Introduction
Acute lymphoblastic leukemia (ALL) is the most
common childhood malignancy, with an incidence of ~2-4 cases per
100,000 children under 15 years of age. Notably, the peak incidence
occurs between 3 and 5 years of age (1). Despite favorable treatment outcomes,
relapsed disease remains the leading cause of mortality in
pediatric ALL patients. Consequently, ongoing research has focused
on identifying improved prognostic markers and treatment
enhancements for ALL management (1). ALL comprises two primary subtypes:
B-cell ALL and T-cell ALL. B-ALL is markedly more prevalent,
constituting ~85% of all cases (2).
Cytogenetic aberrations are common findings in ALL, especially
among pediatric patients and have long been recognized for their
substantial impact on clinical outcomes (3,4).
However, recent advances in sequencing and genomic analysis
technologies have revealed novel alterations at the submicroscopic
scale. These subtle changes play crucial roles in determining
disease aggressiveness and resistance to chemotherapy.
Collectively, these scientific breakthroughs enable the
identification of new ALL subtypes and enhance the precision of
patient prognosis, thereby facilitating more effective risk-adapted
treatment strategies and supportive care (2).
Lysine methyltransferases (KMTs) are proteins that
add methyl marks to lysine residues in both histones and
non-histone proteins. These marks contribute to a wide range of
epigenetic modifications, including the establishment and
propagation of various gene expression patterns. Dysregulation of
KMT activity can cause widespread epigenetic changes that
contribute to cancer development and progression (5). Among these enzymes, SET and MYND
domain-containing protein 2 (SMYD2) is known to play significant
roles in cancer (6,7). In acute lymphoblastic leukemia (ALL),
aberrant SMYD2 expression has been associated with poor
prognosis, associated with unfavorable clinical features such as
advanced age and increased blast counts following chemotherapy
(8,9).
While the role of SMYD2 in ALL has been previously
documented, the contribution of other KMTs to leukemogenesis
remains less well defined. The lysine methyltransferase SET
domain-containing protein 4 (SETD4), has been implicated in the
regulation of various cellular processes, including cell
proliferation, cell cycle regulation, and maintenance of cancer
stem cell (CSC) quiescence (10–14).
Although SETD4 has been studied in the context of breast
cancer, non-small cell lung cancer (NSCLC), and radiation-induced
lymphomagenesis, its potential involvement and clinical
significance in ALL have not yet been investigated.
The present study analyzed the expression pattern of
SETD4 among pediatric ALL patients and non-neoplastic bone
marrow samples and investigated the correlation between
SETD4 transcription changes and the leukemic burden in ALL
patients during chemotherapy and its association with SMYD2
transcription.
Materials and methods
Patient sample collection
The present study was approved by the Ethical
Committee of the Federal District Foundation for Teaching and
Research in Health Sciences (approval no. CEP/FECPS 555/11), with
written informed consent from patients and/or guardians. Bone
marrow aspirates from 83 pediatric ALL patients (40 female and 43
male; mean age at diagnosis, 7.63 years; recruited between June
2008 and October 2011) were collected at José Alencar Children's
Hospital of Brasilia, Federal District, Brazil, during initial
disease presentation as part of routine diagnosis and genetic
analysis of leukemia. B-ALL patients were treated according to the
Brazilian Cooperative Group for Treatment of Childhood Acute
Lymphocytic Leukemia protocol (15)
and T-ALL patients were treated according to the
ALL-Berlin-Frankfurt-Münster (BFM-95) protocol (8). Bone marrow samples from 15 patients
were obtained on the 15th and 29th days of induction chemotherapy.
Additionally, bone marrow samples from eight children with
idiopathic thrombocytopenic purpura were used as non-neoplastic
controls. Blast percentages were confirmed in Wright-Giemsa-stained
smears, with all leukemic samples containing >40% blasts.
Clinical data collection
Clinical characteristics, including sex, age and
white blood cell (WBC) count in peripheral blood at ALL diagnosis,
immunophenotyping of bone marrow blasts, cytogenetic alterations
and bone marrow status at the 15th and 29th days of chemotherapy,
were obtained from medical records and described previously
(9). High-risk patients included
those who were <2 years old or >9 years old, and/or had
peripheral blood WBC counts exceeding 50,000/mm3, and/or
showed infiltration in the central nervous system at the time of
diagnosis and/or unfavorable cytogenetic findings.
RNA isolation, cDNA synthesis and
reverse transcription—quantitative (RT-q) PCR
Bone marrow mononuclear cells were isolated by
Ficoll density centrifugation at 400 × g for 30 min. Total RNA was
extracted from each sample containing 5–10×106 cells
using TRIzol Reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Single-stranded
complementary DNA was generated from total RNA with reverse
transcriptase and random primers, using the High-Capacity cDNA
Reverse Transcription Kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. RT-qPCR was performed on
a StepOnePlus Real-Time PCR System (Applied Biosystems; Thermo
Fisher Scientific, Inc.) using Taq-Man Gene Expression Assays
according to the manufacturer's instructions (Hs00213731_m1; cat.
no. 4331182 for SETD4; Hs00220210_m1, cat. no. 4331182 for
SMYD2; and Hs99999903_m1, cat. no. 4331182 for ACTB;
Thermo Fisher Scientific, Inc.). RT-qPCR assays were carried out in
a final volume of 10 µl in 96-well plates. RT-qPCR was performed in
technical triplicate under the following conditions: 95°C for 2
min, followed by 40 cycles at 95°C for 15 sec and 60°C for 40 sec.
Quantitation cycle (Cq) values were obtained from this experiment
and the 2−ΔΔCq method was applied to these values using
ACTB as a reference gene for input normalization and scaling
all samples by the mean Cq values of non-neoplastic samples
(16). The 3rd quartile of
non-neoplastic samples relative quantification was used as a
threshold to classify a sample as having high or basal SETD4
mRNA expression.
Exploratory analysis of online data
repositories
Microarray expression data for SETD4 were
downloaded from the BloodSpot 3.0 database (https://www.fobinf.com/) as log2-scaled
intensity values for two Affymetrix probe sets, 219482_at and
213989_x_at (17) (Affymetrix;
Thermo Fisher Scientific, Inc.). The dataset comprised
non-neoplastic bone-marrow samples and ALL specimens stratified by
cell lineage (B-ALL and T-ALL, n=47 and n=7) and by cytogenetic
subtype [t(12;21) and t(1;19)]. Data normality was assessed prior
to hypothesis testing: groups with normally distributed values were
compared using one-way ANOVA, whereas non-normally distributed data
were analyzed with the non-parametric Kruskal-Wallis test, with
post hoc Tukey's multiple comparisons test.
Statistical analysis and survival
curves
Descriptive statistics were used to summarize the
data. P<0.05 was considered to indicate a statistically
significant difference. The Mann-Whitney U test was used to compare
SETD4 expression between ALL and non-neoplastic bone marrow
samples and between high-risk and low-risk groups; only samples
with complete clinical information were included in risk
stratification analyses.
Heatmaps were generated using z-scores calculated
from ΔCt values processed in RStudio. Correlations between
SETD4 and SMYD2 mRNA expression levels in 83 ALL
samples were assessed using Spearman's correlation analysis.
Survival curves were estimated using the
Kaplan-Meier method, and patients alive at the last follow-up were
censored. The median follow-up time was 16.6 months (range:
0.3–45.2 months) for overall survival (OS) and 16.5 months (range:
0.1–45.2 months) for event-free survival (EFS). OS and EFS were
both analyzed. Survival outcomes were compared using the log-rank
test. Hazard ratios and 95% confidence intervals were calculated
using univariate Cox proportional hazards regression.
Results
SETD4 transcription is upregulated in
ALL patients
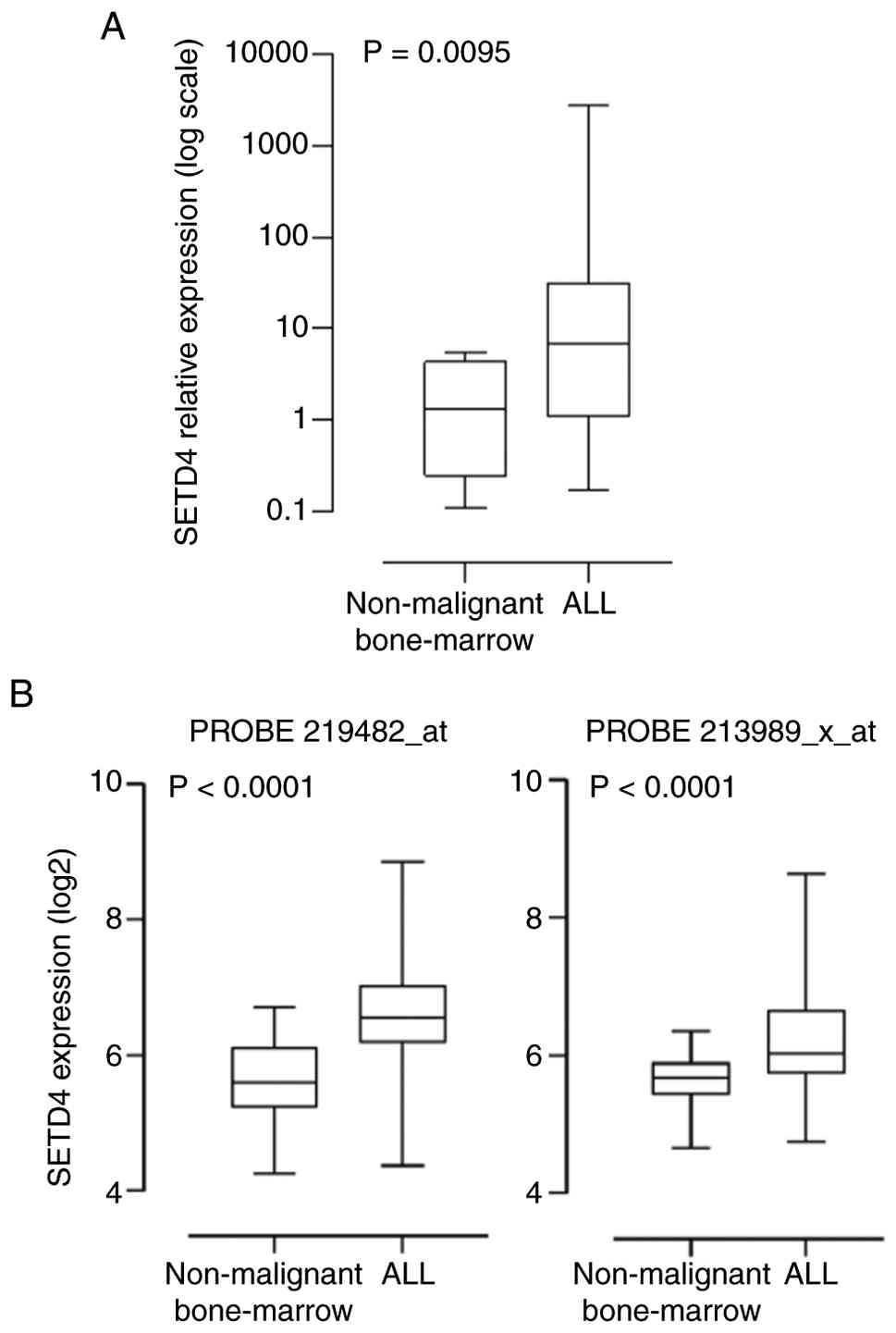
To verify whether SETD4 was differentially
expressed in ALL, RT-qPCR was performed on extracts from bone
marrow (BM) aspirates of ALL patients and of non-malignant BM
samples and evaluated the relative expression using the
housekeeping gene ACTB for normalization. The expression of
SETD4 was significantly greater in malignant samples, with a
median fold-change of 5.14 (95% CI=0.4539–23.74; P=0.0095;
Mann-Whitney U test, Fig. 1A).
Similarly, two distinct microarray datasets with 318
and 317 ALL patients (Affymetrix Probes 219482_at and 23989_X_at,
respectively) available in the BloodSpot database indicated that
SETD4 was highly expressed (P<0.0001) in the ALL samples
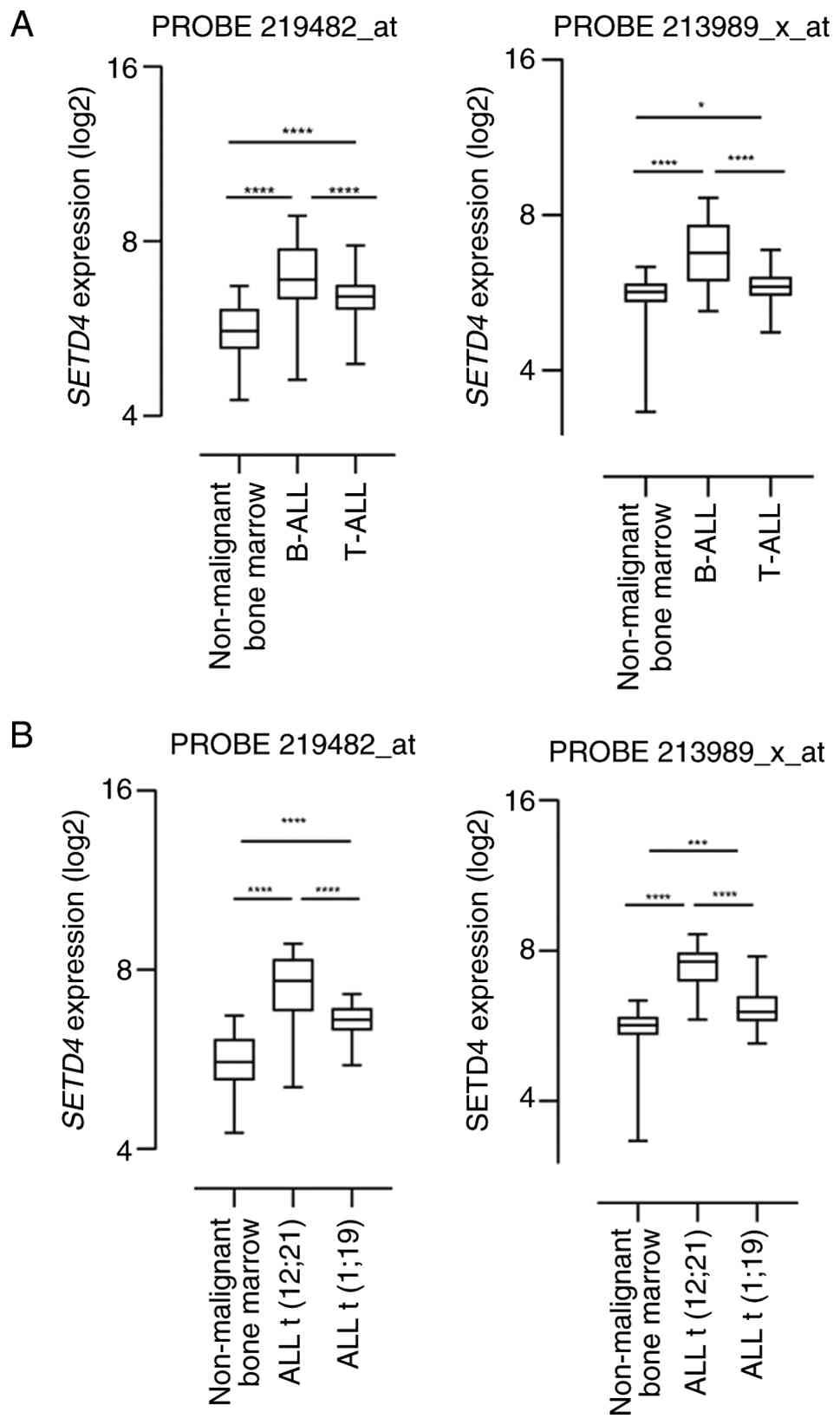
compared with the non-leukemic samples (Fig. 1B). Further analysis of the mRNA
levels using these same probes showed that SETD4 expression
was higher in LLA-B compared with LLA-T and also higher in ALL
t(12;21) in comparison to ALL t(1;19) (Fig. 2A and B). Subtype analyses were not
performed in the cohort due to an insufficient and unbalanced
number of samples in each subgroup.
SETD4 transcription correlates with a
decrease in leukemic burden and SMYD2 transcription during
chemotherapy
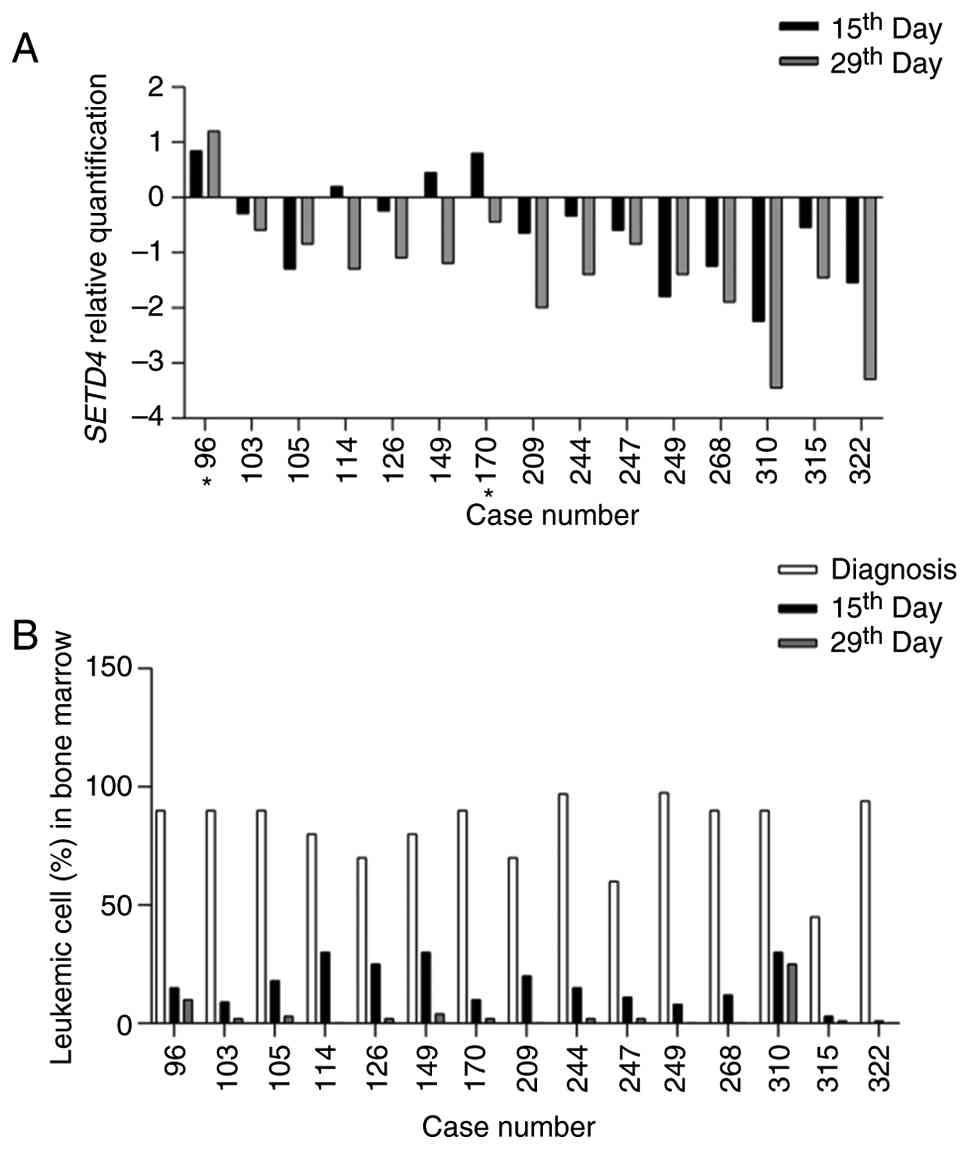
To investigate whether SETD4 is predominantly
transcribed in leukemic cells compared with normal cells, RT- qPCR
we conducted on samples from 15 patients on days 15 and 29 of
chemotherapy and it was assessed whether SETD4 expression
decrease concomitantly with the reduction in leukemic burden. At
the time of diagnosis, 13 patients exhibited high SETD4
expression. Among these, 11 patients demonstrated reduced
SETD4 levels by day 15. Notably, the two patients with basal
SETD4 expression at diagnosis did not exhibit lower levels
at this time point. However, except for patient 96 (from the basal
expression group), all patients showed decreased SETD4
levels by day 29 (Fig. 3A).
Additionally, the present study evaluated the
leukemic burden in the bone marrow of these 15 patients at three
time points: diagnosis, day 15 and 29 after initiating treatment
(Fig. 3B). As expected, all
patients experienced a decline in leukemic cells by days 15 and 29
post-diagnosis, with five patients achieving complete clearance of
leukemic blasts by day 29.
We previously reported that SMYD2
transcription levels are upregulated in ALL patients and is a poor
prognostic factor (9). The present
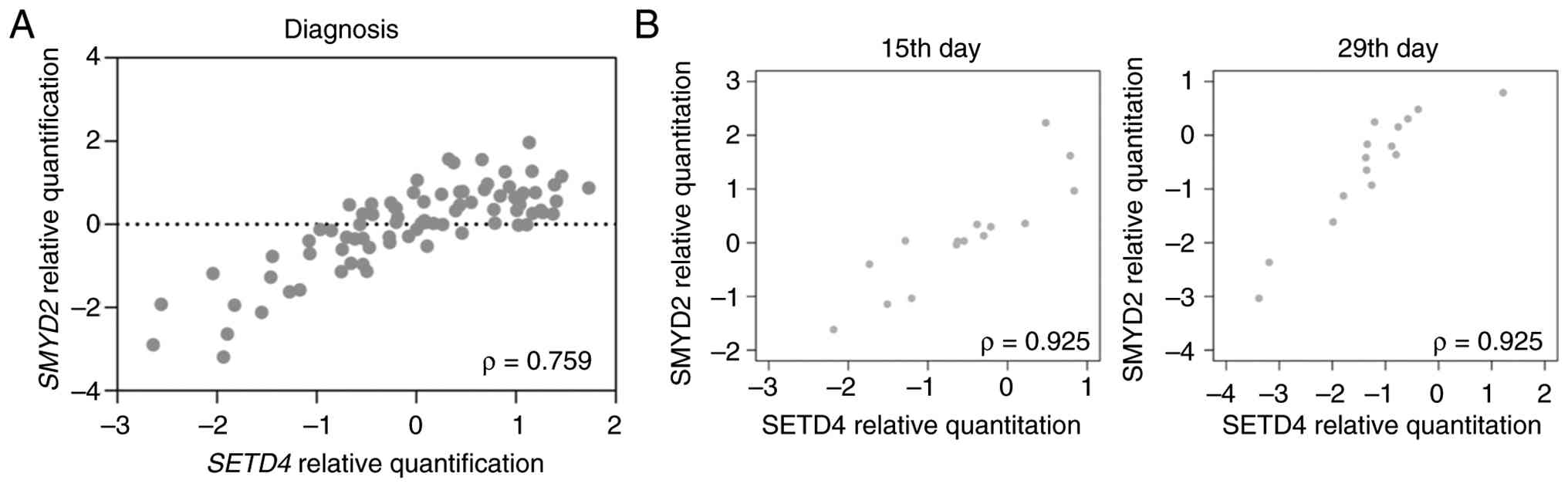
study investigated whether SETD4 and SMYD2 share any
biological relationship in this context. First, it compared the
transcription levels of SETD4 and SMYD2 among all
patients at diagnosis (Fig. 4A).
Next, the transcription levels of SETD4 and SMYD2
were examined on days 15 and 29 of chemotherapy. Surprisingly, a
positive and strong correlation was detected between both genes at
diagnosis (Spearman ρ=0.759; P<0,0001) and on either day of
treatment (Spearman ρ=0.925; P<0.01) (Fig. 4B).
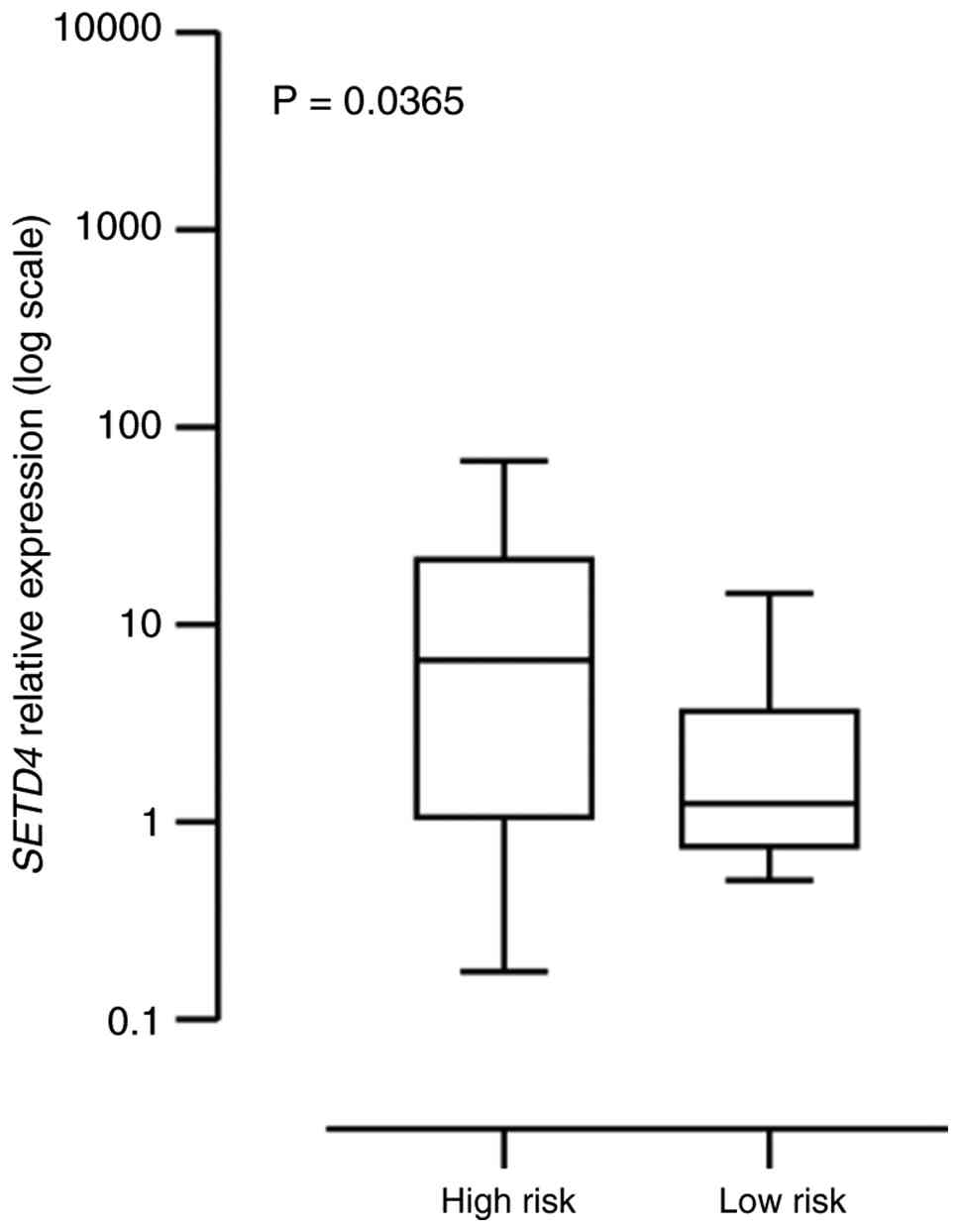
SETD4 transcription levels may be a
marker for risk stratification
To investigate the relationship between SETD4
expression levels and the risk stratification of ALL patients,
samples from high-risk and low-risk groups were compared.
Importantly, high-risk patients presented increased relative
SETD4 mRNA expression (Fig.
5).
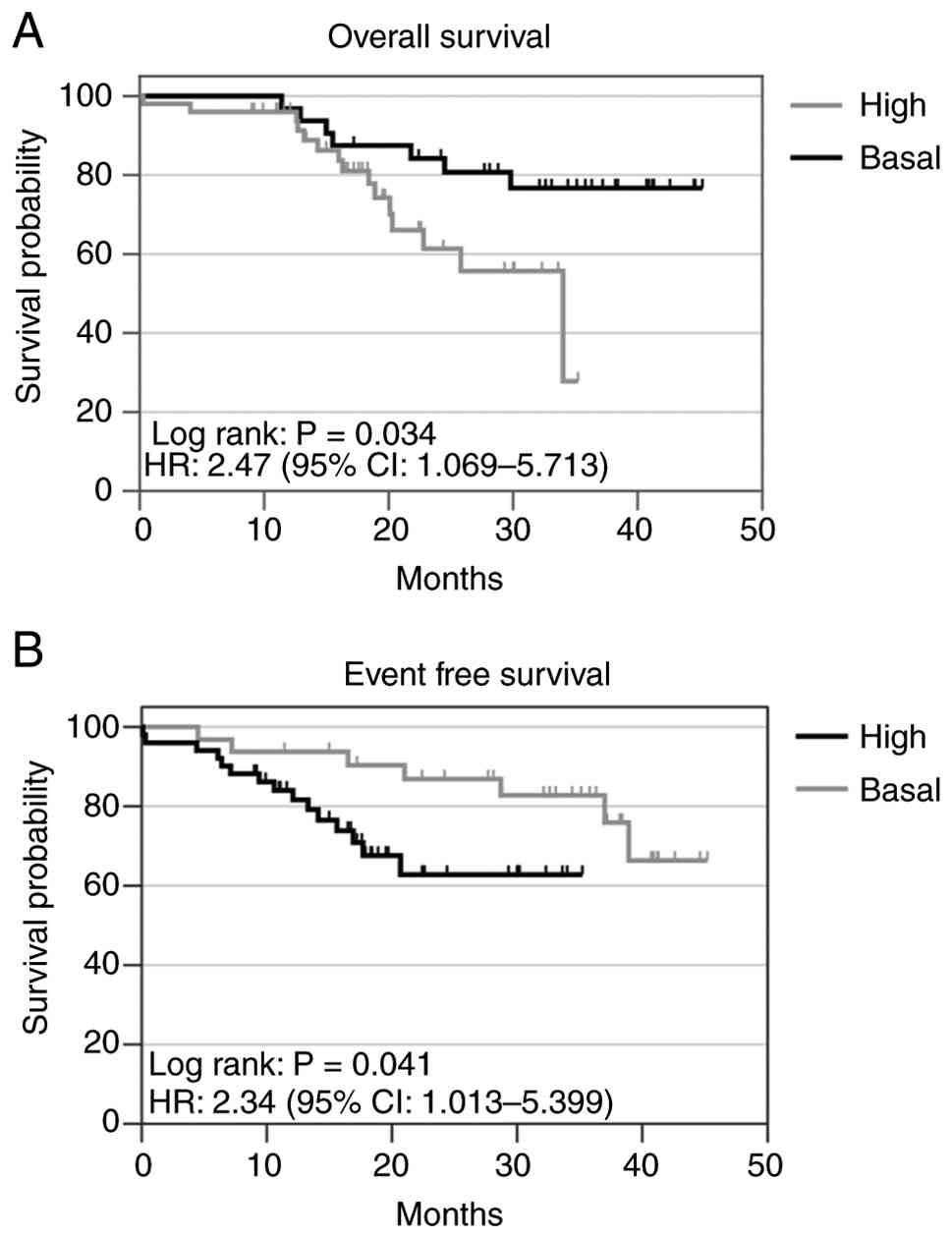
SETD4 overexpression is associated
with a lower overall survival in pediatric ALL patients and a worse
outcome
Since SETD4 is upregulated in most ALL
patients, particularly those classified as high-risk, their
survival was analyzed to determine whether this methyltransferase
could markedly impact survival outcomes. It was observed that high
expression levels of SETD4 were associated with decreased OS
and EFS in this subset of patients.
Kaplan-Meier analysis revealed that the 3-year OS
probability for the group of patients with high SETD4
expression was 27.8%, whereas it was 76.7% for the basal expression
group (P<0.05). Additionally, the 3-year EFS probability for the
basal SETD4 expression group was 82.78%, which was markedly
higher than that of the group with high SETD4 expression
(Fig. 6).
Discussion
Several KMTs have been identified as key players in
leukemogenesis. In mixed lineage leukemia (MLL), the uncontrolled
activities of the KMTs DOT1-like histone lysine methyltransferase
and ASH1-like histone methyltransferase are crucial for abnormal
cell proliferation (18).
Concurrently, rearrangements in MLL, which is also a
methyltransferase, are considered unfavorable prognostic factors
(19–21). Another KMT, Wolf-Hirschhorn syndrome
candidate 1 (WHSC1) (also known as MMSET or NSD2), is aberrantly
highly expressed due to the t(4;14) chromosomal translocation in a
myeloma subtype with a poor prognosis (22). Furthermore, the WHSCH1
p.E1099K mutation is markedly prevalent among patients who
experience relapsed ALL, suggesting its importance in clonal
evolution and the development of drug resistance (23). Additionally, SETD8 has been
shown to regulate the interaction of p53 with Numb through
methylation of the phosphotyrosine-binding domain of the latter
(24). Also, the association
between the rs16917496 polymorphism of the SETD8 gene and
the risk of ALL was significant (25). On the other hand, SETD1A has
been implicated in regulating p53 and its target genes expression
by binding to the Trp53 promoter and inducing specific miRNAs
associated with it (26–28). Moreover, it has been linked to the
process of progenitor B-cell maturation in mice (29).
The first study highlighting the oncogenic
significance of SETD4 was published by Faria et al in
2013 (10). These findings
highlight the role of SETD4 in breast cancer. SETD4 was found in
both the nucleus and cytosol in the breast cancer cell lines
MDA-MB-231, MGSO-3, and MACL-1. Its upregulation was linked to an
ER-negative and triple-negative phenotype. Knockdown of
SETD4 decreased cell proliferation in MDA-MB-231 cells by
affecting the G1/S cell cycle transition, markedly reducing cyclin
D1 levels.
Another study revealed that SETD4 controls
breast CSC quiescence (qCSC) through heterochromatin formation via
trimethylation of H3K20, leading to chemoradiotherapy resistance
and tumor relapse in mice. Moreover, the authors identified SETD4
qCSCs in several cancer types, such as gastric, lung, liver,
ovarian and cervical cancers (11).
In addition, SETD4 upregulation has recently been identified
in advanced-stage NSCLC tissues compared with early-stage tissues,
particularly in the chemoresistant group. Furthermore, SETD4
overexpression facilitated PTEN-mediated inhibition of the
PI3K-mTOR pathway in activated qLCSCs, indicating that SETD4 plays
a role in conferring chemoresistance, tumor progression, and poor
prognosis in NSCLC by regulating the behavior of CSCs (13). Notably, another study revealed that
the knockdown of SETD4 in hepatocellular carcinoma cells
resistant to sorafenib restored their sensitivity to the drug,
leading to a decrease in cell viability. A reduction in
SETD4 expression combined with sorafenib treatment,
downregulated AKT phosphorylation, thereby inducing the death of
HCC cells (30). However, the
involvement of SETD4 in leukemia has not been previously
explored.
Despite the limited cohort size, SETD4 mRNA
levels were consistently higher in diagnostic bone-marrow (BM)
samples from ALL patients than in non-neoplastic BM controls, a
finding validated across public datasets and by independent methods
(RT-qPCR and microarray). Additionally, further investigation in
public databases revealed that SETD4 expression was markedly
higher in B-ALL compared with T-ALL, as well as in ALL t(12;21)
compared with ALL t(1;19).
SETD4 is located on chromosome 21, a region
extensively implicated in leukemogenesis. Alterations involving
chromosome 21, including trisomy 21 and intrachromosomal
amplification of chromosome 21 (iAMP21), are well-established
factors predisposing to leukemogenesis and markers of high-risk
disease in pediatric ALL (31).
Given the relevance of chromosome 21-encoded genes to leukemic
transformation, several genes mapped to this region, such as
RUNX1, ERG, ETS2, and HMGN1, have been extensively
studied and shown to play important clinical and prognostic roles
in ALL (32–35). In this genomic context, the
enrichment of SETD4 expression in B-ALL, particularly in the
t(12;21) subtype observed in public datasets, raises the
possibility that SETD4 may be integrated into
transcriptional networks driven by B-cell-specific oncogenic
programs.
Notably, SETD4 expression levels showed a marked
decrease on treatment days 15 and 29, time points that may reflect
early treatment response and leukemic blast clearance in pediatric
ALL. This dynamic reduction is consistent with the association of
SETD4 expression with leukemic burden states at diagnosis.
However, the interpretation of these treatment-related dynamics is
limited by the availability of paired longitudinal samples, as only
15 patients could be followed during therapy.
As with SETD4, SMYD2 trimethylates histone H3 lysine
4 (H3K4me3) and accelerates the G1/S transition (36–38).
Our previous study showed that SMYD2 is over-expressed in
ALL, negatively associates with OS and EFS, and decreases during
chemotherapy (9). SMYD2 is
therefore considered a therapeutic target in hematological
malignancies and a regulator of cancer-stem-cell quiescence; its
depletion in acute myeloid leukemia reduces chemosensitivity
(39). In the current cohort,
SETD4 and SMYD2 transcript levels were strongly and
positively correlated both at diagnosis and during therapy,
suggesting shared upstream regulators or convergent epigenetic
programs. Future functional studies are warranted to dissect this
relationship and to test whether the combined inhibition of SETD4
and SMYD2 offers therapeutic benefit in ALL.
Additionally, the present study observed that
patients in the high-risk group exhibited elevated SETD4
mRNA expression. Moreover, individuals with high SETD4
levels showed markedly worse overall survival and event-free
survival compared with those with low expression. Although it
remains unclear whether this upregulation represents a driver or
passenger event in leukemogenesis, it is plausible that
SETD4 contributes to the progression of ALL. This hypothesis
is supported by the broader oncogenic potential of SET
domain-containing lysine methyltransferases, which regulate
chromatin dynamics, transcriptional repression, and cellular
differentiation, processes often disrupted in hematological
malignancies (40). Given the
putative role of SETD4 in maintaining stemness and quiescence in
hematopoietic progenitors, its dysregulation may provide a survival
advantage to leukemic stem-like cells, thereby promoting disease
progression and therapeutic resistance.
Despite the likely oncogenic effect of SETD4
on ALL leukemogenesis, its role in cancer development is ambiguous
and appears to be context dependent. SETD4 is mostly
downregulated in prostate cancer cells and tissue samples. A
decrease in the expression of SETD4 is associated with
inferior clinicopathological characteristics, such as pathological
grade, clinical stage and Gleason score (11). Moreover, SETD4 overexpression
inhibited prostate cancer cell proliferation (11). By contrast, the expression of
SMYD2 was elevated in prostate cancer tissues compared with
that in benign prostate tissues, and an increase in SMYD2
expression was linked to a greater risk of biochemical recurrence
following radical prostatectomy. Additionally, reducing
SMYD2 levels suppressed the proliferation of prostate cancer
cells in vivo and in vitro (41).
Importantly, despite the limited sample size, the
data of the present study supports a clinical association between
SETD4 expression and leukemic burden, treatment response, and
survival. One limitation of the present study was the restricted
availability of paired longitudinal samples during therapy, which
constrained further evaluation of SETD4 expression dynamics over
the course of treatment. Future studies with larger longitudinal
cohorts will be important to clarify the temporal role of SETD4 in
ALL.
In addition, although SETD4 and SMYD2
transcript levels were strongly and positively correlated, the
potential interaction between these methyltransferases was not
explored in the present study. Further investigations addressing
this relationship may help elucidate whether SETD4 and SMYD2 act
within shared epigenetic programs and whether their combined
targeting could be therapeutically relevant in ALL.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Council for
Scientific and Technological Development (CNPq), Brazil (Funder ID:
501100003593; grant no. 405618/2025-5) and Fundação de Apoio à
Pesquisa do Distrito Federal (FAPDF; grant no.
00193-00002146/2023-38).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
FP-S, ABM and LHTS were responsible for
conceptualization. LHTS, DARR and FP-S were responsible for
methodology. LHTS managed specimen collection and collected the
data. LAMT performed experiments and contributed to data analysis.
MBL contributed to the experiments presented in the present study.
FP-S, LAMT and MBL wrote and finalized the manuscript. All authors
have read and approved the final manuscript. FP-S, LHTS, LAMT and
MBL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study protocol was approved by the
Ethical Committee of the Federal District Foundation for Teaching
and Research in Health Sciences, Brazil, with written informed
consent from patients and/or guardians, under approval no.
CEP/FEPECS 555/11. The study followed the Declaration of
Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Inaba H and Mullighan CG: Pediatric acute
lymphoblastic leukemia. Haematologica. 105:2524–2539. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tran TH and Hunger SP: The genomic
landscape of pediatric acute lymphoblastic leukemia and precision
medicine opportunities. Semin Cancer Biol. 84:144–152. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carroll AJ, Crist WM, Parmley RT, Roper M
and Finley MD: Pre-B cell leukemia associated with chromosome
translocation 1;19. Blood. 63:721–724. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van den Berghe H, David G, Orshoven ABV,
Louwagie A, Verwilghen R, Daele MCV, Eggermont E and Eeckels R: A
new chromosome anomaly in acute lymphoblastic leukemia (ALL). Hum
Genet. 46:173–180. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Husmann D and Gozani O: Histone lysine
methyltransferases in biology and disease. Nat Struct Mol Biol.
26:880–889. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang J, Perez-Burgos L, Placek BJ,
Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T
and Berger SL: Repression of p53 activity by Smyd2-mediated
methylation. Nature. 444:629–632. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li LX, Zhou JX, Calvet JP, Godwin AK,
Jensen RA and Li X: Lysine methyltransferase SMYD2 promotes triple
negative breast cancer progression. Cell Death Dis. 9:1–17.
2018.PubMed/NCBI
|
|
8
|
Möricke A, Reiter A, Zimmermann M, Gadner
H, Stanulla M, Dördelmann M, Löning L, Beier R, Ludwig WD, Ratei R,
et al: Risk-adjusted therapy of acute lymphoblastic leukemia can
decrease treatment burden and improve survival: Treatment results
of 2169 unselected pediatric and adolescent patients enrolled in
the trial ALL-BFM 95. Blood. 111:4477–4489. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sakamoto LHT, Andrade RV, Felipe MSS,
Motoyama AB and Silva FP: SMYD2 is highly expressed in pediatric
acute lymphoblastic leukemia and constitutes a bad prognostic
factor. Leuk Res. 38:496–502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Faria JAQA, Corrêa NCR, de Andrade C, de
Angelis Campos AC, dos Santos Samuel de Almeida R, Rodrigues TS, de
Goes AM, Gomes DA and Silva FP: SET domain-containing protein 4
(SETD4) is a newly identified cytosolic and nuclear lysine
methyltransferase involved in breast cancer cell proliferation. J
Cancer Sci Ther. 5:58–65. 2013.PubMed/NCBI
|
|
11
|
Wang C, Wang T, Li KJ, Hu LH, Li Y, Yu YZ,
Xie T, Zhu S, Fu DJ, Wang Y, et al: SETD4 inhibits prostate cancer
development by promoting H3K27me3-mediated NUPR1 transcriptional
repression and cell cycle arrest. Cancer Lett. 579:2164642023.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dai L, Ye S, Li HW, Chen DF, Wang HL, Jia
SN, Lin C, Yang JS, Yang F, Nagasawa H and Yang WJ: SETD4 regulates
cell quiescence and catalyzes the trimethylation of H4K20 during
diapause formation in Artemia. Mol Cell Biol. 37:e00453–e00416.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Yu Y, Yang W, Wu L, Yang Y, Lu Q
and Zhou J: SETD4 confers cancer stem cell chemoresistance in
nonsmall cell lung cancer patients via the epigenetic regulation of
cellular quiescence. Stem Cells Int. 2023:73678542023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feng X, Lu H, Yue J, Schneider N, Liu J,
Denzin LK, Chan CS, De S and Shen Z: Loss of Setd4 delays
radiation-induced thymic lymphoma in mice. DNA Repair (Amst).
86:1027542020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brandalise S, Odone V, Pereira W, Andrea
M, Zanichelli M and Aranega V: Treatment results of three
consecutive Brazilian cooperative childhood ALL protocols:
GBTLI-80, GBTLI-82 and −85. ALL Brazilian Group. Leukemia. 7 (Suppl
2):S142–S145. 1993.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bagger FO, Sasivarevic D, Sohi SH, Laursen
LG, Pundhir S, Sønderby CK, Winther O, Rapin N and Porse BT:
BloodSpot: A database of gene expression profiles and
transcriptional programs for healthy and malignant haematopoiesis.
Nucleic Acids Res. 44:D917–D924. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grigsby SM, Friedman A, Chase J, Waas B,
Ropa J, Serio J, Shen C, Muntean AG, Maillard I and
Nikolovska-Coleska Z: Elucidating the importance of DOT1L
recruitment in MLL-AF9 leukemia and hematopoiesis. Cancers (Basel).
13:6422021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
El Chaer F, Keng M and Ballen KK:
MLL-rearranged acute lymphoblastic leukemia. Curr Hematol Malig
Rep. 15:83–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu L, Li Q, Wong SHK, Huang M, Klein BJ,
Shen J, Ikenouye L, Onishi M, Schneidawind D, Buechele C, et al:
ASH1L links histone H3 lysine 36 di-methylation to MLL leukemia.
Cancer Discov. 6:770–783. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aljazi MB, Gao Y, Wu Y, Mias GI and He J:
Histone H3K36me2-specific methyltransferase ASH1L promotes
MLL-AF9-induced leukemogenesis. Front Oncol. 11:7540932021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Issa ME, Takhsha FS, Chirumamilla CS,
Perez-Novo C, Berghe WV and Cuendet M: Epigenetic strategies to
reverse drug resistance in heterogeneous multiple myeloma. Clin
Epigenetics. 9:172017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Narang S, Evensen NA, Saliba J, Pierro J,
Loh ML, Brown PA, Kolekar P, Mulder H, Shao Y, Easton J, et al:
NSD2 E1099K drives relapse in pediatric acute lymphoblastic
leukemia by disrupting 3D chromatin organization. Genome Biol.
24:642023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dhami G, Liu H, Galka M, Voss C, Wei R,
Muranko K, Kaneko T, Cregan SP, Li L and Li SS: Dynamic methylation
of Numb by Set8 regulates its binding to p53 and apoptosis. Mol
Cell. 50:565–576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hashemi M, Sheybani-Nasab M, Naderi M,
Roodbari F and Taheri M: Association of functional polymorphism at
the miR-502-binding site in the 3′ untranslated region of the SETD8
gene with risk of childhood acute lymphoblastic leukemia, a
preliminary report. Tumor Biol. 35:10375–10379. 2014. View Article : Google Scholar
|
|
26
|
Tajima K, Yae T, Javaid S, Tam O, Comaills
V, Morris R, Wittner BS, Liu M, Engstrom A, Takahashi F, et al:
SETD1A modulates cell cycle progression through a miRNA network
that regulates p53 target genes. Nat Commun. 6:82572015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yae T, Tajima K and Maheswaran S: SETD1A
induced miRNA network suppresses the p53 gene expression module.
Cell Cycle. 15:487–488. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ogawa S, Fukuda A, Matsumoto Y, Hanyu Y,
Sono M, Fukunaga Y, Masuda T, Araki O, Nagao M, Yoshikawa T, et al:
SETDB1 inhibits p53-mediated apoptosis and is required for
formation of pancreatic ductal adenocarcinomas in mice.
Gastroenterology. 159:682–96.e13. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tusi BK, Deng C, Salz T, Zeumer L, Li Y,
So CWE, Morel LM, Qiu Y and Huang S: Setd1a regulates progenitor
B-cell-to-precursor B-cell development through histone H3 lysine 4
trimethylation and Ig heavy-chain rearrangement. FASEB J.
29:1505–1515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li GM, Wang YG, Pan Q, Wang J, Fan JG and
Sun C: RNAi screening with shRNAs against histone
methylation-related genes reveals determinants of sorafenib
sensitivity in hepatocellular carcinoma cells. Int J Clin Exp
Pathol. 7:1085–1092. 2014.PubMed/NCBI
|
|
31
|
Gao Q, Ryan SL, Iacobucci I, Ghate PS,
Cranston RE, Schwab C, Elsayed AH, Shi L, Pounds S, Lei S, et al:
The genomic landscape of acute lymphoblastic leukemia with
intrachromosomal amplification of chromosome 21. Blood.
142:711–723. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sood R, Kamikubo Y and Liu P: Role of
RUNX1 in hematological malignancies. Blood. 129:2070–2082. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang J, McCastlain K, Yoshihara H, Xu B,
Chang Y, Churchman ML, Wu G, Li Y, Wei L, Iacobucci I, et al:
Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat
Genet. 48:1481–1489. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fu L, Fu H, Wu Q, Pang Y, Xu K, Zhou L,
Qiao J, Ke X, Xu K and Shi J: High expression of ETS2 predicts poor
prognosis in acute myeloid leukemia and may guide treatment
decisions. J Transl Med. 15:1592017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Page EC, Heatley SL, Rehn J, Thomas PQ,
Yeung DT and White DL: Gain of chromosome 21 increases the
propensity for P2RY8::CRLF2 acute lymphoblastic leukemia via
increased HMGN1 expression. Front Oncol. 13:11778712023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Jin G, Guo Y, Cao Y, Niu S, Fan X
and Zhang J: SMYD2 suppresses p53 activity to promote glucose
metabolism in cervical cancer. Exp Cell Res. 404:1126492021.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cho HS, Hayami S, Toyokawa G, Maejima K,
Yamane Y, Suzuki T, Dohmae N, Kogure M, Kang D, Neal DE, et al: RB1
methylation by SMYD2 enhances cell cycle progression through an
increase of RB1 phosphorylation. Neoplasia. 14:476–486. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang L, Li L, Zhang H, Luo X, Dai J, Zhou
S, Gu J, Zhu J, Atadja P, Lu C, et al: Structure of human SMYD2
protein reveals the basis of p53 tumor suppressor methylation. J
Biol Chem. 286:38725–38737. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zipin-Roitman A, Aqaqe N, Yassin M,
Biechonski S, Amar M, van Delft MF, Gan OI, McDermott SP, Buzina A,
Ketela T, et al: SMYD2 lysine methyltransferase regulates leukemia
cell growth and regeneration after genotoxic stress. Oncotarget.
8:16712–16727. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bennett RL, Swaroop A, Troche C and Licht
JD: The role of nuclear receptor-binding SET domain family histone
lysine methyltransferases in cancer. Cold Spring Harb Perspect Med.
7:a0267082017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Wan F, Zhang J, Zheng S, Yang Y,
Hong Z and Dai B: Targeting SMYD2 inhibits prostate cancer cell
growth by regulating c-Myc signaling. Mol Carcinog. 62:940–950.
2023. View Article : Google Scholar : PubMed/NCBI
|