melanoma (12,38) (Fig.
2).
Elevated OXPHOS drives redox
adaptation through ROS modulation
Beyond the MITF-PGC1α axis, BRAF inhibitors promote
OXPHOS via alternative mechanisms. For instance, while the
BRAFV600E mutation suppresses OXPHOS through constitutive kinase
activation, pharmacological BRAF inhibition reverses this effect
and induces OXPHOS upregulation (21). Following MAPK blockade treatment,
upregulation of the OXPHOS-associated lincRNA enhancer also
promotes metabolic shift from glycolysis to OXPHOS (39).
Enhanced OXPHOS inevitably elevates mitochondrial
ROS production. Rather than merely tolerating high ROS levels,
drug-resistant melanoma cells establish a remodeled redox
homeostasis characterized by moderately elevated ROS. This adapted
state functions not only as a selective survival advantage but also
as a key signaling node that supports resistance. Specifically, ROS
can activate parallel survival pathways, such as the PI3K/AKT
cascade, enabling cell proliferation even under BRAF or MEK
inhibition (12,40). Concurrently, ROS stimulates
compensatory antioxidant responses, including the NFE2 like bZIP
transcription factor 2/antioxidant response element pathway, which
enhances cellular ROS-buffering capacity and limits oxidative
damage (40). Furthermore, ROS
contributes to the epigenetic remodeling of resistant cells through
mechanisms involving DNA methylation and histone modification
(41). Thus, through fine-tuned
regulation of OXPHOS-derived ROS, melanoma cells achieve a
redox-adapted state that sustains proliferation and confers
acquired resistance to targeted therapy.
Altered amino acid metabolism
Increased glutamine utilization via the TCA
cycle
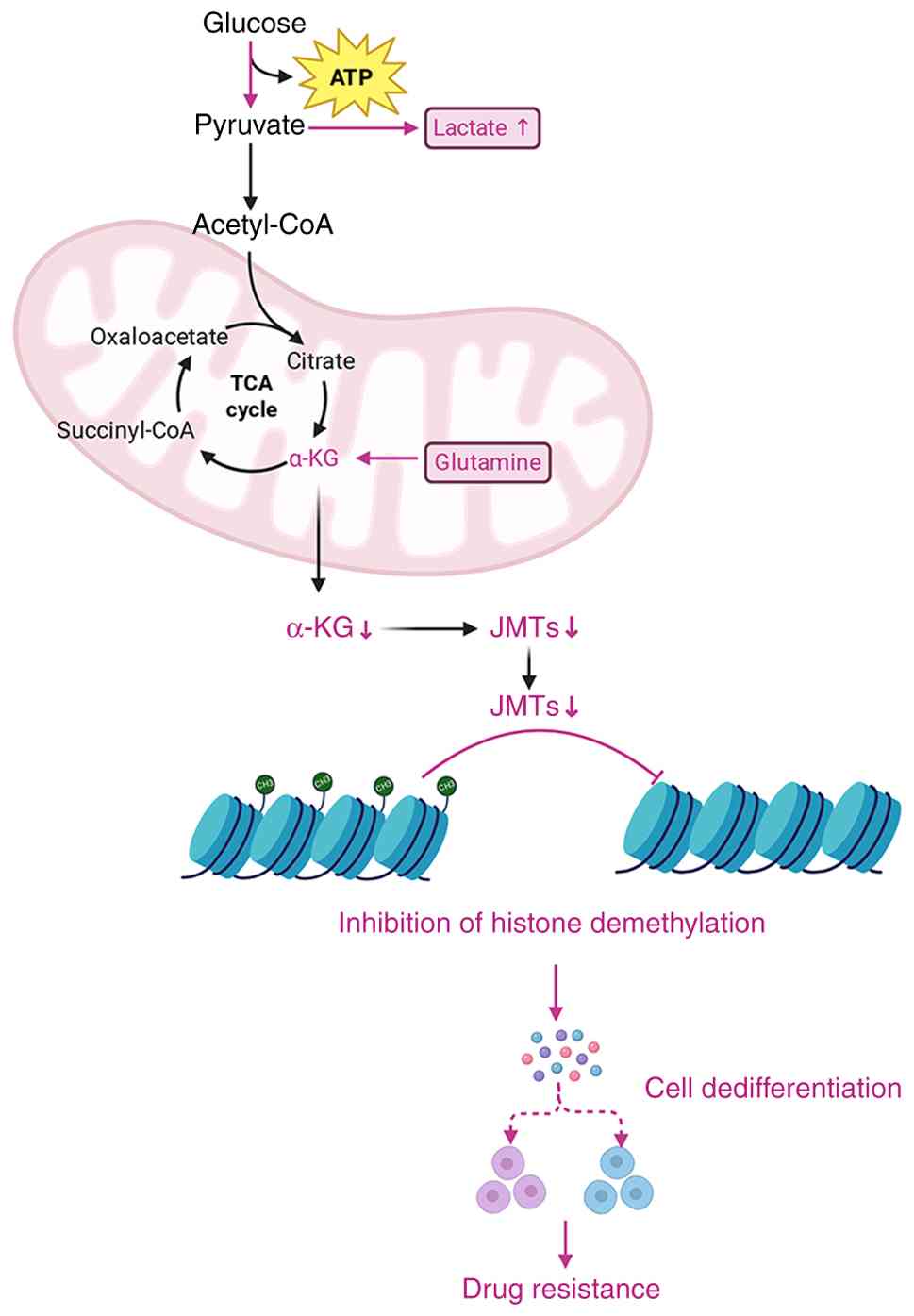
In BRAF-inhibited melanoma cells, since glucose is
primarily converted to lactate, glutamine serves as an alternative
carbon source, fueling the TCA cycle by conversion to
α-ketoglutarate (α-KG), with glutamine consumption leading to
reduced α-KG levels. α-KG serves as a crucial cofactor for Jumonji
C-domain-containing histone demethylases, which rely on α-KG for
catalytic activity to promote histone demethylation and maintain a
differentiated cellular state (42). In the TME, reduced α-KG levels
therefore directly inhibit histone demethylation, subsequently
leading to a hypermethylated epigenetic landscape that drives
cellular dedifferentiation and increased resistance to targeted
therapies in melanoma cells (12).
This creates a dual role for glutamine metabolism in resistance:
While fueling the TCA cycle as an energy source, its consumption
simultaneously depletes a key epigenetic cofactor (α-KG), locking
cells into a resistant, dedifferentiated phenotype (Fig. 3). However, melanoma cells that
acquire resistance to BRAFi exhibit high dependence on glutamine
for proliferation, suggesting that glutamine metabolism may play
context-dependent, seemingly opposing roles at different stages of
therapeutic resistance, acting as both a fuel source and an
epigenetic regulator (12).
Increased glutamine synthesis via the
serine pathway
In BRAF-mutant melanoma following BRAFi treatment,
upregulation of phosphoglycerate dehydrogenase (PHGDH) and
activation of the serine synthesis pathway have been observed
(43). PHGDH is the key enzyme in
de novo serine synthesis, responsible for converting
3-phosphoglycerate into serine (44). Serine indirectly participates in
glutathione (GSH) synthesis by converting into glycine, making
serine supply critical for maintaining intracellular GSH levels
(45). Elevated GSH suppresses
ferroptosis by reducing intracellular ROS production. Tumor cells
can significantly enhance their oxidative stress defense capacity
and increase resistance to targeted therapies by negatively
regulating ferroptosis (46).
Consequently, PHGDH upregulation correlates with resistance to
melanoma targeted therapies and may represent a therapeutic target
(43,44). Notably, the serine synthesis pathway
intersects with glutamine metabolism. Glutamine-derived nitrogen is
utilized in serine and glycine synthesis, and the resulting GSH is
critical for mitigating oxidative stress that may arise from
altered mitochondrial metabolism (e.g., enhanced OXPHOS). Thus, the
co-upregulation of glutamine utilization and serine synthesis forms
an integrated metabolic network that supports redox balance and
epigenetic adaptation, collectively fostering a resistant
phenotype.
Activation of the kynurenine metabolic
pathway
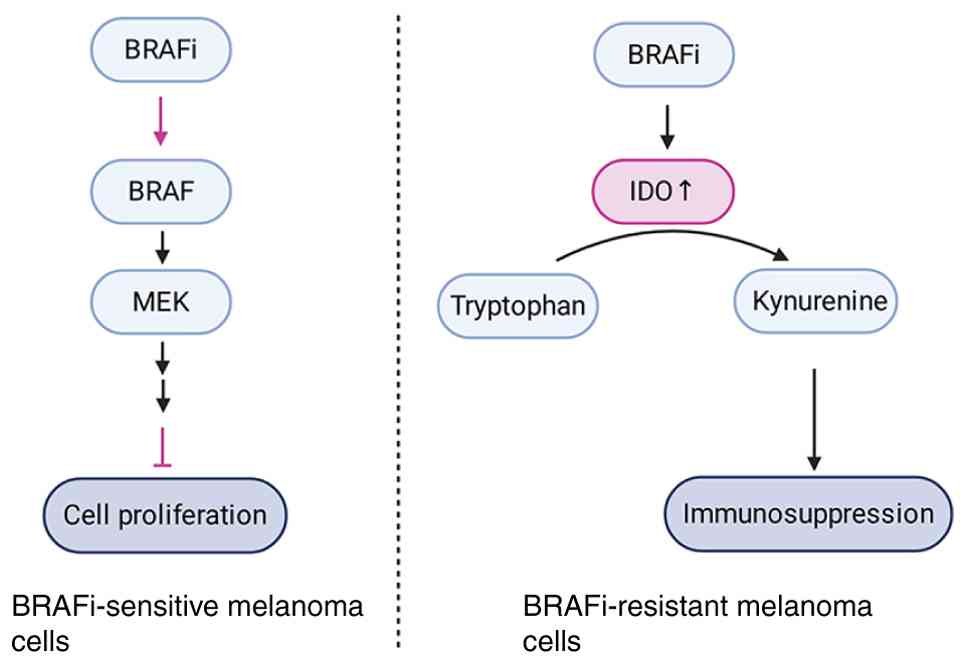
Indoleamine is the primary metabolite of tryptophan,
influencing the TME and immune response through multiple mechanisms
to promote melanoma drug resistance (47). During BRAFi and MEKi therapy, tumor
cells may activate the indoleamine metabolic pathway to enhance
survival and proliferation, thereby diminishing targeted treatment
efficacy (12). In
treatment-sensitive melanoma cells, BRAFi therapy reduces
indoleamine 2,3-dioxygenase (IDO) expression levels. However, IDO
expression may increase in BRAFi-resistant cells. IDO expression is
elevated not only in patient biopsies of primary and metastatic
melanoma but also in IDO mRNA levels during BRAFi resistance
(48). Tryptophan is metabolized
into kynurenine by enzymes such as IDO (49). Research shows that the accumulation
of kynurenine not only promotes the formation of a TME but also
activates the aryl hydrocarbon receptor, thereby inducing the
differentiation of induced regulatory T cells and myeloid-derived
suppressor cells (50). Single-cell
transcriptional profiling further supports that metabolic cues can
reshape key immune-regulatory pathways, such as IL27RA-mediated
immunomodulation (51). Kynurenine
promotes Treg generation, further suppressing immune responses and
thereby favoring tumor cell growth and drug resistance (40) (Fig.
4).
Altered lipid metabolism
Lipid metabolic reprogramming constitutes an
adaptive cellular response to metabolic stressors such as glucose
deprivation and hypoxia. In melanoma, this process is characterized
by enhanced de novo FA synthesis, enabling tumor cells to
dynamically adapt to the evolving TME (52). Lipid metabolic reprogramming also
plays multiple roles in melanoma progression and targeted therapy
resistance. Numerous studies support its role in regulating
melanoma cell differentiation states and promoting resistance to
targeted therapies. Drugs targeting lipid metabolic reprogramming
show strong therapeutic potential in drug-resistant melanoma
(53). Research indicates that
lipid metabolic reprogramming occurs in vemurafenib (BRAFi)-treated
melanoma cells (54), inducing or
maintaining resistance by altering the lipid composition and
biophysical properties of cancer cell membranes (55) (Fig.
5).
Increased FA synthesis
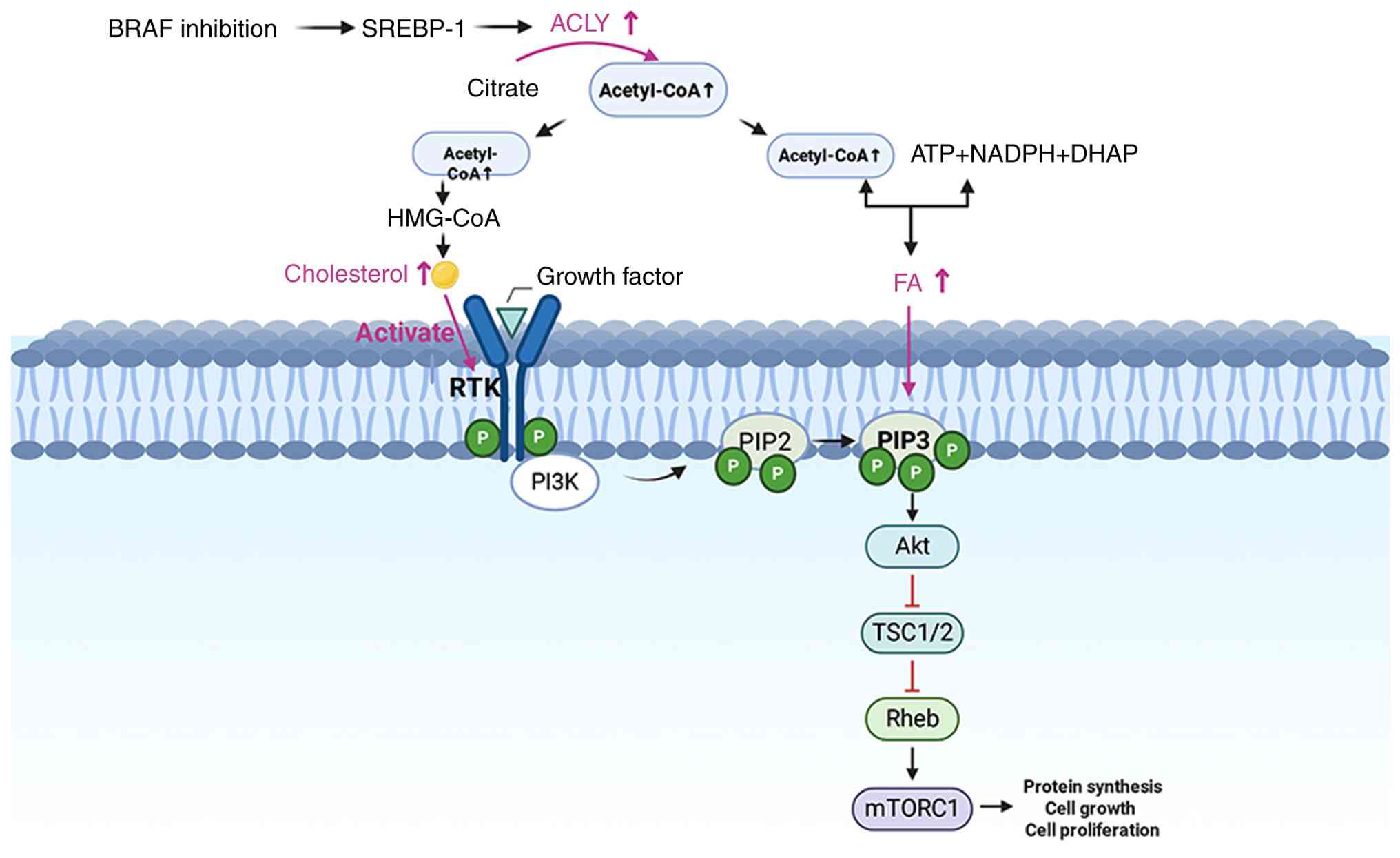
Acetyl-CoA serves as the primary substrate for FA
synthesis. ACLY facilitates the production of oxaloacetate and
cytoplasmic acetyl-CoA, acting as a pivotal enzyme linking
glycolysis and lipid synthesis (56). BRAF-mutant melanomas exhibit
elevated ACLY expression following MAPK inhibition, leading to
increased FA synthesis (37).
FAs can be utilized for synthesizing bioactive
lipids that support cell proliferation and survival by acting as
second messengers in signaling pathways. For instance,
phosphatidylinositol, composed of an inositol ring, glycerol
backbone and two FA chains, is one of the most characteristic
signaling-related lipids (53).
Phosphatidylinositol (3,4,5)-trisphosphate facilitates the
recruitment of AKT to the plasma membrane, where AKT is
subsequently activated by pyruvate dehydrogenase kinase 1 and the
mTOR complex 2 (mTORC2), the latter being hyperactivated downstream
of PI3K signaling. The PI3K-AKT-mTOR pathway is one of the most
common signaling pathways and plays a crucial role in the
acquisition of resistance to targeted therapies in melanoma.
Furthermore, increased expression of various G protein-coupled
bioactive lipid receptors participates in regulating migratory
signaling in melanoma, such as activation of cyclic AMP/PKA, MAPK
and PI3K pathways, which may represent an important mechanism for
the failure of targeted therapy (53,57).
Cholesterol accumulation
Intracellular cholesterol accumulation has been
observed in BRAFV600E-mutant melanoma cells following MAPKi
treatment. Mechanistically, MAPK pathway inhibition promotes the
activation of receptor tyrosine kinases, leading to enhanced
phosphotyrosine (p-Tyr) signaling. This in turn stimulates
downstream pathways such as PI3K-AKT, thereby providing an
alternative survival mechanism that sustains tumor growth under
therapeutic pressure. Notably, dysregulated cholesterol metabolism
appears to drive the activation of these kinases coordinately
(58). The resultant PI3K-AKT
pathway activation is recognized as a key mechanism of adaptive
resistance to targeted therapy in melanoma (12).
Therapeutic targeting of metabolic pathways
in drug-resistant BRAF-mutant melanoma
Targeted metabolic changes combined with MPAKi are
crucial for the treatment of drug-resistant BRAF mutant melanoma.
Below are the corresponding drugs identified for different
metabolic pathways that can improve melanoma treatment (Table I).
 | Table I.Drugs that can be combined with MAPKi
are available for BRAF-mutant melanoma resistant to targeted
therapy. |
Table I.
Drugs that can be combined with MAPKi
are available for BRAF-mutant melanoma resistant to targeted
therapy.
| Drug | Drug type | Target | Metabolic
pathways | (Refs.) |
|---|
| - | LSD inhibitor | LSD1 | Lactic acid
accumulation | (3) |
| Diclofenac | NSAIDs | - | OXPHOS | (4) |
| Lumiracoxib | NSAIDs | - | OXPHOS | (4) |
| AZD8055 | mTORC
inhibitor | mTORC | OXPHOS | (4–6) |
| AZD2014 | mTORC
inhibitor | mTORC | OXPHOS | (4–6) |
| XCT790 | PGC1α
inhibitor | PGC1α | OXPHOS | (4) |
| SR-18292 | PGC1α
inhibitor | PGC1α | OXPHOS | (4) |
| Phenformin | Mitochondrial | Mitochondrial | OXPHOS | (4) |
|
| respiratory complex
I | respiratory |
|
|
|
| inhibitor | complex I |
|
|
| ONC212 | Mitochondria
inhibitor | Mitochondria | OXPHOS | (37) |
| FK866 | NAMPT
inhibitor | NAMPT | OXPHOS | (4) |
| GMX1778 | NAMPT
inhibitor | NAMPT | OXPHOS | (4) |
| - | PHGDH
inhibitor | PHGDH | Serine pathway | (7) |
|
1-methyl-tryptophan | IDO inhibitor | IDO | Kynurenine
pathway | (56,57) |
| Orlistat | FASN inhibitor | FASN | FA synthesis | (8) |
| Physostigmine | HMGCR
inhibitor | HMGCR | Cholesterol
accumulation | (49,59,60) |
Targeting lactic acid
accumulation
LSD1 inhibition (LSD1i)-induced ferroptosis not only
directly eliminates melanoma cells but also enhances host antitumor
immunity against drug-resistant disease. Mechanistically,
therapy-induced lactate accumulation drives lysine lactylation of
LSD1, which in turn suppresses ferroptosis, an iron-dependent form
of regulated cell death. The TFR serves as a key downstream
effector mediating LSD1i-triggered remodeling of the TME.
Consequently, combining LSD1 inhibition with TFR blockade can
overcome acquired resistance to BRAFi/MEKi in melanoma by
establishing a positive feedback loop that restores and amplifies
ferroptotic cell death (29).
Targeting OXPHOS
Combining OXPHOS inhibitors (OPIs) with targeted
therapies can block tumor cell energy metabolism and inhibit
signaling pathways, producing synergistic effects that overcome
melanoma resistance and more effectively kill tumor cells (29).
In BRAF-mutant melanoma, resistance to MEKi is
primarily mediated by elevated OXPHOS activity, which mTORC1i can
counteract. Mechanistically, both BRAF- and NRAS-mutant melanomas
promote MITF expression and upregulate the transcriptional
coactivator PGC1α, leading to enhanced OXPHOS. mTORC1 inhibitors
such as AZD8055 and AZD2014 induce nuclear exclusion of MITF,
thereby suppressing PGC1α expression and OXPHOS activity.
Similarly, direct targeting of PGC1α with compounds such as XCT790
or SR-18292 reduces mitochondrial mass and function, restoring MEKi
sensitivity in OXPHOS-highly resistant melanomas (21).
Other promising OXPHOS-targeting strategies include
novel OPIs and phenformin, as well as nicotinamide
phosphoribosyltransferase (NAMPT) inhibitors (e.g., FK866 and
GMX1778). Given that NAD+ levels are significantly
elevated in BRAFi-resistant melanoma, NAMPTi effectively reduce
NAD+ availability and have been shown to improve
survival in preclinical models (21). Additionally, the
mitochondrial-targeting agent ONC212 exhibits potent
growth-inhibitory effects against vemurafenib-resistant melanoma
cells, supporting its potential for inclusion in multimodal
therapeutic regimens (11).
Of note, non-steroidal anti-inflammatory drugs such
as diclofenac and lumiracoxib also demonstrate OPI activity in
melanoma cells, and their combination with BRAFi has been shown to
delay the onset of BRAFi resistance (21).
Targeting the serine metabolic
pathway
Inhibition of PHGDH acts synergistically with MAPK
pathway blockade in melanoma. By suppressing de novo serine
synthesis, PHGDH targeting reduces GSH production, thereby
increasing oxidative stress in tumor cells. This mechanism restores
sensitivity to MAPKi in resistant cells. Consequently,
co-inhibition of PHGDH and MEK in MEKi-resistant melanoma reduces
cellular oxidative stress tolerance and suppresses proliferation
(43).
Targeting the tyrosinase metabolic
pathway
In vitro studies demonstrate that the IDO1
inhibitor 1-methyl-tryptophan reduces clonogenic capacity in both
parental and BRAF-inhibitor-resistant melanoma cells (48), suggesting that IDO1 inhibition
represents a promising therapeutic strategy for patients with
BRAFi-resistant melanoma (50).
Targeting FA synthesis
FA synthase (FASN), a key multifunctional enzyme
complex, drives de novo lipogenesis, the metabolic process
that converts carbohydrates into FAs. The FASN inhibitor orlistat,
which targets this pathway, is presently under investigation in
combination with BRAFi for the treatment of melanoma (58).
Targeting cholesterol metabolism
In preclinical studies, inhibitors of
3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR)-the rate-limiting
enzyme in the mevalonate pathway that converts HMG-CoA to
mevalonate during cholesterol biosynthesis-have been investigated
for their potential to overcome targeted therapy resistance in
melanoma. Cholesterol depletion disrupts receptor-mediated
endocytosis and endosomal trafficking, and suppresses key survival
pathways such as AKT signaling. Notably, in melanoma cells treated
with MAPK pathway inhibitors, cholesterol depletion abrogates p-Tyr
signaling activation and synergistically enhances cytotoxic effects
both in vitro and in vivo (49,59).
These findings support the combination of MAPKi with
cholesterol-lowering agents to achieve more durable responses in
hyper-MAPK-driven tumors by counteracting adaptive resistance.
Consistent with this, Wang et al (60) reported that physostigmine, an
HMGCR-targeting agent, exerts anticancer activity and sensitizes
melanoma cells to BRAFi.
Discussion
Despite the significant clinical success achieved by
targeted therapies against BRAF-mutant melanoma, the inevitable
emergence of drug resistance continues to be a major barrier to
long-term patient survival. This review systematically synthesizes
evidence illustrating how melanoma cells undergo profound metabolic
reprogramming following MAPK pathway inhibition, thereby developing
resistance. Key adaptations include intracellular lactate
accumulation, enhanced mitochondrial OXPHOS, increased reliance on
glutamine and serine metabolism, activation of the kynurenine
pathway, and alterations in lipid metabolism involving increased FA
synthesis and cholesterol accumulation. These shifts are not merely
correlative; they actively drive resistance by rewiring energy
production, suppressing cell death pathways like ferroptosis,
shaping an immunosuppressive TME and activating bypass survival
signals. A critical appraisal of the literature, however, reveals
complexities and even apparent contradictions. For instance, while
glutamine depletion in the TME is linked to histone
hypermethylation and dedifferentiation, which favor resistance,
other evidence indicates that glutamine addiction itself can be a
metabolic vulnerability in resistant clones. This duality
underscores that the role of specific metabolites may be
context-dependent, varying with the temporal stage of therapy
(early adaptive vs. late acquired resistance) and the particular
tumor ecosystem, necessitating more nuanced, time-resolved
metabolic profiling in future studies. Recent single-cell studies
indicate that metabolic adaptation may also dictate responsiveness
to immune-checkpoint blockade, with transmembrane protein 71
identified as a key immunomodulatory determinant (61). Therefore, understanding how
metabolic pathways interface with immune signaling is essential for
developing more durable therapeutic strategies.
Based on the compiled evidence, several metabolic
targets emerge as particularly promising for therapeutic
intervention. The inhibition of LSD1, whose lactylation is directly
fueled by therapy-induced lactate accumulation, presents a
compelling strategy to reinstate ferroptosis and remodel the TME.
Targeting OXPHOS, a common adaptive node upregulated via the
MITF-PGC1α axis and other mechanisms, holds broad potential,
especially using agents like mTORC1 inhibitors or novel OPIs, which
can reverse the bioenergetic adaptation of resistant cells.
Similarly, PHGDH, the gatekeeper of serine synthesis, represents a
lethal vulnerability when co-inhibited with MAPK blockade, as it
cripples the GSH-mediated antioxidant defense. In the
immunosuppressive landscape, IDO1 inhibition stands out for its
potential to reverse kynurenine-mediated T-cell suppression and
improve immunotherapy or targeted therapy efficacy. Finally,
disrupting lipid metabolism via FASN or HMGCR inhibitors offers a
route to counteract the PI3K-AKT-mTOR pathway activation driven by
increased FAs and cholesterol.
Translating these insights into clinical practice
faces significant hurdles. First, toxicity remains a major concern,
as many metabolic pathways are essential in normal tissues. Second,
tumor heterogeneity and metabolic plasticity may lead to rapid
adaptive resistance. Third, robust predictive biomarkers are
lacking for patient stratification. Future efforts should
prioritize combination strategies, such as pairing metabolic
inhibitors with MAPK or immune checkpoint blockade, and leverage
single-cell multi-omics and metabolic imaging to guide dynamic,
personalized treatment regimens.
Acknowledgements
Not applicable.
Funding
This work was partly supported by the National Natural Science
Foundation of China (grant no. ZR2021MH290) and the Shandong
Provincial Medical and Health Science and Technology Development
Program (grant no. 202104030578).
Availability of data and materials
Not applicable.
Authors' contributions
XZ was involved in the study conceptualization,
literature search and drafting the manuscript. FC contributed to
conceptualization and provided resources. HL was involved in study
supervision and manuscript writing - review and editing. Data
authentication is not applicable. All authors have read and agreed
to the published version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cirillo N: Global epidemiological trends
in the incidence and mortality for melanoma. Skin Health Dis.
5:84–86. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ahmadi F, Karamitanha F and Ramezanpour A:
Clustering trends of melanoma incidence and mortality: A worldwide
assessment from 1995 to 2019. Australas J Dermatol. 63:e206–e217.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu L, Yan F, Qi J, Wang L, Zhou M and Yin
P: Burden of melanoma in China and its provinces from 1990 to 2021:
An analysis for the global burden of disease study 2021. Front
Public Health. 12:14866172024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X, Ma S, Zhu S, Zhu L and Guo W:
Advances in immunotherapy and targeted therapy of malignant
melanoma. Biomedicines. 13:2252025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boutros A, Croce E, Ferrari M, Gili R,
Massaro G, Marconcini R, Arecco L, Tanda ET and Spagnolo F: The
treatment of advanced melanoma: Current approaches and new
challenges. Crit Rev Oncol Hematol. 196:1042762024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang S, Xie R, Zhong A and Chen J:
Targeted therapeutic strategies for melanoma. Chin Med J (Engl).
136:2923–2930. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lopes J, Rodrigues CMP, Gaspar MM and Reis
CP: Melanoma management: From epidemiology to treatment and latest
advances. Cancers (Basel). 14:46522022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chapdelaine AG, Ku GC, Sun G and Ayrapetov
MK: The targeted degradation of BRAF V600E reveals the mechanisms
of resistance to BRAF-targeted treatments in colorectal cancer
cells. Cancers (Basel). 15:58052023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Loftus AW, Zarei M, Kakish H, Hajihassani
O, Hue JJ, Boutros C, Graor HJ, Nakazzi F, Bahlibi T, Winter JM and
Rothermel LD: Therapeutic implications of the metabolic changes
associated with BRAF inhibition in melanoma. Cancer Treat Rev.
129:1027952024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fateeva A, Eddy K and Chen S: Current
state of melanoma therapy and next steps: Battling therapeutic
resistance. Cancers (Basel). 16:15712024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ferraz LS, Costa RTD, Costa CAD, Ribeiro
CAJ, Arruda DC, Maria-Engler SS and Rodrigues T: Targeting
mitochondria in melanoma: Interplay between MAPK signaling pathway
and mitochondrial dynamics. Biochem Pharmacol. 178:1141042020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo W, Wang H and Li C: Signal pathways of
melanoma and targeted therapy. Signal Transduct Target Ther.
6:4242021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang F, Cai F, Dahabieh MS, Gunawardena
K, Talebi A, Dehairs J, El-Turk F, Park JY, Li M, Goncalves C, et
al: Peroxisome disruption alters lipid metabolism and potentiates
antitumor response with MAPK-targeted therapy in melanoma. J Clin
Invest. 133:e1666442023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang J, Joshua AM, Li Y, O'Meara CH,
Morris MJ and Khachigian LM: Targeted therapy, immunotherapy, and
small molecules and peptidomimetics as emerging immunoregulatory
agents for melanoma. Cancer Lett. 586:2166332024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Subbiah V, Puzanov I, Blay JY, Chau I,
Lockhart AC, Raje NS, Wolf J, Baselga J, Meric-Bernstam F, Roszik
J, et al: Pan-cancer efficacy of vemurafenib in BRAF
V600-mutant non-melanoma cancers. Cancer Discov.
10:657–663. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhong J, Yan W, Wang C, Liu W, Lin X, Zou
Z, Sun W and Chen Y: BRAF inhibitor resistance in melanoma:
Mechanisms and alternative therapeutic strategies. Curr Treat
Options Oncol. 23:1503–1521. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cohen JV and Sullivan RJ: Developments in
the space of new MAPK pathway inhibitors for BRAF-mutant melanoma.
Clin Cancer Res. 25:5735–5742. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Doepner M, Lee IY and Ridky TW: Drug
resistant melanoma may be vulnerable to inhibitors of serine
synthesis. J Invest Dermatol. 140:2114–2116. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang H, Shi Y, Ying J, Chen Y, Guo R,
Zhao X, Jia L, Xiong J and Jiang F: A bibliometric and visualized
research on global trends of immune checkpoint inhibitors related
complications in melanoma, 2011–2021. Front Endocrinol (Lausanne).
14:11646922023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robert C, Grob JJ, Stroyakovskiy D,
Karaszewska B, Hauschild A, Levchenko E, Chiarion Sileni V,
Schachter J, Garbe C, Bondarenko I, et al: Five-year outcomes with
dabrafenib plus trametinib in metastatic melanoma. N Engl J Med.
381:626–636. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen D, Zhang L, Li S and Tang L:
Metabolic reprogramming in melanoma therapy. Cell Death Discov.
11:3082025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nong S, Han X, Xiang Y, Qian Y, Wei Y,
Zhang T, Tian K, Shen K, Yang J and Ma X: Metabolic reprogramming
in cancer: Mechanisms and therapeutics. MedComm (2020). 4:e2182023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Wang Z, Zhu B, Deng M, Qiu J, Feng
Y, Ding N and Huang C: Metabolic reprogramming induced by aging
modifies the tumor microenvironment. Cells. 13:17212024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Bai M, Liu M, Zhang Z, Jiang C, Li
K, Chen Y, Xu Y and Wu L: Metabolic reprogramming in lung cancer:
Hallmarks, mechanisms, and targeted strategies to overcome immune
resistance. Cancer Med. 14:e713172025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Q, Shi L, Shi D, Tai N and Wang X:
Targeting metabolic reprogramming to overcome immune tolerance in
melanoma immunotherapy. Front Immunol. 16:15977702025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tangella LP, Clark ME and Gray ES:
Resistance mechanisms to targeted therapy in BRAF-mutant melanoma-A
mini review. Biochim Biophys Acta Gen Subj. 1865:1297362021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pérez-Escuredo J, Van Hée VF, Sboarina M,
Falces J, Payen VL, Pellerin L and Sonveaux P: Monocarboxylate
transporters in the brain and in cancer. Biochim Biophys Acta.
1863:2481–2497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lund J, Aas V, Tingstad RH, Van Hees A and
Nikolić N: Utilization of lactic acid in human myotubes and
interplay with glucose and fatty acid metabolism. Sci Rep.
8:98142018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li A, Gong Z, Long Y, Li Y, Liu C, Lu X,
Li Q, He X, Lu H, Wu K, et al: Lactylation of LSD1 is an acquired
epigenetic vulnerability of BRAFi/MEKi-resistant melanoma. Dev
Cell. 60:1974–1990.e11. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saint-Germain E, Mignacca L, Vernier M,
Bobbala D, Ilangumaran S and Ferbeyre G: SOCS1 regulates senescence
and ferroptosis by modulating the expression of p53 target genes.
Aging (Albany NY). 9:2137–2162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takatani-Nakase T, Ikushima C, Sakitani M
and Nakase I: Regulatory network of ferroptosis and autophagy by
targeting oxidative stress defense using sulfasalazine in
triple-negative breast cancer. Life Sci. 339:1224112024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gill JG, Leef SN, Ramesh V,
Martin-Sandoval MS, Rao AD, West L, Muh S, Gu W, Zhao Z, Hosler GA,
et al: A short isoform of spermatogenic enzyme GAPDHS functions as
a metabolic switch and limits metastasis in melanoma. Cancer Res.
82:1251–1266. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ta N, Jiang X, Zhang Y and Wang H:
Ferroptosis as a promising therapeutic strategy for melanoma. Front
Pharmacol. 14:12525672023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Estrada C, Mirabal-Ortega L, Méry L,
Dingli F, Besse L, Messaoudi C, Loew D, Pouponnot C, Bertolotto C,
Eychène A and Druillennec S: MITF activity is regulated by a direct
interaction with RAF proteins in melanoma cells. Commun Biol.
5:1012022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haq R, Shoag J, Andreu-Perez P, Yokoyama
S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL,
et al: Oncogenic BRAF regulates oxidative metabolism via PGC1α and
MITF. Cancer Cell. 23:302–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Acciardo S, Mignion L, Lacomblez E,
Schoonjans C, Joudiou N, Gourgue F, Bouzin C, Baurain JF, Gallez B
and Jordan BF: Metabolic imaging using hyperpolarized 13
C-pyruvate to assess sensitivity to the B-Raf inhibitor vemurafenib
in melanoma cells and xenografts. J Cell Mol Med. 24:1934–1944.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo W, Ma J, Yang Y, Guo S, Zhang W, Zhao
T, Yi X, Wang H, Wang S, Liu Y, et al: ATP-citrate lyase
epigenetically potentiates oxidative phosphorylation to promote
melanoma growth and adaptive resistance to MAPK inhibition. Clin
Cancer Res. 26:2725–2739. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McQuade JL and Vashisht Gopal Y:
Counteracting oxidative phosphorylation-mediated resistance of
melanomas to MAPK pathway inhibition. Mol Cell Oncol.
2:e9916102015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gambi G, Mengus G, Davidson G, Demesmaeker
E, Cuomo A, Bonaldi T, Katopodi V, Malouf GG, Leucci E and Davidson
I: The LncRNA LENOX interacts with RAP2C to regulate metabolism and
promote resistance to MAPK inhibition in melanoma. Cancer Res.
82:4555–4570. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen W, Geng D, Chen J, Han X, Xie Q, Guo
G, Chen X, Zhang W, Tang S and Zhong X: Roles and mechanisms of
aberrant alternative splicing in melanoma-implications for targeted
therapy and immunotherapy resistance. Cancer Cell Int. 24:1012024.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fufa TD, Baxter LL, Wedel JC, Gildea DE;
NISC Comparative Sequencing Program, ; Loftus SK and Pavan WJ: MEK
inhibition remodels the active chromatin landscape and induces
SOX10 genomic recruitment in BRAF(V600E) mutant melanoma cells.
Epigenetics Chromatin. 12:502019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jiang J, Srivastava S, Liu S, Seim G,
Claude R, Zhong M, Cao S, Davé U, Kapur R, Mosley AL, et al:
Asparagine starvation suppresses histone demethylation through iron
depletion. iScience. 26:1064252023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nguyen MQ, Teh JLF, Purwin TJ, Chervoneva
I, Davies MA, Nathanson KL, Cheng PF, Levesque MP, Dummer R and
Aplin AE: Targeting PHGDH upregulation reduces glutathione levels
and resensitizes resistant NRAS-mutant melanoma to MAPK kinase
inhibition. J Invest Dermatol. 140:2242–2252.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jasani N, Xu X, Posorske B, Kim Y, Wang K,
Vera O, Tsai KY, DeNicola GM and Karreth FA: PHGDH induction by
MAPK is essential for melanoma formation and creates an actionable
metabolic vulnerability. Cancer Res. 85:314–328. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shunxi W, Xiaoxue Y, Guanbin S, Li Y,
Junyu J and Wanqian L: Serine metabolic reprogramming in
tumorigenesis, tumor immunity, and clinical treatment. Adv Nutr.
14:1050–1066. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Y, Fan Z, Hu S, Lu C, Xiang Y and
Liao S: Ferroptosis: A new strategy for cancer therapy. Front
Oncol. 12:8305612022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oldan JD, Giglio BC, Smith E, Zhao W,
Bouchard DM, Ivanovic M, Lee YZ, Collichio FA, Meyers MO, Wallack
DE, et al: Increased tryptophan, but not increased glucose
metabolism, predict resistance of pembrolizumab in stage III/IV
melanoma. Oncoimmunology. 12:22047532023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sandri S, Watanabe LRM, Oliveira EA,
Faião-Flores F, Migliorini S, Tiago M, Felipe-Silva A, Vazquez VL,
da Costa Souza P, Consolaro MEL, et al: Indoleamine 2,3-dioxygenase
in melanoma progression and BRAF inhibitor resistance. Pharmacol
Res. 159:1049982020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu XY, Shi XJ, Hu A, Wang JQ, Ding Y,
Jiang W, Sun M, Zhao X, Luo J, Qi W and Song BL: Feeding induces
cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature.
588:479–484. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hubková B, Valko-Rokytovská M, Čižmárová
B, Zábavníková M, Mareková M and Birková A: Tryptophan: Its
metabolism along the kynurenine, serotonin, and indole pathway in
malignant melanoma. Int J Mol Sci. 23:91602022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Y, Anwar M, Wang X, Zhang B and Ma B:
Integrative transcriptomic and single-cell analysis reveals IL27RA
as a key immune regulator and therapeutic indicator in breast
cancer. Discov Oncol. 16:9772025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Delgado-Goñi T, Galobart TC, Wantuch S,
Normantaite D, Leach MO, Whittaker SR and Beloueche-Babari M:
Increased inflammatory lipid metabolism and anaplerotic
mitochondrial activation follow acquired resistance to vemurafenib
in BRAF-mutant melanoma cells. Br J Cancer. 122:72–81. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang R, Yan Q, Liu X and Wu J: Unraveling
lipid metabolism reprogramming for overcoming drug resistance in
melanoma. Biochem Pharmacol. 223:1161222024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Talebi A, Dehairs J, Rambow F, Rogiers A,
Nittner D, Derua R, Vanderhoydonc F, Duarte JAG, Bosisio F, Van den
Eynde K, et al: Sustained SREBP-1-dependent lipogenesis as a key
mediator of resistance to BRAF-targeted therapy. Nat Commun.
9:25002018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vergani E, Beretta GL, Aloisi M,
Costantino M, Corno C, Frigerio S, Tinelli S, Dugo M, Accattatis
FM, Granata A, et al: Targeting of the lipid metabolism impairs
resistance to BRAF kinase inhibitor in melanoma. Front Cell Dev
Biol. 10:9271182022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Granchi C: ATP-citrate lyase (ACLY)
inhibitors as therapeutic agents: A patenting perspective. Expert
Opin Ther Pat. 32:731–742. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wu X, Yu J, Yan J, Dai J, Si L, Chi Z,
Sheng X, Cui C, Ma M, Tang H, et al: PI3K/AKT/mTOR pathway
inhibitors inhibit the growth of melanoma cells with mTOR H2189Y
mutations in vitro. Cancer Biol Ther. 19:584–589. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Stamatakos S, Beretta GL, Vergani E, Dugo
M, Corno C, Corna E, Tinelli S, Frigerio S, Ciusani E, Rodolfo M,
et al: Deregulated FASN expression in BRAF inhibitor-resistant
melanoma cells unveils new targets for drug combinations. Cancers
(Basel). 13:22842021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang XD, Kim C, Zhang Y, Rindhe S, Cobb MH
and Yu Y: Cholesterol regulates the tumor adaptive resistance to
MAPK pathway inhibition. J Proteome Res. 20:5379–5391. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang HY, Yu P, Chen XS, Wei H, Cao SJ,
Zhang M, Zhang Y, Tao YG, Cao DS, Qiu F and Cheng Y: Identification
of HMGCR as the anticancer target of physapubenolide against
melanoma cells by in silico target prediction. Acta Pharmacol Sin.
43:1594–1604. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li B, Lin R, Hua Y, Ma B and Chen Y:
Single-cell RNA sequencing reveals TMEM71 as an immunomodulatory
biomarker predicting immune checkpoint blockade response in breast
cancer. Discov Oncol. 16:12562025. View Article : Google Scholar : PubMed/NCBI
|