Introduction
Acute lymphoblastic leukemia (ALL) is the most
common malignant hematological cancer in children with an globally
incidence of ~3 cases per 100,000 individuals per year (1,2). The
majority of cases exhibit various chromosomal alterations, which
are associated with the survival and evolution of the disease. This
is complemented by genomic studies that together have provided key
information regarding the diagnosis, prognosis and development of
novel therapeutic strategies (1,3).
Specifically, 80–85% of ALL cases are B-cell precursor (BCP) ALL
(B-ALL/BCP-ALL), with the remaining cases involving T-cells
(1).
It has been reported that one of the most common
chromosomal abnormalities identified in malignant hematological
processes is the presence of an extra chromosome 21, which is
identified in ~15% of ALL cases worldwide. Children with regular
trisomy 21 are 20, 150 and 400–600 times more likely to develop
ALL, acute myeloid leukemia (AML) and acute megakaryoblastic
leukemia, respectively. Notably, trisomy 21 is the most common
acquired aneuploidy in patients with any type of leukemia (4). Despite being the chromosome with the
lowest gene density, chromosome 21 has been reported to contain a
notable number of genes associated with the development of
leukemia, majority of which have a locus within the Down syndrome
(DS) critical region (DSCR) (5).
In 2003, the United Kingdom National Cancer Research
Institute Childhood Leukemia Working Party reported a novel
cytogenetic abnormality termed the intrachromosomal amplification
of chromosome 21 (iAMP21). This rare alteration occurs in only 2–5%
of cases of childhood BCP lymphoblastic leukemia, majority of which
are older children with a low white blood cell count at diagnosis
(3,6,7).
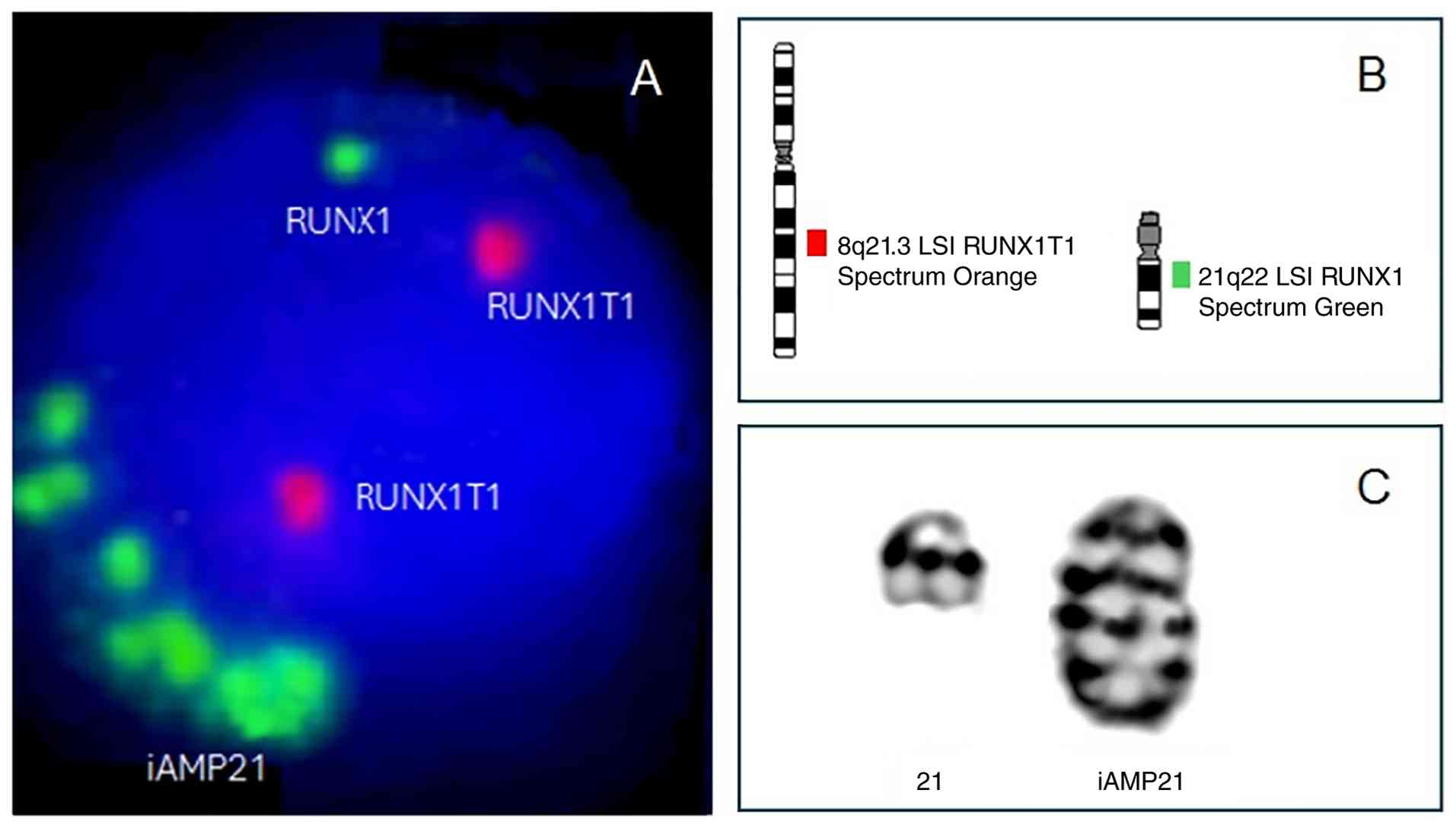
iAMP21 was first observed using fluorescence in situ
hybridization (FISH), when several signals corresponding to the
Runt-related transcription factor 1 (RUNX1) gene were
identified during the analysis of the t(8;21)(q22;q22)
translocation in patients with AML. At present, iAMP21 is commonly
defined as ‘three or more extra copies of RUNX1 on a single
abnormal chromosome 21 (a total of five or more RUNX1
signals per cell) (Fig. 1)
(8,9).
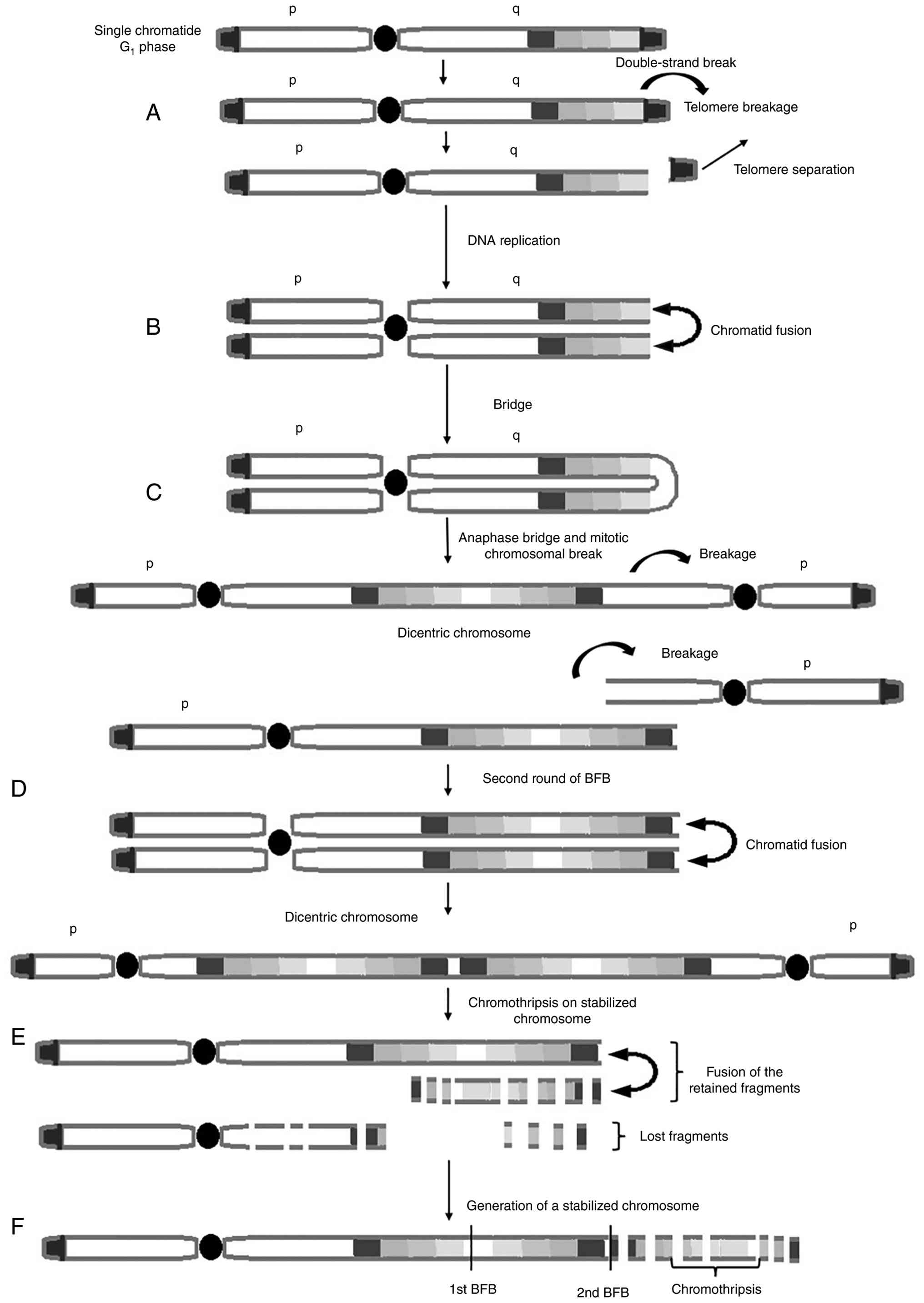
The abnormal chromosome 21 presents a complex
structure that comprises multiple regions of gain, amplification,
inversion and deletion (8). It has
been proposed that the origin of the derivative chromosome may be
the result of a series of events including an initial double-strand
break, followed by fusion and bridge formation, with the final
presence of chromothripsis (Fig. 2)
(10,11). As the RUNX1 gene locus is
located within the region primarily responsible for chromosome 21
amplification, an easy and accessible method to identify iAMP21-ALL
is to erythroblast transformation-specific (ETS) variant
transcription factor 6 (ETV6)::RUNX1 fusion probe
mark this gene via FISH (12).
Therefore, majority of cases of iAMP21-ALL have been identified by
FISH. However, chromosome 21 can also be structurally
heterogeneous, which can only be evaluated using chromosome banding
analysis (7). The structure of the
chromosome is described as an addition, duplication or simply a
derivative of chromosome 21 (11).
While iAMP21 arises through chromosomal instability, the last
chromosome remains stable and is unique for each case (Fig. 1C) (9).
Cytogenetics and whole genome sequencing enabled the
identification of iAMP21, which constituted a novel, distinct
genetic subgroup within ALL (13,14).
Notably, children with iAMP21 do not respond adequately to
traditional treatment. Thus, the early detection of chromosome
amplification is crucial, as this subgroup requires intensive
treatment. (3,6,10,15).
There is debate regarding the relationship between
iAMP21 and the development of leukemia. Amplification can be
heterogeneous, in particular at the molecular level where there is
notable gene destabilization, depending on the structural
rearrangement of chromosome 21 (16). Multiple genomic studies have
delineated the common region of amplification in iAMP21, which
ranges from 1.57 to 20.77 Mb depending on the cohort and
methodology employed. Comprehensive array-comparative genomic
hybridization and multiplex ligation dependent probe amplification
analyses have consistently identified the amplification of genes
including chromatin assembly factor 1 subunit B (CHAF1B),
dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A
(DYRK1A), ETS-related gene (ERG), high mobility group
nucleosome binding domain 1 (HMGN1) and RUNX1, with
ripply transcriptional repressor 3 (RIPPLY3/DSCR6) recently
identified as the most highly upregulated gene within the minimal
1.57 Mb region that overlaps with the DSCR. These genes are
differentially expressed in iAMP21-ALL compared with non-iAMP21-ALL
and represent candidates for coordinated involvement in
leukemogenesis (1,10,11,15–18).
Ofverholm et al (19)
characterized the genomic and transcriptional landscape of iAMP21,
and identified differentially expressed genes, including DYRK1A,
CHAF1B and SON, which had a notable association with
iAMP21-ALL. Due to the large size of the region involved in
amplification and the deregulation of several genes, including
RUNX1, DYRK1A, CHAF-1 and ERG, a coordinated action
of these genes has been hypothesized to occur for the development
of iAMP21-ALL (16).
The present review describes the biological role of
the primary genes associated with iAMP21-ALL and their relationship
with disease development.
CHAF1B
The CHAF1B, also termed CAF1, MPP7, chromatin
assembly factor I (CAF-I), CAF1A, CAF1P60, CAF-IP60 or MPHOSPH7, is
part of CAF-I and is required for the assembly of histone octamers
onto newly replicated DNA. This subunit is typically located in the
nucleus and relocated into the cytoplasm during mitosis.
CHAF1B is differentially phosphorylated in a cell
cycle-dependent manner. Furthermore, CHAF1B is a member of
the WD-repeat histone regulator 1 family, may also be involved in
DNA repair (20) and is located
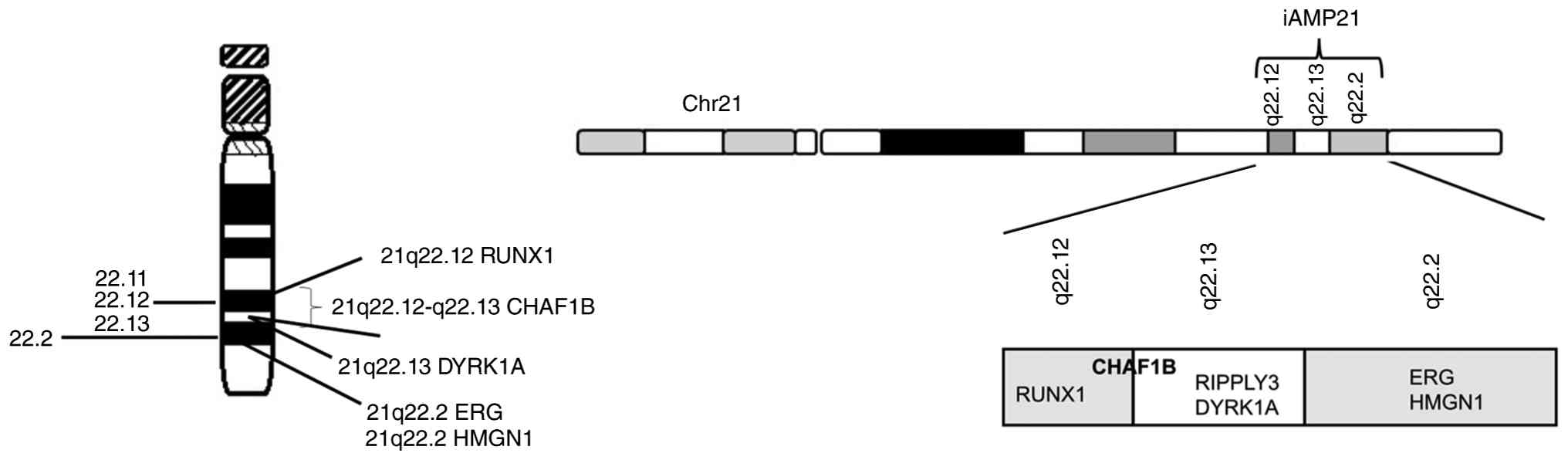
within the DSCR of chromosome 21 (21q22.12-q22.13; Fig. 3) (21,22).
Remodeling the chromatin structure after replication
is a key process in maintaining gene regulation (23) and epigenetic information, and the
regulation of the alteration of chromatin states. Consisting of
three subunits, CAF1 participates in nucleosome assembly at the
beginning of the S phase of the cell cycle. CAF1 acts as a histone
chaperone and interacts with the Proliferating Cell Nuclear Antigen
(PCNA). Furthermore, CHAF1B serves a key role in epigenetic
silencing, as well as in the replication of centromeric
heterochromatin by interacting with the heterochromatin protein. It
is well established that CAF1 is key to forming tetramers of
histones H3 and H4 at the replication fork, as well as nucleosomes,
during the S phase (24). The
participation of CHAF1B in these processes is key to the
cell cycle and any impairment in its function affects normal cell
division. CHAF1B was found to be altered in various cancer
types such as high-grade gliomas, melanomas, endometrial, renal,
cervical and prostate cancer, likely due to its key role in
chromatin replication (21,23–25).
Furthermore, another key function of CHAF1B is in DNA repair
during interphase through the ATP-dependent nucleotide excision
mechanism and under the action of PCNA (21).
CHAF1B is highly expressed in the bone marrow
and testes (19) and typically
serves a role in hematopoiesis. However, the upregulation of
CHAF1B can lead to leukemia. When CHAF1B binds
specifically to chromatin, it can have a pro-leukemic effect by
affecting the binding of transcription factors such as
CCAAT/enhancer-binding protein α (CEPBA), which are key to blood
cell differentiation, particularly in the myeloid lineage (24). The process of leukemogenesis is
promoted when the expression levels of different transcription
factors, such as friend leukemia integration 1 (FLI1),
CCAAT/enhancer-binding protein β and RUNX1, are suppressed
due to the upregulation of CHAF1B. An increase in DNA
methyltransferase activity leads to hypermethylation of the FLI1
promoter and a notable reduction in its expression, and
RUNX1 indicated no change in expression despite
amplification of the gene, possibly as an effect of CHAF1B
upregulation (21). Furthermore,
CHAF1B is upregulated in several solid tumors and has been
associated with increased tumor grade and aggressiveness (25). Various neoplasms, including
melanomas, endometrial tumors, prostate cancer and gliomas, have
been associated with CHAF1B upregulation. However, the
mechanisms underlying this association are not fully understood
(23–25). Specifically, there is controversy
regarding the association of tumorigenesis with CHAF1B
upregulation and it has been hypothesized that this may simply be
due to the high proliferation of abnormal cells (23). Notably, it has been observed that
CHAF1B is highly expressed in patients with DS and acute
megakaryocytic leukemia.
A previous study conducted in mice confirmed the key
role of CHAF1B in hematopoiesis and cell viability through
the conditional elimination of CHAF1B in
interferon-sensitive cells. The results also demonstrated that
CHAF1B upregulation increases hematopoietic stem and
progenitor cell proliferation. After confirming the association
between CHAF1B upregulation and leukemia development,
researchers suggested that targeting the CAF1-dependent pathway in
the nucleosome assembly process could represent an innovative
treatment approach (23).
CHAF1B is key to DNA replication-coupled
nucleosome assembly. Therefore, its loss or reduced activity may
lead to a decrease in the proliferation of leukemic cells. Li et
al (23) identified that
CHAF1B serves a key role in silencing genes involved in
myeloid cell differentiation. The study reported that reducing the
dose of CHAF1B induced the differentiation of human AML
cells and murine mixed lineage leukemia-ALL fused gene from
chromosome 9 cells, and the same conclusion was reached when
studying the transcriptome of cells that overexpressed or deleted
the gene. Since CHAF1B shares a genomic locus with CEPBA,
FLI1 and RUNX1, it interacts with these transcription
factors at the chromatin level. These transcription factors are key
regulators of hematopoietic differentiation and any increase in the
dose of CHAF1B influences the silencing of these genes key
to leukemic cell differentiation (23).
The overall summary of the physiological roles of
the main genes involved in the common region of chromosome 21,
which is affected in iAMP21, is presented in Table I, as well as the genetic alterations
associated with the development of hematological malignancies.
 | Table I.Physiological roles and genetic
alterations in hematological malignancies of key genes located in
the commonly affected region of chromosome 21 in iAMP21. |
Table I.
Physiological roles and genetic
alterations in hematological malignancies of key genes located in
the commonly affected region of chromosome 21 in iAMP21.
| Function | CHAF1B | DYRK1A | ERG | HMGN1 | RUNX1 | RIPPLY3 |
|---|
| Physiological
roles | Encodes p60
required for chromatin assembly, regulates DNA transcription,
repair and replication; regulation of gene expression; normal
hematopoiesis | Phosphorylates
diverse regulatory proteins; regulation of cell proliferation,
differentiation and growth; DNA damage repair | Regulator of cell
proliferation, differentiation and apoptosis; regulates
hematopoiesis, differentiation and maturation of megakaryocytic
cells | Binds nucleosomal
DNA; associated with transcriptionally active chromatin; modulates
gene expression; DNA damage repair | Transcription
factor key in hematopoiesis and embryonic development; cell
division and proliferation regulator | Transcriptional
repressor that regulates the transcriptional activity of TBX1 and
STAT3; involved in negative regulation of transcription by RNA
polymerase II |
| Genetic alterations
and roles in hematological malignancies | Genomic instability
that increases the risk of mutations associated with cancer;
upregulation promotes leukemia by suppressing the expression levels
of TFs in myeloid differentiation, including RUNX1 | Dysregulation is
associated with leukemia by disruption of cell cycle control; it
has both oncogenic and tumor suppressor functions; upregulation
deactivates CDK1, which positively regulates mitosis and inhibits
the cell cycle | Involved in CTs,
resulting in different fusion gene products such as FUS::ERG in
AML; elevated ERG and c-Myc expression levels are necessary for
pre-B cells clonal expansion and leukemia development; ERG
deletions can be identified in 3–7% of BCP ALL cases | Methylase activity
of HMGN1 is associated with increased transcriptional activation
and development of leukemia; is associated with increased chromatin
accessibility, expression, and histone H3K27 acetylation of l oci
necessary for HSCS and leukemia | CTs associated with
several types of leukemia (>50 affect RUNX1: t(12;21) in
pediatric ALL; RUNX1 can act as a tumor promoter or suppressor in
hematological malignancies; somatic mutations are observed in AML,
ALL and MDS | Upregulated in the
iAMP21 cases studied; no associations with hematological
malignancies have been reported yet |
DYRK1A
DYRK1A, also termed MNB, DYRK, HP86, MNBH,
MRD7 or DYRK1, encodes a member of the DYRK family (20). DYRK1A is a member of a
conserved family of CMGC kinases, named from the first letter of
each family member, including cyclin-dependent kinases (CDK),
mitogen-activated protein kinases, glycogen synthase kinase 3 and
CDK-like kinases (26). They are
structurally characterized by the presence of a protein kinase
domain, a leucine zipper motif and a series of 13 highly conserved
histidine repeats (20). The DYRK
family consists of five members divided into two classes, with the
first class including DYRK1A and DYRK1B, and the
second class including DYRK2, DYRK3 and DYRK4. Of
these five variants, only DYRK1A (21q22.13; Fig. 3) is located on chromosome 21, whilst
DYRK1B is on 19q13.2, DYRK2 is on 12q15, DYRK3
is on 1q32.1 and DYRK4 is on 12p13.32 (22).
DYRK1A increases its activity through
autophosphorylation of its activation loop and participates in the
phosphorylation of several regulatory proteins, such as STAT3 and
FOXO1 (26). DYRK1A
regulates cell proliferation and brain development by participating
in the Notch signaling pathway. It has been hypothesized that
DYRK1A is associated with learning disabilities in
individuals with DS, autism and Alzheimer's disease (26). During transcription, this gene
encodes at least 5 isoforms that differ in the 5′-UTR or in the
3′-coding regions and has ubiquitous expression in bone marrow and
testes (20).
DYRK1A is a kinase that participates in key
biological processes, including chromatin transcription, mRNA
splicing, signal transduction, DNA repair, cell cycle control and
neuronal function development. DYRK1A is dysregulated in
various types of malignant neoplasms, including lung and pancreatic
cancer, glioblastoma, melanoma and leukemia, as well as in other
diseases such as type 1 and type 2 diabetes mellitus and heart
disease (26–28). The DYRK1A protein appears to
serve a key role in the development of these conditions and its
modulation may lead to cell cycle dysregulation control (27). A notable development in the current
understanding of the role of DYRK1A in these processes has
been the study of patients with DS, who are at an increased risk of
developing various congenital disorders, as well as leukemia,
lymphoma and retinoblastoma (26–28).
These conditions have been associated with different functions of
the DYRK1A gene. Therefore, it has been suggested that there
is an association between these alterations and the presence of
constitutional trisomy 21. However, the role of DYRK1A
remains to be elucidated, as there are several studies indicating
that it has both oncogenic and tumor suppressor functions (27,28).
It has been reported that upregulation of
DYRK1A deactivates CDK1. This cyclin-dependent kinase serves
a role in regulating mitosis and cell cycle inhibition.
Furthermore, the protein levels of DYRK1A upregulation are
positively associated with those of STAT3,
cellular-mesenchymal-epithelial transition factor (MET) and
EGFR, as it has been reported that DYRK1A small
interfering RNA can suppress the levels of EGFR and MET
receptor tyrosine kinases in lung and pancreatic cancer cells
(27).
Children with trisomy 21 who have a GATA-binding
factor 1 mutation associated with DYRK1A upregulation are at
an increased risk of developing acute megakaryoblastic leukemia.
DYRK1A affects the regulation of transcription factors of
the nuclear factor of activated T cells, which are key to
megakaryopoiesis (26).
In the normal process of lymphopoiesis,
DYRK1A phosphorylates cyclin D3, marking this protein for
degradation. This inactivates lymphocytes and promotes their
differentiation. Bhansali et al (28) studied both B-ALL cell lines and
patient samples, including those with poor prognosis such as those
associated with the Philadelphia chromosome, and reported that
DYRK1A was overexpressed.
The association between the development of leukemia
and the involvement of DYRK1A is well established. Analysis
of a human pediatric ALL database revealed DYRK1A
amplification or mRNA upregulation in 10.83% of all cases.
Autoimmune diseases and malignant B-cell neoplasms can develop when
B-cell activating factor (BAFF) becomes dysregulated, and the
survival of both mature and immature T2 B cells depends directly on
BAFF. Notable reductions in both cell types have been associated
with DYRK1A deficiency, as DYRK1A acts as a key
mediator in the BAFF signaling pathway. It facilitates B-cell
survival and the activation of the non-canonical NF-kB pathway.
When this process is dysregulated, it contributes to the
development of B-ALL. It has been suggested that the integration of
a DYRK1A inhibitor in the treatment of ALL may improve
outcomes (29).
Therapeutic targets for DYRK1A have been
evaluated in B-ALL cell lines treated with ETH 1610, a potent
inhibitor of DYRK1A, which demonstrated a dose-dependent
decrease in cell number. The effects were most notable in trisomy
21 or hyperdiploid cells. In patients with DS and B-ALL, synergy is
observed between EHT 1610 and multiple chemotherapeutic agents,
such dexamethasone, cytarabine and methotrexate (26).
ERG
The ERG gene encodes a member of the ETS
family of transcriptions factors; it is also termed p55,
ERG−3 or LMPHM14. These transcription factors serve a role
in regulating processes such as embryonic development, cell
proliferation and differentiation, angiogenesis, inflammation and
apoptosis. The ERG gene encodes a protein with a DNA-binding
ETS domain and a pointed domain that exhibits nuclear activity and
associates with chimeric oncoproteins. ERG not only serves a
key role in hematopoietic process regulation through platelet
adhesion and vascular cell remodeling but also contributes to the
differentiation and maturation of megakaryocytic cells. AML that
involves a chromosomal translocation (16;21)(p11;q22) is associated
with poor prognosis and resistance to treatment. This type of
leukemia involves the FUS::ERG gene fusion. Other ERG
fusions include TMPSMR2::ERG and NDRG1::ERG in
prostate cancer, as well as EWS::ERG in Ewing's sarcoma. It
has been reported that the ERG gene has several alternative
promoters that generate >24 variants via alternative splicing.
However, the function of these variants has not yet been fully
established (20). ERG
(21q22.2) is a key transcription factor that is extensively
expressed in multiple tissues (whole blood, brain, cortex, spinal
cord, heart, kidney) (Fig. 3)
(22).
It has been reported that ERG and
c-Myc work in conjunction in a regulatory network in ALL,
which is triggered by the fusion of breakpoint cluster region
(BCR)::ABL1 genes. This association regulates the expression
levels of genes involved in ribosome formation and RNA polymerase I
subunit regulation. Clonal expansion of pre-B cells and subsequent
development of leukemia require increased expression levels of
ERG and c-Myc in B-ALL cells with the BCR::ABL
fusion. By contrast, in human BCR::ABL1 B-ALL cell lines,
reduction of ERG or c-Myc levels limited the
expansion of leukemia cells and delayed the development of leukemia
in transplanted mice. It has also been suggested that ERG
and c-Myc are co-expressed in other B-ALL subtypes, in
addition to B-ALL with BCR::ABL gene fusion (30).
ERG (ETS transcription factor) gene deletions
can be identified in 3–7% of cases of pediatric BCP-ALLs (31,32).
ERG gene deletions occurs almost exclusively in other B-ALL
subtypes, a heterogeneous subset comprising 20–25% of pediatric
BCP-ALL cases, defined by the absence of cytogenetic alterations
identified by chromosome banding analysis. Previous studies have
reported that the negative prognostic impact is reduced when
simultaneous deletions occur in ERG and Ikaros family zinc
finger 1 (31,33).
The processes of hematopoietic differentiation,
megakaryopoiesis and the development of megakaryoblastic leukemia
in patients with DS are all associated with the direct involvement
of ERG (32). As
aforementioned, ERG is frequently upregulated in prostate
carcinoma. This rarely occurs in acute leukemia because another
type of damage is more prevalent (33). However, cases of AML with poor
prognosis have been described as they show upregulation of the gene
(33). Although it has been
suggested that B lymphoid cell development may be regulated when
ERG is enhanced, its association with ALL development
remains to be elucidated. Notably, there are a few reported cases
in which ERG is deleted, suggesting that ERG
deregulation is a notable but secondary event in leukemogenesis
(31,32). However, if it is identified during
the initial phases of leukemogenesis, it may manifest as a clonal
event. Previous studies have demonstrated that dysregulation can be
complex and affect both alleles in majority of cases of ERG
deletion. Therefore, the concept of a single gene alteration may
not be applicable and the idea of a single-copy gene being
inactivated (haploinsufficiency) may not fully explain the
biological impact (31,32).
ERG, along with other transcription factors
such as double homeobox 4 (DUX4), may be deregulated and exhibit
differential gene expression, constituting a specific subtype of
B-cell progenitor ALL. The genomic alterations described for
ERG, which present atypical gene expression manifested as
the upregulation or downregulation of normal transcripts or the
production of chimeric transcripts, are associated with different
mechanisms. These can include shorter transcripts, have different
splicing patterns, start translation from unusual codons or be
hybrid genes that produce transcripts with both coding and
non-coding functions. An example of this is the expression of the
novel ERG coding transcript, ERG isoform 1
(ERGalt) (33). By contrast,
clonal or subclonal deletions of ERG are also observed in
majority of cases and ERG deregulation by DUX4 is similar to
the deregulation of other ETS genes in solid tumors such prostate
cancer and Ewing's sarcoma (32,33).
As in myeloid cells, ERG expression is
preferential and strong in both B lymphocytes and immature T cells.
Identifying emerging markers in different signaling pathways will
be key to designing novel therapeutic alternatives in the future.
Certain pathways associated with ERG upregulation, such as
the Wnt/β-catenin, p53 and PI3K/AKT/PTEN pathways, are associated
with hyperactive or defective kinases or altered kinase expression
levels, as is the case with casein kinase 2 (CK2), leading to
resistance to kinase inhibitors. CK2 upregulation has been observed
in hematological malignancies such as acute and chronic leukemias,
including T-cell ALL (T-ALL), B-ALL and AML, and has been
associated with poor clinical outcomes (34). Furthermore, an increase in
ERG expression has been associated with poor relapse-free
survival in patients with T-ALL. By contrast, low ERG
expression has been associated with high white blood cell counts
and poor relapse-free survival rates in pediatric patients with
B-ALL. The expression levels of CK2, Myc and ERG in
pediatric patients with ALL have not been fully studied. Therefore,
it is proposed that characterizing these genes could lead to the
identification of novel disease markers and/or therapeutic targets
in this type of leukemia (34).
HMGN1
HMGN1 (21q22.2; Fig 3) (22), also termed HMG14, is a nucleosome
remodeling protein. HMGN1 is expressed in nearly all cell
and tissue types in the body; however it is particularly notable in
the bone marrow and lymph nodes (20). The main function of HMGN1 is
to act as a nucleosome-binding protein, which regulates chromatin
structure and function. This facilitates access of regulatory
factors to DNA, thereby affecting gene expression, development and
the cellular response to damage (20). HMGN1 exhibits notable
demethylating activity, which increases transcriptional activity
and could be associated with the development of leukemia (5).
HMGN1 upregulation suppresses the
trimethylation of lysine 27 on histone H3 (H3K27). Furthermore, it
simultaneously promotes the proliferation of B cells and B-ALL
cells in vitro and in vivo, respectively (35). HMGN1 amplification and H3K27
acetylation are both epigenetic mechanisms that promote chromatin
opening. This process can affect hematopoietic stem cells (HSCs),
which impacts myeloid differentiation and leads to leukemia. The
upregulation of HMGN1, in conjunction with the presence of
the fusion oncoprotein AML-ETO9a, can produce a cellular
environment that promotes genomic instability and the dysregulation
of signaling pathways that are key to the development of myeloid
cells and the onset of leukemia. The factors that modulate
chromatin accessibility in HSCs and leukemia stem cells (LSCs) are
key to gene expression. This allows HSCs to differentiate into
blood cells and maintain homeostasis. However, in LSCs,
dysregulation of these factors promotes uncontrolled cell
proliferation, which is a hallmark of leukemia. By modifying
chromatin structure and increasing histone acetylation,
HMGN1 promotes blast immaturity and the persistence of LSCs.
Furthermore, its ability to decompress chromatin allows
transcription factors and coactivators, such as the histone
acetyltransferase (HAT) CREB-binding protein (CBP)7p300, to bind
more easily. This makes it a potential therapeutic alternative for
AML (4).
Competition between histone H1 and
nucleosome-binding proteins belonging to the HMGN family promotes
chromatin decondensation, increased accessibility and locally
enhanced transcription. HMGN interacts with nucleosomes, regardless
of the DNA sequence, through the recognition of histones H2A and
H2B7 by a specific nucleosome-binding domain. It has a specific
location on regulatory marks in active promoters, enabling it to
influence the organization of nucleosomes, DNase hypersensitivity
patterns and post-translational histone marks. As HMGN1
controls access to functional elements in chromatin, it can
influence myeloid differentiation by increasing gene expression,
which blocks differentiation and promotes the accumulation of
leukemic stem cells, alongside H3K27 acetylation and AML oncogenes.
HMGN1 could be a therapeutic target with the inhibition of
HAT, as it is dependent on CBP and p300 (4). In cases of ALL with an extra
chromosome 21, iAMP21 or DS, an increase of gene expression in the
amplification of DSCR, including HMGN1, has been identified.
This was particularly observed in cases with a deletion in the
Xp22p22 or Yp11p11 region that generates the purinergic receptor
P2Y, G-protein coupled, 8::cytokine receptor-like factor 2
(CRLF2) fusion and causes CRLF2 upregulation and
activation of the Janus kinase-STAT pathway, a common process in
B-ALL. This fusion is most prevalent in patients with ALL and DS,
followed by those with iAMP21, and it is rare in patients (only
5–16%) with ALL without an extra chromosome 21 (36).
In general, it is well established that the
upregulation of HMGN1 reduces chromatin compaction and
modulates epigenomic control globally (5).
RUNX1
RUNX1, also termed AML1, CBFA2, EVI-1,
AMLCR1, PEBP2aB, CBF2α, AML1-EVI-1 or PEBP2α, is
extensively expressed in the appendix, bone marrow and 24 other
tissues (20).
RUNX1 (21q22.12; Fig. 3) (22) forms a complex with its associated
protein, core binding factor β, which is key to efficient DNA
binding and complex stability. As a member of the Runt-related
transcription factor family, RUNX1 serves a key role in
various biological processes, including hematopoietic stem cell
differentiation and normal development. However, it is also
involved in the progression of cancer types, such as leukemia
(20). Various alterations to this
gene have been identified, including mutations, upregulation and
increased copy number. Translocations have been particularly well
documented and are associated with different types of leukemia
(37). RUNX1 has been
associated with >50 different translocations. In hematological
malignancies, the most notable are the t(12;21) translocation
involving the ETV6::RUNX1 fusion gene, which is identified
in pediatric ALL, and the t(8;21) and t(3;21) translocations
involving the AML1::MTG8 and RUNX1::MECOM fusion
genes, respectively, which are identified in AML. The latter fusion
results in the upregulation of the EVI1 factor, which acts as an
oncogene (38).
The mammalian RUNX family of transcription factors
comprises three members: i) RUNX1; ii) RUNX2; and
iii) RUNX3. These proteins serve key roles in a variety of
biological processes, such as cell differentiation, proliferation,
apoptosis and embryonic development. Each exhibits distinct tissue
expression patterns and functions. For instance, RUNX1 is
involved in hematopoiesis, RUNX2 in osteogenesis and
RUNX3 in neurogenesis and lymphopoiesis (39). The diversity of RUNX transcripts is
due to the presence of two different promoters for each of the
three RUNX genes (RUNX1, RUNX2 and RUNX3), which
result in alternative splicing and the formation of different
N-terminal amino acid sequences in the resulting isoforms. These
transcripts produce various mRNA molecules that serve specific
roles in regulating gene expression and cell differentiation in
different tissues and developmental stages (38).
Previous studies have reported that RUNX1 is
frequently involved in the progression of acute leukemia and serves
a key role in other types of cancer like pancreatic adenocarcinoma,
uterine endometrioid carcinoma, lung adenocarcinoma and colorectal
adenocarcinoma (39,40). Depending on its expression level,
RUNX1 can act as a tumor promoter or suppressor in
hematological malignancies (40).
Mutations in this gene are relatively frequent and its study is of
key importance in hematological malignancies. For example,
RUNX1 proteins have been found to fuse with other genes in
patients with AML. RUNX1-ETO fusion is found in 10–20% of
these cases. In children with ALL, TEL-RUNX1 fusion is found
in 20–25% of cases (41).
Germline mutations in RUNX1 cause familial
platelet disorder with associated myeloid malignancies. In
particular, somatic mutations of the gene are observed in different
types of leukemia such as AML, ALL, myelodysplastic syndromes and
chronic myelomonocytic leukemia. Due to the involvement of
RUNX1 in these processes, it is considered a key element for
the development of potential therapeutic strategies (38,42).
Whether due to germline or somatic alterations, the inactivation of
RUNX1 is considered a determining factor in the development
of hematological neoplasms (41).
Furthermore, transcriptomic studies have reported
that RUNX1 in particular is differentially expressed in
patients with shorter survival times. Both increased and decreased
levels of RUNX1 gene expression have been described in
different types of leukemia, as well as in different neoplasms
(37). Somatic mutations in the
RUNX1 gene are commonly observed in hematological
malignancies (39), including AML
and myelodysplastic syndromes. These mutations often worsen the
prognosis and are associated with a poor response to standard
chemotherapy (37). It has been
suggested that RUNX1 works with other transcription factors
to promote leukemia development. This is evident in its interaction
with Notch1, a key element in signaling pathways that regulates
cell development. By behaving as an oncogene, Notch1 promotes the
growth and survival of cancer cells, particularly in acute T cell
lymphoblastic leukemia. This association in turn promotes the
expression levels of various proto-oncogenes, including Notch3, Myc
and DTX1, and contributes to chromatin reprogramming, generating
abnormal gene expression, which is key in cell development and
malignancy (37).
By contrast, most chromosomal translocations lead
to gene fusion, which is the initial event in leukemia onset that
confers self-renewal properties to HSCs or lymphoid progenitors
(40). For example, translocation
t(12;21)(p13;q22) is the most common abnormality in childhood ALL
with an approximate frequency of 25% and creates the fusion of the
ETV6::RUNX1 genes (43).
Although it is associated with a favorable prognosis in majority of
cases, it may be associated with late relapses associated with the
appearance of additional alterations (44). These additional alterations are
predominantly caused by rare genomic rearrangements mediated by
aberrant recombination-activating gene (RAG) activity. Jakobczyk
et al (44) demonstrated
that RAG1 transcripts are directly upregulated by
ETV6::RUNX1 from the −1,200 bp RAG1 enhancer and by
RUNX1 from the −80bp RAG1 promoter in human pre-B cells. The
study proposed a multistep mechanism, where the translocation
usually starts in utero. Recent studies have reported that
both RUNX1 and the ETV6::RUNX1 fusion binds directly
to the promoter and enhancers of the RAG1 gene. This causes the
upregulation of RAG1, which appears to be a key process in the
pathogenesis of childhood B-cell ALL (44,45).
RIPPLY3
RIPPLY3 (21q22,13; Fig. 3) (22), a transcriptional repressor that acts
by repressing the activity of other genes, was among the relevant
genes reported to be amplified in iAMP-ALL. Notably, it was the
most upregulated gene in all of the iAMP21 cases studied.
RIPPLY3 was identified in 2000; however, it has been
infrequently explored in ALL. The reported expression levels
suggest a probable association between RIPPLY3 and iAMP21.
However, prior to establishing this association, the role of
RIPPLY3 in the development of neoplastic processes,
particularly leukemia, has not been well established (15). The notable similarity in
transcriptional signatures suggests that RIPPLY3 activity
serves a key role in the development of iAMP21-ALL. It has been
hypothesized that RIPPLY3 is associated with other
neighboring genes, as demonstrated by patients with iAMP21-ALL who
have low RUNX1 amplification (15).
RIPPLY3, also termed DSCR6, has been studied
in other types of neoplasms. The role of RIPPLY3 in cell
proliferation suggests that it serves a notable role in regulating
proliferation and division, as demonstrated in the thyroid gland,
where an association has been observed between the presence of
papillary thyroid carcinoma and abnormal development of the thyroid
gland (46).
Other previous studies have reported that abnormal
methylation patterns in key DNA sequences in the early stages of
carcinogenesis can be used to detect certain neoplasms, such as
ovarian cancer. A total of 11 candidate DNA methylation markers,
including RIPPLY3, were identified that exhibited high
sensitivity and specificity to distinguish women with ovarian
cancer from those without it (47,48).
Lastly, it should be noted that at the time of
diagnosis or confirmation, patients receive standard treatment,
which is mainly based on the use of steroids, such as prednisone,
dexamethasone or methylprednisolone, either on their own or in
combination with systemic chemotherapy or intrathecal therapy.
During induction therapy, four drugs are used: i) Steroids; ii)
vincristine; iii) L-asparaginase; and iv) anthracyclines (49,50).
At this stage, any changes and modifications are made based on
patient response. This is mainly due to the genetic heterogeneity
of patients and provides a unique leukemia profile. These
malignancies are characterized by a combination of chromosomal
abnormalities, oncogenic mutations and epigenetic deregulation
(51). As is the case with the
presence of iAMP21, which is associated with a poor prognosis.
Children presenting with iAMP21-ALL tend to be older (with an
average age of 9 years), have a common or pre-B immunophenotype and
have low platelet and white blood cell counts. Treatment should
therefore be intensified in these cases to reduce the risk of
relapse (52,53).
It is key to consider that the presence or absence
of specific sentinel genetic lesions in leukemic blasts serves a
key role in determining prognosis and in the stratification of ALL
therapy (53). Data from the UK ALL
trial demonstrated that patients with iAMP21-ALL had a 10-year
event-free survival of only 15%. However, the overall survival was
markedly higher at 71%, indicating that these patients responded
well to more intensive post-relapse therapy (54). Other studies reported a 5-year
event-free survival of 26% and 5-year overall survival of 69% for
28 patients with iAMP21, and the Austrian and German ALL
Berlin-Frankfurt-Munster (ALL-BFM) reported a 6-year EFS of 38% and
overall survival of 66% for 29 iAMP21 patients (52).
To facilitate the development of possible targets
for novel therapies and reduce the toxicity of current high-risk
treatments, studies that investigate the role of these genes on
chromosome 21 and decipher the genomic complexity of the iAMP21
chromosome should be carried out in the future. The aim is to have
more options in addition to early stem cell transplantation and to
implement methodologies that allow the molecular profile to be
broadened.
Discussion
iAMP21 defines a biologically distinct subtype of
childhood BCP-ALL, characterized by poor prognosis under standard
treatment protocols but improved outcomes with intensified therapy.
Despite the structural heterogeneity of the derivative chromosome
21, the common amplified region encompasses functionally related
genes involved in chromatin assembly (CHAF1B), cell cycle
regulation (DYRK1A), transcriptional control (ERG and
RUNX1) and epigenetic modulation (HMGN1), suggesting
that leukemogenesis in iAMP21-ALL results from the coordinated
dysregulation of these pathways rather than a single oncogenic
driver. Recent identification of RIPPLY3 as the most
upregulated gene within the minimal common region opens novel
research avenues, although its precise role in leukemic
transformation remains to be elucidated.
Further functional characterization of genes within
the amplified region, validation in prospective clinical cohorts
and integration of cytogenetic, genomic and transcriptomic data are
key to refining risk stratification and identifying therapeutic
vulnerabilities specific to iAMP21-ALL. Comprehensive genetic
evaluation including FISH or microarray analysis alongside targeted
gene panels for CHAF1B, DYRK1A, ERG, HMGN1, RUNX1 and
RIPPLY3 should be incorporated into diagnostic algorithms
for pediatric BCP-ALL, particularly in cases with adverse
prognostic features or suboptimal treatment responses. Further
understanding of the molecular mechanisms converging in iAMP21 may
potentially guide the development of biology-driven therapeutic
strategies tailored to the unique genetic landscape of this
high-risk ALL subtype in the future.
Acknowledgements
Not applicable.
Funding
The present review was funded by the Department of Science,
Humanities, Technology and Innovation (Secihti) scholarship (CVU:
633121).
Availability of data and materials
Not applicable.
Authors' contributions
CEUG and ASC conceived the present review, and
wrote, reviewed and edited the manuscript. JAV and HMZ wrote,
reviewed and edited the manuscript. All authors read and approved
the final manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mroczkowska A and Lejman M:
Intrachromosomal amplification of chromosome 21 in childhood acute
lymphoblastic leukemia: Study of 3 cases. Case Rep Oncol.
14:592–598. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marrapodi MM, Di Paola A, Di Feo G, Di
Domenico O, Di Martino M, Argenziano L, Falcone M, Di Pinto D,
Rossi F and Pota E: Novel therapeutic approaches in pediatric acute
lymphoblastic leukemia. Int J Mol Sci. 26:113622025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garcia DR, Arancibia AM, Ribeiro RC, Land
MG and Silva ML: Intrachromosomal amplification of chromosome 21
(iAMP21) detected by ETV6/RUNX1 FISH screening in childhood acute
lymphoblastic leukemia: A case report. Rev Bras Hematol Hemoter.
35:369–371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mowery CT, Reyes JM, Cabal-Hierro L, Higby
KJ, Karlin KL, Wang JH, Kimmerling RJ, Cejas P, Lim K, Li H, et al:
Trisomy of a down syndrome critical region globally amplifies
transcription via HMGN1 overexpression. Cell Rep. 25:1898–1911.e5.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lane AA, Chapuy B, Lin CY, Tivey T, Li H,
Townsend EC, van Bodegom D, Day TA, Wu SC, Liu H, et al:
Triplication of a 21q22 region contributes to B cell transformation
through HMGN1 overexpression and loss of histone H3 Lys27
trimethylation. Nat Genet. 46:618–623. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang M, Yi ES, Kim HJ, Yoo KH, Koo HH and
Kim SH: Intrachromosomal amplification of chromosome 21 in Korean
pediatric patients with B-cell precursor acute lymphoblastic
leukemia in a single institution. Blood Res. 52:100–105. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harrison CJ, Moorman AV, Schwab C, Carroll
AJ, Raetz EA, Devidas M, Strehl S, Nebral K, Harbott J,
Teigler-Schlegel A, et al: An international study of
intrachromosomal amplification of chromosome 21 (iAMP21):
Cytogenetic characterization and outcome. Leukemia. 28:1015–1021.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fuka G, Farias-Vieira TM, Hummel L, Blunck
CB, Santoro JC, Terra-Granado E, Barbosa TC, Emerenciano M and
Pombo-de-Oliveira MS: Evaluation of multiplex ligation dependent
probe amplification (MLPA) for identification of acute
lymphoblastic leukemia with an intrachromosomal amplification of
chromosome 21 (iAMP21) in a Brazilian population. Mol Cytogenet.
8:352015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harrison CJ: Blood Spotlight on iAMP21
acute lymphoblastic leukemia (ALL), a high-risk pediatric disease.
Blood. 125:1383–1386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duployez N, Boudry-Labis E, Decool G,
Grzych G, Grardel N, Abou Chahla W, Preudhomme C and
Roche-Lestienne C: Diagnosis of intrachromosomal amplification of
chromosome 21 (iAMP21) by molecular cytogenetics in pediatric acute
lymphoblastic leukemia. Clin Case Rep. 3:814–816. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim J, Lyu CJ, Shin S, Lee ST and Choi JR:
Frequency and clinical characteristics of intrachromosomal
amplification of chromosome 21 in Korean childhood B-lineage acute
lymphoblastic leukemia. Ann Lab Med. 36:475–480. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koleilat A, Smadbeck JB, Zepeda-Mendoza
CJ, Williamson CM, Pitel BA, Golden CL, Xu X, Greipp PT, Ketterling
RP, Hoppman NL, et al: Characterization of unusual iAMP21
B-lymphoblastic leukemia (iAMP21-ALL) from the Mayo Clinic and
Children's Oncology Group. Genes Chromosomes Cancer. 61:710–719.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garniasih D, Susanah S, Sribudiani Y and
Hilmanto D: Intrachromosomal amplification of chromosome 21 in
pediatric patients with B-cell precursor acute lymphoblastic
leukemia: A systematic review and meta-analyses. Eur Chem Bull.
12:136–145. 2023.
|
|
14
|
Brady SW, Roberts KG, Gu Z, Shi L, Pounds
S, Pei D, Cheng C, Dai Y, Devidas M, Qu C, et al: The genomic
landscape of pediatric acute lymphoblastic leukemia. Nat Genet.
54:1376–1389. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hormann FM, Hoogkamer AQ, Boeree A,
Sonneveld E, Escherich G, den Boer ML and Boer JM: Integrating copy
number data of 64 iAMP21 BCP-ALL patients narrows the common region
of amplification to 1.57 Mb. Front Oncol. 13:11285602023.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao Q, Ryan SL, Iacobucci I, Ghate PS,
Cranston RE, Schwab C, Elsayed AH, Shi L, Pounds S, Lei S, et al:
The genomic landscape of acute lymphoblastic leukemia with
intrachromosomal amplification of chromosome 21. Blood.
142:711–723. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Strefford JC, van Delft FW, Robinson HM,
Worley H, Yiannikouris O, Selzer R, Richmond T, Hann I, Bellotti T,
Raghavan M, et al: Complex genomic alterations and gene expression
in acute lymphoblastic leukemia with intrachromosomal amplification
of chromosome 21. Proc Natl Acad Sci USA. 103:8167–872. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wrona E, Braun M, Pastorczak A, Taha J,
Lejman M, Kowalczyk J, Fendler W and Młynarski W: MLPA as a
complementary tool for diagnosis of chromosome 21 aberrations in
childhood BCP-ALL. J Appl Genet. 60:347–355. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ofverholm I, Zachariadis V, Taylan F,
Marincevic-Zuniga Y, Tran AN, Saft L, Nilsson D, Syvänen AC,
Lönnerholm G, Harila-Saari A, et al: Overexpression of chromatin
remodeling and tyrosine kinase genes in iAMP21-positive acute
lymphoblastic leukemia. Leuk Lymphoma. 61:604–613. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
HAF1B chromatin assembly factor 1 subunit
B [Homo sapiens (human)]-Gene-NCBI [Internet], . Nih.gov.
2026.[cited 2026 Mar 23]. Available from. https://www.ncbi.nlm.nih.gov/gene/8208
|
|
21
|
Volk A and Crispino JD: The role of the
chromatin assembly complex (CAF-1) and its p60 subunit (CHAF1b) in
homeostasis and disease. Biochim Biophys Acta. 1849:979–986. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Database G. GeneCards-Human Genes|Gene
Database|Gene Search. Genecards.org. 2019.Available from:.
https://www.genecards.org
|
|
23
|
Li Q, Zhang X and Zhang Z: CHAF1B
Overexpression: A brake for the differentiation of leukemia cells.
Cancer Cell. 34:693–694. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Volk A, Liang K, Suraneni P, Li X, Zhao J,
Bulic M, Marshall S, Pulakanti K, Malinge S, Taub J, et al: A
CHAF1B-dependent molecular switch in hematopoiesis and leukemia
pathogenesis. Cancer Cell. 34:707–723.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Polo SE, Theocharis SE, Grandin L,
Gambotti L, Antoni G, Savignoni A, Asselain B, Patsouris E and
Almouzni G: Clinical significance and prognostic value of chromatin
assembly factor-1 overexpression in human solid tumours.
Histopathology. 57:716–724. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim JH, Li L and Resar LM: Doubling up on
function: Dual-specificity tyrosine-regulated kinase 1A (DYRK1A) in
B cell acute lymphoblastic leukemia. J Clin Invest.
131:e1426272021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deboever E, Fistrovich A, Hulme C and
Dunckley T: The omnipresence of DYRK1A in human diseases. Int J Mol
Sci. 23:93552022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhansali RS, Rammohan M, Lee P, Laurent
AP, Wen Q, Suraneni P, Yip BH, Tsai YC, Jenni S, Bornhauser B, et
al: DYRK1A regulates B cell acute lymphoblastic leukemia through
phosphorylation of FOXO1 and STAT3. J Clin Invest. 131:e1359372021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Xie X, Jie Z, Zhu L, Yang JY, Ko CJ,
Gao T, Jain A, Jung SY, Baran N, et al: DYRK1A mediates
BAFF-induced noncanonical NF-kB activation to promote autoimmunity
and B-cell leukemogenesis. Blood. 138:2360–2371. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Behrens K, Brajanovski N, Xu Z, Viney EM,
DiRago L, Hediyeh-Zadeh S, Davis MJ, Pearson RB, Sanij E, Alexander
WS and Ng AP: ERG and c-MYC regulate a critical gene network in
BCR::ABL1-driven B cell acute lymphoblastic leukemia. Sci Adv.
10:eadj88032024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zaliova M, Potuckova E, Hovorkova L,
Musilova A, Winkowska L, Fiser K, Stuchly J, Mejstrikova E,
Starkova J, Zuna J, et al: ERG deletions in childhood acute
lymphoblastic leukemia with DUX4 rearrangements are mostly
polyclonal, prognostically relevant and their detection rate
strongly depends on screening method sensitivity. Haematologica.
104:1407–1416. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clappier E, Auclerc MF, Rapion J, Bakkus
M, Caye A, Khemiri A, Giroux C, Hernandez L, Kabongo E, Savola S,
et al: An intragenic ERG deletion is a marker of an oncogenic
subtype of B-cell precursor acute lymphoblastic leukemia with a
favorable outcome despite frequent IKZF1 deletions. Leukemia.
28:70–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang J, McCastlain K, Yoshihara H, Xu B,
Chang Y, Churchman ML, Wu G, Li Y, Wei L, Iacobucci I, et al:
Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat
Genet. 48:1481–1489. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lo Nigro L, Arrabito M, Andriano N,
Iachelli V, La Rosa M and Bonaccorso P: Characterization of CK2,
MYC and ERG expression in biological subgroups of children with
acute lymphoblastic leukemia. Int J Mol Sci. 26:10762025.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cabal-Hierro L, van Galen P, Prado MA,
Higby KJ, Togami K, Mowery CT, Paulo JA, Xie Y, Cejas P, Furusawa
T, et al: Chromatin accessibility promotes hematopoietic and
leukemia stem cell activity. Nat Commun. 11:14062020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Page EC, Heatley SL, Rehn J, Thomas PQ,
Yeung DT and White DL: Gain of chromosome 21 increases the
propensity for P2RY8::CRLF2 acute lymphoblastic leukemia via
increased HMGN1 expression. Front Oncol. 13:11778712023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Islam R, Jenkins CE, Cao Q, Wong J,
Bilenky M, Carles A, Moksa M, Weng AP and Hirst M: RUNX1 colludes
with NOTCH1 to reprogram chromatin in T cell acute lymphoblastic
leukemia. iScience. 26:1067952023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sood R, Kamikubo Y and Liu P: Role of
RUNX1 in hematological malignancies. Blood. 129:2070–2082. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin TC: RUNX1 and cancer. Biochim Biophys
Acta Rev Cancer. 1877:1887152022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fu L, Fu H, Tian L, Xu K, Hu K, Wang J,
Wang J, Jing H, Shi J and Ke X: High expression of RUNX1 is
associated with poorer outcomes in cytogenetically normal acute
myeloid leukemia. Oncotarget. 7:15828–15839. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yi H, He Y, Zhu Q and Fang L: RUNX
proteins as epigenetic modulators in cancer. Cells. 11:36872022.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yokota A, Huo L, Lan F, Wu J and Huang G:
The clinical, molecular and mechanistic basis of RUNX1 mutations
identified in hematological malignancies. Mol Cells. 43:145–152.
2020.PubMed/NCBI
|
|
43
|
Aydin C, Cetin Z, Manguoglu AE, Tayfun F,
Clark OA, Kupesiz A, Akkaya B and Karauzum SB: Evaluation of
ETV6/RUNX1 fusion and additional abnormalities involving ETV6
and/or RUNX1 genes using FISH technique in patients with childhood
acute lymphoblastic leukemia. Indian J Hematol Blood Transfus.
32:154–161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jakobczyk H, Jiang Y, Debaize L, Soubise
B, Avner S, Sérandour AA, Rouger-Gaudichon J, Rio AG, Carroll JS,
Raslova H, et al: ETV6-RUNX1 and RUNX1 directly regulate RAG1
expression: one more step in the understanding of childhood B-cell
acute lymphoblastic leukemia leukemogenesis. Leukemia. 36:549–554.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jepsen VH, Hanel A, Picard D, Bhave R,
Hasselmann R, Mehtonen J, Schliehe-Diecks J, Kath CJ, Suppiyar V,
Prasad Y, et al: H1-0 is a specific mediator of the repressive
ETV6::RUNX1transcriptional landscape in preleukemia and B cell
acutelymphoblastic leukemia. Hemasphere. 9:e701162025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhong LK, Deng XY, Shen F, Cai WS, Feng
JH, Gan XX, Jiang S, Liu CZ, Zhang MG, Deng JW, et al:
Identification of a 3-Gene prognostic index for papillary thyroid
carcinoma. Front Mol Biosci. 9:8079312022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Marinelli LM, Kisiel JB, Slettedahl SW,
Mahoney DW, Lemens MA, Shridhar V, Taylor WR, Staub JK, Cao X,
Foote PH, et al: Methylated DNA markers for plasma detection of
ovarian cancer: Discovery, validation, and clinical feasibility.
Gynecol Oncol. 165:568–576. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kontic M and Markovic F: Use of DNA
methylation patterns for early detection and management of lung
cancer: Are we there yet? Oncol Res. 33:781–793. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Moreno Lorenzana D, Juárez Velázquez MDR,
Reyes León A, Martínez Anaya D, Hernández Monterde A, Salas Labadía
C, Navarrete Meneses MDP, Zapata Tarrés M, Juárez Villegas L,
Jarquín Ramírez B, et al: CRLF2 and IKZF1 abnormalities in Mexican
children with acute lymphoblastic leukemia and recurrent gene
fusions: Exploring surrogate markers of signaling pathways. J
Pathol Clin Res. 7:410–421. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Duffy C, Graetz DE, Lopez AMZ, Carrillo
AK, Job G, Chen Y, Devidas M, Leon SA, Bonzi SA, Flores PC, et al:
Retrospective analysis of outcomes for pediatric acute
lymphoblasticleukemia in South American centers. Front Oncol.
13:12542332013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Albu CC, Bica F, Nan L, Bubulac L,
Bogdan-Andreescu CF, Şerbanică IV, Poalelungi CV, Cadar E,
Bănățeanu AM and Burcea A: B-cell acute lymphoblastic leukemia in a
child with down syndrome and High-risk genomic lesions. Curr Issues
Mol Biol. 47:7042025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Heerema NA, Carroll AJ, Devidas M, Loh ML,
Borowitz MJ, Gastier-Foster JM, Larsen EC, Mattano LA Jr, Maloney
KW, Willman CL, et al: Intrachromosomal amplification of chromosome
21 is associated with inferior outcomes in children with acute
lymphoblastic leukemia treated in contemporary standard-risk
children's oncology group studies: A report from the children's
oncology group. J Clin Oncol. 31:3397–402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ryan SL, Matheson E, Grossmann V, Sinclair
P, Bashton M, Schwab C, Towers W, Partington M, Elliott A, Minto L,
et al: The role of the RAS pathway in iAMP21-ALL. Leukemia.
30:1824–1831. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schwab C and Harrison CJ: Advances in
B-cell precursor acute lymphoblastic leukemia genomics. Hemasphere.
2:e532018. View Article : Google Scholar : PubMed/NCBI
|