Introduction
Ovarian cancer is the most lethal gynecological
malignancy, characterized by extensive peritoneal metastasis and
the frequent development of chemoresistance, which necessitates the
exploration of novel therapeutic strategies (1). Amplification or upregulation of HER2
has been documented in a subset of ovarian cancer, particularly in
the aggressive subtypes lacking effective targeted therapies,
making it a compelling candidate for targeted intervention
(2). Despite the marked success of
chimeric antigen receptor (CAR)-T cell therapy in hematological
malignancies, its clinical application in solid tumors such as
ovarian cancer has been hampered by the immunosuppressive tumor
microenvironment (TME) and the physical barriers posed by the
fibrous stroma that restrict T-cell infiltration (3). Against this backdrop, novel cell
therapies based on CAR engineering platforms have been proposed and
are undergoing continuous optimization (4).
Developed based on the innate biological properties
of macrophages, CAR-macrophage (CAR-M) technology represents an
innovative approach in the field of immunotherapy (5–7). This
strategy involves genetically modifying macrophages to express
specific CARs, thereby enabling targeted recognition of
tumor-associated antigens and enhancing their phagocytic activity
(1). In contrast to the clinically
established CAR-T cell therapy, CAR-M therapy exhibits distinct
antitumor mechanisms: Engineered macrophages are capable of not
only phagocytosing and presenting antigens, but also potently
initiating immune cascade responses (8). This innovative methodology markedly
augments the capacity of the immune system to identify and
eliminate malignant cells, offering novel pathways for cancer
treatment. However, the antitumor efficacy of first-generation
CAR-Ms remains limited, and its sustained in vivo
performance lacks robust support from extensive preclinical and
clinical evidence. In light of these limitations, novel engineering
strategies are being explored to enhance the functionality and
potency of CAR-Ms (9).
During the development of CAR immune cell therapies,
such as CAR-T and CAR-natural killer (NK) cells, researchers have
demonstrated that co-expression of cytokines alongside the CAR
construct notably enhances therapeutic efficacy (10,11).
This strategy not only improves the biological functions and in
vivo persistence of CAR immune cells, but also concurrently
activates both innate and adaptive immune responses. It facilitates
the release of multiple inflammatory mediators and chemokines,
promotes immune cell infiltration into solid tumors, and enhances
tumor recognition and clearance (12).
In the context of CAR-M therapy, co-expression of
IFN-γ has been shown to augment phagocytic activity and promote a
pro-inflammatory polarization, thereby converting immunologically
‘cold’ tumors into ‘hot’ tumors and strengthening antitumor
responses (13). Moreover, key
regulatory genes have been identified that modulate these
processes. For example, aconitate decarboxylase 1 (ACOD1) in
macrophages acts as a crucial metabolic regulator involved in
inflammatory regulation. Wang et al (14) demonstrated that ACOD1 knockout
increased reactive oxygen species production, enhanced
phagocytosis, improved tumor-killing capacity and prolonged
survival in ovarian tumor-bearing mice. In addition, optimization
of CAR structure remains a central strategy for enhancing immune
cell function. In CAR-M research, including several products
currently undergoing clinical studies (such as CT-0508 and SY001),
the intracellular domain design based on CD3ζ is still employed.
This signaling molecule, originally applied in CAR-T cell therapy,
can still function in macrophages by inducing spleen associated
tyrosine kinase activation. However, to fully realize the
therapeutic potential of CAR-Ms, it is essential to move beyond the
constraints of conventional T cell signaling and integrate
macrophage-specific intracellular domains to achieve optimal
efficacy. For example, tandem Toll/interleukin-1
receptor/resistance protein and CD3ζ domains have been shown to
induce potent phagocytosis and inflammatory polarization (15). Beyond direct CAR modification and
genetic regulation, combination strategies integrating cell
therapies with immune checkpoint inhibitors (ICIs) represent
another highly promising approach (16).
The combination of ICIs with cell-based therapies
has been increasingly validated in clinical practice, markedly
improving the durability and efficacy of cellular treatments. In
CAR-T cell therapy for hematological malignancies, the
incorporation of ICIs [programmed cell death protein 1 (PD-1) or
programmed death ligand 1 (PD-L1) monoclonal antibodies (mAbs] has
demonstrated notable benefits, including higher response rates and
manageable toxicity profiles, supported by robust data from
multiple clinical trials (17,18).
By contrast, clinical outcomes in solid tumors remain modest
despite ongoing investigations, with some studies reporting
conflicting results (19,20). While CAR-M therapy has emerged as a
promising modality for solid tumors, its combination with ICIs,
particularly those targeting phagocytosis checkpoints, has been
underexplored. Due to the dependence of CAR-Ms on phagocytic
function and the limited antitumor activity of first-generation
constructs, blockade of phagocytosis checkpoints may represent a
viable strategy to enhance CAR-M efficacy. The present study
focused on evaluating the combined use of a CD47 mAb and CAR-M
therapy, with the aim of developing a novel augmentation strategy
based on phagocytosis checkpoint inhibition.
Materials and methods
Cells and animals
The cell lines and their culture media used in the
present study, namely SKOV3, A2780 and THP-1, were obtained from
Procell Life Science & Technology Co., Ltd. THP-1 and A2780
cells were cultured in RPMI-1640 medium (cat. no. 01-100-1ACS;
Shanghai Basalmedia Technologies Co., Ltd.) supplemented with 10%
fetal bovine serum (FBS; cat. no. 087-150; Wisent Biotechnology),
whereas SKOV3 cells were maintained in McCoy's 5A medium (cat. no.
L630KJ; Shanghai Basalmedia Technologies Co., Ltd.) containing 10%
FBS. All cell lines were incubated at 37°C under a humidified
atmosphere containing 5% CO2. The identity of each cell
line was confirmed by short tandem repeat profiling, and regular
testing was conducted to ensure they were free from mycoplasma
contamination.
Wild-type female BALB/c nude mice (age, 6–8 weeks)
were acquired from GemPharmatech Co., Ltd. and housed under
specific pathogen-free (SPF) conditions. The animals were provided
with SPF-grade water and diet, and maintained under a 12-h
light/dark cycle. All experimental procedures were approved by the
Animal Ethics Committee of Anhui Medical University (Hefei, China;
approval no. PZ-2025-029).
In vivo tumor model
To establish a xenograft tumor model, tumor cells in
the logarithmic growth phase were harvested, resuspended in
pre-cooled PBS and inoculated subcutaneously into nude mice.
Specifically, 1×106 SKOV3 cells were injected into the
subcutaneous space of the flank region. A total of 4 h after
inoculation, 2×106 anti-HER2 CAR-Ms or empty
vector-transduced macrophages were administered via subcutaneous
injection. Once the average tumor volume reached 50 mm3,
the mice treated with anti-HER2 CAR-Ms were randomly divided into
two groups, one of which received an intravenous injection of CD47
mAb (cat. no. E16HU047; EnoGene Biotech Co, Ltd.) via the tail
vein. Tumor growth was monitored regularly, with measurements taken
every 3 days. When the maximum tumor volume approached 1,500
mm3, euthanasia was performed on the tumor-bearing mice
using inhalational anesthetics. For euthanasia, animals were deeply
anesthetized with 5% isoflurane for 15 min in a closed chamber
until respiration ceased completely. Death was confirmed by absence
of spontaneous breathing for >5 min and absence of heartbeat. No
additional drugs were used.
CAR-M production
The CAR construct was cloned into a
replication-deficient type 5 adenoviral vector backbone under the
control of a CMV promoter, and high-titer adenovirus was
subsequently packaged by OBiO Technology (Shanghai) Corp., Ltd.
Anti-HER2 CAR-Ms and vector control cells were generated by
transducing THP-1-derived macrophages with the adenovirus,
according to the manufacturer's instructions. Transduced cells were
digested with Accutase (cat. no. A6964-100ML; Sigma-Aldrich; Merck
KGaA) cell dissociation solution and collected. Subsequently, the
expression levels of green fluorescent protein (GFP) in the cells
were analyzed using flow cytometry to determine CAR expression.
Flow cytometry
After collection, the cells were resuspended in PBS
and labeled with corresponding flow cytometry antibodies. The
mixture was incubated at 4°C for 30 min, followed by washing and
processing on a flow cytometer. For intracellular proteins
requiring fixation and permeabilization, the cells were first
incubated with fixation buffer (cat. no. 00-5223-57; Thermo Fisher
Scientific, Inc.) at 20°C for 10–20 min, followed by
permeabilization buffer (cat. no. 00-5223-57; Thermo Fisher
Scientific, Inc.) at 20°C for 10–15 min before antibody labeling.
The antibodies used included anti-HER2 (cat. no. MA5-60199;
Invitrogen; Thermo Fisher Scientific, Inc.), anti-CD86 (cat. no.
374207; BioLegend, Inc.), anti-CD206 (cat. no. 321125; BioLegend,
Inc.) and anti-Ki-67 (cat. no. 350514; BioLegend, Inc.). The
experiment was conducted using a flow cytometer (CytoFLEX; Beckman
Coulter, Inc.) and the results were analyzed with CytExpert
software (version 2.6.0.105; Beckman Coulter, Inc.).
Analysis of phagocytosis by flow
cytometry
Briefly, 1 µl CMPTX dye (cat. no. C34552;
Invitrogen; Thermo Fisher Scientific, Inc.) was added to a
1-ml/1×106 SKOV3 or A2780 cell suspension and incubated
in a cell culture incubator at 37°C for 20–30 min, after which, the
cells were centrifuged at 1,500 rpm (125.7 × g) for 5 min at 4°C,
the supernatant was discarded and the cells were washed with PBS to
remove residual dye. THP-1M cells or anti-HER2 CAR-Ms, each
expressing GFP, were co-cultured with tumor cells (SKOV3 or A2780)
labeled with CMPTX (detected via the ECD channel) dye at a 1:1
ratio (1×105 cells each) in a 37°C incubator for 4 h.
After co-culture, all cells were collected, washed with PBS and
subjected to flow cytometry. None of the macrophages received prior
M1 stimulation. The percentage of CMPTX+ cells within
the GFP+ macrophage population was used to quantify
phagocytosis of target cells by macrophages. Flow cytometry was
performed on a CytoFLEX instrument (CytoFLEX; Beckman Coulter,
Inc.) and the data were analyzed using CytExpert software.
Analysis of the effects of CD47 mAb on
the phagocytic and cytotoxic activities of CAR-Ms
Co-culture systems were established in three
experimental groups: Empty control + SKOV3; anti-HER2 CAR-M +
SKOV3; and anti-HER2 CAR-M + SKOV3 + CD47 mAb. For the phagocytosis
assay, 1×105 GFP+ THP-1Ms or anti-HER2 CAR-Ms
were co-cultured with 1×105 CMPTX-labeled SKOV3 cells
(pretreated with CD47 mAb) at 37°C for 4 h. Following co-culture,
all cells were collected, washed with PBS and subjected to flow
cytometric analysis. Please note that none of the macrophages
received prior M1 stimulation. The percentage of CMPTX+
cells within the GFP+ macrophage population was
quantified to evaluate phagocytic activity.
In the cytotoxicity assay, unlabeled tumor cells
were co-cultured with GFP+ CAR-Ms for 24 h. The residual
tumor cells were then identified as the GFP− cell
population and normalized to the control group. All flow cytometry
data were acquired using a CytoFLEX flow cytometer and analyzed
with CytExpert software.
ELISA
The concentrations of IFN-γ (cat. no. ELH-IFNg;
RayBio, Inc.), TNF-α (cat. no. ELH-TNFα; RayBio, Inc.), IL-6 (cat.
no. KIT10395A; Sino Biological, Inc.) and IL-1β (cat. no. EL-H0149;
Wuhan Elabscience Biotechnology Co., Ltd.) in the supernatant of
the co-culture system were detected by ELISA kits, according to the
manufacturer's instructions. The results were analyzed using a
microplate reader (Infinite M1000 Pro; Tecan Group, Ltd.).
H&E staining and
immunohistochemistry
Immunohistochemical experiments were performed
according to standard protocols (21), and antigen retrieval was performed
as per the requirements of the primary antibodies, including
proliferating cell nuclear antigen (PCNA; cat. no. ab92552; Abcam)
and α-smooth muscle actin (SMA; cat. no. ab5694; Abcam). Briefly,
isolated tumor tissues were fixed with 4% paraformaldehyde (cat.
no. BL539A; Biosharp Life Sciences) at room temperature for 24 h,
then embedded in paraffin and cut into 5-µm sections. After
dewaxing, the sections were subjected to antigen retrieval by
heating in citrate buffer (pH 6.0) at 100°C for 15 min, followed by
rehydration with 0.5% Triton X-100 (cat. no. X100-100ML;
MilliporeSigma), followed by blocking with 1.5% BSA (from a
two-step detection kit; cat. no. PV-9000; OriGene Technologies,
Inc.) at 37°C for 30 min. The primary antibody was diluted at 1:200
and incubated with the sections overnight at 4°C. Subsequently, a
horseradish peroxidase-conjugated secondary antibody (from a
two-step detection kit; cat. no. PV-9000; OriGene Technologies,
Inc.) was diluted at 1:500 and incubated with the sections at 37°C
for 30 min, and staining was developed with DAB (cat. no. BB-3503;
BestBio, Inc.). Images of the sections were then captured using a
brightfield microscope (Leica Microsystems, Inc.).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 8.0 (Dotmatics). Data normality and homogeneity of variances
were assessed using the Shapiro-Wilk test and Levene's test,
respectively. For comparisons between two independent groups, an
unpaired two-tailed Student's t-test was applied. For comparisons
among three or more groups, one-way analysis of variance followed
by Tukey's post hoc test for multiple comparisons was used. All
experiments were performed independently, at least in triplicate,
and data are presented as the mean ± standard error. P<0.05 was
considered to indicate a statistically significant difference.
Results
Anti-HER2 CAR-Ms can specifically
target HER2-positive ovarian cancer cells
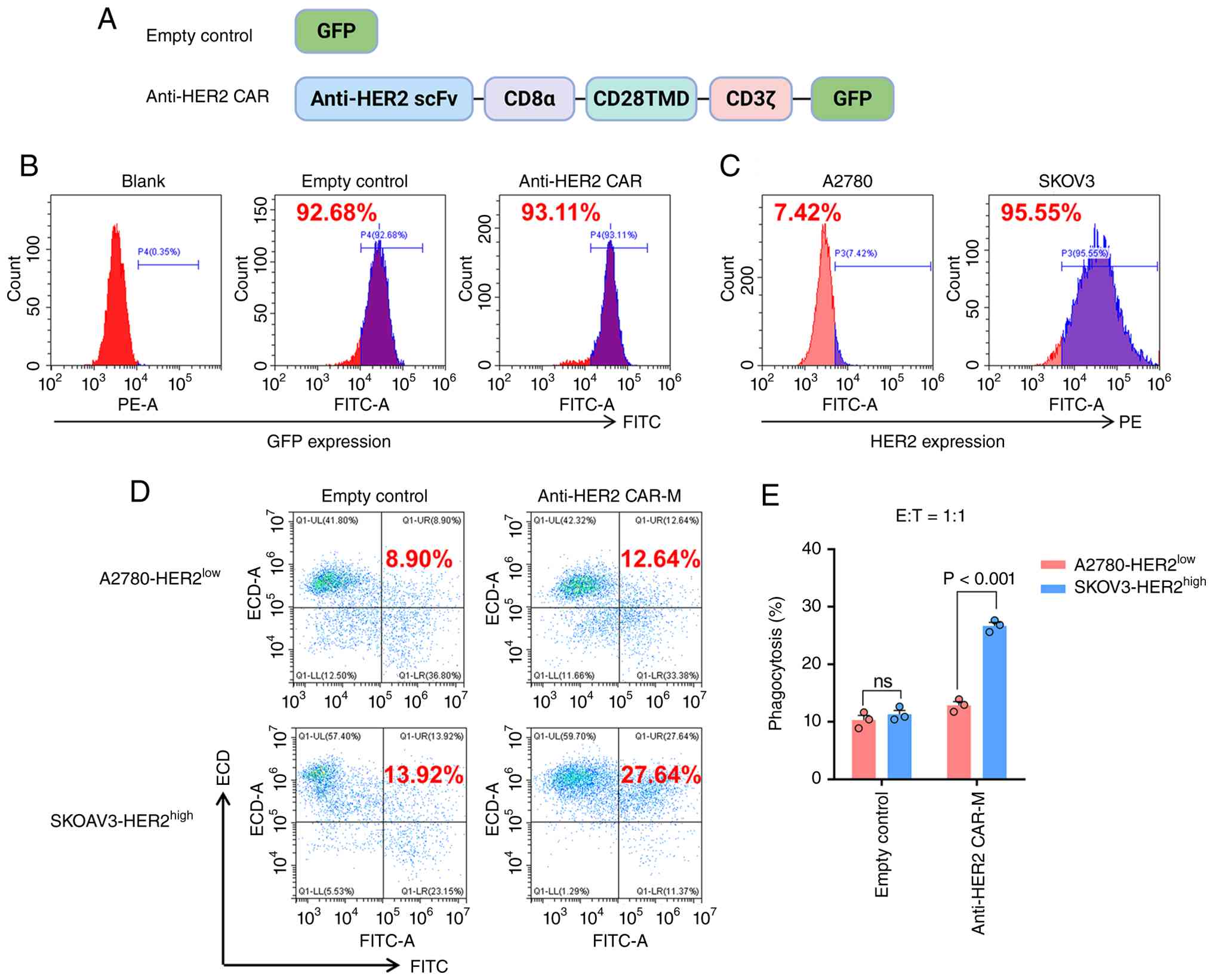
An anti-HER2 CAR was constructed (Fig. 1A), in which the extracellular
antigen-binding domain consisted of an anti-HER2 single-chain
variable fragment (scFv) that specifically recognizes HER2. CD8α
was utilized as a hinge region to connect the antigen-binding
domain to the transmembrane domain derived from CD28. The
intracellular signaling domain was composed of CD3ζ, which contains
multiple immunoreceptor tyrosine-based activation motifs
responsible for activating macrophage phagocytosis. Additionally,
GFP was incorporated as a reporter gene to monitor CAR expression
and subcellular localization. Control macrophages were transduced
with an empty vector.
The GFP tag was used to indicate the transduction of
the CAR molecule. Flow cytometric analysis demonstrated high GFP
expression in both the control and anti-HER2 CAR-Ms (Figs. 1B and S1). To evaluate the target specificity of
anti-HER2 CAR-Ms, two ovarian cancer cell lines were selected:
SKOV3, which exhibits high HER2 expression; and A2780, with low
HER2 expression, as confirmed by flow cytometry (Fig. 1C). Upon co-culture with CAR-Ms,
significant phagocytosis of SKOV3 cells was observed, whereas only
baseline phagocytic activity was detected against A2780 cells
(Fig. 1D and E). These results
indicate that anti-HER2 CAR-Ms were successfully constructed and
can specifically target and phagocytose HER2-positive ovarian
cancer cells.
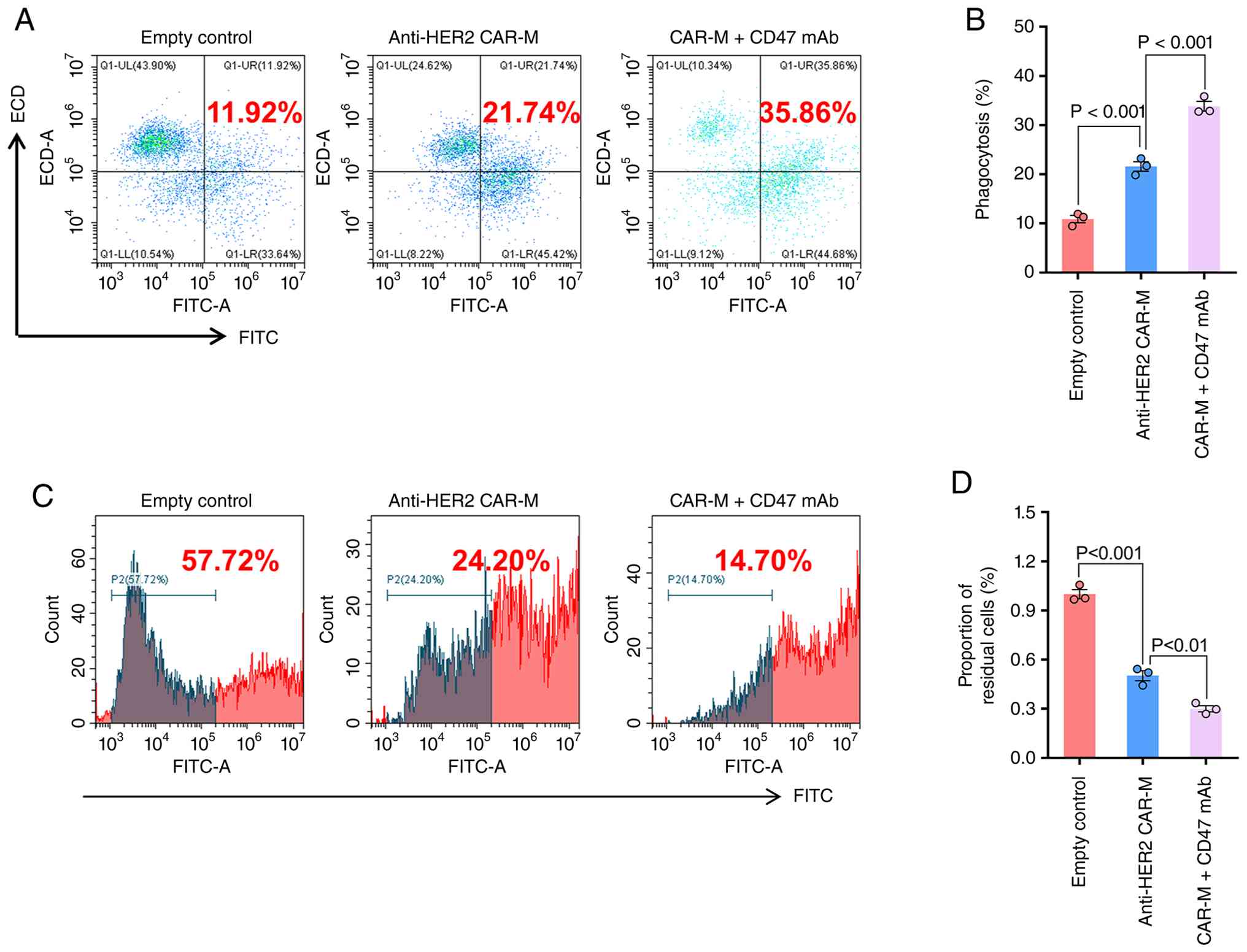
CD47 mAb enhances the anti-ovarian
cancer activity of anti-HER2 CAR-Ms in vitro
To investigate the potential synergistic effect
between CD47 mAb and CAR-Ms, anti-HER2 CAR-Ms were co-cultured with
tumor cells in the presence or absence of CD47 mAb. Given that the
targeting specificity of the CAR molecule has been validated by
co-culture experiments, here co-culture was performed only with
HER2-positive SKOV3 cells. Flow cytometric analysis revealed that
the presence of the CD47 mAb enhanced the phagocytic ability of
anti-HER2 CAR-Ms against SKOV3 cells (Figs. 2A and B, S1). Furthermore, the effect of CD47 mAb
was evaluated on the cytotoxic function of anti-HER2 CAR-Ms. The
results demonstrated that CD47 mAb potentiated the elimination of
HER2-positive ovarian cancer cells by anti-HER2 CAR-Ms (Fig. 2C and D). Collectively, these
findings indicated that CD47 mAb significantly augments the
antitumor activity of anti-HER2 CAR-Ms against target
antigen-positive ovarian cancer cells.
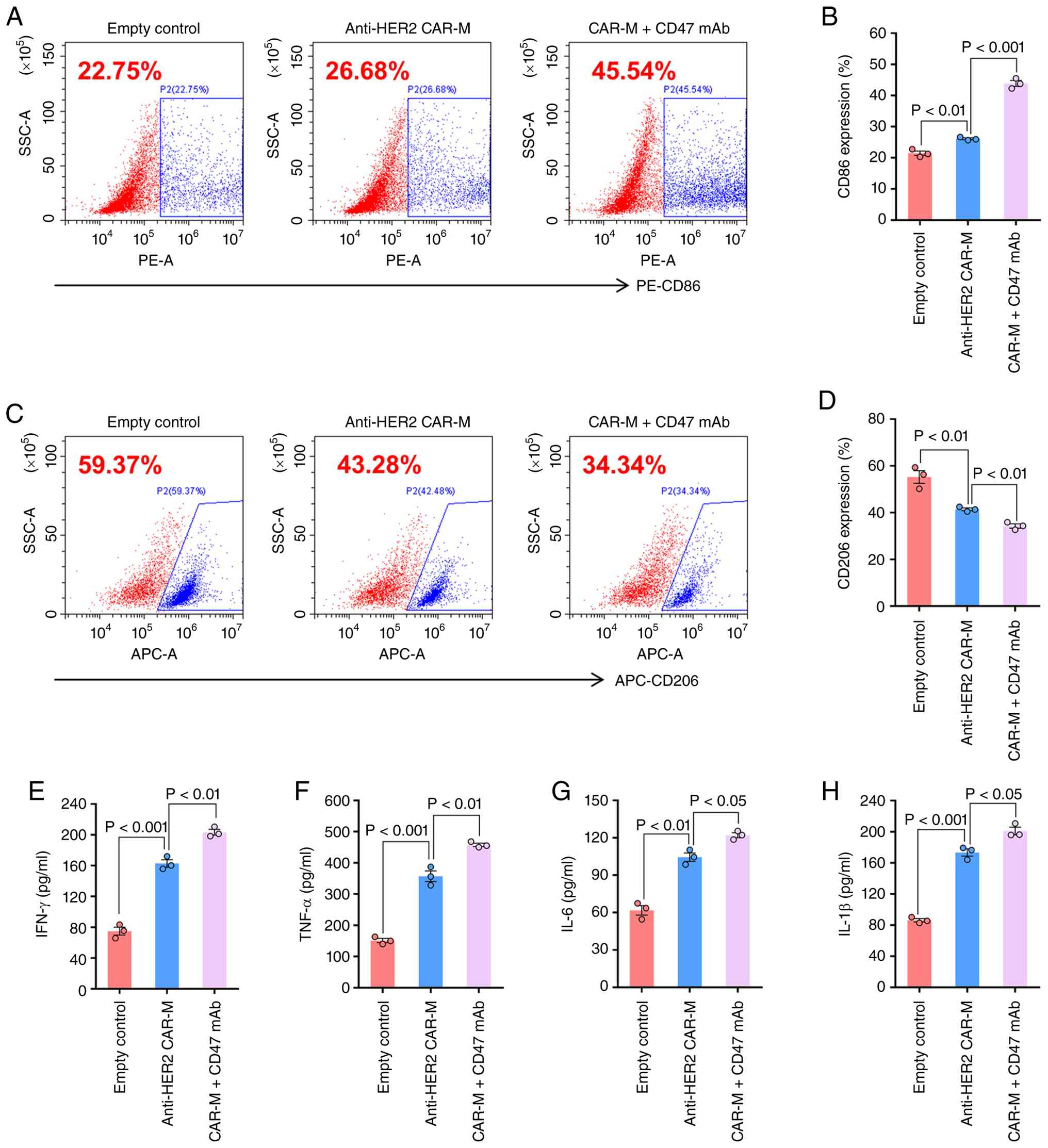
CD47 mAb enhances the pro-inflammatory
polarization of anti-HER2 CAR-Ms in vitro
Beyond their phagocytic capability, the inflammatory
polarization of CAR-Ms represents a key mechanism through which
they counteract the immunosuppressive TME of solid malignancies and
is regarded as a critical determinant of their antitumor efficacy
(22). Using flow cytometry, the
expression levels of the surface markers CD206 and CD86 were
evaluated on macrophages following co-culture with SKOV3 cells. The
results indicated that, relative to the empty vector control group,
CAR-Ms exhibited upregulation of CD86 and downregulation of CD206
(Figs. 3A-D, S1). This shift toward a pro-inflammatory
phenotype was further enhanced in the presence of a CD47 mAb.
Subsequent analysis of inflammatory cytokines in the co-culture
supernatant revealed increased secretion of IFN-γ, TNF-α, IL-6 and
IL-1β compared with in CAR-Ms alone (Fig. 3E-H), supporting the conclusion that
CD47 mAb augments CAR-M-mediated inflammatory activation.
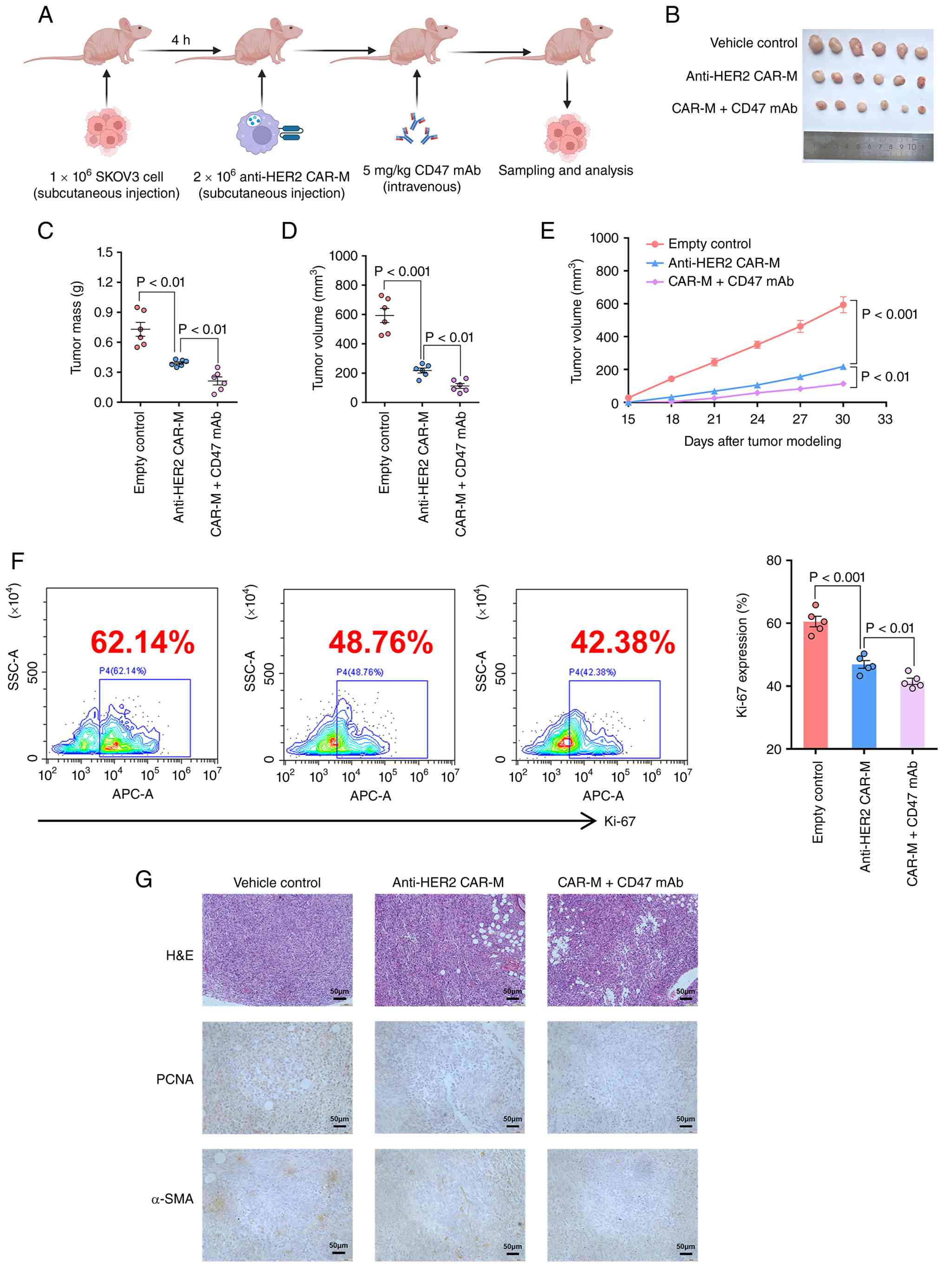
CD47 mAb enhances the antitumor
activity of CAR-Ms in a subcutaneous tumor model in vivo
To further evaluate whether CD47 mAb augments the
antitumor efficacy of CAR-Ms in vivo, a subcutaneous tumor
model was established and CAR-Ms were administered in combination
with CD47 mAb. A total of 4 h after subcutaneous implantation of
SKOV3 cells, a single dose of macrophages was injected
subcutaneously, followed by intravenous injection of CD47 mAb
(Fig. 4A). The results demonstrated
that anti-HER2 CAR-Ms significantly suppressed ovarian tumor growth
compared with that in the control group, and this inhibition was
further enhanced by the addition of CD47 mAb (Fig. 4B-E). Moreover, flow cytometric
analysis revealed that the combination treatment group exhibited
the lowest levels of Ki-67 expression in the tumor tissues among
all groups (Fig. 4F). Similarly,
immunohistochemical staining showed comparable trends for PCNA and
α-SMA (Fig. 4G), indicating a
synergistic antitumor effect between CD47 mAb and CAR-Ms in
suppressing ovarian cancer progression.
Discussion
HER2 is a proto-oncogene that exhibits amplification
or upregulation in various solid tumors, including ovarian, breast,
gastric and lung cancer (23–25),
making it a valuable prognostic and predictive biomarker (26). In recent decades, HER2 has emerged
as a key target for drug development, involving therapeutic
modalities such as tyrosine kinase inhibitors, antibody-drug
conjugates, bispecific antibodies and cell therapy (27,28).
However, successful treatments remain limited, with trastuzumab
being the first approved first-line targeted therapy for cancer,
demonstrating favorable efficacy in the treatment of advanced-stage
malignancies (29).
CAR-Ms represent an emerging therapeutic modality
within the field of adoptive cell therapy. Leveraging the
established CAR technology platform, this approach provides a novel
strategy to selectively activate macrophages for targeting a broad
spectrum of malignancies. Furthermore, CAR-M therapy demonstrates
distinct advantages over other CAR immune therapies, particularly
in the context of solid tumor treatment (30,31).
One rationale for developing macrophages as a novel cellular
therapy is their inherent tumor-homing capacity. Unlike
hematological malignancies, which are characterized by fluid
dispersion, solid tumors present dense physical barriers composed
of fibroblasts and stromal matrix that restrict immune cell
infiltration. This limitation often leads to the failure of T cell
or NK cell therapies (3). By
contrast, macrophages efficiently penetrate solid tumor tissues,
enabling CAR-Ms to directly engage with and eliminate malignant
cells. The antitumor mechanisms of CAR-M platforms involve
phagocytosis and subsequent downstream effects, such as potent
activation of adaptive immunity and macrophage-specific cytokine
secretion. A previous study has demonstrated that CAR-Ms can
secrete TNF-α to induce tumor cell apoptosis, and enhance the
infiltration of CD8+ T cells and NK cells (15).
An initial Phase I clinical trial of a CAR-M therapy
(NCT04660929) has reported safety and clinical efficacy outcomes.
Among 14 treated patients, no dose-limiting toxicities, severe
cytokine release syndrome (CRS) or immune effector cell-associated
neurotoxicity syndrome were observed. Some patients developed grade
1–2 CRS, which resolved within 3 days. Sequential tumor biopsies
revealed that adoptively transferred anti-HER2 CAR-Ms migrated into
the TME, prompting its remodeling, enhancing CD8+ T-cell
expansion and boosting broad antitumor immunity. To the best of our
knowledge, these data provide the first clinical evidence
supporting the efficacy, safety and tolerability of CAR-M therapy
(3). Nevertheless, the long-term
effects of CAR-Ms remain unassessed. Strategies to improve in
vivo persistence and antitumor activity include further
engineering and combination therapies, such as with PD-1/PD-L1
inhibitors. Due to the dependence of CAR-Ms on phagocytic function,
targeting phagocytosis checkpoints may represent a more effective
and suitable approach (32). For
example, Zeller et al (33)
demonstrated that dual blockade of CD47 and LILRB1 enhances CD20
antibody-dependent phagocytosis of lymphoma cells by macrophages.
Furthermore, Li et al (34)
revealed that targeting the CD200R1-CD200 axis represents a
promising macrophage immune checkpoint blockade strategy, where
blocking or eliminating CD200R1 in macrophages or CD200 in tumor
cells enhances phagocytosis and suppresses tumor growth.
Another pivotal issue is whether CAR-Ms can elicit
sustained antitumor responses, which hinges upon their controlled
elimination and interactions within the TME. Available evidence has
indicated that adoptively transferred macrophages exhibit tropism
in vivo (35). The majority
of infused CAR-Ms accumulate in the liver, with a smaller
proportion infiltrating tumor tissues (36). Current investigations have focused
primarily on the persistence of CAR-Ms within tumors. In animal
models, these cells demonstrate extended survival; however, their
numbers diminish rapidly within the first few days
post-administration. This decline suggests that non-proliferative
CAR-Ms undergo regulated elimination in vivo, thereby
supporting long-term safety profiles (7).
A major challenge in CAR-M therapy lies in the
immunosuppressive nature of the solid TME. Circulating monocytes
and macrophages recruited into tumor sites are often educated by
immunosuppressive signals to adopt a pro-tumor M2 phenotype
(37). Although CAR-M activation
promotes an M1-like phenotype, the sustainability of this
polarization remains unclear. A previous study employed engineered
adenoviruses or exogenous cytokine stimulation to induce M1
polarization, while others propose that first-generation CAR
constructs alone may suffice (38).
Nevertheless, the durability of these interventions remains
compromised by the immunosuppressive tumor milieu. Theoretically,
in addition to enhancing the phagocytic activity of CAR-Ms and
lowering their activation threshold, blocking phagocytosis
checkpoints can reprogram tumor-associated macrophages (TAMs). This
reprogramming not only eliminates the functional impairments that
TAMs impose on CAR-M therapy but also converts this major
immunosuppressive cell population into antitumor effector cells.
Through this dual-pronged mechanism, a sustained innate immune
response can be established by alleviating immunosuppression while
enhancing cytotoxic activity (39).
Nevertheless, further studies are required to determine whether
phagocytosis checkpoint blockade can consistently achieve
CAR-M-mediated reprogramming of TAMs within the complex TME, and
whether the potential safety risks associated with excessive
macrophage activation are controllable (40).
The present study demonstrated that blockade of CD47
using a mAb enhanced CAR-M-mediated phagocytosis and tumor cell
killing while concurrently promoting inflammatory polarization.
Mechanistically, CD47 functions as a ‘don't eat me’ signal by
engaging signal regulatory protein α (SIRPα) on the macrophage
surface, which recruits SHP-1 and SHP-2 phosphatases to the
intracellular domain of SIRPα, thereby inhibiting phagocytic
synapse formation and suppressing cytoskeletal rearrangement.
Blockade of CD47 with a monoclonal antibody disrupts this
CD47-SIRPα interaction, relieving the inhibitory constraint on
macrophage phagocytosis (41). In
the context of CAR-M therapy, this disinhibition likely lowers the
threshold for CAR-mediated activation, permitting more robust
ITAM-dependent signaling through the CD3ζ intracellular domain and
consequently enhancing both phagocytic and cytotoxic effector
functions. Furthermore, the enhanced phagocytosis of tumor cells
may provide additional pro-inflammatory stimuli through the
exposure of damage-associated molecular patterns, thereby promoting
the M1-like polarization and increased secretion of inflammatory
cytokines observed in the current study. These mechanistic insights
support the synergistic potential of combining phagocytosis
checkpoint inhibitors with CAR-based macrophage therapy.
Several limitations of the present study should be
acknowledged. First, the in vivo experiments were conducted
in immunodeficient BALB/c nude mice, which lack functional T cells
and NK cells. This model does not recapitulate the intact immune
system and therefore precludes assessment of the secondary effects
of CD47 blockade on adaptive immune activation, such as antigen
cross-presentation by CAR-Ms and subsequent T-cell priming
(42). Future studies employing
syngeneic tumor models or humanized immune system mice are
warranted to fully evaluate the immunomodulatory consequences of
combining CAR-Ms with CD47 mAb. In addition, only a subcutaneous
xenograft model was used, which does not reflect the peritoneal
dissemination pattern typical of clinical ovarian cancer.
Orthotopic or patient-derived xenograft models would provide more
clinically relevant insights. Moreover, the CAR-Ms were derived
from the THP-1 cell line, and it remains to be determined whether
primary human monocytes or induced pluripotent stem cell-derived
macrophages with a similar engineering strategy could achieve
comparable efficacy and safety profiles. The long-term persistence,
biodistribution and eventual fate of adoptively transferred CAR-Ms
with or without CD47 mAb were not investigated. Longitudinal
imaging and terminal tissue analysis are needed to assess these
pharmacokinetic parameters. Finally, only a single dose of CD47 mAb
was administered; optimization of dosing schedule, duration of
treatment and potential biomarkers of response requires further
exploration. Addressing these limitations through comprehensive
preclinical studies will be essential to advance this combination
strategy toward clinical translation.
In summary, to the best of our knowledge, the
present study was the first to evaluate the combination of CAR-M
therapy with phagocytic checkpoint inhibitors in vitro and
in vivo. The results demonstrated that an anti-CD47 mAb
enhanced the antitumor efficacy of CAR-Ms by promoting phagocytosis
and modulating their inflammatory activation. Given that CAR-M
therapy elicits broad innate and adaptive immune responses,
combining CAR-Ms with other types of ICIs represents a promising
strategy (43). In the future,
next-generation CAR-M designs may incorporate intracellular CD47
scFv domains, thereby integrating phagocytic checkpoint blockade
directly into the CAR-M construct (44). Such an approach could facilitate
localized inhibitor delivery via tumor-infiltrating CAR-Ms or
through in vivo CAR-M programming, potentially mitigating
the systemic toxicity associated with checkpoint inhibition
(45).
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by the Open Fund of Key
Laboratory of Anti-inflammatory and Immune Medicine, Ministry of
Education (grant no. KFJJ-2024-05).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JQia, QK and JQiu designed the study. JQia and QK
performed the experiments. JQia drafted the manuscript. QK and JQiu
revised the manuscript. JQia and JQiu confirm the authenticity of
all the raw data. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Animal Ethics Committee of Anhui Medical University (approval no.
PZ-2025-029).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CAR
|
chimeric antigen receptor
|
|
CAR-M
|
chimeric antigen receptor
macrophage
|
|
CRS
|
cytokine release syndrome
|
|
FBS
|
fetal bovine serum
|
|
ICIs
|
immune checkpoint inhibitors
|
|
monoclonal antibody
|
mAb
|
|
scFv
|
single-chain variable fragment
|
|
TME
|
tumor microenvironment
|
References
|
1
|
Caruso G, Weroha SJ and Cliby W: Ovarian
cancer: A review. JAMA. 334:1278–1291. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meric-Bernstam F, Makker V, Oaknin A, Oh
DY, Banerjee S, González-Martín A, Jung KH, Ługowska I, Manso L,
Manzano A, et al: Efficacy and safety of trastuzumab deruxtecan in
patients with HER2-Expressing solid tumors: Primary results from
the DESTINY-PanTumor02 phase II trial. J Clin Oncol. 42:47–58.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xin Q, Chen Y, Sun X, Li R, Wu Y and Huang
X: CAR-T therapy for ovarian cancer: Recent advances and future
directions. Biochem Pharmacol. 226:1163492024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, Zhou L, Chen X, Wang S, Chen W, Li
Z, Qiu J, Li R, Tu J and Lin N: Optimizing next-generation
CAR-macrophages against solid tumors: Challenges and potential
strategies. J Hematol Oncol. 10:302026. View Article : Google Scholar
|
|
5
|
Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang
Y, Han D, Hong W, Wei W and Tu J: CAR-macrophage: A new
immunotherapy candidate against solid tumors. Biomed Pharmacother.
139:1116052021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Y, Xin Q, Zhu M, Qiu J, Luo Y, Li R,
Wei W and Tu J: Exploring CAR-macrophages in non-tumor diseases:
Therapeutic potential beyond cancer. J Adv Res. 77:481–496. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reiss KA, Angelos MG, Dees EC, Yuan Y,
Ueno NT, Pohlmann PR, Johnson ML, Chao J, Shestova O, Serody JS, et
al: CAR-macrophage therapy for HER2-overexpressing advanced solid
tumors: A phase 1 trial. Nat Med. 31:1171–1182. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Hu R, Xie X and Li Y: Expanding
the frontier of CAR therapy: Comparative insights into CAR-T,
CAR-NK, CAR-M, and CAR-DC approaches. Ann Hematol. 104:4305–4317.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li N, Geng S, Dong ZZ, Jin Y, Ying H, Li
HW and Shi L: A new era of cancer immunotherapy: Combining
revolutionary technologies for enhanced CAR-M therapy. Mol Cancer.
23:1172024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steffin D, Ghatwai N, Montalbano A, Rathi
P, Courtney AN, Arnett AB, Fleurence J, Sweidan R, Wang T, Zhang H,
et al: Interleukin-15-armoured GPC3 CAR T cells for patients with
solid cancers. Nature. 637:940–946. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van den Eynde A, Gehrcken L, Verhezen T,
Lau HW, Hermans C, Lambrechts H, Flieswasser T, Quatannens D, Roex
G, Zwaenepoel K, et al: IL-15-secreting CAR natural killer cells
directed toward the pan-cancer target CD70 eliminate both cancer
cells and cancer-associated fibroblasts. J Hematol Oncol. 17:82024.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang L, Pan S, Wei X, Xu X and Wei Q:
Arming CAR-T cells with cytokines and more: Innovations in the
fourth-generation CAR-T development. Mol Ther. 31:3146–3162. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kang M, Lee SH, Kwon M, Byun J, Kim D, Kim
C, Koo S, Kwon SP, Moon S, Jung M, et al: Nanocomplex-mediated in
vivo programming to chimeric antigen Receptor-M1 macrophages for
cancer therapy. Adv Mater. 33:e21032582021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Su S, Zhu Y, Cheng X, Cheng C,
Chen L, Lei A, Zhang L, Xu Y, Ye D, et al: Metabolic Reprogramming
via ACOD1 depletion enhances function of human induced pluripotent
stem cell-derived CAR-macrophages in solid tumors. Nat Commun.
14:57782023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lei A, Yu H, Lu S, Lu H, Ding X, Tan T,
Zhang H, Zhu M, Tian L, Wang X, et al: A second-generation
M1-polarized CAR macrophage with antitumor efficacy. Nat Immunol.
25:102–116. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uslu U, Castelli S and June CH: CAR T cell
combination therapies to treat cancer. Cancer Cell. 42:1319–1325.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chong EA, Alanio C, Svoboda J, Nasta SD,
Landsburg DJ, Lacey SF, Ruella M, Bhattacharyya S, Wherry EJ and
Schuster SJ: Pembrolizumab for B-cell lymphomas relapsing after or
refractory to CD19-directed CAR T-cell therapy. Blood.
139:1026–1038. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hirayama AV, Kimble EL, Wright JH,
Fiorenza S, Gauthier J, Voutsinas JM, Wu Q, Yeung CCS, Gazeau N,
Pender BS, et al: Timing of anti-PD-L1 antibody initiation affects
efficacy/toxicity of CD19 CAR T-cell therapy for large B-cell
lymphoma. Blood Adv. 8:453–467. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bagley SJ, Binder ZA, Lamrani L, Marinari
E, Desai AS, Nasrallah MP, Maloney E, Brem S, Lustig RA, Kurtz G,
et al: Repeated peripheral infusions of anti-EGFRvIII CAR T cells
in combination with pembrolizumab show no efficacy in glioblastoma:
A phase 1 trial. Nat Cancer. 5:517–531. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi C, Liu C, Gong J, Liu D, Wang X, Zhang
P, Qin Y, Ge S, Zhang M, Peng Z, et al: Claudin18.2-specific CAR T
cells in gastrointestinal cancers: Phase 1 trial final results. Nat
Med. 30:2224–2234. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ramos-Vara JA: Principles and methods of
immunohistochemistry. Methods Mol Biol. 1641:115–128. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huo Y, Zhang H, Sa L, Zheng W, He Y, Lyu
H, Sun M, Zhang L, Shan L, Yang A and Wang T: M1 polarization
enhances the antitumor activity of chimeric antigen receptor
macrophages in solid tumors. J Transl Med. 21:2252023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ricci AD, Rizzo A, Rojas Llimpe FL, Di
Fabio F, De Biase D and Rihawi K: Novel HER2-directed treatments in
advanced gastric carcinoma: AnotHER paradigm shift? Cancers
(Basel). 13:16642021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Astore M, Fabbri L, Monte A, Deiana C,
Rizzo A, Tavolari S, Deserti M, Brandi G, Palloni A and Frega G:
Molecular alterations and pathways in intrahepatic
cholangiocarcinoma: Available evidence and new perspectives. Int J
Mol Sci. 26:119612025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lamberti G, Andrini E, Sisi M, Rizzo A,
Parisi C, Di Federico A, Gelsomino F and Ardizzoni A: Beyond EGFR,
ALK and ROS1: Current evidence and future perspectives on newly
targetable oncogenic drivers in lung adenocarcinoma. Crit Rev Oncol
Hematol. 156:1031192020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rizzo A, Ricci AD, Bonucci C, Tober N,
Palloni A, Frega G and Brandi G: Experimental HER2-targeted
therapies for biliary tract cancer. Expert Opin Investig Drugs.
30:389–399. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sahin TK, Makasheva B, Guven DC, Aksoy S,
Di Lauro V and Rizzo A: Elacestrant in metastatic breast cancer:
Current advancements and future perspectives. Expert Rev Anticancer
Ther. December 28–2025.(Epub ahead of print).
|
|
28
|
Sahin TK, Rizzo A, Guven DC and Aksoy S:
Post-progression treatment options after CDK4/6 inhibitors in
hormone receptor-positive, HER2-negative metastatic breast cancer.
Cancer Treat Rev. 135:1029242025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smyth EC and Sundar R: Combining
chemotherapy, trastuzumab, and immune-checkpoint inhibitors in
HER2-positive gastro-oesophageal cancer. Lancet. 402:2168–2170.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Klichinsky M, Ruella M, Shestova O, Lu XM,
Best A, Zeeman M, Schmierer M, Gabrusiewicz K, Anderson NR, Petty
NE, et al: Human chimeric antigen receptor macrophages for cancer
immunotherapy. Nat Biotechnol. 38:947–953. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li J, Chen P and Ma W: The next frontier
in immunotherapy: Potential and challenges of CAR-macrophages. Exp
Hematol Oncol. 13:762024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li D, Xiong W, Wang Y, Feng J, He Y, Du J,
Wang J, Yang M, Zeng H, Yang YG, et al: SLAMF3 and SLAMF4 are
immune checkpoints that constrain macrophage phagocytosis of
hematopoietic tumors. Sci Immunol. 7:eabj55012022.PubMed/NCBI
|
|
33
|
Zeller T, Lutz S, Münnich IA, Windisch R,
Hilger P, Herold T, Tahiri N, Banck JC, Weigert O, Moosmann A, et
al: Dual checkpoint blockade of CD47 and LILRB1 enhances CD20
antibody-dependent phagocytosis of lymphoma cells by macrophages.
Front Immunol. 13:9293392022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Wang Z, Qin X, Zhong MC, Tang Z,
Qian J, Dou J, Hussell T, King PD, Nunès JA, et al: CD200R1-CD200
checkpoint inhibits phagocytosis differently from SIRPα-CD47 to
suppress tumor growth. Nat Commun. 16:51452025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang P, Zhang G and Wan X: Challenges and
new technologies in adoptive cell therapy. J Hematol Oncol.
16:972023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng H, Yang X, Huang N, Yuan S, Li J,
Liu X, Jiang Q, Wu S, Ju Y, Kleeff J, et al: Chimeric antigen
receptor macrophages targeting c-MET(CAR-M-c-MET) inhibit
pancreatic cancer progression and improve cytotoxic
chemotherapeutic efficacy. Mol Cancer. 23:2702024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y, Zhu X, Liu H, Wang C, Chen Y, Wang
H, Fang Y, Wu X, Xu Y, Li C, et al: The application of HER2 and
CD47 CAR-macrophage in ovarian cancer. J Transl Med. 21:6542023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hadiloo K, Taremi S, Heidari M and
Esmaeilzadeh A: The CAR macrophage cells, a novel generation of
chimeric antigen-based approach against solid tumors. Biomark Res.
11:1032023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Madsen CØ, Hulen TM, Ormhøj M, Hadrup SR,
Svane IM and Met Ö: Driving CAR therapies beyond T cells. Trends
Cancer. S2405-8033(26)00039-7. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nadella V and Sharma A: Targeting
macrophages in immunotherapy: The ascent of CAR-Macrophages. Int J
Mol Sci. 27:12922026. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ahvati H, Roudi R, Sobhani N and Safari F:
CD47 as a potent target in cancer immunotherapy: A review. Biochim
Biophys Acta Rev Cancer. 1880:1892942025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Y, Zhu X, Chen Y, Yang Z, Shen Z,
Chen M, Liu C, Zhou Y, Wang H, Zhu M, et al: IL-21 promotes the
anti-tumor effect of anti-CD47 chimeric antigen receptor
macrophages in ovarian cancer. Commun Biol. 9:632025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang
J, Zhao P, Fang L, Shi Y and Wang P: Emerging phagocytosis
checkpoints in cancer immunotherapy. Signal Transduct Target Ther.
8:1042023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rafiq S, Yeku OO, Jackson HJ, Purdon TJ,
van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al:
Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances
anti-tumor efficacy in vivo. Nat Biotechnol. 36:847–856. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jing W, Han M, Wang G, Kong Z, Zhao X, Fu
Z, Jiang X, Shi C, Chen C, Zhang J, et al: An in situ engineered
chimeric IL-2 receptor potentiates the tumoricidal activity of
proinflammatory CAR macrophages in renal cell carcinoma. Nat
Cancer. 6:838–853. 2025. View Article : Google Scholar : PubMed/NCBI
|