Introduction
Acute myeloid leukemia (AML) is a clonal malignant
proliferative disease originating from myeloid progenitor cells in
the hematopoietic system. AML is characterized by a high relapse
rate, with a 3- to 5-year overall survival (OS) rate of <30%
(1). For patients with high-risk or
relapsed/refractory (R/R) AML, allogeneic hematopoietic stem cell
transplantation (allo-HSCT) remains the only potentially curative
treatment option (2). However, its
clinical application is often complicated by serious adverse events
such as graft-vs.-host disease (GVHD) (3). With the growing understanding of the
leukemia microenvironment and immune landscape, various
immunotherapies including immune checkpoint inhibitors,
antibody-drug conjugates (ADCs) and cellular therapies have entered
clinical trials (3). Nevertheless,
these approaches still face limitations such as resistance and
disease relapse, highlighting the urgent need for novel therapeutic
strategies.
Chimeric antigen receptors (CARs) are synthetic
receptor molecules engineered through gene editing technologies.
They endow effector T cells with the ability to specifically
recognize target antigen epitopes, thereby enhancing T lymphocyte
recognition and activation toward tumor antigens (4). A CAR construct generally comprises
four main components: i) An extracellular antigen recognition
domain, typically composed of a single-chain variable fragment
(scFv), which enables the specific targeting of tumor-associated
antigens or tumor-specific antigens, conferring high selectivity to
CAR-T cells; ii) a hinge or spacer region, which provides
structural flexibility and connects to the scFv; iii) a
transmembrane domain, often derived from molecules such as CD3ζ,
CD4, CD8 or CD28, serving as a structural bridge between
extracellular and intracellular regions; and iv) an intracellular
signaling domain, primarily composed of CD3ζ, responsible for
initiating T cell activation and downstream signaling cascades that
are important for the cytotoxic function of CAR-T cells (5). To date, CAR-T cell therapy has
achieved breakthrough success in treating B cell hematologic
malignancies, such as acute lymphoblastic leukemia (ALL) (6). However, its application in AML remains
notably limited due to various challenges.
The present review summarizes advances in CAR-T cell
therapy for AML within a conceptual framework that separates
fundamental biological barriers from technical and engineering
limitations. From a biological perspective, AML poses unique
challenges including the lack of leukemia-specific antigens, clonal
heterogeneity, antigen escape, persistence of leukemia stem cells
and a profoundly immunosuppressive tumor microenvironment (TME).
From an engineering perspective, current CAR-T approaches are
further constrained by limited in vivo persistence,
treatment-related toxicities, insufficient functional control and
manufacturing complexity. By integrating these two dimensions, the
present review aims not only to summarize current progress, but
also to highlight unresolved questions, areas of controversy and
future directions for the rational development of next-generation
AML CAR-T therapies.
Advancements in CAR-T cell therapy for
AML
CD33
CD33, also known as sialic acid-binding
immunoglobulin-like lectin 3, is a differentiation-associated
antigen predominantly expressed on myeloid progenitor cells and
broadly distributed across various stages of myeloid lineage
development (7). In AML, CD33 is
highly expressed on leukemic progenitor cells, whereas its
expression on normal hematopoietic stem and progenitor cells
(HSPCs) is relatively low (8). This
differential expression profile renders CD33 a compelling target
for AML-directed immunotherapy.
Among CD33-targeted strategies, ADCs have achieved
notable clinical progress. In 2017, the U.S. Food and Drug
Administration approved gemtuzumab ozogamicin (GO) for the
treatment of CD33+ AML, marking the first approved ADC
for AML and representing a major milestone in precision medicine
for hematologic malignancies (9).
The clinical success of GO has verified the potential of CD33 as an
effective therapeutic target and has also promoted the development
of subsequent cell-based immunotherapies, including CAR-T cell
therapy.
As CD33-directed CAR-T cells advance into clinical
evaluation, a primary research objective is to enhance antileukemic
efficacy while minimizing on-target, off-tumor toxicity. To improve
therapeutic safety, researchers have developed CD33-targeted
CAR-engineered natural killer (NK) cells, which have shown potent
cytotoxicity against AML cells both in vitro and in
vivo, with limited cytotoxic effects on normal HSPCs in phase I
trials (10). In parallel,
‘controllable CAR systems’ have emerged as a promising
safety-enhancing strategy. By integrating inducible safety switches
such as inducible caspase-9 (iCasp9) into the CAR construct, these
systems enable the selective elimination of CAR-T cells upon
administration of small-molecule dimerizers such as AP1903, thereby
mitigating prolonged myelosuppression and reducing the risk of
severe adverse events (11).
CD33-targeted CAR-T therapy has now entered multiple
phase I/II clinical trials. In the dose-escalation clinical trial,
NCT03971799, conducted in adolescents and young adults with R/R
AML, cytokine release syndrome (CRS), an acute systemic
inflammatory toxicity caused by CAR-T-cell activation and cytokine
release, occurred in 68% of patients, with grade ≥3 CRS reported in
21% (12). This highlights that,
despite promising antileukemic activity, CD33-directed CAR-T
therapy in AML may be associated with substantial treatment-related
immune toxicity. However, cross-trial comparison of toxicity rates
should be approached with caution due to differences in study
population, dose level and trial design as these may substantially
affect the observed safety profile. Despite these toxicities,
promising efficacy was observed: In the highest dose level cohort,
40% of patients achieved complete remission (CR) with minimal
residual disease (MRD) negativity (12) (Table
I). To further improve the safety profile, the use of
donor-derived CD33 CAR-T cells (such as VCAR33) in the
post-hematopoietic stem cell transplantation (HSCT) setting has
been explored. In a phase I/II trial, 41.7% (5/12) of patients
achieved bone marrow CR, with four attaining MRD negativity.
Additionally, patients with central nervous system (CNS) leukemia
also showed MRD clearance following CAR-T infusion. The treatment
was generally well tolerated, with most patients experiencing only
grade 1–2 CRS and no cases of immune effector cell-associated
neurotoxicity syndrome (ICANS). Some patients developed transient
hepatic dysfunction and infections, which were manageable with
supportive care. As of June 2024, six patients had either undergone
a second HSCT or remained in remission following CD33 CAR-T therapy
(13).
 | Table I.Contextual summary of representative
single-target CAR-T strategies in acute myeloid leukemia. |
Table I.
Contextual summary of representative
single-target CAR-T strategies in acute myeloid leukemia.
| Target | Expression
characteristics | Therapeutic
advantages | Major
challenges | Efficacy index (CR
Rate) | Toxic
reactions(≥grade 3) | (Refs.) |
|---|
| CD33 | Highly expressed in
AML progenitor cells, low expression in normal hematopoietic
cells | Validated as an ADC
target; CAR-NK reduces myeloid toxicity | Off-target
toxicity; CRS (21% ≥grade 3) | 40% in DL4
cohort | CRS (21%), bone
marrow suppression | (8,10,12,13) |
| CD123 | Highly expressed in
AML blasts and LSCs, low expression in normal HSCs | High specificity;
suitable for MRD monitoring | Targeting normal
monocytes, requiring optimization of iCasp9 | ≤81.8% | Monocytopenia
(15%) | (24,26,28–30) |
| CLL-1 | Expressed in 80% of
adult AML cases and the majority of pediatric AML cases, not
expressed in normal HSCs | Strong specificity;
suitable for MRD monitoring | Delayed monocyte
recovery (28% of patients) | >50% in
pediatric cohort | No dose-limiting
toxicity | (14,16–18) |
| CD7 | Ectopically
expressed in 20–35% of AML cases, commonin high-risk subtypes | Gene editing
relieves fratricide; suitable for specific subtypes | Fratricide;
immunosuppression | Not disclosed | No severe ICANS
reported | (19,21,22) |
| FLT3 | Mutated (ITD/TKD)
in >50% of AML cases, regulating cell proliferation and
differentiation | Strong
mutation-driven property; suitable for combination targeted
therapy |
|
|
| (30,31) |
C-type lectin domain family 12 member
A (CLEC12A)
CLEC12A, also known as C-type lectin-like molecule-1
(CLL-1), is a type II transmembrane glycoprotein that is highly
expressed on leukemia precursor cells and leukemia stem cells
(LSCs) of AML, while its expression is extremely low on normal
HSPCs, making it a potential target for AML treatment (14). This differential expression of CLL-1
confers high specificity for targeted therapy and reduces the risk
of off-target effects. Moreover, the expression of CLL-1 remains
relatively stable during disease progression, suggesting it has
value in the application of MRD monitoring and has potential as a
reliable prognostic biomarker (15). CLL-1 is still present in AML
subtypes with low or absent expression of CD33/CD34 (16), which expands the treatment coverage
and overcomes the limitations associated with traditional single
antigen targets. Multicenter clinical trials have suggested that
the CLL-1 targeted CAR-T cell therapy can achieve encouraging
therapeutic effects. In a clinical trial of pediatric patients with
R/R AML, the CR rate was >50%, and the safety profile appeared
favorable. Specifically, all patients developed only grade 1–2
cytokine release syndrome (CRS) and no lethal events were reported
(17).
The preliminary results of early clinical trials
(NCT04219163 and ChiCTR1900027684) further corroborate the efficacy
and safety of the CLL-1 targeted CAR-T therapy in pediatric and
adult patients with AML, with the highest CR rate reaching 81.8%,
and only 5% of patients experienced ≥3 grade CRS (17,18)
(Table II). Nevertheless, these
findings should be interpreted within the context of the individual
study settings, as differences in patient population, disease
status and trial design may limit direct comparison with other
CAR-T studies.
| Table II.Contextual summary of representative
clinical trials of targeted CAR-T therapies in AML. |
Table II.
Contextual summary of representative
clinical trials of targeted CAR-T therapies in AML.
| Trial no. | Target | Sample size | Patient
population | Key findings | Toxicity management
strategies |
|---|
| NCT03971799 | CD33 | 30 | R/R AML
(adolescents) | 40% CR rate in DL4
cohort; 68% patients experienced CRS | No safety switch
used |
|
ChiCTR1900027684 | CLEC12A | 42 | Adult R/R AML | 81.8% CR rate; no
dose-limiting toxicity | Not mentioned |
| NCT04219163 | CLEC12A | 28 | Pediatric R/R
AML | >50% CR rate;
delayed monocyte recovery | Supportive
care |
| NCT04581390 | CD7 | 15 | CD7+ R/R
AML | Gene-edited CAR-T
reduced fratricide | CRISPR-mediated
knockout of CD7/TCR |
| NCT04601529 | CD123 | 20 | FLT3+
AML | iCasp9 safety
switch reduces toxicity | Inducible caspase-9
system |
CD7
CD7 is a transmembrane glycoprotein belonging to the
immunoglobulin superfamily, primarily expressed on T cells and NK
cells. In AML, 20–35% of patients display ectopic expression of
CD7, particularly among those with cytogenetically high-risk
subtypes (19), CD7+ AML
is frequently associated with resistance to chemotherapy, increased
disease aggressiveness and worse clinical outcomes (20). Therefore, despite its relatively
limited expression prevalence, CD7 represents a compelling
therapeutic target for selected AML subgroups.
CD7-targeted CAR-T cells have demonstrated potent,
antigen-specific cytotoxicity against CD7+ AML cells in
preclinical studies (21). However,
two major challenges hinder their clinical translation: i)
Fratricide: Since CAR-T cells inherently express CD7, they may
attack each other during ex vivo expansion, compromising
their viability and persistence; and ii) off-target toxicity: The
ubiquitous expression of CD7 on normal T cells and NK cells
increases the risk of profound immunosuppression and related
complications (22).
To address these limitations, CRISPR/Cas9
gene-editing technology has been applied to eliminate the
expression of CD7, T cell receptor and human leukocyte antigen
(HLA) class II molecules, thereby generating ‘universal’ CD7 CAR-T
cells. These engineered cells exhibit enhanced proliferative
capacity, prolonged functional persistence and reduced
immunotoxicity (23). In addition,
a study has explored the transient use of tyrosine kinase
inhibitors, such as ibrutinib and dasatinib, to reversibly suppress
CAR signaling. This pharmacological approach mitigates fratricide
during cell manufacturing while preserving the cytolytic function
of CAR-T cells (22). In animal
models, this strategy has yielded durable antileukemic responses,
and early-phase clinical trials are currently underway (22).
Collectively, CD7-directed CAR-T cell therapy offers
a promising treatment modality for patients with CD7+
AML. Continued advancements in gene editing and pharmacologic
modulation are expected to further enhance the safety and
therapeutic efficacy of this approach.
CD123
CD123, the α subunit of the IL-3 receptor α, is
highly expressed on leukemic blasts and LSCs in AML, while its
expression is either absent or minimal in normal hematopoietic stem
cells (HSCs). Several therapeutic approaches targeting CD123, such
as bispecific antibodies and ADCs, are currently in clinical
development (24). As early as
2002, Testa et al (25)
conducted a systematic analysis of CD123 expression in AML and
reported that ~45% of cases exhibited high levels of CD123. This
upregulation was associated with increased blast proliferation,
elevated white blood cell counts and hypersensitivity to IL-3
signaling, all of which were associated with worse prognosis. These
observations have since been consistently validated in subsequent
studies (26,27).
Clinically, CD123 has proven useful for risk
stratification and prognostic assessment in AML. In pediatric
cohorts, CD123 expression was stratified into quartiles, and
patients in the highest-expression quartile (Q4) were notably
enriched for adverse genetic alterations, including FMS-like
tyrosine kinase 3 (FLT3)-internal tandem duplications (ITD) and
lysine methyltransferase 2A rearrangements (26). By contrast, favorable genetic
markers such as t(8;21), inv (16)
and CCAAT enhancer binding protein α mutations were more frequently
observed in the low-expression group. Patients with high CD123
expression demonstrated markedly shorter OS and event-free
survival, establishing CD123 as an independent biomarker of worse
prognosis (26). To address the
on-target/off-tumor toxicity associated with CD123-targeted CAR-T
therapy, several research groups have incorporated safety switches
into CAR constructs. These include inducible systems such as iCasp9
and CD20, which can be externally activated in response to severe
treatment-related toxicity (28,29).
Activation of these switches enables rapid and selective
elimination of CAR-T cells, thereby minimizing non-specific damage
to normal tissues and improving the overall safety profile of the
therapy.
FLT3
FLT3 is a glycosylated protein belonging to the
class III receptor tyrosine kinase family. It is predominantly
expressed on HSCs and myeloid progenitors, where it plays a key
role in regulating cell survival, proliferation and
differentiation. FLT3 mutations are detected in >50% of patients
with AML (30). These mutations are
primarily categorized into two major types: ITDs within the
juxtamembrane domain, accounting for ~25% of cases, and point
mutations in the tyrosine kinase domain, which occurs in 6–8% of
patients (31); the current
standard of care for FLT3-mutated AML involves induction
chemotherapy in combination with midostaurin, followed by allo-HSCT
(31). A recent clinical study
demonstrated that gilteritinib, when administered in conjunction
with allo-HSCT, can notably extend relapse-free survival in
patients with FLT3-ITD+ AML (32). Additionally, quizartinib has shown
promise as a post-transplant maintenance therapy, sustaining FLT3
inhibition to reduce MRD and prevent relapse (33). While the majority of research to
date has focused on FLT3-ITD+ AML, the therapeutic
potential of FLT3 targeting in patients that are
FLT3-ITD− remains largely unexplored. Further
prospective studies are needed to elucidate the role of FLT3 in
non-canonical signaling pathways and to assess its broader
applicability as an immunotherapeutic target in diverse AML
subtypes.
The clinical outcomes summarized in Tables I and II are derived from heterogeneous studies
and are not directly comparable. Differences in trial phase, sample
size, patient age, disease status, target antigen selection,
dose-escalation design and response criteria may substantially
influence the reported CR rates and toxicity profiles. Therefore,
these tables are intended to provide a contextual overview of the
current clinical landscape rather than to indicate the superiority
of one CAR-T target over another.
Target specificity and persistence of CAR-T
therapy in AML
The barriers limiting CAR-T therapy in AML can be
broadly categorized into two dimensions. The first comprises
fundamental biological barriers intrinsic to AML, such as antigen
overlap with normal hematopoietic cells, clonal and phenotypic
heterogeneity, antigen escape, leukemia stem cell persistence and
suppressive bone marrow microenvironment. The second involves
technical and engineering limitations of CAR-T therapy itself,
including suboptimal CAR construct design, inadequate persistence,
restricted controllability, treatment-related toxicities and
manufacturing constraints. Although these categories are
analytically distinct, they are biologically interconnected and
together shape the limited efficacy of CAR-T therapy in AML
(5).
Despite the success of CAR-T cell therapy in B cell
malignancies (6), its application
in AML remains limited by the unique biological characteristics of
this disease. AML is highly heterogeneous, with leukemic cells
exhibiting substantial immunophenotypic variability across
different patients, disease stages and clonal subtypes. Compared
with CD19, a highly specific and consistently expressed antigen in
B-ALL, AML lacks a universally reliable, leukemia-specific target
antigen (34,35). Frequently explored targets in AML,
such as CD33 and CD123, are abundantly expressed on AML blasts
(36); however, they are also
present at varying levels on normal HSPCs (37). This overlap contributes to notable
on-target/off-tumor toxicity. For instance, CAR-T therapies
targeting CD33 or CD123 have been associated with hematologic
toxicities, including myelosuppression, cytopenias and prolonged
marrow suppression, which may result in serious hematologic
complications (38). In addition to
target specificity challenges, CAR-T cells in patients with AML
often display suboptimal in vivo expansion and worse
persistence, leading to transient therapeutic responses and high
relapse rates, reported to range from 60 to 80%. Enhancing the
specificity and durability of CAR-T cells in AML thus remains a key
objective in ongoing translational research.
Immunosuppressive TME
The TME of AML is a major obstacle to the efficacy
of CAR-T cell therapy. This immunosuppressive microenvironment is
characterized by the presence of various inhibitory cells,
molecules and metabolic limitations, which work together to weaken
the antileukemic efficacy of CAR-T cells and lead to treatment
resistance and failure (39). As
illustrated in Fig. 1,
AML-associated immunosuppression involves both extrinsic inhibitory
signals from the TME, such as suppressive myeloid cells, regulatory
T cells, tumor-associated macrophages and inhibitory cytokines, and
intrinsic functional consequences in CAR-T cells, including
progressive exhaustion marked by reduced cytokine production and
increased checkpoint expression. A thorough understanding of these
complex inhibitory mechanisms is important for developing more
effective CAR-T cell strategies (40). Importantly, recent clinical evidence
has reinforced the central role of the AML microenvironment in
determining CAR-T treatment outcomes. In a landmark study, Bhagwat
et al (41) provided the
first in-human demonstration that cytokines released from
AML-associated myeloid cells such as granulocyte-macrophage colony
stimulating factor, IL-3 and FLT3-L activate JAK/STAT-dependent
pro-survival signaling and induce exhaustion-related
transcriptional programs in leukemic blasts. These
microenvironment-driven alterations directly impair CAR-T cell
expansion and accelerate functional exhaustion, highlighting that
resistance in AML arises not solely from antigenic factors but
predominantly from TME-mediated immunologic and transcriptional
reprogramming. This concept is further summarized in Fig. 1, which associates TME-derived
suppressive signaling to the transition from activated CAR-T cells
to an exhausted functional state.
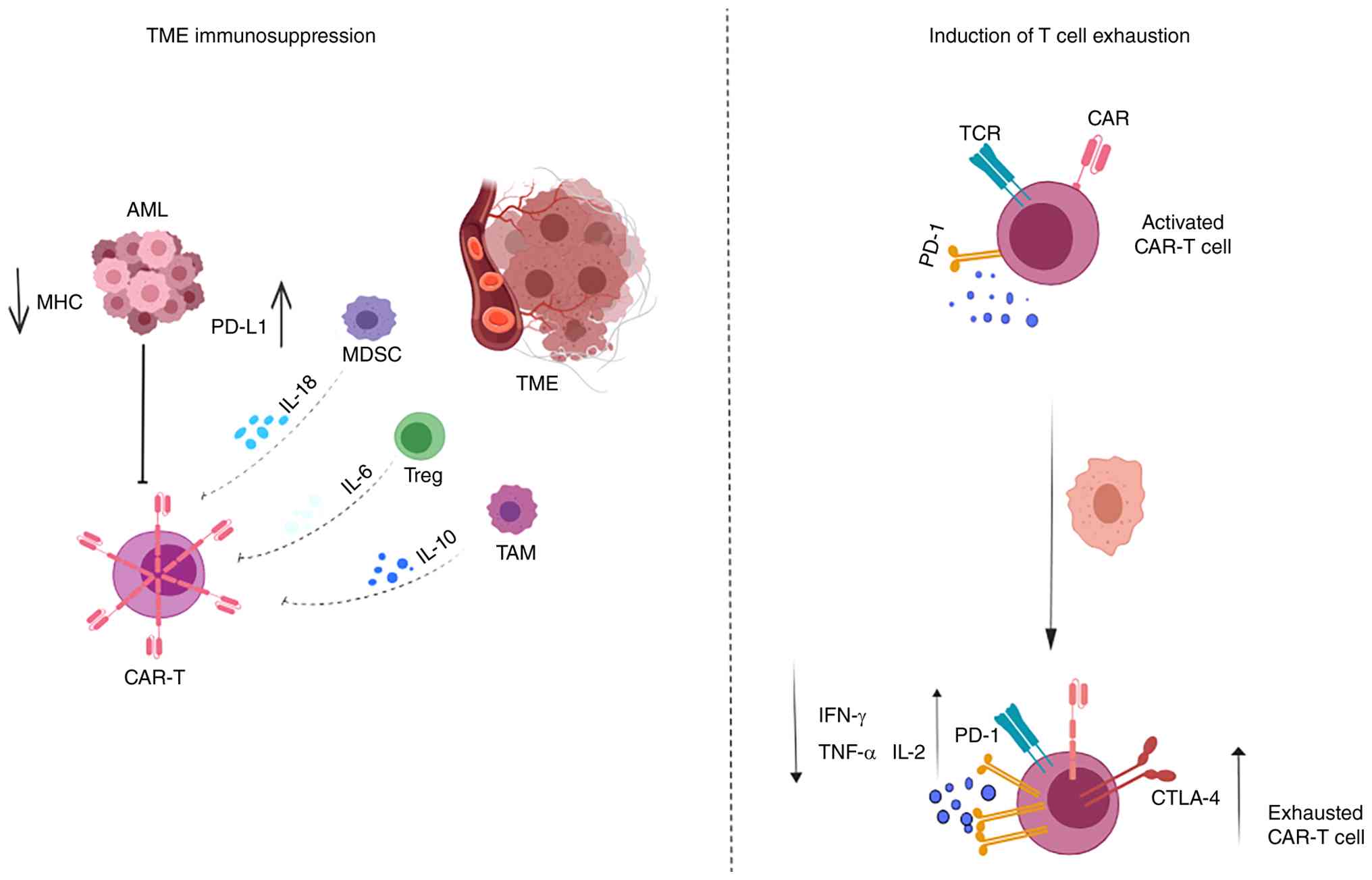 | Figure 1.CAR-T cell dysfunction in AML. The
AML microenvironment suppresses CAR-T cells through PD-L1
expression, MHC downregulation and immunosuppressive cytokines such
as IL-6 and IL-10, and chronic stimulation induces exhaustion via
PD-1 and CTLA-4 upregulation. CAR-T, chimeric antigen receptor T
cell; AML, acute myeloid leukemia; MHC, major histocompatibility
complex; PD-L1, programmed death-ligand 1; MDSC, myeloid-derived
suppressor cells; TME, tumor microenvironment; TAM,
tumor-associated macrophage; TCR, T cell receptor; Treg, regulatory
T cell; CTLA-4, cytotoxic T-lymphocyte associated protein 4. |
The formation of an oxygen-depleted microenvironment
is a direct consequence of the rapid proliferation of cancer cells
and insufficient angiogenesis. This oxygen deficiency not only
directly impairs the metabolic activities and survival of CAR-T
cells, but also further exacerbates the inhibition of CAR-T cells
by inducing adaptive responses of cancer cells (42,43).
Under hypoxic conditions, hypoxia-inducible factor-1 plays a
central role in cancer cells, mediating their adaptation to the
hypoxic environment, for instance, by promoting angiogenesis and
regulating the expression of cancer stem cell markers to enhance
survival (44). The TME reshapes
its metabolic mechanisms and transcriptome profile through the
hypoxic response, leading to challenges such as nutrient
competition and accumulation of metabolic waste for CAR-T cells,
thereby damaging their proliferation, persistence and effector
functions (39), Furthermore, the
2025 consensus guidance by Naik et al (40) reinforces these observations by
systematically outlining how TME-derived cytokines, suppressive
myeloid populations and metabolic deprivation converge to blunt
CAR-T cell activity in AML. This guideline emphasizes the clinical
necessity of incorporating TME-targeted interventions such as
JAK/STAT blockade, myeloid-directed therapies or metabolic
reprogramming into future CAR-T design to overcome
microenvironment-driven resistance. Therefore, a deep understanding
and targeting of hypoxia and its related metabolic pathways are
important for overcoming metabolic limitations in the TME and
enhancing the efficacy of CAR-T cells. As summarized in Fig. 2, these barriers have driven the
development of multiple optimization strategies, including
precision genome editing, checkpoint modulation, enhancement of T
cell fitness and combinatorial engineering approaches, which are
discussed in later sections of the present review.
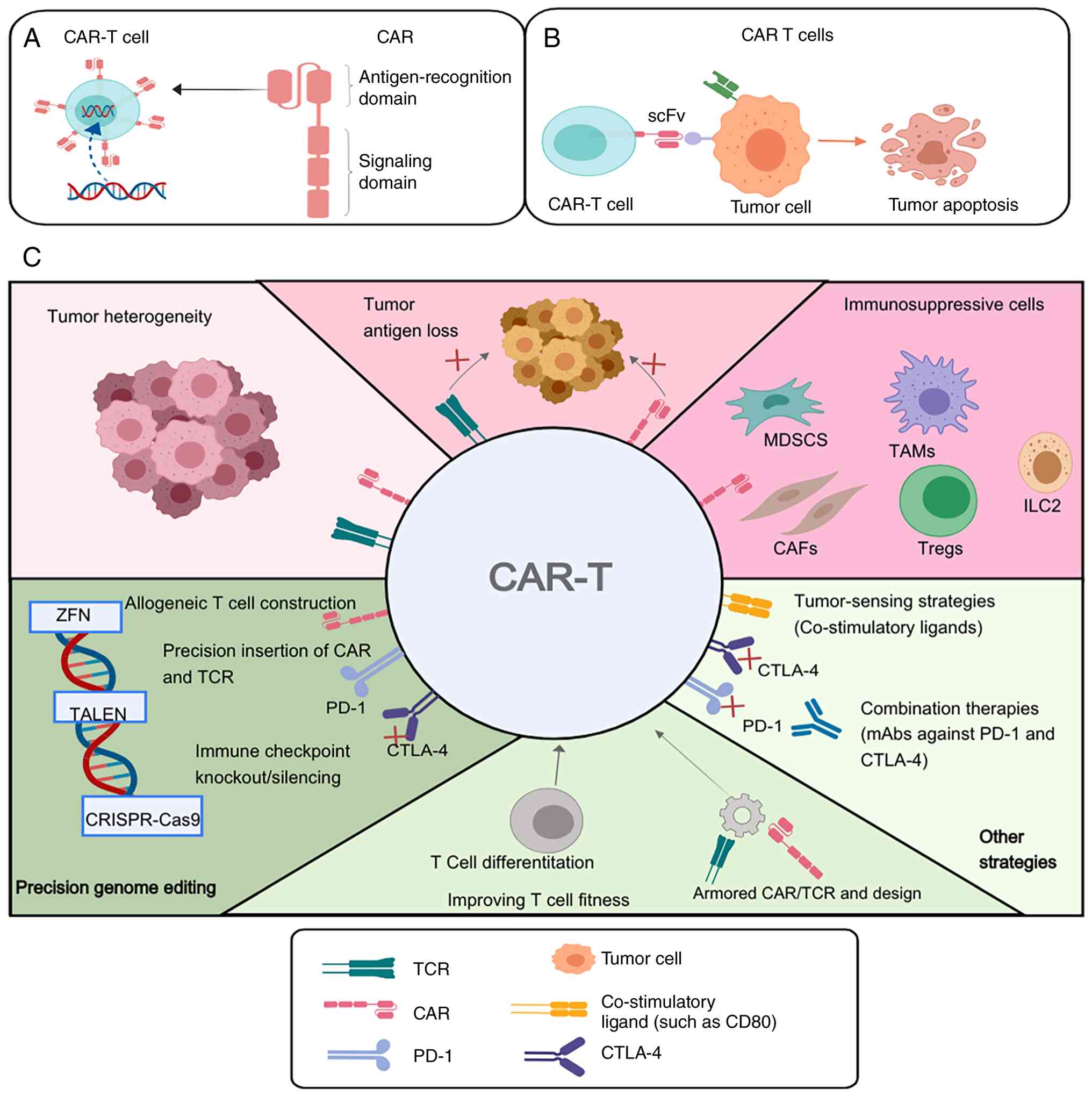 | Figure 2.Limitations and optimization
strategies of CAR-T therapy in AML. (A) Schematic illustration of
the basic structure of a CAR, including the antigen-recognition
domain and signalling domains. (B) Schematic illustration of
CAR-T-cell recognition of tumor cells and the induction of tumor
apoptosis. (C) Major biological and technical barriers affecting
CAR-T therapy in AML and representative strategies to overcome
them, including tumor heterogeneity, immunosuppressive cells,
precision genome editing, improving T cell fitness and other
combinatorial engineering approaches. CAR, chimeric antigen
receptor; CAR-T, chimeric antigen receptor T cell; CTLA-4;
cytotoxic T-lymphocyte associated protein 4; MDSC, myeloid-derived
suppressor cells; CAFs, cancer-associated fibroblasts; Treg,
regulatory T cell; PD-1, programmed death-ligand 1; TCR, T cell
receptor; ZFN, zinc-finger nuclease; scFv, single-chain variable
fragment; mAbs, monoclonal antibodies. |
Toxicities and adverse effects
CAR-T cell therapy is associated with notable
toxicities that limit its broader application in AML. CRS is the
most common and potentially life-threatening adverse event, arising
from extensive T cell activation and the subsequent release of
pro-inflammatory cytokines. Clinical manifestations of CRS range
from mild symptoms such as fever and fatigue to severe conditions
including shock and multi-organ failure (45). Another notable complication is
ICANS, which frequently occurs following CRS and is considered to
result from blood-brain barrier disruption and increased expression
of vascular activation markers such as angiopoietin-2 (24). Currently, tocilizumab effectively
mitigates CRS but exhibits limited penetration of the CNS,
rendering it less effective for treating ICANS. Corticosteroids are
commonly employed to manage neurotoxicity; however, their
immunosuppressive effects may impair CAR-T cell expansion and
antitumor activity, posing a clinical challenge in balancing
therapeutic efficacy and toxicity management (46).
Opportunities and challenges of CAR-T cell
therapy in the treatment of AML
Novel target strategies
Dual-targeted CAR-T cell therapies have emerged as
promising strategies to overcome the limitations of single-antigen
targeting in AML. For instance, Ma et al (47) developed Loop33 × 123 CAR-T cells, a
bispecific CAR-T design incorporating tandem antigen-recognition
domains that enables simultaneous targeting of the two myeloid
antigens CD33 and CD123. This dual targeting effectively eliminates
both AML blasts and LSCs. In vitro assays demonstrated that
Loop33 × 123 CAR-T cells exhibited superior cytotoxicity compared
with single-target CAR-T cells, while an in vivo study
showed notable prolongation of survival in murine models without
evident toxicity (48).
Similarly, dual-target CAR-T cells targeting CD123
and CLL-1, designed either via a bicistronic vector or tandem scFv
architecture, have been developed to achieve precise recognition
and elimination of diverse AML subtypes. A study reported that
CD123/CLL-1 dual-targeted CAR-T therapy exerted robust antileukemic
effects in animal models and reduced the likelihood of antigen
escape (49). Furthermore,
comparative analyses of two tandem CAR constructs (CD123/CLL-1 vs.
CLL-1/CD123) revealed that dual-targeted CAR-T cells possessed
enhanced tumor-killing efficacy and improved resistance to antigen
escape relative to single-target counterparts (50). Beyond these, other dual-target CAR
designs such as CD33/CLL-1 are also under investigation, further
broadening the immunotherapeutic target landscape in AML.
While these findings highlight the potential of
dual-target strategies, their translational relevance in AML
remains uncertain because the available evidence is still largely
preclinical. Expanding antigen coverage may help reduce immune
escape, but it may also increase the risk of hematopoietic toxicity
when target expression overlaps with normal cells. In addition,
greater structural complexity may introduce further challenges for
signaling coordination, persistence and manufacturing consistency
(5). Whether these theoretical
advantages can be translated into a clinically meaningful
improvement in efficacy without compromising safety will require
validation in future clinical studies.
CAR-T cell optimization
strategies
Beyond target selection, Fig. 2 highlights the importance of
engineering strategies to improve T cell fitness, controllability
and resistance to immunosuppressive cues in AML CAR-T therapy.
Universal chimeric antigen receptor T
cell (UCAR-T) technology
Conventional CAR-T therapy relies on autologous T
cells harvested from patients with hematologic malignancies, which
entails challenges such as prolonged manufacturing times, high
costs and considerable product heterogeneity (51). UCAR-T cells, derived from healthy
donors and genetically engineered via CRISPR/Cas9 or TALEN
technologies to disrupt T cell receptor and HLA genes, represent
‘off-the-shelf’ universal CAR-T products designed to minimize the
risk of GVHD. Recent preclinical studies have demonstrated that
optimized UCAR-T cells exhibit a defined memory phenotype and
dose-dependent antitumor efficacy, along with favorable safety
profiles in xenograft models (52).
UCAR-T is attractive because it may simplify
production and reduce delays associated with autologous
manufacturing. However, its broader clinical application is still
constrained by several unresolved issues, including alloreactivity,
host immune rejection, limited persistence after infusion and the
need for robust large-scale manufacturing. Gene editing may help
address some of these barriers, but it also raises additional
concerns regarding genomic stability and regulatory oversight
(53,54). Therefore, the clinical value of
UCAR-T will depend on whether these products can achieve
reproducible safety and durable efficacy in patients.
Logic-gated CAR-T technology
Logic-gated CAR-T cells regulate T cell activation
and cytotoxicity by integrating multiple cellular signals, enabling
precise discrimination between malignant and normal cells. Key
design strategies include: i) OR gate CARs, which recognize
multiple antigens simultaneously and are thus referred to as
multi-antigen CARs (55); ii) gate
CARs, which require co-expression of two target antigens (such as
CD19 and CD22) to become fully activated (56); and iii) NOT gate CARs, which detect
markers expressed on healthy cells (such as CD34) and trigger
inhibitory signaling pathways to protect normal tissues and reduce
off-tumor toxicity (55).
These designs are conceptually attractive as they
offer a more selective way to distinguish leukemic cells from
normal tissues. Their clinical application, however, may be more
complicated than their design principle suggests. Because
logic-gated systems depend on coordinated multi-signal responses,
their activity may vary with antigen density, dynamic target
expression and the surrounding leukemic microenvironment. Greater
circuit complexity may also complicate vector construction,
manufacturing workflows and quality control (57,58).
Whether this added precision is sufficient to justify the increased
complexity remains an important translational question.
Clinical translation, standardized
manufacturing and artificial intelligence (AI)
The clinical translation of CAR-T therapy is
advancing rapidly in parallel with technological innovations that
integrate manufacturing processes with AI. Leading pharmaceutical
companies including Gilead, Novartis, Johnson & Johnson and
Bristol-Myers Squibb have successfully reduced CAR-T cell
manufacturing timelines from >30 days to ~14 days, with ongoing
efforts aimed at achieving a 7-day production cycle. This
acceleration is primarily driven by the adoption of automated, good
manufacturing practice-compliant platforms and streamlined, rapid
quality control procedures (51).
For instance, fully automated, closed-loop manufacturing systems
such as CliniMACS Prodigy, coupled with expedited 24-h production
workflows, have demonstrated feasibility in preclinical and early
clinical settings while preserving key naïve and memory T cell
subsets essential for therapeutic efficacy (59). Concurrently, AI has emerged as a
transformative tool in CAR-T development across multiple domains:
i) CAR design and target recognition optimization, where machine
learning algorithms predict antigen affinity, structural stability
and intracellular signaling activation to inform the engineering of
enhanced CAR constructs; ii) intelligent manufacturing process
control, leveraging digital twin models and reinforcement learning
to enable real-time monitoring and regulation of bioreactor
parameters, thereby improving process stability and product yield;
and iii) quantitative prediction of therapeutic efficacy and
toxicity through the integration of multi-omics, imaging and
clinical datasets, providing robust support for dose optimization
and personalized treatment strategies.
Collectively, these innovations herald the emergence
of a ‘digital pharmaceutical’ paradigm in CAR-T therapy, offering
promising avenues to enhance safety, manufacturing efficiency and
patient accessibility. From an engineering perspective, these
optimization strategies have substantially expanded the design
space of AML CAR-T therapy, however, technical sophistication does
not necessarily guarantee clinical feasibility. For dual-target
CARs, universal CAR-T platforms and logic-gated systems alike, the
central translational challenge is whether theoretical advantages
can be converted into reproducible clinical benefit without
introducing excessive toxicity, manufacturing complexity or
regulatory barriers. Addressing these issues will be essential for
translating next-generation CAR-T strategies into meaningful and
durable clinical benefit.
Conclusions and future perspectives
Despite substantial progress, the central challenge
of AML CAR-T therapy remains unresolved: Whether therapeutic
failure is driven primarily by insufficient target specificity or
by microenvironment-induced functional suppression. In practice,
these mechanisms are likely intertwined. The absence of truly
leukemia-restricted antigens increases the risk of
on-target/off-tumor toxicity, whereas the AML microenvironment
simultaneously limits CAR-T expansion, persistence and
cytotoxicity. This suggests that further improvements in target
selection alone may be insufficient unless they are accompanied by
strategies that restore T cell fitness within the leukemic
niche.
Another area of ongoing debate is whether
increasingly sophisticated engineering solutions such as
dual-target CARs, logic-gated circuits, universal CAR-T platforms
and safety-switch systems will translate into meaningful clinical
benefit in AML. Although these approaches are highly promising, the
majority of supporting evidence remains preclinical and their true
feasibility, scalability and safety in patients are still
uncertain. Thus, the future of AML CAR-T therapy will likely depend
not on any single innovation, but on the rational integration of
biologically informed target selection, engineering optimization
and microenvironment-directed combination strategies.
To fully realize the therapeutic potential of CAR-T
cell therapy in AML, several key barriers must be overcome: i)
Identification of highly specific and immune escape-resistant
antigens to minimize on-target/off-tumor effects; ii) structural
optimization of CARs to improve their in vivo persistence,
proliferative capacity and functional control; and iii) integration
of combination strategies that modulate the TME such as targeting
immunosuppressive cellular components and incorporating immune
checkpoint blockade, to restore effective antileukemic immune
responses. With continued advances in molecular biology,
gene-editing technologies and systems immunology, CAR-T therapy is
expected to evolve from a novel experimental intervention into a
mainstream therapeutic modality for AML. Sustained efforts in
translational and clinical research will be essential for
overcoming current barriers and establishing CAR-T cell therapy as
a cornerstone of AML treatment.
Acknowledgements
The figures were generated using MedPeer (https://www.medpeer.cn/product/index/product).
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
JZ was responsible for manuscript preparation and
figure creation. YC provided guidance on the overall approach and
contributed to the manuscript revisions. FW, DS and ZH conducted
the literature review. All authors have read and approved the final
version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CAR
|
chimeric antigen receptor
|
|
AML
|
acute myeloid leukemia
|
|
OS
|
overall survival
|
|
R/R
|
relapsed/refractory
|
|
HSCT
|
hematopoietic stem cell
transplantation
|
|
allo-HSCT
|
allogeneic hematopoietic stem cell
transplantation
|
|
GVHD
|
graft-vs.-host disease
|
|
ADCs
|
antibody-drug conjugates
|
|
scFv
|
single-chain variable fragment
|
|
ALL
|
acute lymphoblastic leukemia
|
|
HSCs
|
hematopoietic stem cells
|
|
HSPCs
|
hematopoietic stem and progenitor
cells
|
|
iCasp9
|
inducible caspase-9
|
|
CRS
|
cytokine release syndrome
|
|
CR
|
complete remission
|
|
DL4
|
dose level cohort
|
|
MRD
|
minimal residual disease
|
|
ICANS
|
immune effector cell-associated
neurotoxicity syndrome
|
|
CLL-1
|
C-type lectin-like molecule-1
|
|
LSCs
|
leukemia stem cells
|
|
TME
|
tumor microenvironment
|
|
HLA
|
human leukocyte antigen
|
|
CNS
|
central nervous system
|
|
AI
|
artificial intelligence
|
|
UCAR-T
|
universal chimeric antigen receptor T
cell
|
|
FLT3
|
FMS-like tyrosine kinase 3
|
|
ITD
|
internal tandem duplications
|
|
GO
|
gemtuzumab ozogamicin
|
|
CLEC12A
|
C-type lectin domain family 12 member
A
|
|
NK
|
natural killer
|
References
|
1
|
Almotiri A: CAR T-cell therapy in acute
myeloid leukemia. Saudi Med J. 45:1007–1019. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thol F, Döhner H and Ganser A: How I treat
refractory and relapsed acute myeloid leukemia. Blood. 143:11–25.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gao C, Li X, Xu Y, Zhang T, Zhu H and Yao
D: Recent advances in CAR-T cell therapy for acute myeloid
leukaemia. J Cell Mol Med. 28:e183692024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cao Y, Efetov SK, He M, Fu Y, Beeraka NM,
Zhang J, Zhang X, Bannimath N and Chen K: Updated clinical
perspectives and challenges of chimeric antigen receptor-T cell
therapy in colorectal cancer and invasive breast cancer. Arch
Immunol Ther Exp (Warsz). 71:192023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Damiani D and Tiribelli M: CAR-T cells in
acute myeloid leukemia: Where do we stand? Biomedicines.
12:11942024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Short NJ and Jabbour E: The present and
future of CAR T-cell therapy for adult B-cell ALL. Blood.
145:1485–1497. 2025. View Article : Google Scholar
|
|
7
|
Taylor VC, Buckley CD, Douglas M, Cody AJ,
Simmons DL and Freeman SD: The Myeloid-Specific Sialic Acid-binding
Receptor, CD33, Associates with the Protein-tyrosine Phosphatases,
SHP-1 and SHP-2*. J Biol Chem. 274:11505–11512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y, Wang S, Schubert ML, Lauk A, Yao H,
Blank MF, Cui C, Janssen M, Schmidt C, Göllner S, et al:
CD33-directed immunotherapy with third-generation chimeric antigen
receptor T cells and gemtuzumab ozogamicin in intact and
CD33-edited acute myeloid leukemia and hematopoietic stem and
progenitor cells. Int J Cancer. 150:1141–1155. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Swaminathan M and Cortes JE: Update on the
role of gemtuzumab-ozogamicin in the treatment of acute myeloid
leukemia. Ther Adv Hematol. 14:204062072311547082023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang R, Wang X, Yan H, Tan X, Ma Y, Wang
M, Han X, Liu J, Gao L, Gao L, et al: Safety and efficacy of
CD33-targeted CAR-NK cell therapy for relapsed/refractory AML:
Preclinical evaluation and phase I trial. Exp Hematol Oncol.
14:12025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kvorjak M, Ruffo E, Tivon Y, So V, Parikh
A, Deiters A and Lohmueller J: Conditional control of universal CAR
T cells by cleavable off-switch adaptors. ACS Synth Biol.
12:2996–3007. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Balestra T, Arenas Merizalde C, Chen RK,
Roach J, Bowser B, Salomon R, Welch A, Yates B, Schaller K,
Verneris MR, et al: Autologous CD33CART for children and
adolescents/young adults with relapsed/refractory AML: Phase 1/2
clinical trial correlative biology analyses demonstrate potent in
vitro, in vivo, and ex vivo anti-leukemia activity. Blood. 144
(Suppl 1):S3692024. View Article : Google Scholar
|
|
13
|
Lin Y, Zhao D, Deng B, Liu D, Yan H, Li B,
Xia Y, Zheng R, Wu T and Tong C: The safety and efficacy of CD33
CAR-T therapy for RR AML after HSCT. Blood. 144 (Suppl
1):S34672024. View Article : Google Scholar
|
|
14
|
Shao RN, Xin HL and Shi XF: Target
selection for CAR-T therapy in acute myeloid leukemia-review.
Zhongguo Shi Yan Xue Ye Xue Za Zhi. 32:965–969. 2024.(In Chinese).
PubMed/NCBI
|
|
15
|
Soleimani Samarkhazan H, Zehtabcheh S,
Seraji HR, Beqaj SH, Tayefeh S, Mohammadi MH and Aghaei M:
Unveiling the potential of CLL-1: A promising target for AML
therapy. Biomarker Res. 13:282025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Wang W, Chen H, Li W, Huang T,
Zhang W, Ling W, Lai P, Wang Y, Geng S, et al: C-type lectin-like
molecule-1 as a biomarker for diagnosis and prognosis in acute
myeloid leukemia: A preliminary study. Biomed Res Int.
2021:66439482021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin X, Zhang M, Sun R, Lyu H, Xiao X,
Zhang X, Li F, Xie D, Xiong X, Wang J, et al: First-in-human phase
I study of CLL-1 CAR-T cells in adults with relapsed/refractory
acute myeloid leukemia. J Hematol Oncol. 15:882022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang H, Bu C, Peng Z, Li G, Zhou Z, Ding
W, Zheng Y, He Y, Hu Z, Pei K, et al: Characteristics of anti-CLL1
based CAR-T therapy for children with relapsed or refractory acute
myeloid leukemia: The multi-center efficacy and safety interim
analysis. Leukemia. 36:2596–604. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu Y, Liu Y, Wen S, Kuang N, Zhang X, Li J
and Wang F: Naturally selected CD7 CAR-T therapy without genetic
editing demonstrates significant antitumour efficacy against
relapsed and refractory acute myeloid leukaemia (R/R-AML). J Transl
Med. 20:6002022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lv K, Cai C, Chen J, Xu M, Wan L, Zhou M,
Du Y, Ma X, Wu X, Tang X, et al: Prognostic value of lymphoid
marker CD7 expression in acute myeloid leukemia patients undergoing
allogeneic hematopoietic cell transplantation in first
morphological complete remission. Int J Hematol. 114:464–471. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gomes-Silva D, Atilla E, Atilla PA, Mo F,
Tashiro H, Srinivasan M, Lulla P, Rouce RH, Cabral JMS, Ramos CA,
et al: CD7 CAR T cells for the therapy of acute myeloid leukemia.
Mol Ther. 27:272–280. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Watanabe N, Mo F, Zheng R, Ma R, Bray VC,
van Leeuwen DG, Sritabal-Ramirez J, Hu H, Wang S, Mehta B, et al:
Feasibility and preclinical efficacy of CD7-unedited CD7 CAR T
cells for T cell malignancies. Mol Ther. 31:24–34. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu Y, Zhou Y, Zhang M, Zhao H, Wei G, Ge
W, Cui Q, Mu Q, Chen G, Han L, et al: Genetically modified
CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy
for relapsed/refractory CD7-positive hematological malignancies: A
phase I clinical study. Cell Res. 32:995–1007. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pelosi E, Castelli G and Testa U: CD123 a
therapeutic target for acute myeloid leukemia and blastic
plasmocytoid dendritic neoplasm. Int J Mol Sci. 24:27182023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Testa U, Riccioni R, Militi S, Coccia E,
Stellacci E, Samoggia P, Latagliata R, Mariani G, Rossini A,
Battistini A, et al: Elevated expression of IL-3Ralpha in acute
myelogenous leukemia is associated with enhanced blast
proliferation, increased cellularity, and poor prognosis. Blood.
100:2980–2988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lamble AJ, Eidenschink Brodersen L, Alonzo
TA, Wang J, Pardo L, Sung L, Cooper TM, Kolb EA, Aplenc R, Tasian
SK, et al: CD123 expression is associated with high-risk disease
characteristics in childhood acute myeloid leukemia: A report from
the children's oncology group. J Clin Oncol. 40:252–261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arai N, Homma M, Abe M, Baba Y, Murai S,
Watanuki M, Kawaguchi Y, Fujiwara S, Kabasawa N, Tsukamoto H, et
al: Impact of CD123 expression, analyzed by immunohistochemistry,
on clinical outcomes in patients with acute myeloid leukemia. Int J
Hematol. 109:539–544. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peters DT, Savoldo B and Grover NS:
Building safety into CAR-T therapy. Hum Vaccin Immunother.
19:22754572023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Naik S, Madden R, Lipsitt A, Lockey T,
Bran J, Rubnitz JE, Klco J, Shulkin B, Patil SL, Schell S, et al:
Safety and anti-leukemic activity of CD123-CAR T cells in pediatric
patients with AML: Preliminary results from a phase 1 trial. Blood.
140 (Suppl 1):S4584–S4585. 2022. View Article : Google Scholar
|
|
30
|
Nitika Wei J and Hui AM: Role of
biomarkers in FLT3 AML. Cancers (Basel). 14:11642022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Negotei C, Colita A, Mitu I, Lupu AR,
Lapadat ME, Popovici CE, Crainicu M, Stanca O and Berbec NM: A
Review of FLT3 kinase inhibitors in AML. J Clin Med. 12:64292023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levis MJ, Hamadani M, Logan B, Jones RJ,
Singh AK, Litzow M, Wingard JR, Papadopoulos EB, Perl AE, Soiffer
RJ, et al: Gilteritinib as post-transplant maintenance for AML with
internal tandem duplication mutation of FLT3. J Clin Oncol.
42:1766–1775. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cortes J: Quizartinib: A potent and
selective FLT3 inhibitor for the treatment of patients with
FLT3-ITD-positive AML. J Hematol Oncol. 17:1112024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cai F, Zhang J, Gao H and Shen H: Tumor
microenvironment and CAR-T cell immunotherapy in B-cell lymphoma.
Eur J Haematol. 112:223–235. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Subklewe M, Rutella S and Curti A: The
immunotherapy landscape in AML: Defining knowledge gaps toward
rational combinatorial strategies. Semin Hematol. 62:209–217. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ehninger A, Kramer M, Röllig C, Thiede C,
Bornhäuser M, von Bonin M, Wermke M, Feldmann A, Bachmann M, et al:
Distribution and levels of cell surface expression of CD33 and
CD123 in acute myeloid leukemia. Blood Cancer J. 4:e2182014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Taussig DC, Pearce DJ, Simpson C,
Rohatiner AZ, Lister TA, Kelly G, Luongo JL, Danet-Desnoyers GH and
Bonnet D: Hematopoietic stem cells express multiple myeloid
markers: Implications for the origin and targeted therapy of acute
myeloid leukemia. Blood. 106:4086–4092. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kenderian SS, Ruella M, Shestova O,
Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, Song D, Porter
DL, et al: CD33-specific chimeric antigen receptor T cells exhibit
potent preclinical activity against human acute myeloid leukemia.
Leukemia. 29:1637–1647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tiwari A, Trivedi R and Lin SY: Tumor
microenvironment: Barrier or opportunity towards effective cancer
therapy. J Biomed Sci. 29:832022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Naik S, Aplenc R, Baumeister SHC, Becilli
M, Bhagwat AS, Bonifant CL, Budde LE, Chien CD, Curran KJ, Daniyan
AF, et al: International consensus guidelines for the conduct and
reporting of CAR T-cell clinical trials in AML. Blood Adv.
9:6047–6058. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kankeu Fonkoua LA, Sirpilla O, Sakemura R,
Siegler EL and Kenderian S: CAR T cell therapy and the tumor
microenvironment: Current challenges and opportunities. Mol Ther
Oncolytics. 19:69–77. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wicks EE and Semenza GL: Hypoxia-inducible
factors: Cancer progression and clinical translation. J Clin
Invest. 132:e1598392022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khanolkar RA and Dudley JC: CAR T cell
therapy and the tumor microenvironment: Current challenges and
opportunities. Mol Ther Oncolytics. 28:58–77. 2023.
|
|
44
|
Schito L and Semenza GL: Hypoxia-inducible
factors: Master regulators of cancer progression. Trends Cancer.
2:758–770. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fu YS, Li SX, Jiang H and Lin Z: Mechanism
and preclinical evaluation of cytokine release syndrome induced by
monoclonal antibodies. Chin J New Drugs. 33:1442–1448. 2024.(In
Chinese).
|
|
46
|
Boucher JC, Shrestha B, Vishwasroa P,
Leick M, Cervantes EV, Ghafoor T, Reid K, Spitler K, Yu B, et al:
Bispecific CD33/CD123 targeted chimeric antigen receptor T cells
for the treatment of acute myeloid leukemia. Mol Ther Oncolytics.
31:1007512023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ma H, Yan Z, Gu R, Xu Y, Qiu S, Xing H,
Tang K, Tian Z, Rao Q, Wang M and Wang J: Loop33 × 123 CAR-T
targeting CD33 and CD123 against immune escape in acute myeloid
leukemia. Cancer Immunol Immunother. 74:202024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hossain N, Sahaf B, Abramian M, Spiegel
JY, Kong K, Kim S, et al: Bispecific CD33/CD123 targeted chimeric
antigen receptor T cells for improved selective preclinical
treatment of acute myeloid leukemia. Leukemia. 38:127–138.
2024.
|
|
49
|
Xie D, Jin X, Sun R, Zhang M, Lu W, Cao X,
Guo R, Zhang Y and Zhao M: Bicistronic CAR-T cells targeting CD123
and CLL1 for AML to reduce the risk of antigen escape. Transl
Oncol. 34:1016952023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang XY, Bian MR, Lin GQ, Yu L, Zhang YM
and Wu DP: Tandem bispecific CD123/CLL-1 CAR-T cells exhibit
specific cytolytic effector functions against human acute myeloid
leukaemia. Eur J Haematol. 112:83–93. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dias J, Garcia J, Agliardi G and Roddie C:
CAR-T cell manufacturing landscape-lessons from the past decade and
considerations for early clinical development. Mol Ther Methods
Clin Dev. 32:1012502024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pavlovic K, Carmona-Luque M, Corsi GI,
Maldonado-Pérez N, Molina-Estevez FJ, Peralbo-Santaella E,
Cortijo-Gutiérrez M, Justicia-Lirio P, Tristán-Manzano M,
Ronco-Díaz V, et al: Generating universal anti-CD19 CAR T cells
with a defined memory phenotype by CRISPR/Cas9 editing and safety
evaluation of the transcriptome. Front Immunol. 15:14016832024.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lonez C and Breman E: Allogeneic CAR-T
therapy technologies: has the promise been met? Cells. 13:1462024.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu Z, Wang Y, Jin X and Wang L: Universal
CAR cell therapy: Challenges and expanding applications. Transl
Oncol. 51:1021472025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cheever A, Kang CC, O'Neill KL and Weber
KS: Application of novel CAR technologies to improve treatment of
autoimmune disease. Front Immunol. 15:14651912024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Anderson GSF, Walker I, Roy JP and Chapman
MA: And-gate CAR T-cells to improve tumour specificity and
targeting of low-expression antigens in multiple myeloma. Blood.
142 (Suppl 1):S7512023. View Article : Google Scholar
|
|
57
|
Nolan-Stevaux O and Smith R: Logic-gated
and contextual control of immunotherapy for solid tumors:
Contrasting multi-specific T cell engagers and CAR-T cell
therapies. Front Immunol. 15:14909112024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hamieh M, Mansilla-Soto J, Rivière I and
Sadelain M: Programming CAR T cell tumor recognition: Tuned antigen
sensing and logic gating. Cancer Discov. 13:829–843. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ahmadi M, Putnam N, Dotson M, Hayoun D,
Padilla J, Fatima N, Bhanap P, Nonterah G, de Mollerat du Jeu X and
Ji Y: Accelerating CAR T cell manufacturing with an automated
next-day process. Curr Res Transl Med. 73:1034892025.PubMed/NCBI
|














