Introduction
Breast cancer (BC) remains the most commonly
diagnosed cancer among women worldwide. According to the most
recent GLOBOCAN 2022 estimates from the International Agency for
Research on Cancer, BC accounted for 2,296,840 new cases and
666,103 deaths worldwide in 2022 (1). Notably, the global BC incidence is
expected to increase by >40% by 2040 (2), with China exhibiting the highest
incidence rate in Asia (3). BC is a
heterogeneous disease that is classified by the expression of key
receptors, such as the estrogen receptor (ER), progesterone
receptor (PR) and HER2 (4).
Furthermore, ~75% of BC cases are ER− and/or
PR+ (5), making adjuvant
endocrine therapy (AET) a standard treatment that notably reduces
recurrence and mortality and improves overall survival (6–8). The
American Society of Clinical Oncology recommends 5–10 years of AET
(9); however, its prolonged use is
often associated with adverse effects, including hot flashes, night
sweats, fatigue, sleep disturbance, sexual dysfunction, joint
stiffness, joint dysfunction and joint pain, all of which may
adversely affect patients' quality of life (10). In addition, 15–25% of BC cases are
characterized by HER2 overexpression or gene amplification
(11,12), which is associated with more
aggressive disease, higher histological grade, early metastasis and
worse prognosis (12–14).
RTA-408 (omaveloxolone) is a synthetic oleanane
triterpenoid with broad biological activity owing to its ability to
modulate redox-sensitive transcriptional pathways. It binds to
kelch-like ECH-associated protein 1 (KEAP1) at a critical cysteine
residue, thereby stabilizing nuclear factor erythroid 2-related
factor 2 (Nrf2) and inducing the expression of antioxidant and
phase II detoxification genes, including sulfiredoxin, thioredoxin,
thioredoxin reductase, glutathione reductase, peroxiredoxin,
glutamate cysteine ligase and quinone reductase (15). Nrf2 activation bolsters cellular
antioxidant defenses and exerts anti-inflammatory effects by
attenuating reactive oxygen species-driven inflammatory signaling
(15). RTA-408 also inhibits IKK,
blocking NF-κB activation and consequently reducing
pro-inflammatory gene expression while promoting apoptosis in
cancer cells (15). In addition, it
affects the heme-regulated transcriptional repressor BTB and CNC
homolog 1 (BACH1); in models of acute lung injury, RTA-408 prevents
ferroptotic cell death by suppressing BACH1 activity (16). These mechanisms translate into
cytoprotective outcomes in diverse preclinical models. RTA-408
mitigates oxidative tissue damage and inflammation and inhibits
tumor growth in xenograft cancer models (15). Clinically, a first-in-human phase I
trial in patients with stage IV relapsed/refractory melanoma or
non-small cell lung cancer showed that oral RTA-408 was well
tolerated at biologically active doses (with evidence of Nrf2
target engagement and minimal toxicity), although no confirmed
tumor responses were observed (15). In a pivotal placebo-controlled trial
for Friedreich's ataxia (a neurodegenerative disease),
omaveloxolone treatment improved neurological function compared
with placebo, as demonstrated by a 2.40±0.96-point
placebo-corrected improvement in modified Friedreich's Ataxia
Rating Scale (mFARS) score at week 48, with improvements observed
across mFARS components, including bulbar function, upper-limb
coordination, lower-limb coordination and upright stability, most
notably upright stability (17).
This benefit, along with a manageable safety profile, has resulted
in the approval of omaveloxolone as the first-line therapy for
Friedreich's ataxia (18). Ongoing
research is exploring the therapeutic potential of omaveloxolone in
other conditions, such as inflammatory bowel disease, by leveraging
its antioxidant, anti-inflammatory and immunomodulatory properties
(18). The efficacy of
omaveloxolone in BC remains unexplored; therefore, the present
study aimed to investigate the antitumor efficacy and underlying
mechanisms of RTA-408 in BC.
Materials and methods
Cell culture
Human BC cell lines, MCF-7 (ER+) and
MDA-MB-231 (triple-negative), were obtained from the Bioresource
Collection and Research Center. MCF-7 cells were cultured in
RPMI-1640 (cat. no. 31800-022; Gibco; Thermo Fisher Scientific,
Inc.), whereas MDA-MB-231 cells were cultured in DMEM (cat. no.
31600-034; Gibco; Thermo Fisher Scientific, Inc.). The culture
medium included 10% heat-inactivated FBS (cat. no. FC926;
MilliporeSigma; Merck KGaA), 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C in a humidified 5% CO2 incubator.
Cultures were passaged 2–3 times per week and used within 20
passages.
Drug preparation and treatment
RTA-408 (cat. no. HY-12212; MedChemExpress) and
SP600125 (cat. no. HY-12041; MedChemExpress) were dissolved in DMSO
to prepare 10 mM stock solutions and stored at −20°C. Working
solutions were freshly prepared in complete medium; the final DMSO
concentration did not exceed 0.1%. For JNK blockade, cells were
pre-incubated for 1 h at 37°C with 10 µM SP600125 before the
addition of RTA-408. Vehicle controls received an equivalent volume
of DMSO.
Cell-viability assay
Exponentially growing cells were seeded in 24-well
plates at a density of 3×104 cells/well. After overnight
attachment, the cultures were exposed to RTA-408 at 0–1,000 nM for
72 h at 37°C. Viability was quantified using an MTT assay (cat. no.
L11939.06; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Formazan crystals were dissolved in
200 µl DMSO, and absorbance was read at 570 nm on an ELISA reader
(MULTISKAN FC; Thermo Fisher Scientific, Inc.). The results are
expressed as a percentage of the untreated control. Each condition
was performed in triplicate in six independent experiments.
Apoptosis flow cytometry
Apoptosis was analyzed using a Muse®
Annexin V & Dead Cell Kit (cat. no. MCH100105; Cytek
Biosciences) according to the manufacturer's instructions. After
treatment, 3×105 cells were collected, washed twice with
PBS and resuspended in 100 µl of the staining reagent supplied with
the Kit. Following a 15 min incubation (room temperature, in the
dark), samples were diluted to 500 µl and immediately analyzed on a
flow cytometer (MUSE; Cytek Biosciences). For each sample, 20,000
events were recorded.
Western blotting
Cells were lysed in RIPA buffer supplemented with
protease (cat. no. 04693159001; Roche Diagnostics GmbH) and
phosphatase inhibitors (cat. no. 04906845001; Roche Diagnostics
GmbH). Equal amounts of protein (50 µg) were separated by 10–12%
SDS-PAGE gel electrophoresis and electro-transferred to PVDF
membranes (cat. no. NEF1002001PK; PerkinElmer, Inc.). After
blocking with 5% non-fat milk for 1 h at room temperature,
membranes were incubated overnight at 4°C with primary antibodies
against phosphorylated-JNK (p-JNK; 1:500; cat. no. 4668; Cell
Signaling Technology, Inc.), total JNK (1:1,000; cat. no. 9252;
Cell Signaling, phosphorylated-p38 (p-p38; 1:500; cat. no 9211;
Cell Signaling Technology, Inc.), total p38 (1:1,000; cat. no.
9212; Cell Signaling Technology, Inc.), phosphorylated ERK1/2
(p-ERK1/2; 1:500; cat. no. 9101; Cell Signaling Technology, Inc.),
total ERK1/2 (1:2,000; cat. no. 9102; Cell Signaling Technology,
Inc.), beclin-1 (1:1,000; cat. no. 11306-1-AP; Proteintech Group,
Inc.), light-chain 3B (LC3B; 1:1,000; cat. no. 3868; Cell Signaling
Technology, Inc.), p62 (1:1,000; cat. no. 18420-1-AP; Proteintech
Group, Inc.), poly(ADP-ribose) polymerase (PARP; 1:1,000; cat. no.
13371-1-AP; Proteintech Group, Inc.) and β-actin (1:20,000; cat.
no. A5441; MilliporeSigma). HRP-conjugated secondary antibodies
(goat anti-rabbit antibody: cat. no. AP132P; 1:5,000;
MilliporeSigma; and goat anti-mouse antibody: cat. no. AP124P;
1:5,000; MilliporeSigma; Merck KGaA) were applied for 1 h at room
temperature and signals were visualized using Western
Lightning™ Plus-ECL substrate (Revvity, Inc.) on a
ChemiDoc MP imaging system (8.0.2.0/21; Wuhan Servicebio Technology
Co., Ltd.). Band intensities were quantified using ImageJ (‘Fiji’
package; version 2.9.0; National Institutes of Health) and
normalized to β-actin.
Statistical analysis
Data are presented as the mean ± SD from ≥6
independent experiments. Statistical significance was assessed
using one-way ANOVA, followed by Tukey's post hoc test (GraphPad
Prism; version 9; Dotmatics). P<0.05 was considered to indicate
a statistically significant difference, with *P<0.05,
**P<0.01 and ***P<0.001 vs. the control and
#P<0.05 and ##P<0.01 vs. the
corresponding RTA-408 group.
Results
Dose-dependent reduction of cell
viability in MCF-7 and MDA-MB-231 BC cells following RTA-408
treatment
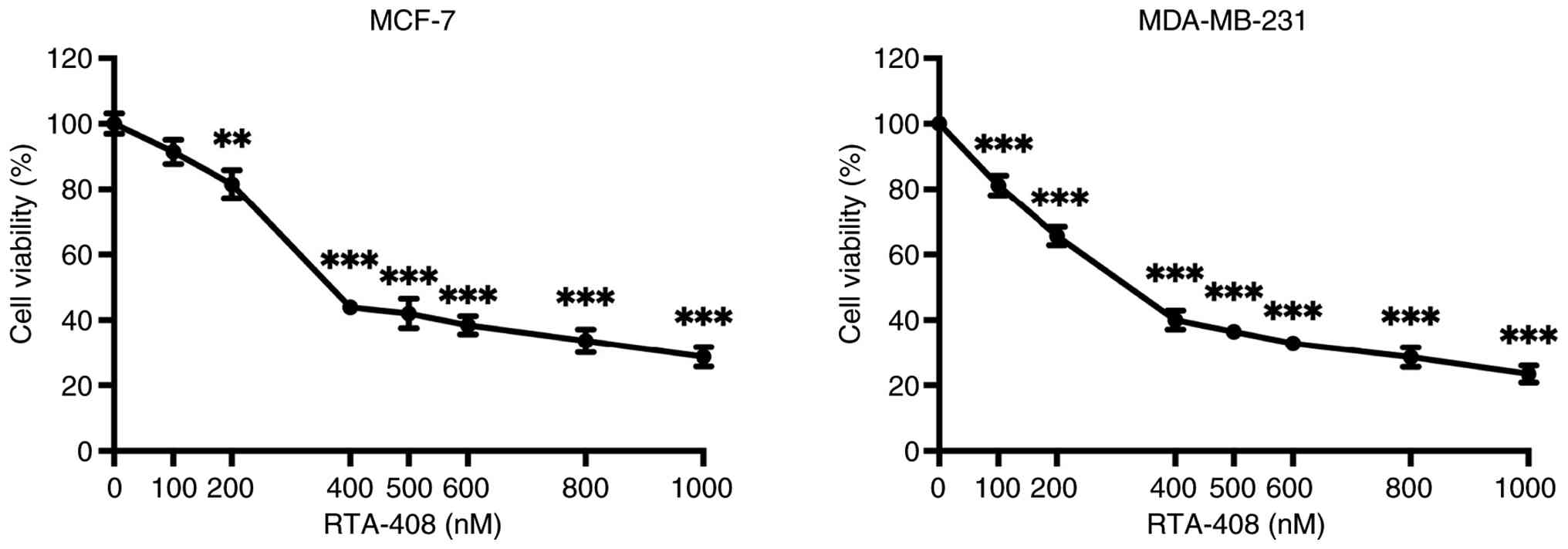
To assess concentration-dependent cytotoxicity,
MCF-7 BC cells were exposed to 0–1,000 nM RTA-408 for 72 h, after
which cell viability was quantified using an MTT assay (Fig. 1). Cell survival remained close to
baseline at 100 nM and decreased to 81.52±12.40% at 200 nM
(P<0.01 vs. control). A sharper decline was observed at 400 nM,
where viability decreased to 43.93±6.18% and further reductions
were recorded between 500–600 nM (42.08±13.49% and 38.41±8.54%,
respectively), with all values from 400 nM onward achieving
P<0.001. At the highest doses of 800 and 1,000 nM, viability
decreased by 33.61±10.51% and 28.74±8.95%, respectively
(P<0.001).
Under identical treatment conditions, MDA-MB-231
cells exhibited a monotonic loss of viability (Fig. 1). Viability remained near 80% at 100
nM (P<0.001), followed by a reduction to 65.65±8.39% survival at
200 nM (P<0.001). Exposure to 400 nM lowered viability to
40.0±8.40%, with a gradual decline to 36.48±6.34% and 28.64±8.59%
at 600 and 800 nM, respectively (P<0.001). The maximal
concentration of 1,000 nM reduced viability to 23.42±7.90%
(P<0.001). These data demonstrate that RTA-408 induced a robust,
dose-dependent cytotoxic response in both HR+ and
triple-negative BC cell models, supporting its potential as a
broadly effective antiproliferative agent.
Dose-dependent induction of apoptosis
and autophagy by RTA-408 in BC cells
Quantification of Annexin V-positive cells revealed
a dose-dependent increase in total apoptosis after 72 h of RTA-408
treatment in both MCF-7 and MDA-MB-231 cells (Fig. 2A). In MCF-7 cells, the apoptotic
fraction increased progressively with increasing drug concentration
and reached statistical significance at 400 nM (P<0.001),
whereas the increases at 100 and 200 nM did not reach statistical
significance. In MDA-MB-231 cells, RTA-408 also produced a clear
dose-dependent increase in apoptosis, with significant elevations
observed at 100 and 200 nM (both P<0.01) and at 400 nM
(P<0.001).
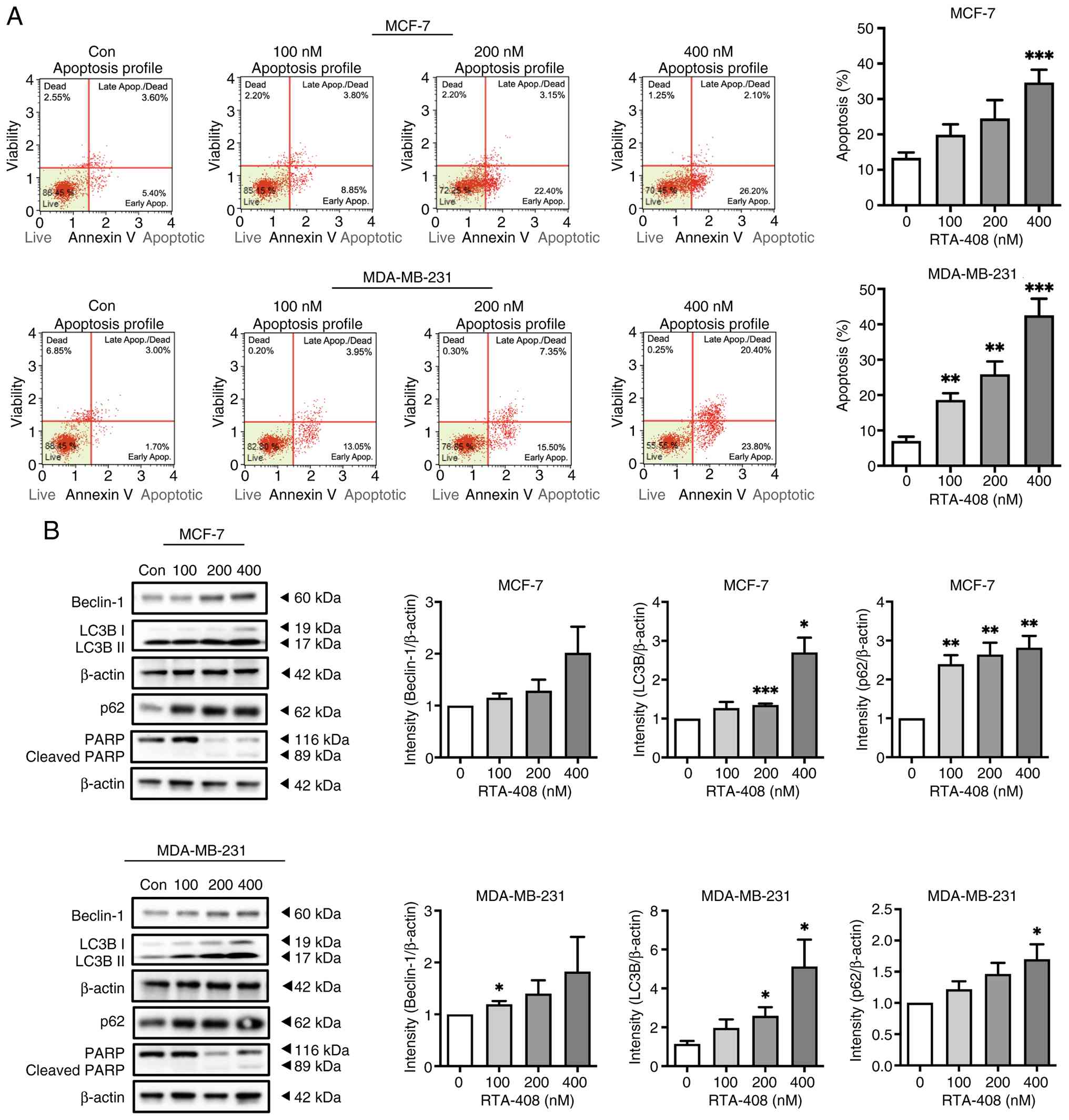 | Figure 2.RTA-408 induces apoptosis and
increases autophagy-associated marker accumulation in breast cancer
cells. (A) Representative Annexin V & Dead Cell Kit flow
cytometry dot plots of MCF-7 and MDA-MB-231 cells following 72-h
exposure to vehicle Con or RTA-408 at 100, 200 or 400 nM. Quadrants
denote live, early apoptotic, late apoptotic/dead and dead
populations. Bar graphs to the right summarize total apoptosis
(early + late apoptotic populations) as a percentage of total
events. Data are presented as the mean ± SD (n=6). (B)
Representative western blots of Beclin-1, LC3B-I/LC3B-II, p62, PARP
and β-actin in MCF-7 and MDA-MB-231 cells after 72 h treatment with
the indicated concentrations of RTA-408. Molecular weight markers
are indicated. The PARP blot shows full-length PARP (116 kDa) and
cleaved PARP (89 kDa). Right-side bar graphs show densitometric
quantification of beclin-1, LC3B-IIand p62 normalized to β-actin
and expressed relative to the control group. Data are presented as
the mean ± SD (n=6). Statistical significance was determined by
one-way ANOVA, followed by Tukey's post hoc test: *P<0.05,
**P<0.01 and ***P<0.001 vs. control. Con, control; Apop.,
apoptosis; LC3B, microtubule-associated protein 1 light chain 3B;
PARP, poly (ADP-ribose) polymerase. |
PARP cleavage was additionally examined by western
blotting as a biochemical indicator of apoptosis. A lower band
consistent with cleaved PARP was detectable, particularly at higher
RTA-408 concentrations; however, the cleaved PARP signal was
relatively weak compared with the robust increase in Annexin
V-positive apoptotic fractions observed by flow cytometry (Fig. 2B). Therefore, PARP cleavage was
interpreted as complementary biochemical evidence, whereas flow
cytometry served as the primary quantitative assessment of
apoptosis.
Western blotting analysis was further used to
examine the autophagy-related markers Beclin-1, LC3B-II and p62
(Fig. 2B). In MCF-7 cells, Beclin-1
showed a moderate increase after RTA-408 treatment, although this
increase did not reach statistical significance. LC3B-II was
significantly increased at 200 nM (P<0.001) and 400 nM
(P<0.05), whereas p62 was significantly elevated at 100, 200 and
400 nM (all P<0.01). In MDA-MB-231 cells, Beclin-1 reached
statistical significance at 100 nM (P<0.05), while the increases
at 200 and 400 nM did not reach statistical significance. LC3B-II
was significantly increased at 200 and 400 nM (both P<0.05) and
p62 reached statistical significance at 400 nM (P<0.05).
Collectively, these findings indicate that RTA-408 induces
apoptosis and is accompanied by accumulation of autophagy-related
markers in breast cancer cells.
Dose-dependent activation of MAPK
signaling cascades by RTA-408 in BC cells
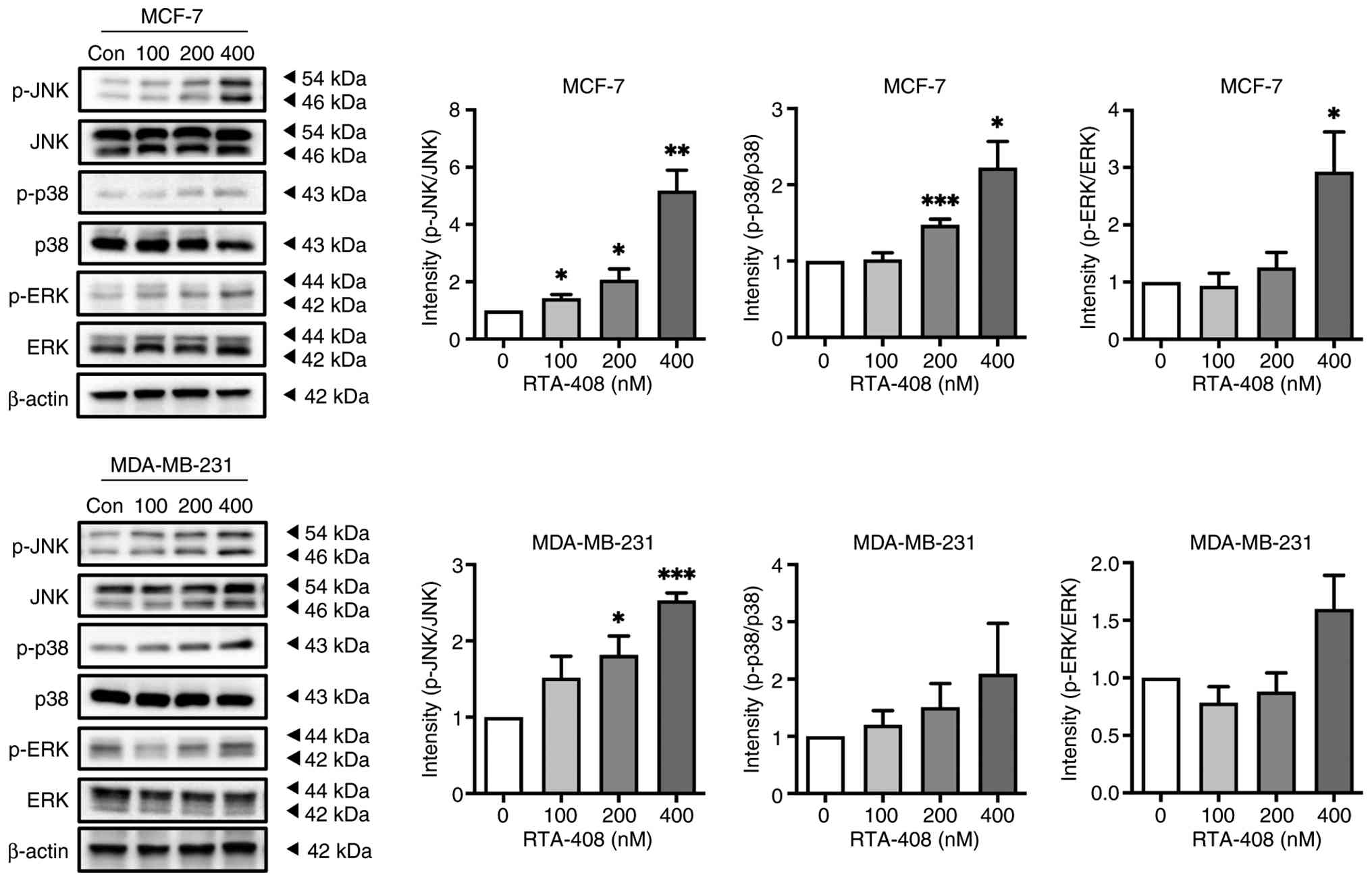
To elucidate whether RTA-408 modulates
stress-activated MAPK signaling, the phosphorylation of JNK, p38
and ERK were quantified after 72 h of exposure to 0–400 nM. In
MCF-7 cells, p-JNK increased by ~2-fold at 100 nM and ~6-fold at
400 nM, whereas total JNK remained unchanged, indicating robust
pathway activation (Fig. 3). p-p38
exhibited a modest yet significant increase at 400 nM and p-ERK
displayed a significant 3-fold increase at the highest dose,
determining the concurrent engagement of multiple MAPK branches
without altering the total p38 or ERK abundance (Fig. 3). In MCF-7 cells, p-JNK was
significantly increased at 100 and 200 nM (both P<0.05) and at
400 nM (P<0.01), p-p38 was significantly increased at 200 nM
(P<0.001) and 400 nM (P<0.05), whereas p-ERK reached
statistical significance only at 400 nM (P<0.05). In MDA-MB-231
cells, p-JNK reached statistical significance at 200 nM (P<0.05)
and 400 nM (P<0.001), whereas p-p38 and p-ERK did not reach
statistical significance at the tested concentrations. These
findings demonstrate dose-dependent stimulation of JNK and ERK and,
to a lesser extent, p38 signaling cascades by RTA-408, implicating
MAPK activation in its downstream cytotoxic mechanisms (Fig. 3).
JNK inhibitor SP600125 inhibits
RTA-408-induced JNK phosphorylation in BC cells
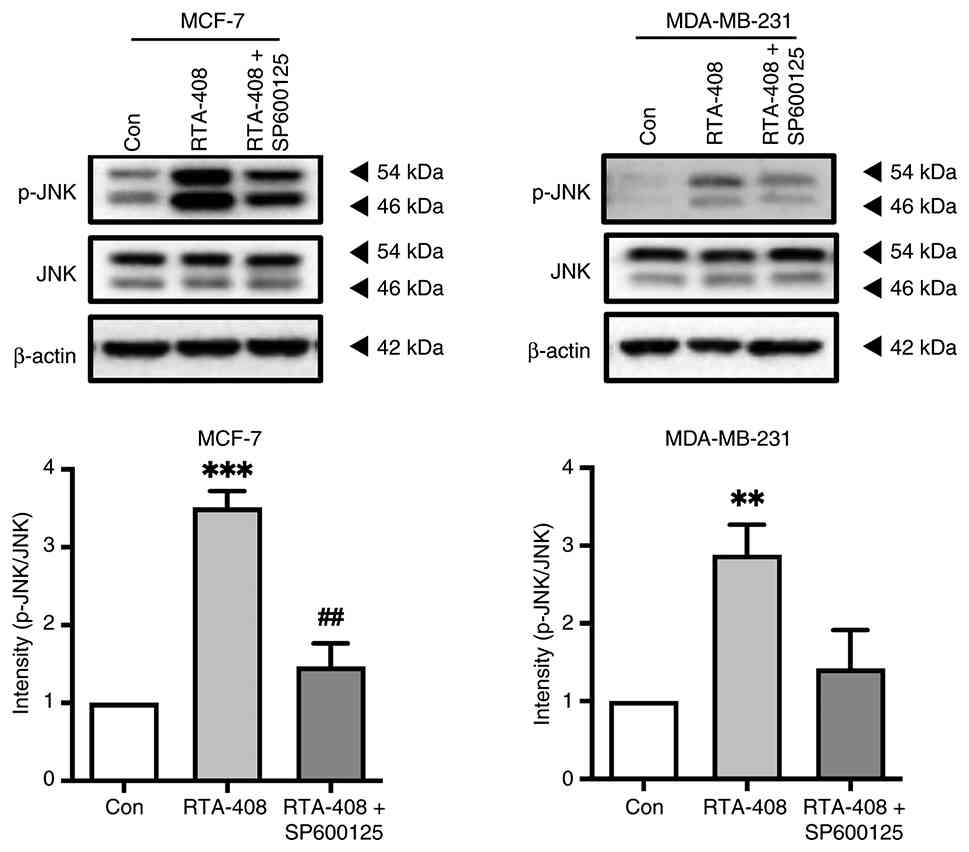
To build upon the JNK phosphorylation demonstrated
in Fig. 3, the pharmacologic JNK
inhibitor SP600125 was used as an initial tool to probe JNK pathway
involvement. To determine whether JNK activation was a direct
downstream target of RTA-408, both BC cell lines were treated with
RTA-408 alone or in combination with SP600125 and the ratio of
phosphorylated to total JNK was quantified. In MCF-7 cells, RTA-408
alone exhibited an ~3.5-fold increase in p-JNK relative to the
control (P<0.001), whereas co-treatment with SP600125 reduced
this increase to ~1.5-fold (P<0.01 vs. RTA-408 alone). An
analogous pattern was observed in MDA-MB-231 cells; RTA-408
increased p-JNK by ~3-fold (P<0.01) and inhibition with SP600125
reduced phosphorylation to baseline. Total JNK and β-actin remained
unchanged, indicating that RTA-408 increased JNK phosphorylation
and that this effect was attenuated by pharmacologic inhibition
(Fig. 4).
JNK inhibitor SP600125 inhibits
RTA-408-induced apoptosis and autophagy in BC cells
To evaluate whether JNK signaling contributes to the
RTA-408-induced phenotype, cells were treated with RTA-408 in the
absence or presence of the pharmacologic JNK inhibitor SP600125.
Annexin V & Dead Cell Kit flow cytometric analysis showed that
RTA-408 significantly increased total apoptosis in both MCF-7 and
MDA-MB-231 cells (Fig. 5A).
Co-treatment with SP600125 reduced the apoptotic fraction in both
cell lines; however, in MCF-7 cells, this decrease represented only
a downward trend and did not reach statistical significance,
whereas in MDA-MB-231 cells, the attenuation was statistically
significant (Fig. 5A).
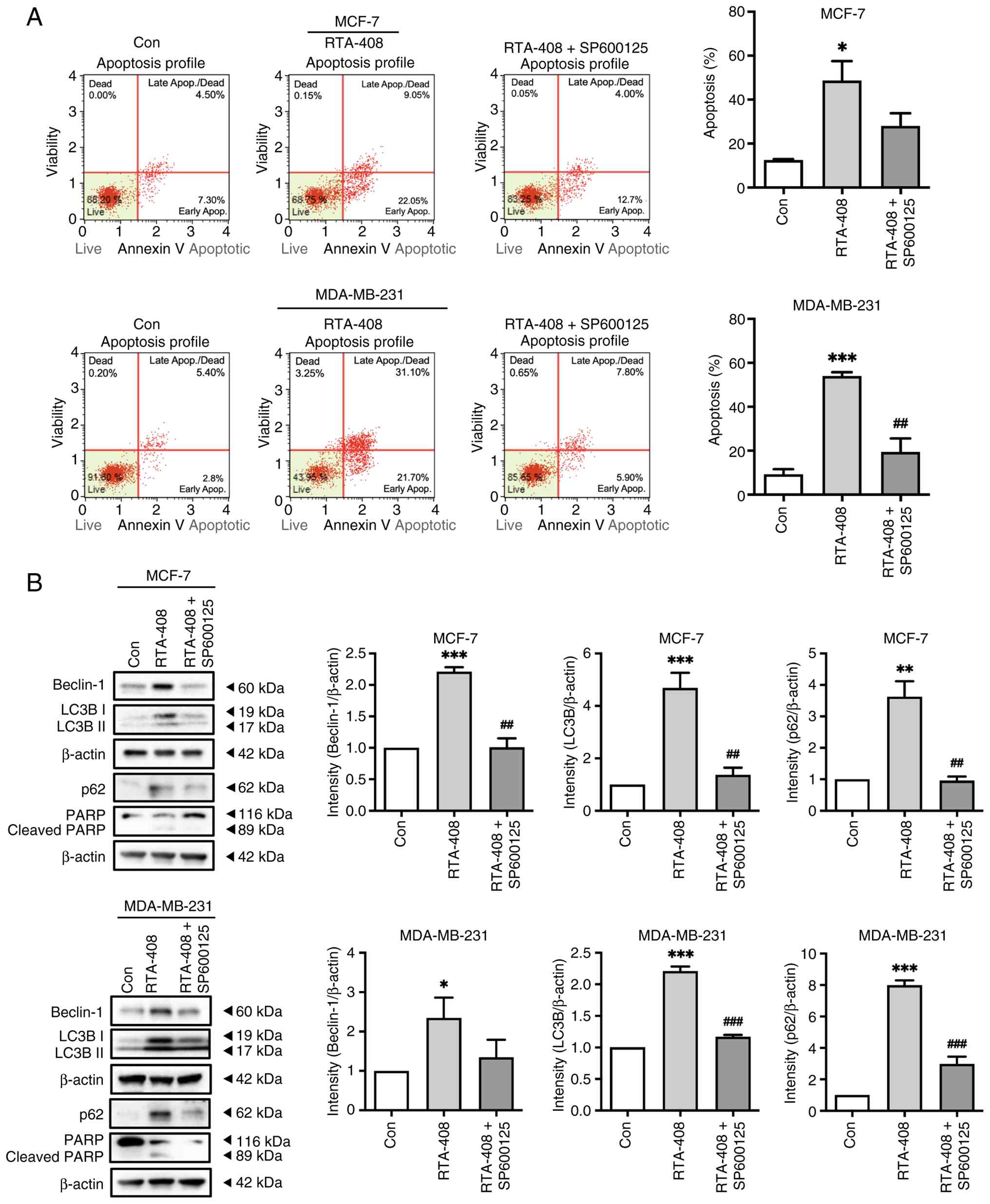 | Figure 5.Pharmacologic JNK inhibition
attenuates RTA-408-induced apoptosis and autophagy-associated
marker accumulation. (A) Representative Annexin V & Dead Cell
Kit flow cytometry plots of MCF-7 and MDA-MB-231 cells after 72 h
treatment with vehicle control (Con), RTA-408 (400 nM) or RTA-408 +
SP600125 (10 µM; 1 h pretreatment followed by co-treatment). Bar
graphs to the right summarize total apoptosis (early + late
apoptotic populations) as a percentage of total events. RTA-408
significantly increased apoptotic fractions in both cell lines.
SP600125 reduced RTA-408-induced apoptosis in both cell lines; this
reduction did not reach statistical significance in MCF-7 cells but
was significant in MDA-MB-231 cells. (B) Representative western
blots of beclin-1, LC3B-I/LC3B-II, p62, PARP and β-actin in MCF-7
and MDA-MB-231 cells under the same treatment conditions. Molecular
weight markers are indicated. The PARP blot shows full-length PARP
(116 kDa) and cleaved PARP (89 kDa). Right-side histograms show
densitometric quantification of beclin-1, LC3B-II and p62
normalized to β-actin and expressed relative to the control group.
SP600125 attenuated the RTA-408-induced increases in beclin-1,
LC3B-II and p62 in MCF-7 cells and reduced LC3B-II and p62
accumulation in MDA-MB-231 cells, with a decreasing trend in
beclin-1. Data are presented as the mean ± SD (n=6). Statistical
significance was determined by one-way ANOVA followed by Tukey's
post hoc test: *P<0.05, **P<0.01 and ***P<0.001 vs.
control; ##P<0.01 and ###P<0.001 vs.
RTA-408 alone. Con, control; Apop., apoptosis; LC3B,
microtubule-associated protein 1 light chain 3B; PARP, poly
(ADP-ribose) polymerase. |
To further support apoptosis biochemically, PARP
cleavage was examined by western blotting. RTA-408 increased
cleaved PARP, and this effect was reduced in the presence of
SP600125, consistent with the flow cytometric findings (Fig. 5B). Western blotting analysis further
showed that RTA-408 increased beclin-1, LC3B-II and p62 in both
cell lines and these changes were attenuated by SP600125 (Fig. 5B). In MCF-7 cells, SP600125
treatment significantly reduced the RTA-408-induced increases in
beclin-1, LC3B-II and p62. In MDA-MB-231 cells, SP600125 treatment
significantly reduced LC3B-II and p62, whereas beclin-1 showed a
decreasing trend without statistical significance. Collectively,
these findings support that JNK signaling contributes, at least in
part, to RTA-408-induced apoptosis, and that the accumulation of
autophagy-related markers was attenuated, at least in part, by
pharmacologic JNK inhibition.
Discussion
RTA-408 is a synthetic triterpenoid antioxidant and
inflammatory modulator with broad antitumor activity (19). Probst et al (19) first reported that RTA-408 inhibited
the growth of numerous human tumor cells at nanomolar
concentrations, including eight tumor cell lines: Melanoma,
non-small cell lung cancer, breast adenocarcinoma, colorectal
cancer, renal cancer and pancreatic cancer. In these cells, RTA-408
notably induced caspase activation and substrate cleavage,
demonstrating a pro-apoptotic effect. Previous studies have
explored the broad antitumor potential of RTA-408 (19). For example, in a drug-resistant lung
cancer model, RTA-408 effectively overcame cisplatin resistance by
downregulating WW domain-containing E3 ubiquitin-protein ligase 1,
thereby blocking the ubiquitination and degradation of nuclear
receptor coactivator 4 and inducing ferritinophagy and ferroptosis,
demonstrating that RTA-408 may reverse chemotherapy resistance in
lung cancer (20). Similarly, in
the context of malignant brain tumors, RTA-408 demonstrated potent
inhibitory effects against advanced/drug-resistant glioblastoma
multiforme cells, effectively inhibiting the proliferation and
colony formation of temozolomide-resistant glioblastoma cells and
inducing G1 cell cycle arrest and dose-dependent
apoptosis (21). These findings
highlight the broad-spectrum inhibitory efficacy of RTA-408 across
a number of cancer cell types and states and its potential for
clinical application. To the best of our knowledge, the present
study provides the first detailed mechanistic characterization of
RTA-408 in both ER-positive/hormone receptor-positive and
triple-negative BC cell models, demonstrating nanomolar,
dose-dependent antiproliferative effects together with
apoptosis-associated responses, autophagy-related marker
accumulation and JNK pathway involvement. These findings build on a
previous report showing that RTA-408 inhibited tumor-cell growth
and induced caspase activity/apoptosis in a broad human tumor-cell
panel, including melanoma, non-small cell lung cancer, breast
adenocarcinoma, colorectal carcinoma, renal cell adenocarcinoma and
pancreatic cancer (19). Its
antitumor activity has also been extended to resistant tumor
settings, including cisplatin-resistant lung cancer and
temozolomide-resistant glioblastoma, through mechanisms involving
ferritinophagy/ferroptosis, apoptosis and cell-cycle arrest, as
well as to glioblastoma models through CDC20 downregulation
(20–22). Furthermore, the first-in-human phase
I evaluation of oral omaveloxolone in patients with stage IV
relapsed/refractory melanoma or non-small cell lung cancer,
together with recent evidence of JNK-mediated antitumor and
radiosensitizing activity in glioblastoma models, supports its
broader translational relevance (15,23).
In the present study, a significant increase was
observed in the expression of both apoptosis- and
autophagy-associated markers in BC cells after RTA-408 treatment.
For example, the number of cells positive for Annexin
V/7-amino-actinomycin D (7-AAD) double staining increased, along
with elevated expression of the autophagy-associated proteins
Beclin-1, LC3B-II and p62. It should also be noted that although
Annexin V/7-AAD-based flow cytometry demonstrated a clear
dose-dependent increase in apoptotic cells, the cleaved PARP signal
was relatively weak in the western blot analysis. This discrepancy
may reflect differences in assay sensitivity or the timing of PARP
cleavage detection. Therefore, PARP cleavage was considered
supportive biochemical evidence, whereas Annexin V/7-AAD-based flow
cytometry was used as the primary quantitative readout of apoptosis
in the present study. This phenomenon of concurrent apoptosis and
autophagy-associated marker accumulation has also been reported in
other studies. For example, the small-molecule compound
1,3-dibutyl-2-thiooxo-imidazolidine-4,5-dione (C1) was shown to
induce non-canonical autophagy and apoptosis in multiple human
cancer cell lines through ROS-dependent ERK and JNK activation,
while curcumin induced both apoptosis and autophagy in osteosarcoma
MG63 cells (22,23). However, the functional role of
autophagy in determining cell fate remains unclear. Autophagy may
serve as a cellular stress defense response, promoting cell
survival and delaying apoptosis, however, under specific
circumstances, autophagy can also serve as a specialized pathway
that promotes cell death (24). For
example, a previous study showed that complete inhibition of
autophagy with 3-methyladenine markedly enhanced curcumin-induced
apoptosis in osteosarcoma cells, suggesting that autophagy serves a
protective role under these conditions, potentially counteracting
apoptosis (25). Therefore, it is
currently hypothesized that autophagy may serve either a pro-death
or a companion (protective) role in different contexts, with
complex cross-regulation between autophagy and apoptotic signaling
(26). The concurrent increase in
apoptosis and autophagy-associated markers detected in the present
study suggests that RTA-408 induces apoptosis and is associated
with the accumulation of these markers. However, because autophagic
flux was not directly assessed using lysosomal inhibitors, these
findings do not distinguish between enhanced autophagosome
formation from impaired autophagosome degradation.
In the present study, phosphorylation of JNK, p38
and ERK was assessed at a single 72 h endpoint after RTA-408
treatment. At this measured time point, JNK showed the largest
increase in phosphorylation among the MAPK pathways examined,
whereas p38 changes were more modest and ERK phosphorylation was
increased mainly at the highest concentration. These findings
support the involvement of MAPK signaling, particularly JNK, in the
cellular response to RTA-408. However, because no time-course
analysis was performed, the activation kinetics of these pathways
cannot be determined from the present data.
To determine that the aforementioned cell death
effects were indeed dependent on JNK signaling, the pharmacologic
inhibitor SP600125 was used to block JNK activity. The results
showed that the addition of SP600125 significantly reversed
RTA-408-induced apoptosis and autophagy. Specifically, treatment
with SP600125 reduced annexin V positivity and PARP cleavage while
also inhibiting or abolishing the increase in autophagy markers
LC3B-II and beclin-1, demonstrating that both RTA-408-induced
apoptosis and autophagy are JNK-dependent (25). Similar evidence has been previously
reported. For example, the JNK-specific inhibitor SP600125
effectively blocked curcumin-induced autophagy in osteosarcoma
cells, demonstrating that the JNK pathway serves a key role in
curcumin-induced autophagy (23).
Furthermore, SP600125 has been shown to simultaneously inhibit
resveratrol-induced autophagy and apoptosis, further supporting the
idea that JNK is a common upstream regulator of both cell death
processes (27). These results
validate the mechanism proposed in the present study: RTA-408
simultaneously activates intracellular apoptosis and autophagy by
triggering the JNK pathway.
Previous research regarding RTA-408 and associated
triterpenoid derivatives (such as
2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid) has primarily
focused on the Nrf2 antioxidant and NF-κB anti-inflammatory
pathways (19). As an
antioxidant-inflammatory modulator, RTA-408 binds to KEAP1 and
stabilizes Nrf2 at low concentrations, inducing the expression of
downstream antioxidant genes [such as NAD(P)H quinone dehydrogenase
1 (NQO1) and heme oxygenase 1 (HO-1)], while inhibiting the
expression of pro-inflammatory mediators, including nitric oxide
synthase 2, prostaglandin-endoperoxide synthase 2, C-C motif
chemokine ligand 2 and C-C motif chemokine ligand 5 (19). By increasing Nrf2 activity, RTA-408
can reduce oxidative stress and inflammation in the tumor
microenvironment, thereby reversing tumor-induced immune evasion
and inhibiting tumor growth and metastasis (19). Previous studies have shown that
RTA-408 modulates several oncogenic signaling pathways at
antiproliferative concentrations. In a broad human tumor-cell
panel, RTA-408 inhibited NF-κB signaling in a manner consistent
with IKKβ inhibition, decreased cyclin D1 levels, increased
CDKN1A/p21 expression and enhanced JNK phosphorylation at
concentrations that inhibited tumor-cell growth and induced caspase
activity/apoptosis (19). More
recent studies further reported that RTA-408/omaveloxolone induced
G1 cell-cycle arrest, apoptosis and caspase-3 activation in
temozolomide-resistant glioblastoma cells as well as suppressed
glioblastoma growth in vitro and in vivo through
cell-cycle arrest associated with CDC20 downregulation (21,22).
In addition, JNK inhibition has been shown to reverse
RTA-408-mediated reductions in cell viability, apoptosis induction,
migration inhibition and radiosensitization in glioblastoma models,
supporting functional involvement of JNK signaling in another
cancer context (23).
The novelty of the present study lies in extending
the mechanistic understanding of RTA-408 in BC cells beyond its
previously described Nrf2-centered antioxidant/anti-inflammatory
functions and antitumor effects in other cancer models (15,19–23).
The present findings show that, in ER-positive/hormone
receptor-positive and triple-negative BC cell models, RTA-408
increases JNK phosphorylation and is associated with annexin
V-defined apoptosis, PARP cleavage and accumulation of
autophagy-related markers. These results suggest that
MAPK-JNK-associated pro-death signaling contributes to the cellular
response to RTA-408 in BC cells and may provide a basis for further
evaluating this compound in BC. However, since Nrf2 activity and
autophagic flux were not directly assessed, the relationship among
oxidative-stress modulation, JNK activation and
autophagy-associated marker accumulation remains to be clarified.
Despite this, a number of limitations in the present study should
be acknowledged. First, the present study was conducted exclusively
in vitro using two-dimensional monolayer cultures, which did
not recapitulate the tumor microenvironment, stromal and immune
interactions or the pharmacokinetic and metabolic constraints
present in vivo; therefore, the efficacy and safety of
RTA-408 cannot be directly extrapolated to the clinical setting.
Second, although SP600125 attenuated the effects of RTA-408, JNK
involvement was evaluated solely with this pharmacologic inhibitor.
In the absence of genetic approaches, such as siRNA/shRNA-mediated
knockdown or dominant-negative JNK constructs, definitive
mechanistic specificity cannot be established and off-target
effects remain possible. Third, Nrf2 signaling was not directly
assessed in the present study, as nuclear Nrf2 accumulation and
canonical downstream targets such as NQO1 and HO-1 were not
measured. Consequently, the association between Nrf2 activation and
JNK signaling could be determined from the present dataset, and the
possibility that RTA-408 exerts dose-dependent cytoprotective and
cytotoxic effects through distinct pathways remains to be
clarified. In addition, although RTA-408 increased beclin-1,
LC3B-II and p62 accumulation, autophagic flux was not directly
assessed using lysosomal inhibitors; therefore, the present data
cannot distinguish between increased autophagosome formation and
impaired autophagosome degradation. Fourth, non-malignant mammary
epithelial cells were not included; thus, tumor selectivity and
safety could not be inferred from the present study. In addition,
the development of drug resistance, the reversibility of growth
inhibition after prolonged exposure or the potential interaction of
RTA-408 with standard chemotherapeutic, endocrine or HER2-targeted
therapies were not evaluated. These issues represent important gaps
that warrant further mechanistic, translational and in vivo
investigation.
In conclusion, the present study found that RTA-408
exerted dose-dependent antiproliferative effects in both MCF-7 and
MDA-MB-231 BC cells. RTA-408 increased annexin V-defined apoptosis,
PARP cleavage and JNK phosphorylation, and was accompanied by the
accumulation of the autophagy-associated markers beclin-1, LC3B-II
and p62. Pharmacologic inhibition with SP600125 attenuated JNK
phosphorylation, reduced apoptotic responses and diminished
autophagy-related marker accumulation, supporting that JNK
signaling contributes, at least in part, to these effects. Given
the lack of direct autophagic flux analysis, genetic validation of
JNK and inclusion of normal mammary epithelial cell controls, these
findings should be interpreted cautiously and support further
mechanistic and in vivo evaluation of RTA-408 in BC
models.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from An-Nan Hospital,
China Medical University, Taiwan (grant nos. ANHRF114-17 and
ANHRF114-27).
Availability of data and materials
The datasets generated in the present study may be
requested from the corresponding author.
Authors' contributions
YJC and HPT conceived and designed the present
study. YJC performed the majority of the experiments and collected
the data. MYC, IHC, WCC, TTT and CYW assisted with data acquisition
and interpretation. HPT and JHL supervised the research and
analyzed the data. HPT and JHL confirm the authenticity of all the
raw data. YJC and HPT drafted the manuscript. JHL critically
revised the manuscript for important intellectual content. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript or to generate images, and subsequently,
the authors revised and edited the content produced by the
artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Arnold M, Morgan E, Rumgay H, Mafra A,
Singh D, Laversanne M, Vignat J, Gralow JR, Cardoso F, Siesling S
and Soerjomataram I: Current and future burden of breast cancer:
Global statistics for 2020 and 2040. Breast. 66:15–23. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao W, Chen HD, Yu YW, Li N and Chen WQ:
Changing profiles of cancer burden worldwide and in China: A
secondary analysis of the global cancer statistics 2020. Chin Med J
(Engl). 134:783–791. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grann VR, Troxel AB, Zojwalla NJ, Jacobson
JS, Hershman D and Neugut AI: Hormone receptor status and survival
in a population-based cohort of patients with breast carcinoma.
Cancer. 103:2241–2251. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Early Breast Cancer Trialists' and
Collaborative Group (EBCTCG), . Davies C, Godwin J, Gray R, Clarke
M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, et al: Relevance
of breast cancer hormone receptors and other factors to the
efficacy of adjuvant tamoxifen: patient-level meta-analysis of
randomised trials. Lancet. 378:771–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Daly B, Olopade OI, Hou N, Yao K,
Winchester DJ and Huo D: Evaluation of the quality of adjuvant
endocrine therapy delivery for breast cancer care in the United
States. JAMA Oncol. 3:928–935. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roberts K, Rickett K, Greer R and Woodward
N: Management of aromatase inhibitor induced musculoskeletal
symptoms in postmenopausal early breast cancer: A systematic review
and meta-analysis. Crit Rev Oncol Hematol. 111:66–80. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamirisa N and Hunt KK: Neoadjuvant
chemotherapy, endocrine therapy, and targeted therapy for breast
cancer: ASCO guideline. Ann Surg Oncol. 29:1489–1492. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan CWH, Tai D, Kwong S, Chow KM, Chan
DNS and Law BMH: The effects of pharmacological and
non-pharmacological interventions on symptom management and quality
of life among breast cancer survivors undergoing adjuvant endocrine
therapy: A systematic review. Int J Environ Res Public Health.
17:29502020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ross JS, Fletcher JA, Linette GP, Stec J,
Clark E, Ayers M, Symmans WF, Pusztai L and Bloom KJ: The Her-2/neu
gene and protein in breast cancer 2003: Biomarker and target of
therapy. Oncologist. 8:307–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Piccart-Gebhart MJ, Procter M,
Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga
J, Bell R, Jackisch C, et al: Trastuzumab after adjuvant
chemotherapy in HER2-positive breast cancer. N Engl J Med.
353:1659–1672. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lower EE, Glass E, Blau R and Harman S:
HER-2/neu expression in primary and metastatic breast cancer.
Breast Cancer Res Treat. 113:301–306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Slamon D, Eiermann W, Robert N, Pienkowski
T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, et
al: Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J
Med. 365:1273–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Creelan BC, Gabrilovich DI, Gray JE,
Williams CC, Tanvetyanon T, Haura EB, Weber JS, Gibney GT,
Markowitz J, Proksch JW, et al: Safety, pharmacokinetics, and
pharmacodynamics of oral omaveloxolone (RTA 408), a synthetic
triterpenoid, in a first-in-human trial of patients with advanced
solid tumors. Onco Targets Ther. 10:4239–4250. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu Y, Zhang Y, Ge L, He S, Zhang Y, Chen
D, Nie Y, Zhu M and Pang Q: RTA408 alleviates
lipopolysaccharide-induced acute lung injury via inhibiting
Bach1-mediated ferroptosis. Int Immunopharmacol. 142:1132502024.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lynch DR, Chin MP, Delatycki MB, Subramony
SH, Corti M, Hoyle JC, Boesch S, Nachbauer W, Mariotti C, Mathews
KD, et al: Safety and efficacy of omaveloxolone in friedreich
ataxia (MOXIe Study). Ann Neurol. 89:212–225. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dasgupta D, Tripathi A, Griffard-Smith R,
Yellapu N, Dhorajiya P and Pyaram K: Therapeutic potential of NRF2
activating drug RTA-408 in suppressing T cell effector responses
and inflammatory bowel disease. J Immunol. 214:1951–1968. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Probst BL, Trevino I, McCauley L,
Bumeister R, Dulubova I, Wigley WC and Ferguson DA: RTA 408, a
novel synthetic triterpenoid with broad anticancer and
anti-inflammatory activity. PLoS One. 10:e01229422015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang X, Liu T, Fei Y, Zhang S, Yang Y,
Chen Z, Zhu R, Deng S, Zhang T, Wu D and Xu Y: RTA-408 overcomes
cisplatin-resistant lung cancer by inhibiting WWP1-mediated NCOA4
ubiquitination to induce ferritinophagy and ferroptosis. Free Radic
Biol Med. 238:595–610. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee KT, Lieu AS, Lin CL, Hsu YC and Tsai
TH: Synthetic oleanolic acid derivative, RTA-408, overcome in
TMZ-resistant glioblastoma cells by inducing apoptosis and G1 cell
cycle arrest. Med Oncol. 42:3532025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee KT, Hsu YC, Lieu AS, Lin CL and Tsai
TH: Omaveloxolone suppresses cell growth and causes cell cycle
arrest by downregulating CDC20 expression in glioblastoma cells
both in vitro and in vivo. J Cell Mol Med. 29:e706072025.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsai HP, Qin H, Chong YB, Chen IH, Kuo SH,
Tseng TT and Lieu AS: RTA-408 enhances radiosensitivity and
inhibited tumor progression via JNK pathway in glioblastoma.
Kaohsiung J Med Sci. e701422025.(Epub ahead of print). PubMed/NCBI
|
|
24
|
Wong CH, Iskandar KB, Yadav SK, Hirpara
JL, Loh T and Pervaiz S: Simultaneous induction of non-canonical
autophagy and apoptosis in cancer cells by ROS-dependent ERK and
JNK activation. PLoS One. 5:e99962010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Chen P, Hong H, Wang L, Zhou Y
and Lang Y: JNK pathway mediates curcumin-induced apoptosis and
autophagy in osteosarcoma MG63 cells. Exp Ther Med. 14:593–599.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Ping J, Jiang N and Xu L:
Resveratrol inhibits hepatic stellate cell activation by regulating
autophagy and apoptosis through the SIRT1 and JNK signaling
pathways. J Food Biochem. 46:e144632022. View Article : Google Scholar : PubMed/NCBI
|