Introduction
Over the last decade, liquid biopsy have attracted
increasing attention in modern oncology due to its potential
clinical applications. Circulating tumor cells, circulating tumor
nucleic acids [such as circulating tumor DNA (ct-DNA) and
circulating tumor RNA], exosomes, and tumor proteins are considered
as possible biomarkers for characterizing the molecular
characteristics of malignant tumors, thus providing insights into
both the genetic and phenotypic features of cancers, eventually
facilitating the development of personalized medicine (1,2).
ct-DNA is a fraction of total cell-free DNA found in
blood plasma. Under normal conditions, plasma DNA originates from
hematopoietic cells and is present at low concentrations in healthy
individuals. DNA enters the bloodstream through several processes,
such as cell death, necrosis, or apoptosis (3). However, inflammation, intense
exercise, or severe stress can also enhance the content of
circulating free DNA. In patients with cancer, ct-DNA fragments
encompass the same somatic mutations as those found in the primary
tumor. On average, ct-DNA fragments are ~143–145 base pairs in
length-shorter than normal cell-free DNA-and exhibit a short
half-life of ~2 h. The liver serves as the primary site for ct-DNA
clearance from the circulation (3,4).
It has been reported that plasma ct-DNA levels can
be associated with a patient's tumor burden, with advanced cancers
typically displaying higher concentrations of ct-DNA in the
bloodstream. The amount of ct-DNA released into the circulation
also depends on several factors, including tumor location, cell
proliferation, and necrosis. Bettegowda et al (5) demonstrated that patients with
localized disease or primary central nervous system tumors rarely
exhibited detectable ct-DNA levels, while those with metastatic
colon, pancreatic, bladder, or ovarian cancers commonly did.
Furthermore, the short half-life of ct-DNA can also enable
real-time monitoring of tumor clonal heterogeneity during the
disease course.
Pre-analytical and analytical variables can
significantly affect ct-DNA analysis. Factors such as the type of
blood collection tubes used, volume of plasma processed, storage
conditions, and specimen handling can markedly affect the accuracy
of ct-DNA results. The DNA extraction method should therefore be
carefully considered to ensure optimal reproducibility (6,7).
Beyond technical aspects, several clinical and biological factors,
including the timing of sample collection, disease stage, prior or
ongoing treatments, and coexisting medical conditions, can also
affect the proportion of detectable ct-DNA fraction in plasma.
However, the application of highly sensitive analytical techniques,
such as next-generation sequencing (NGS) or digital PCR, has
significantly improved the detection threshold for ct-DNA in
patients with cancer (8,9).
ct-DNA serves as a liquid biopsy biomarker with
unique advantages that help overcome the limitations of
conventional ‘gold standard’ tissue biopsies. Blood collection is
simple, minimally invasive, and safe, posing minimal risk compared
with interventional tissue sampling (10). Additionally, rabid analysis of
tumor-derived DNA can offer a minimally invasive method,
particularly in cases where conventional biopsy is difficult to
perform, unsafe, or when tissue samples are insufficient. The
ability to obtain serial blood samples also eliminates the need for
repeated tissue biopsies, thus providing dynamic insight into the
molecular profile of the tumor over time. This approach enables
real-time monitoring of spatial and temporal tumor heterogeneity,
facilitates the assessment of treatment response, and aids the
early detection of resistant clones, ultimately supporting more
precise clinical decision-making (11,12).
Colorectal cancer (CRC) is one of the most prevalent
types of cancer worldwide and remains a leading cause of
cancer-related mortality (13). The
therapeutic landscape of metastatic CRC has evolved significantly
over the past decade, with the introduction of targeted therapies,
immunotherapies, and advanced chemotherapy regimens, alongside
continuous efforts to improve overall survival. According to
current clinical guidelines, the assessment of microsatellite
instability (MSI) and RAS and BRAF mutations is a
critical step prior therapy initiation. The detection of RAS
or BRAF mutations excludes the use of anti-epidermal growth
factor receptor (EGFR)-targeted therapy, directing patients instead
toward antiangiogenic and chemotherapeutic combinations. For
patients initially treated with chemotherapy plus anti-EGFR
therapy, switching to alternative regimens upon disease progression
is recommended (14).
However, CRC is a biologically heterogeneous disease
showing both spatial and temporal diversity, and acquired anti-EGFR
resistance mechanisms can emerge over time. In this context, liquid
biopsy represents a valuable tool for capturing the molecular
characteristics of metastatic CRC throughout the course of
treatment, thus guiding therapeutic decision-making (15). Recently, the ESMO Precision Medicine
Working Group issued recommendations supporting the integration of
ct-DNA analysis into clinical practice, identifying CRC as one of
the major areas of application (16).
The present study aimed to assess ct-DNA in patients
with metastatic CRC undergoing therapy, using NGS to identify
tumor-related mutations in both tissue samples and cell-free plasma
DNA.
Materials and methods
Patients
In the current study, a total of 45 patients who
attended the University Hospital of Ioannina (Ioannina, Greece)
between September 2018 and November 2022 were enrolled. All
participants were required to have a new diagnosis of metastatic
CRC and an adequate tumor tissue sample available for molecular
profiling. In addition, blood samples were collected from patients
at multiple time points during the course of treatment.
Specifically, blood collection for ct-DNA isolation and analysis
was performed at diagnosis (prior to therapy initiation) and at the
time of first and second disease progression. Both tumor tissues
and plasma samples were subjected to NGS for comprehensive
mutational profiles. All patients provided written informed consent
prior to enrollment and the study protocol was approved by the
Ethics Committee of the University Hospital of Ioannina (approval
no. 11/18-04-2018).
Specifically, the inclusion and exclusion criteria
were as follows. Eligible participants were adults aged 18 years or
older with a new diagnosis of metastatic colorectal adenocarcinoma
who had adequate FFPE tumor tissue available for molecular
profiling, were able to provide serial blood samples at baseline
and during follow-up, and provided written informed consent for
participation and molecular testing. Patients were excluded if they
had received prior systemic treatment for metastatic disease before
baseline sampling, lacked sufficient tumor tissue for NGS analysis,
were unable to provide the required blood samples for ctDNA
testing, or had a coexisting malignancy or medical condition that
could interfere with proper sample collection or reliable NGS
analysis.
Cell-free total nucleic acids (cfTNA)
isolation
Peripheral blood samples were collected into
Cell-Free DNA BCT tubes (Streck LLC.), particularly designed to
preserve cfTNA. Plasma was obtained through two sequential
centrifugations at 447 × g for 10 min at 4°C to ensure complete
removal of cellular components. cfTNA was then extracted from 2 ml
plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen,
Inc.), according to the manufacturer's instructions. To minimize
pre-analytical variability, all blood samples were processed within
2 h of venipuncture. Plasma aliquots were stored at −80°C, and
repeated freeze-thaw cycles were avoided to preserve cfTNA
integrity.
Tissue selection and DNA
extraction
Formalin-fixed, paraffin-embedded (FFPE) tumor
biopsy sections were stained with hematoxylin and eosin to ensure
that the tumor cell content exceeded 75% whenever feasible. A
pathologist identified and marked the representative tumor areas
for DNA extraction. Genomic DNA was then isolated from 10 µm-thick
unstained FFPE sections using the QIAamp FFPE Tissue Kit (Qiagen,
Inc.). DNA concentration and purity were determined using a
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
To ensure consistency, DNA quality and yield were further verified
by Qubit fluorometric analysis prior library preparation.
Preparation of NGS libraries
For the analysis of FFPE-derived DNA samples, a
custom Ion AmpliSeq panel was employed, based on the Ion AmpliSeq
Colon and Lung Cancer Research Panel v2 (Thermo Fisher Scientific,
Inc.). This panel was modified to include one additional amplicon
targeting exon 14 skipping mutations in the MET gene and two
additional amplicons covering exons 2 and 3 of the HRAS
gene. Fusion RNA transcript analysis was performed using the Ion
AmpliSeq™ RNA Fusion Lung Cancer Research Panel (Thermo Fisher
Scientific, Inc.). A detailed list of the 23 targeted genes is
available upon request. The analyzed genes included AKT1, ALK,
BRAF, CTNNB1, DDR2, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3,
KRAS, MAP2K1, MET, NOTCH, NRAS, PIK3CA, PTEN, SMAD4, STK11,
TP53, and HRAS. The DNA concentrations from FFPE samples
were measured using the Qubit™ 2.0 Fluorometer and the Qubit™ dsDNA
HS Assay Kit (Thermo Fisher Scientific, Inc.). Libraries were
prepared from 10 ng of FFPE-derived DNA or 6 µl of ct-DNA using the
Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Amplicon amplification
was achieved with the Ion AmpliSeq™ HiFi Master Mix (Thermo Fisher
Scientific, Inc.), and the amplified products were digested with
FUPA reagent followed by barcoding with the IonCode™ Barcode
Adapters 1–384 Kit (Thermo Fisher Scientific, Inc.). Libraries were
purified using the Agencourt AMPure XP PCR purification system
(Beckman Coulter, Inc.).
For total cfTNA analysis, the Oncomine™ Pan-Cancer
Cell-Free Assay (Thermo Fisher Scientific, Inc.) was utilized. This
assay incorporates unique molecular identifiers (UMIs) in the form
of random molecular tags to label each molecule prior library
amplification, thus enabling the distinction of true variants from
random sequencing errors, thereby enhancing accuracy and minimizing
false-positive results. The assay targeted hotspot mutations,
including single-nucleotide variants (SNVs) and small insertions or
deletions, in genes such as AKT1, ALK, AR, ARAF, BRAF, CHEK2,
CTNNB1, DDR2, EGFR, ERBB2, ERBB3, ESR1, FGFR1-4, FLT3, GNA11, GNAQ,
GNAS, HRAS, IDH1, IDH2, KIT, KRAS, MAP2K1, MAP2K2, MET, MTOR, NRAS,
NTRK1, NTRK3, PDGFRA, PIK3CA, RAF1, RET, ROS1, SF3B1, SMAD4,
and SMO. Copy number variations were also assessed in
CCND1-3, CDK4, CDK6, EGFR, ERBB2, FGFR1-3, MET, and
MYC, and RNA fusions involving 12 key driver genes,
including ALK, BRAF, ERG, ETV1, FGFR1-3, MET, NTRK1, NTRK3,
RET, and ROS1, were also evaluated. The analytical
sensitivity of the cfTNA assay was validated at ~0.1–0.5% mutant
allele fraction (MAF) for SNVs/indels, ~1% for gene fusions, and
≥1.4-2-fold change for copy number variation (CNV) detection. For
FFPE-derived DNA, the Ion AmpliSeq workflow reliably detected
mutations with a limit of detection of ~5% MAF, applying a minimum
coverage threshold of 500× for confident variant calling. cfTNA
concentrations were quantified using the Qubit™ 2.0 Fluorometer and
the Qubit™ ssDNA Assay Kit (Thermo Fisher Scientific, Inc.), while
libraries were prepared according to the manufacturer's
instructions. Briefly, reverse transcription of cfTNA was carried
out using the SuperScript™ VILO™ Master Mix. Subsequently, the
resulting cDNA was subjected to a two-cycle PCR to attach random
sequence tags and A/P1 adaptors to both fragment ends, followed by
purification using the Agencourt AMPure XP PCR system (Beckman
Coulter, Inc.). An additional 18-cycle PCR was then performed to
amplify the tagged fragments and introduce a unique barcode for
each sample using the Tag Sequencing Barcode Set (Thermo Fisher
Scientific, Inc.). Purification and size selection of the barcoded
libraries were carried out with Agencourt AMPure XP beads, and
library concentrations were subsequently quantified by real-time
PCR using the Ion Library TaqMan® Quantitation Kit
(Thermo Fisher Scientific, Inc.).
For clonal amplification, 100 pmol of DNA or cfTNA
libraries were pooled and amplified on Ion Sphere™ Particles (ISPs)
via emulsion PCR using the Ion OneTouch™ 2 instrument and the Ion
540™ Kit-OT2, following the manufacturer's guidelines. Quality
control was then performed with the Ion Sphere™ Quality Control Kit
to ensure that ~10–30% of ISPs were template-positive. Enrichment
of template-positive ISPs was carried out on the Ion OneTouch™ ES
instrument. The enriched particles were then loaded onto an Ion
540™ Chip and sequenced on the Ion GeneStudio™ S5 Prime System
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions.
Analysis of NGS data
NGS data analysis was performed using Ion Reporter™
software version 5.10.1.0 integrated with Torrent Suite™ version
5.10.1 (Thermo Fisher Scientific, Inc.). The sequencing results
were subsequently reviewed and further analyzed using Sequence
Pilot (version 4.3.0; JSI Medical Systems GmbH). Coverage analysis
was conducted with the Coverage Analysis plug-in (version 5.0.4.0),
providing quality metrics for each library within the sequencing
run. For FFPE DNA libraries, CNV analysis was carried out using Ion
Reporter™ software, which determines CNVs based on relative copy
number compared with a reference control sample. CNVs were assigned
confidence scores, with values >10 indicating high-confidence
amplifications, serving as the threshold for defining copy number
gains. For cfTNA samples, mutation detection, RNA fusion
identification, and CNV analysis were conducted according to the
manufacturer's workflow using Ion Reporter™ Oncomine TagSeq
Pan-Cancer Liquid Biopsy v2.1-Single Sample software (Thermo Fisher
Scientific, Inc.). Furthermore, the distribution of MAFs in both
plasma and tissue samples was also evaluated. All sequencing runs
incorporated internal quality controls, including coverage metrics
and spike-in control samples, to ensure reproducibility and
instrument stability across all experiments.
Statistical analysis
The primary objective of the current study was to
evaluate the concordance between mutation detection in tumor tissue
DNA and cell-free plasma DNA. To accomplish this, tumor mutations
were identified and compared in both sample types, and Cohen's κ
statistic was then performed to assess the level of agreement. More
particularly, all statistical analyses were conducted using the R
programming language (R Core Team: R: A Language and Environment
for Statistical Computing. Vienna, Austria: Foundation for
Statistical Computing; http://www.r-project.org/). Continuous variables are
expressed as the mean ± standard deviation and median with
interquartile range (IQR), while categorical variables are
presented as counts (n) and percentages (%). Categorical variables
were compared using either the χ2 test or Fisher's exact
test, as appropriate, whereas paired nominal data, defined as
categorical measurements obtained from the same patient in tumor
tissue and plasma samples, were analyzed using the McNemar test.
The normality of continuous variables was assessed by
Kolmogorov-Smirnov test. Based on data distribution, comparisons of
continuous data between independent (unpaired) groups were
performed using the Mann-Whitney U test for non-normally
distributed data. KRAS MAF values did not follow a normal
distribution; therefore, comparisons between tissue and plasma
samples were conducted using the Mann-Whitney U test. Correlations
between continuous variables were evaluated using Pearson's or
Spearman's rank correlation coefficients, as appropriate.
Sensitivity (Se), specificity (Sp), positive predictive value
(PPV), negative predictive value (NPV), and overall agreement were
estimated with 95% confidence intervals (CIs) using binomial
distribution. Agreement represents the degree of concordance
between the two methods, and Cohen's κ was employed to quantify
intermethod reliability. Two-tailed P-values and 95% CIs calculated
using Fisher's r-to-z transformation. P<0.05 was considered to
indicate a statistically significant difference. For Spearman's
rank correlation analyses, two-tailed P-values and 95% confidence
intervals were calculated using Fisher's r-to-z transformation,
with standard error estimated according to the method of Fieller,
Hartley and Pearson.
Results
Patient characteristics
Among the 45 patients enrolled in this study, 18
were female and 27 were male, all diagnosed with metastatic
colorectal adenocarcinoma. The primary tumor was located in the
left colon in 32 patients, the right colon in nine patients, and
the transverse colon in three patients, while one patient had an
unknown primary tumor site. The most common metastatic sites were
the liver, lungs, and peritoneum. A total of eight patients had
previously received adjuvant chemotherapy with 5-fluorouracil,
either alone or in combination with oxaliplatin, following an
earlier diagnosis of stage III CRC. The remaining patients
presented with de novo metastatic disease at initial
diagnosis. MSI testing was performed in a total of 33 patients.
Among them, 2 patients showed loss of mismatch repair protein
expression, consistent with MSI, while the remaining 31 patients
had microsatellite-stable (MSS) tumors. All patients were treated
with first-line therapy for metastatic CRC, according to the
current clinical guidelines and the treating physician's
discretion. More specifically, 9 patients received first-line
therapy with mFOLFOX6 combined with an anti-EGFR agent, seven
received mFOLFOX6 plus bevacizumab, and one patient was treated
with mFOLFOX6 alone. Fourteen patients received XELOX plus
bevacizumab, one received capecitabine plus bevacizumab, and one
received XELOX alone. In addition, 3 patients were treated with
capecitabine monotherapy, 4 received FOLFIRI plus an anti-EGFR
agent, 2 received FOLFIRI plus bevacizumab, 2 were treated with
FOLFOXIRI plus bevacizumab, and 1 received FOLFIRI alone. The
demographic and clinical characteristics of the study population
are summarized in Table SI.
Tissue NGS results
Among the 45 tissue samples analyzed by NGS, 24
samples harbored KRAS mutations. The most common variants
detected were KRAS c.35G>A (p.G12D) in 6 samples,
KRAS c.38G>A (p.G13D) in 6 samples, KRAS
c.35G>T (p.G12V) in 3 samples, and KRAS c.34G>A
(p.G12S) in 3 samples. Less common variants included KRAS
c.64C>A (p.Q22K), KRAS c.436G>A (p.A146T), KRAS
c.183A>C (p.Q61H), and KRAS Q61R in 1 patient each, and
KRAS c.35G>C (p.G12A) in 2 patients. In addition, 2
patients carried NRAS mutations, and more specifically
NRAS c.38G>T (p.G13V) and NRAS c.181C>A
(p.Q61K). BRAF V600E mutations were detected in three
patients, while 1 patient carried a BRAF G466E mutation.
Notably, KRAS, NRAS, and BRAF mutations were mutually
exclusive in all tissue samples. During tissue profiling with NGS,
among the remaining genes included in the sequencing panel, 27
patients carried TP53 mutations, 6 PIK3CA mutations,
1 patient harbored an APC mutation, 2 SMAD4
mutations, and two FBXW7 mutations. Overall, mutations in
genes other than KRAS, NRAS, and BRAF were identified
in 35 of the 45 tissue samples analyzed.
Baseline plasma ct-DNA results
Among the 45 baseline plasma samples analyzed by
NGS, 20 harbored KRAS mutations, including KRAS
c.35G>A (p.G12D) mutation in 5 samples, KRAS c.38G>A
(p.G13D) in 6 samples, KRAS c.35G>T (p.G12V) in 2
samples, KRAS c.34G>A (p.G12S) in 3 samples, KRAS
c.35G>C (p.G12A) in 1 sample, KRAS c.183A>C (p.Q61H)
in 2 samples, while 1 sample carried a KRAS c.436G>A
(p.A146T) mutation. In addition, 2 patients carried NRAS
mutations, specifically NRAS c.38G>T (p.G13V) and
NRAS c.181C>A (p.Q61K). Furthermore, BRAF V600E
mutations were identified in 3 samples. As observed in tissue
samples, KRAS, NRAS, and BRAF mutations were mutually
exclusive in plasma samples. Beyond the aforementioned mutations
detected in plasma samples after ct-DNA isolation, additional
changes were identified in other genes included in the NGS panel.
Specifically, 17 samples carried TP53 mutations, four
harbored PIK3CA mutations, two had APC mutations, and
PTEN, MET, FBXW7, and AKT mutations were identified
in one sample each. Overall, 24 plasma samples tested positive for
alterations in genes other than KRAS, NRAS, and
BRAF.
Following first-line treatment and at first disease
progression, blood samples were again collected from 33 patients
and processed for ct-DNA isolation and mutation analysis. Among
them, 10 patients harbored KRAS mutations, including
KRAS c.35G>A (p.G12D) in four patients, KRAS
c.35G>T (p.G12V) in 2 patients, KRAS c.38G>A (p.G13D)
in 2 patients, KRAS Q61R in one patient, while 1 patient
carried co-occurring KRAS c.38G>A (p.G13D) and
KRAS c.34G>A (p.G12S) mutations. NRAS mutations
were detected in 2 samples, specifically NRAS c.38G>T
(p.G13V) and NRAS c.183A>T (p.Q61H). In addition, 3
samples harbored a BRAF V600E mutation. No samples showed
concurrent KRAS, NRAS, and BRAF mutations. At first
disease progression, additional mutations were also identified in
other genes. Particularly, APC mutations were found in four
samples, TP53 mutations in 13 samples, and SMAD4
mutations in two samples. Furthermore, copy number amplifications
were detected in EGFR (one sample) and FGFR1 (one
sample). Overall, among the 33 available samples at disease
progression, 18 carried mutations in genes other than KRAS,
NRAS, and BRAF.
At second disease progression, 2 blood samples were
collected. Both samples harbored KRAS c.38G>A (p.G13D)
mutations, while remaining wild-type for NRAS and
BRAF. Among them, one sample carried an APC mutation,
and the other harbored a TP53 mutation.
Concordance between tissue and
baseline plasma ct-DNA
The results of the statistical analysis demonstrated
a high level of concordance between each mutation tested. More
specifically, for KRAS mutations, the overall agreement rate
between tissue and plasma samples was 87%, with a PPV of 95% and a
NPV of 80.8% [Cohen's κ=0.74 (0.55–0.93); Se=79.2%; Sp=95.5%]. The
combined analysis of KRAS and NRAS mutations also
showed an agreement rate of 87%, with PPV=95.5% and NPV=79.2%
[Cohen's κ=0.74 (0.55-.93); Se=80.8%; Sp=95%). For BRAF
mutations, concordance was even higher, reaching 97.8%, with
PPV=100% and NPV=97.8% [Cohen's κ=0.85 (0.55–1.00); Se=75%;
Sp=100%]. For other, less frequent mutations, a positive agreement
of 67.4% was also observed, with PPV=40.9% and NPV=67.4% [Cohen's
κ=0.33 (0.09–0.57); Se=62.9%; Sp=81.8%). The McNemar test verified
these findings, showing no statistically significant differences in
the detection of KRAS, NRAS, and BRAF mutations
between tissue and plasma samples (P>0.05; Table SII).
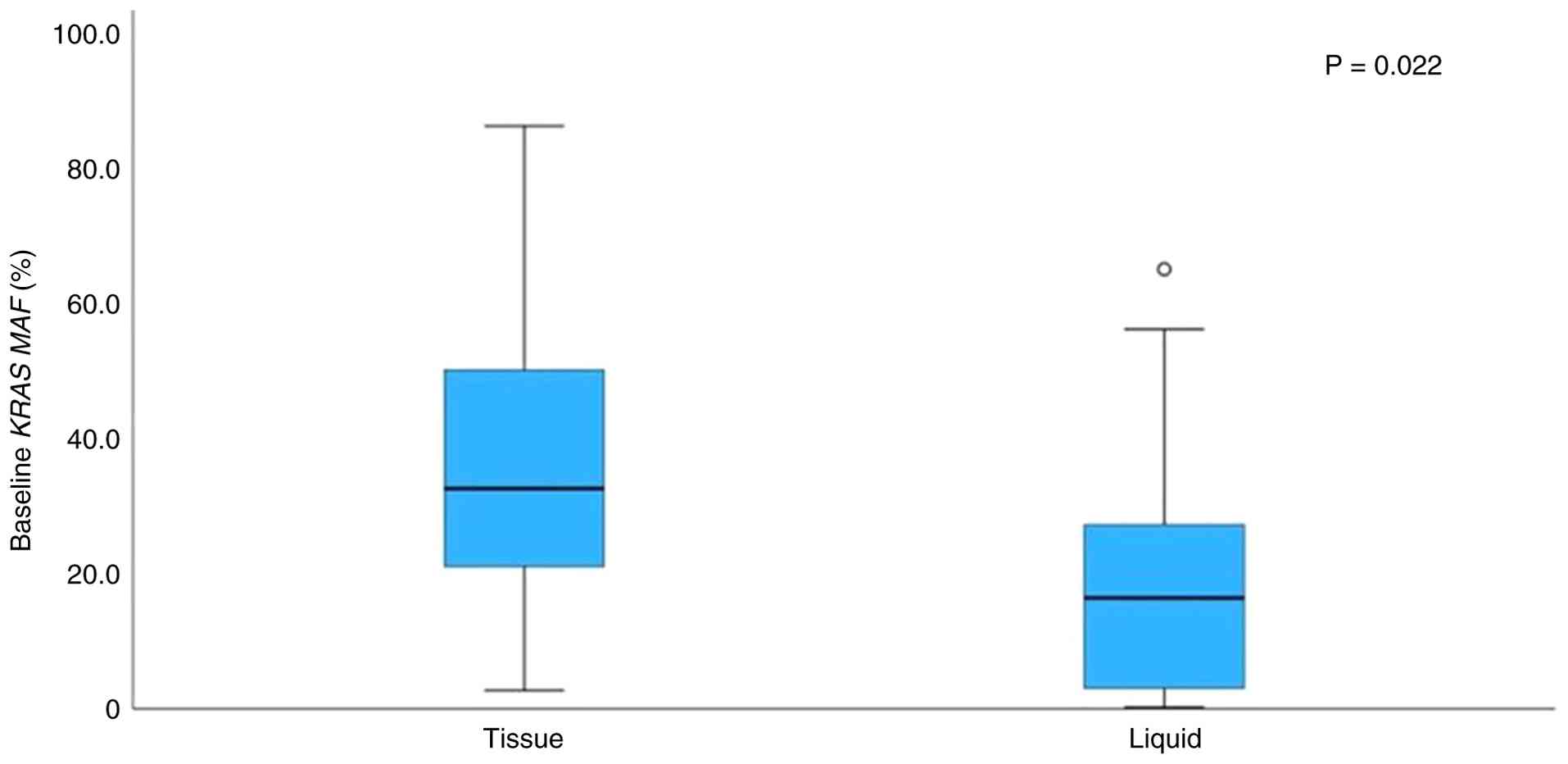
Regarding KRAS, the median MAF in tissue
samples was significantly higher compared with plasma samples
(Mann-Whitney test, P=0.022; Fig.
1). However, Spearman's correlation analysis showed no
significant association between tissue and plasma KRAS MAF
values (Spearman's ρ=0.25; P=0.313). In several cases, MAF values
were higher in plasma compared with tissues, and vice versa. The
above data are summarized in Table
SIII.
Change in mutational status with
serial ct-DNA monitoring
The assessment of changes in mutational status
during disease progression is of great importance. At progression,
one patient who was initially KRAS/NRAS wild-type developed
a novel KRAS/NRAS mutation, while seven patients who
harbored KRAS/NRAS mutations at baseline lost these
mutations at progression. Similar findings were observed in plasma
samples. More particularly, two patients acquired new
KRAS/NRAS mutations at progression, while six patients lost
their baseline KRAS/NRAS mutations during progression. By
contrast, the BRAF mutational status remained largely stable
throughout the disease course. All patients who were BRAF
wild-type at baseline maintained their wild-type status during
progression, while only one patient with a BRAF mutation at
baseline reverted to wild-type status during progression. However,
no changes in BRAF mutational status were detected in liquid
biopsy samples between baseline and progression. Among other genes
included in the sequencing panel, six patients developed new
mutations at progression compared with baseline tissue samples,
while 12 patients lost previously detected mutations. Consistently,
in baseline plasma samples, 10 patients developed new mutations,
and eight patients lost prior mutations during progression. The
detailed mutational status for KRAS, BRAF, and other genes
is summarized in Table SIV.
Overall, 62.5% of patients with KRAS or NRAS
mutations at baseline retained these mutations during progression,
whereas 37.5% lost them. Conversely, 11.8% of patients who were
wild-type KRAS/NRAS at baseline developed new
mutations at progression, while 88.2% remained wild-type (Fig. 2).
Discussion
Understanding the molecular profile of cancer is
essential for guiding therapy decisions, personalizing treatment
strategies, and ultimately improving patient outcomes. However,
tumor tissue samples are commonly insufficient for performing all
required molecular analyses and are often difficult to obtain for
re-evaluation during disease progression, particularly in cases
requiring fine-needle aspiration biopsies. Therefore, a faster and
less invasive technique is urgently needed. In this context,
detecting and analyzing ct-DNA and tumor mutational status through
liquid biopsy represents a promising approach (5,17).
ct-DNA analysis can enable the assessment of
molecular heterogeneity and provide novel insights into the overall
mutational landscape of the disease. It further allows real-time
monitoring of tumor dynamics, reflecting the evolving nature of
cancer progression. Since the molecular profile of cancer can
change over time, liquid biopsy offers a convenient, accurate and
repeatable alternative to repeated conventional tissue biopsies.
Furthermore, ct-DNA serves as a valuable prognostic biomarker, thus
aiding in the early detection of disease progression and in
identifying patients at higher risk of poor outcomes (5,18).
Tie et al (19) demonstrated that ct-DNA could serve
as a strong indicator for predicting recurrence after surgery,
independent of adjuvant treatment. In the context of targeted
therapy, integrating liquid biopsy into clinical practice is
crucial for detecting ct-DNA, eventually allowing the early
identification of resistance-related mutations and timely
therapeutic adjustments. Liquid biopsies also play a key role in
real-time evaluation of resistance mechanisms, thus improving
prognostic assessment and identifying patients at higher risk of
recurrence. Importantly, it has been reported that ct-DNA levels
are associated with tumor burden, thereby offering clinicians a
more comprehensive understanding of disease progression and
treatment efficacy (17,20).
Conversely, Siravegna et al (21) emphasized the challenges associated
with liquid biopsy, including the need for standardization of blood
collection and processing procedures to maintain sample stability
at room temperature and to minimize pre-analytical variability.
Ongoing efforts aim to develop standardized methods for ct-DNA
quantification, optimizing isolation protocols to enhance ct-DNA
yield, and improving the sensitivity of ct-DNA detection. The
aforementioned advancements are particularly significant for rare
molecular alterations, which are commonly critical for the early
detection of drug resistance.
Herein, ct-DNA and mutational profiling was
performed in 45 patients with metastatic CRC using both tissue and
plasma samples. The most commonly identified mutations involved
KRAS, NRAS, and BRAF, along with additional mutations
in several other genes. When KRAS, NRAS, and BRAF
mutations were excluded, 78% of samples carried mutations in other
genes. The analysis revealed strong concordance between mutations
detected in tissue and plasma samples, thus supporting the
increased clinical feasibility of liquid biopsy during therapy.
Furthermore, dynamic changes in mutational status throughout
disease progression were detected, with some wild-type genes
acquiring mutations and others reverting to wild type. Therefore,
liquid biopsy could provide an accessible and reliable alternative
for genetic testing, thus enabling more informed and timely
treatment decisions.
In our cohort, among the 26 patients with KRAS/NRAS
mutations detected in baseline tumor tissue, 62.5% (15/24) of those
with available serial plasma samples retained their mutation at
first progression, whereas 37.5% (9/24) demonstrated complete
clearance in circulating DNA. Conversely, among baseline KRAS/NRAS
wild-type patients, 11.8% (2/17) developed a new RAS mutation at
progression. Descriptively, patients with persistent KRAS/NRAS
mutations were more likely to experience radiologic progression at
the corresponding time point, while those with mutation clearance
more frequently achieved longer intervals of disease control.
Although the sample size limits definitive statistical conclusions,
these numerical trends support the potential prognostic value of
ct-DNA mutation dynamics.
Particularly, KRAS, NRAS, and BRAF are
key components of the rat sarcoma viral oncogene homolog
(RAS)/rapidly accelerated fibrosarcoma/mitogen-activated protein
kinase (MAPK) signaling pathway, which is known to regulate cell
growth, proliferation, and survival. Mutations in KRAS and
NRAS, both encoding members of the RAS family of proteins,
can result in constitutive activation of downstream signaling
cascades. Consistently, mutations in BRAF, a
serine/threonine-protein kinase acting further downstream, drive
aberrant pathway activation. These molecular alterations can
collectively promote uncontrolled tumor cell proliferation,
contribute to oncogenesis, and play a critical role in therapeutic
resistance. Clinically, KRAS and NRAS mutations are
well-established predictive biomarkers of resistance to anti-EGFR
therapies, whereas the BRAF V600E mutation is associated
with aggressive tumor biology, poor prognosis, and limited
responsiveness to standard chemotherapy. Understanding the
biological functions of these genes is therefore essential for
guiding personalized therapy, interpreting the current findings,
and reinforcing the utility of liquid biopsy in detecting these
alterations to enable timely treatment decisions (6,15,18,22).
An important finding of the present study was the
presence of mutational shifts between baseline tumor profiles and
subsequent plasma ct-DNA analyses during disease progression. These
shifts are clinically relevant, as they often reflect adaptive
mechanisms of therapeutic resistance. For example, the emergence of
KRAS or NRAS mutations following treatment with
anti-EGFR antibodies has been well-documented and represents a
classic mechanism of on-target resistance, rendering continued EGFR
blockade ineffective. Similarly, the acquisition of MET
amplification or HER2 alterations can act as bypass
mechanisms, maintaining MAPK or phosphatidylinositol 3-kinase
signaling activation despite upstream inhibition.
From a therapeutic perspective, the detection of
such resistance-associated alterations in plasma underscores the
potential of liquid biopsy as a dynamic tool to guide treatment
adaptation. Patients who develop RAS mutations during
anti-EGFR therapy could benefit from switching to anti-vascular
endothelial growth factor-based regimens, while identification of
MET amplification or HER2 overexpression could
promote the use of targeted inhibitors against these factors.
Furthermore, the detection of acquired gene fusions or rare
activating mutations could facilitate patient enrollment in
biomarker-driven clinical trials investigating novel agents against
emerging resistance mechanisms.
Equally important was the observation of mutational
clearance or persistence in plasma during therapy. Clearance of
tumor-specific variants could indicate a favorable treatment
response and improved prognosis, whereas persistence or
re-emergence could suggest minimal residual disease and early
relapse, often preceding radiographic evidence of progression.
These findings underscored the significant value of serial ct-DNA
monitoring into clinical practice-not only for identifying
resistance but also for prognostic assessment and early therapeutic
intervention. In the current cohort, preliminary descriptive
analysis suggested that patients with persistent KRAS/NRAS
mutations were more likely to experience disease progression, while
those demonstrating clearance achieved longer disease control.
Although the limited sample size could constrain the statistical
power of these associations, the observed trends could reinforce
the prognostic relevance of ct-DNA dynamics.
Although this study was not designed or powered to
draw definitive conclusions regarding clinical outcomes,
preliminary descriptive correlations suggested that patients with
persistent KRAS/NRAS mutations were more likely to
experience disease progression compared with those who achieved
mutational clearance during therapy. Future large-scale studies
integrating molecular and clinical data are warranted to verify and
further explore these findings.
The present study provided valuable insights but
also has several limitations. The relatively small cohort of 45
patients could constrain the generalizability of the findings, and
serial plasma samples were not available at all-time points of
disease progression. Subgroup analyses, such as comparisons between
MSI-high and MSS tumors or between left- and right-sided CRC, could
not be conducted due to limited sample size. Additionally, minor
technical or pre-analytical variations could affect the results,
and genomic alterations outside the targeted panel were not
assessed. Despite these limitations, the results revealed a strong
concordance between tissue- and plasma-based testing and
successfully captured dynamic mutational changes during treatment.
Larger, prospective studies integrating molecular dynamics with
clinical outcomes are essential to validate the prognostic and
therapeutic value of ct-DNA.
Looking ahead, the integration of ct-DNA analysis
into routine clinical workflows holds considerable promise. ct-DNA
testing could serve as a complementary approach to imaging and
traditional tissue biopsies, particularly for monitoring minimal
residual disease, anticipating relapse, and optimizing therapeutic
sequencing, such as considering anti-EGFR rechallenge following
clearance of RAS mutations. Beyond its clear clinical
relevance, liquid biopsy also offers notable practical advantages,
thus providing a minimally invasive, repeatable, and rapid
alternative to tissue re-biopsy, eventually enhancing its
suitability for real-world clinical practice.
A deeper understanding of ct-DNA biology can further
advance its clinical application; however, the results of this
study already underscored its significant value in metastatic CRC
as a minimally invasive biomarker for capturing tumor
heterogeneity, monitoring therapeutic resistance, and guiding
personalized treatment strategies throughout the disease course
(23,24).
In summary, the current study demonstrated that
liquid biopsy using ct-DNA analysis represents a reliable and
minimally invasive approach for the detection of tumor mutations in
metastatic CRC. A high level of concordance was observed between
plasma and tissue samples, particularly for KRAS, NRAS, and
BRAF. In addition, dynamic changes in mutational status were
detected during disease progression, thus highlighting the
potential of ct-DNA for real-time monitoring of tumor evolution.
These findings supported its role as a complementary tool to
conventional tissue biopsy in guiding personalized treatment
strategies. However, larger, prospective studies are warranted to
validate these results and further establish the integration of
liquid biopsy into routine clinical practice for patients with
metastatic CRC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The sequencing data generated in the present study
may be found in the Zenodo repository at the following URL:
https://doi.org/10.5281/zenodo.17742406. Raw
instrument-level sequencing files (FASTQ or BAM) were not generated
or retained during the Ion AmpliSeq workflow, as sequencing
analysis and variant calling were performed directly within the
Torrent Suite™ and Ion Reporter™ pipelines. Consequently, only
processed variant-level data (mutation calls, allele frequencies,
and summary tables) are available and represent the full extent of
the retained sequencing output. All analyses were performed for
research and experimental purposes only and were not used for
clinical decision-making; therefore, no formal clinical diagnostic
reports were generated or archived. The other data generated in the
present study may be requested from the corresponding author.
Authors' contributions
GZ was involved in the conceptualization and design
of the study, data interpretation and drafting of the manuscript.
MY participated in data collection, literature review and
manuscript drafting. PN contributed to data collection,
methodology, statistical analysis and manuscript editing. AT
performed the laboratory analyses, validated the molecular testing
results and contributed to data interpretation. IP performed the
laboratory experiments, provided technical support and was involved
in result validation. NT contributed to data collection, literature
review and manuscript drafting. KD carried out statistical
analyses, visualized data and critically reviewed the manuscript.
GN contributed to the study design, interpretation of molecular and
genetic data, and critically revised the manuscript for important
intellectual content. EK contributed to the interpretation of
clinical and genetic findings, provided input into study design,
and critically revised the manuscript for important intellectual
content. GP provided supervision, contributed to the conception of
the study, and critically revised the final manuscript. GZ and MY
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All patients provided written informed consent for
participation in this study. The study was conducted in accordance
with the principles of The Declaration of Helsinki and was approved
by the Institutional Review Board of the University Hospital of
Ioannina (approval no. 11/18-04-2018).
Patient consent for publication
Written informed consent was obtained from the
patients for the publication of this study and any accompanying
images or clinical information. The patients reviewed the final
version of the manuscript and agreed to its publication. Every
effort has been made to ensure anonymity, and no identifying
information is included in the publication.
Competing interests
GZ reports receiving personal fees as an invited
speaker from Amgen, Ipsen, Leo Pharma and Merck. GP serves as the
Chief Medical Officer for the European Society for Medical Oncology
and is a member of the American Society of Clinical Oncology, the
Hellenic Cooperative Oncology Group and the Hellenic Society of
Medical Oncology. All other authors declare that they have no
competing interests.
References
|
1
|
Heitzer E, Haque IS, Roberts CES and
Speicher MR: Current and future perspectives of liquid biopsies in
genomics-driven oncology. Nat Rev Genet. 20:71–88. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siravegna G, Mussolin B, Venesio T,
Marsoni S, Seoane J, Dive C, Papadopoulos N, Kopetz S, Corcoran RB,
Siu LL and Bardelli A: How liquid biopsies can change clinical
practice in oncology. Ann Oncol. 30:1580–1590. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thierry AR, El Messaoudi S, Gahan PB,
Anker P and Stroun M: Origins, structures, and functions of
circulating DNA in oncology. Cancer Metastasis Rev. 35:347–376.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mouliere F, Robert B, Arnau Peyrotte E,
Del Rio M, Ychou M, Molina F, Gongora C and Thierry AR: High
fragmentation characterizes tumour-derived circulating DNA. PLoS
One. 6:e234182011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bettegowda C, Sausen M, Leary RJ, Kinde I,
Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al:
Detection of circulating tumor DNA in early- and late-stage human
malignancies. Sci Transl Med. 6:224ra242014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diaz LA Jr and Bardelli A: Liquid
biopsies: Genotyping circulating tumor DNA. J Clin Oncol.
32:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kastrisiou M, Zarkavelis G, Kougioumtzi A,
Sakaloglou P, Kostoulas C, Georgiou I, Batistatou A, Pentheroudakis
G and Magklara A: Development and validation of a targeted ‘Liquid’
NGS panel for treatment customization in patients with metastatic
colorectal cancer. Diagnostics (Basel). 11:23752021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Corcoran RB and Chabner BA: Application of
Cell-free DNA analysis to cancer treatment. N Engl J Med.
379:1754–1765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang CC, Du M and Wang L: Bioinformatics
analysis for circulating cell-free DNA in cancer. Cancers (Basel).
11:8052019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tivey A, Church M, Rothwell D, Dive C and
Cook N: Circulating tumour DNA - looking beyond the blood. Nat Rev
Clin Oncol. 19:600–612. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
486:532–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dasari A, Morris VK, Allegra CJ, Atreya C,
Benson AB III, Boland P, Chung K, Copur MS, Corcoran RB, Deming DA,
et al: ct-DNA applications and integration in colorectal cancer: An
NCI colon and rectal-anal task forces whitepaper. Nat Rev Clin
Oncol. 17:757–770. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
14
|
Cervantes A, Adam R, Roselló S, Arnold D,
Normanno N, Taïeb J, Seligmann J, De Baere T, Osterlund P, Yoshino
T, et al: Metastatic colorectal cancer: ESMO Clinical Practice
Guideline for diagnosis, treatment and follow-up. Ann Oncol.
34:10–32. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mauri G, Vitiello PP, Sogari A, Crisafulli
G, Sartore-Bianchi A, Marsoni S, Siena S and Bardelli A: Liquid
biopsies to monitor and direct cancer treatment in colorectal
cancer. Br J Cancer. 127:394–407. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pascual J, Attard G, Bidard FC, Curigliano
G, De Mattos-Arruda L, Diehn M, Italiano A, Lindberg J, Merker JD,
Montagut C, et al: ESMO recommendations on the use of circulating
tumour DNA assays for patients with cancer: A report from the ESMO
Precision Medicine Working Group. Ann Oncol. 33:750–768. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heitzer E, Ulz P and Geigl JB: Circulating
tumor DNA as a liquid biopsy for cancer. Clin Chem. 61:112–123.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wan JCM, Massie C, Garcia-Corbacho J,
Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R and Rosenfeld N:
Liquid biopsies come of age: Towards implementation of circulating
tumour DNA. Nat Rev Cancer. 17:223–238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tie J, Cohen JD, Wang Y, Christie M and
Simons K: Circulating tumor DNA analyses as markers of recurrence
risk and benefit of adjuvant therapy for stage III colon cancer.
JAMA Oncol. 5:1710–1717. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reinert T, Henriksen TV, Christensen E,
Sharma S, Salari R, Sethi H, Knudsen M, Nordentoft I, Wu HT, Tin
AS, et al: Analysis of plasma cell-free DNA by ultradeep sequencing
in patients with stages I to III colorectal cancer. JAMA Oncol.
5:1124–1131. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Siravegna G, Marsoni S, Siena S and
Bardelli A: Integrating liquid biopsies into the management of
cancer. Nat Rev Clin Oncol. 14:531–548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Douillard JY, Oliner KS, Siena S,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Panitumumab-FOLFOX4 treatment and RAS mutations
in colorectal cancer. N Engl J Med. 369:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD,
Robitaille S, et al: K-ras mutations and benefit from cetuximab in
advanced colorectal cancer. N Engl J Med. 359:1757–1765. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bardelli A and Pantel K: Liquid biopsies,
what we do know (yet). Cancer Cell. 31:172–179. 2017. View Article : Google Scholar : PubMed/NCBI
|