Introduction
5-Fluorouracil (5-FU), a potent antitumor drug
(1), is used for treatment of
tumors such as colorectal (2),
breast (3) and liver carcinomas
(4). The main cytotoxic effects are
likely to be mediated by the inhibition of thymidylate synthetase,
a key enzyme that promotes the de novo synthesis of
thymidylic acid, which leads to the formation of desoxythymidine
triphosphate (dTTP) (5).
Furthermore, 5-FU influences the processing of mRNA, rRNA and the
interaction of t-RNA with the DNA primase (6). More recent studies have revealed
another p53-dependent mechanism of 5-FU action. 5-FU treatment
enhances expression of the ferredoxin reductase gene and protein,
and this contributes to oxidative stress-mediated apoptosis
(7,8).
Ferredoxin reductase (protein, FR; gene, FDXR) is
part of an electron transport system and is involved in the
synthesis of steroid hormones by mitochondrial cytochrome P450scc.
Here, FR catalyzes the rate limiting step namely the conversion of
cholesterol to pregnenolone. The required electrons (9,10) are
transferred from NADPH to the flavoprotein ferredoxin reductase and
the iron-sulfur protein adrenodoxin which mediates the electron
transport to all mitochondrial forms of cytochrome P450 (11,12).
Further studies indicate that the mitochondrial P450 systems can
leak electrons and produce O2-derived free radicals.
Reactive oxygen species (ROS), such as H2O2
or superoxide radicals, are known to be harmful to cells by
damaging nucleic acids and proteins (13). Several groups have proposed that a
feed forward loop beginning with 5-FU causes ROS-mediated cell
damage and the stabilization of p53 as a mechanism for apoptosis.
Subsequently, p53 induces expression of the FDXR mRNA and formation
of ferredoxin reductase (FR), which sensitizes cells to
ROS-mediated apoptosis (7). It was
found that FDXR can be up-regulated by a p53 mutant that is
competent in inducing apoptosis, but not by a tumor-derived p53
mutant that is defective in transactivation, which suggests that
FDXR is a potential mediator of p53-dependent apoptosis (8).
These studies clearly emphasize that, in addition to
the biosynthesis of steroid hormones, FR sensitizes cancer cells to
ROS-induced apoptosis. Two variants of the FDXR mRNA are known. The
difference is based on a 18-bp deletion, which arises from a
passive non-regulated alternative splicing at the 5′-end of exon 7
(9). This region is directly
adjacent to the NADPH binding site located in exon 6 (14). Reductase activity is associated only
with the −18-variant (15), since
the additional 6 amino acids probably disrupt the second β-sheet of
the ferredoxin reductase βαβ-structure (14). The inactive variant (+18-variant) is
less abundant, and constant ratios (100:1) have been reported for a
variety of organs, such as fetal and adult adrenal as well as the
testis. In tissues such as term placenta, fetal liver, kidney,
lung, brain, muscle and heart, the +18-form is barely detectable.
An impact of chemotherapeutic drugs such as 5-FU on this ratio has
not been reported, neither for cell lines nor for tissues.
Therefore, we studied the impact of 5-FU on the splice variants of
FDXR in normal intestinal and tumor tissue from 40 advanced
colorectal cancer patients which had been treated with the 5-FU
prodrug doxifluridine (5′DFUR).
Materials and methods
Forty patients (n=24 males, n=16 females; mean age
65 years, range 46–75 years) with advanced colorectal carcinoma but
without serious complications were treated with 5′DFUR for 14 days,
as described (16). Briefly, the
administration of 5′DFUR (800–12,000 mg/day) was halted the day
before surgery. The average dose administered to each patient was
1.3 g 5′DFUR. Two weeks after the curative surgery, treatment with
5′DFUR (600 mg/day) was restarted and continued for 2 years or for
the remaining life span of the patient.
Tissue collection
Samples for histological diagnosis and RNA
extraction were taken from tumor and the surrounding normal tissue
of each patient prior to and after treatment. Specimens for RNA
isolation were immersed immediately in Isogen
(phenol/guanidine-isothiocyanate) and stored at −80°C until RNA
extraction.
RNA extraction and synthesis of cDNA by
reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted (17), and the RNA content was quantified
spectrophotometrically. The integrity of the RNA was judged by
electrophoresis on 1.2% denaturing formaldehyde gels. The mixture
(50 μl) for the cDNA included 2 μg total RNA, 3 μg/μl random
hexamers, 60 units ribonuclease inhibitor, 10 mM dNTP mix, 0.1 M
DTT, 250 mM Tris-HCl and 100 units Mu-MLV reverse transcriptase
(Gibco, Rockville, MD, USA). Synthesis was performed at 37°C for 60
min, and the cDNA was stored in aliquots at −20°C.
Quantitative real-time RT-PCR
(q-PCR)
For quantitative real-time RT-PCR, analysis was
performed using glass capillaries in the LightCycler™ instrument
(Roche Diagnostics, Mannheim, Germany). The mRNA levels for the
splice variants (FDXR-18, FDXR+18) and dihydropyrimidine
dehydrogenase (DPD) were estimated by q-PCR. cDNA (diluted 1:10) (5
μl), equivalent to 20 ng of the initial RNA template, was used in
each PCR. Two independent RT reactions were prepared for each RNA,
and for each cDNA product, real-time PCRs were carried out in
duplicate for a total of 4 readings. Each run included reaction mix
controls with water in place of the template. As an additional
negative control, quantifications of reactions prepared in parallel
without addition of the RT enzyme were also run at least once for
each set of replicate RT reactions. For quantification of each
splice variant, cDNA (diluted 1:10) was amplified in a total volume
of 10 μl containing 3 mM MgCl2, specific 5′ and 3′
oligonucleotide primers (FDXR-18: 12.5 pmol each, forward: 5′-GAA
CGT GGC TCT GGA CGT, reverse 5′-TTC GTG ATG TCC GTT CT; FDXR+18: 25
pmol each, forward 5′-GAA CGT GGC TCT GGA CGT, reverse 5′ TGT CCG
TTC TCT GGC ACA A; DPD: 4 pmol each, forward 5′-CAG GAT CCA GAG CTG
GTG CG, reverse 5′-TCT TGC GAT GCT CAC AAT ATC AGT GAC) synthesized
by TIB MOLBIOL (Berlin, Germany) and 1 μl LightCycler-FastStart DNA
Master SYBR Green® I Kit, according to the
manufacturer's instructions. To reduce sample-to-sample
variability, results were calculated as ratios to DPD as a
housekeeping gene.
PCR grade water was added to a total volume of 20
μl. The temperature profiles consisted of an initial denaturation
step at 95°C (FDXR-18 for 10 min, FDXR+18 for 20 min, DPD for 1
min) followed by 40 amplification cycles [FDXR-18: denaturation at
95°C for 0 sec, annealing at 56°C for 5 sec, elongation at 72°C for
5 sec; FDXR+18: denaturation at 95°C for 10 sec, annealing at 64°C
for 10 sec, elongation at 72°C for 10 sec; DPD: denaturation at
95°C for 0 sec, annealing at 58°C for 15 sec, elongation at 72°C
for 15 sec. Ramp rates were set to 20°C/sec (except DPD elongation
at 3°C/sec)]. Melting curves (FDXR: 95°C for 0 sec, 45°C for 10
sec, 95°C for 0 sec; DPD: 95°C for 0 sec, 58°C for 30 sec, 95°C for
0 sec) were analyzed to confirm the specificity (FDXR-18: 85.1°C,
FDXR+18: 86.2°C, DPD: 67.6°C) of the PCR amplification. External
standards were used for the quantification of the unknown amounts.
Standards were generated by PCR amplification from cDNA templates
with the above-mentioned primers as follows. In a total volume of
50 μl, 5 μl cDNA, 10X PCR buffer containing 15 mM MgCl2,
4 μl 150 μM dNTPs, 40 pmol 5′ and 3′ oligonucleotides (each) and 1
unit Taq Gold Polymerase (Applied Biosystems, Weiterstadt, Germany)
were mixed. The experimental protocol included 12 min of initial
denaturation at 95°C, followed by 35 amplification cycles at 95°C
for 30 sec, 65°C for 30 sec, 72°C for 30 sec and a final period of
a 3-min elongation. After amplification, the DNA fragments were
purified with MicroSpin™ S-300 HR columns (Amersham Biosciences,
New York, NY, USA). The concentration of the respective DNA was
determined by measuring absorbance in a spectral photometer at 260
nm. Serial dilutions of the known DNA were used for standard
curves. We tested the efficiency of the primers used for
quantification of the splice variants with a second set of primers,
and found that the primer pair of the +18-variant was 3.72-fold
more efficient, resulting in a correction factor of 0.27. FDXR-18
and FDXR+18 mRNA levels were calculated as DPD ratios. Total FDXR
was determined from the amounts of the splice variants.
Histological evaluation of treatment
Histological evaluation of the effect of 5′DFUR
treatment was described by Tanaka-Nozaki et al (16). Survival was determined at time of
follow-up examination.
Statistical analysis
Significant differences between total FDXR, both
splice variants and between subgroups were estimated by using the
Student's t-test (SigmaPlot version 8.0). A significance level of
5% was established. Relations between total FDXR, −18-form and
+18-form, the ratio −18/+18 and pathological effects were
calculated by using correlation coefficients.
Results
Total FDXR mRNA levels and the
distribution of the two alternative spliced variants in intestinal
tissue
FDXR mRNA levels including the two splice variants
were measured in normal intestinal tissue and tumor tissue of 40
cancer patients before and after 5′DFUR treatment using a sensitive
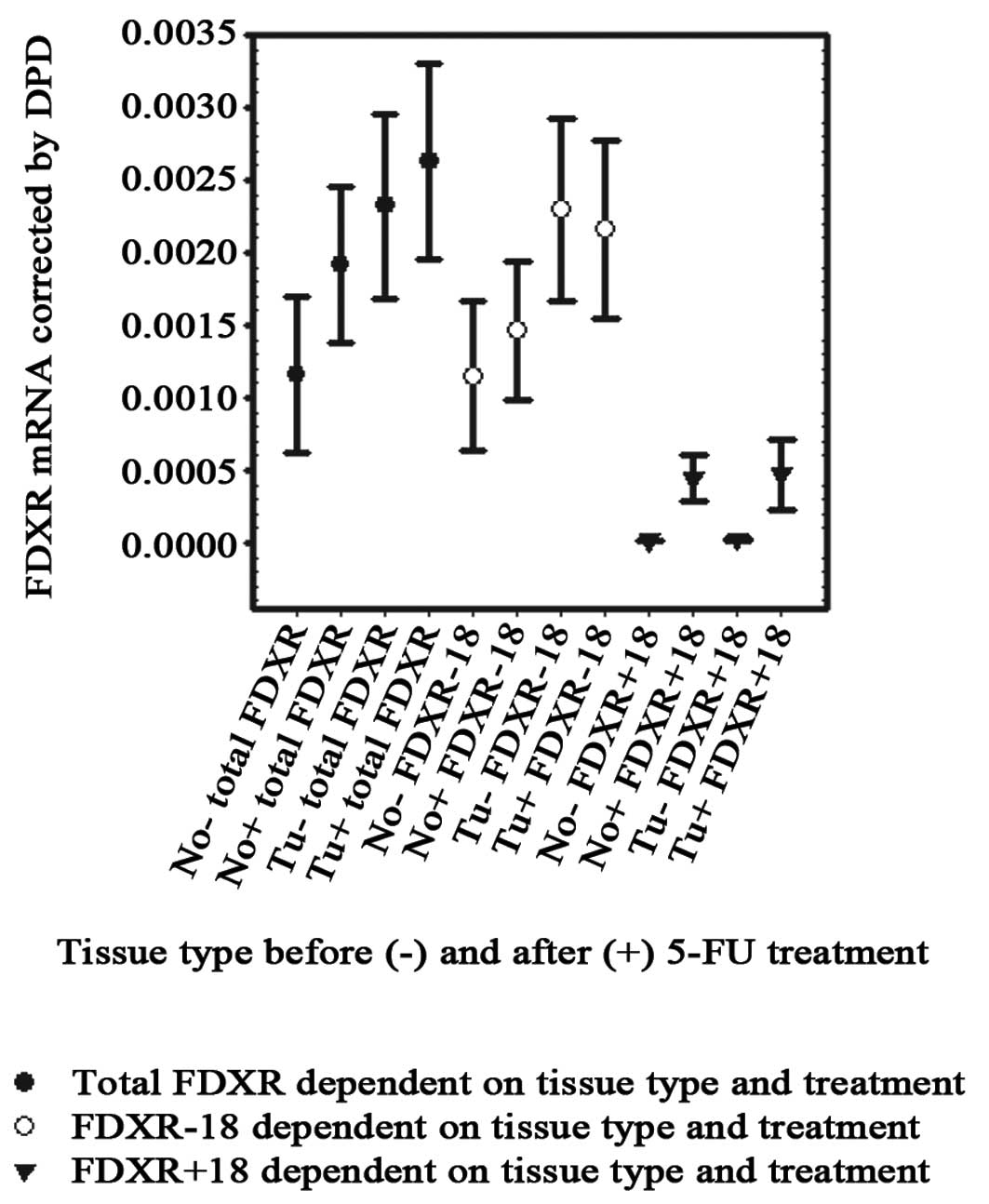
and specific quantitative RT-PCR assay. Fig. 1 summarizes the results. Total FDXR
mRNA levels (sum of variants) were 2-fold higher in tumor compared
to normal tissue. This nearly statistically significant difference
(p=0.051) was based on greater prominence of the −18-variant in
tumor tissue (mean 2.29×10−3) compared to normal tissue
surrounding the tumor (1.15×10−3, p=0.049). Similar to
reported results from other tissue, the +18-form was barely
detectable in intestinal tissue. Nevertheless, we found similar
trends for the invariant +18-form, reaching 1.85-fold higher levels
in tumors (p=0.053), while levels in non-treated tissues were
almost equal to the detection limits. After 5′DFUR treatment, the
levels for total FDXR mRNA were only slightly enhanced in normal
tissue (1.64-fold, p=0.181) while total levels in tumors were not
significantly increased (1.13-fold, p=0.638).
Earlier studies reported a constant 100:1 ratio in
favor of the −18-variant for various tissues (9). Using our more sensitive technique, we
evaluated this finding in tissue prior to treatment and found in
normal tissue (n=16 cases) and additionally confirmed in tumor
tissue (n=18 cases) even higher ratios (up to 1000:1). After 5′DFUR
treatment, the catalytic active −18-variant increased 1.28-fold in
normal tissue and decreased slightly (0.94-fold) in tumors. The
individual analysis clearly demonstrated that the variances were
based on 8 individual cases (n=37). In contrast, levels of the
+18-variant were clearly enhanced after 5′DFUR treatment; 31-fold
in normal tissue (p=0.019) and 17-fold in tumor tissue (p=0.071).
Thus, 5′DFUR treatment for 14 days changed the ratio towards the
+18-variant (Fig. 1), reaching
significant differences only in tumor tissue (p=0.0118 vs. p=0.0521
for normal tissue). Thus, the proposed 100:1 ratio was now found
only in 4 of 37 cases based on evaluation of normal tissue and in 6
of 38 cases based on tumor evaluation, while prior to treatment the
−18- and +18-variants differed significantly in normal (p=0.0356)
and tumor (p=0.0006) tissue.
Association of the FDXR steady state mRNA
level with treatment success and survival
FDXR enzyme activity sensitizes cells to ROS-induced
apoptosis. Thus, we speculated from our results that induction of
+18 mRNA splice variants might be associated with less successful
treatment, based on the histological evaluation of the tissue
sections or reduced survival time. Due to the small numbers in this
study, only preliminary data could be gained but the predicted
association (r=−0.62) between +18-variant mRNA induction and
treatment in a subgroup of female patients was found. Furthermore,
the increase in the +18-variant in normal tissue was also
associated with no or limited treatment success (5 cases).
Inversely, 8 of 14 successfully treated cases showed no or very
limited induction in tumor tissue (<1.3-fold), while 4 of 5
patients with a strong induction of this variant had no or only
minor treatment success.
Discussion
5-Fluorouracil (5-FU), one of the most useful
antitumor agents (1), is well-known
for its strong impact on different mRNAs (6). More recent in vitro data
(7) indicate that p53-dependent
apoptosis induced by 5-FU treatment is mediated by ferredoxin
reductase (FR). FR is located at the inner mitochondrial membrane
and involved in an electron shuttle via ferredoxin, which is free
in solution, to cytochrome P450 for the cholesterol sidechain
cleavage. Electrons might leave this system and are transferred to
other acceptors (18), thereby
generating reactive oxygen species (O2-radicals,
H2O2) followed by induction of apoptosis.
These data indicate that 5-FU induces overexpression of FDXR
mediated by p53 or via a p53-independent manner which sensitizes
tumor cells to ROS-induced apoptosis thereby overcoming apoptosis
inhibition commonly found in tumors (19).
To extend this knowledge to in vivo
circumstances, we estimated FDXR mRNA levels including the
bioactive −18 splice-variant and the inactive +18-variant in
colorectal tissue of 40 patients prior to and after 5-FU (5′DFUR)
treatment for 14 days. Treatment resulted in higher mRNA levels in
tumor (1.13-fold) and normal tissue (1.64-fold), but these trends
did not reach statistical significance. Hwang et al studied
the impact of 5-FU on FDXR expression in the colorectal cancer cell
line HCT 116 (7). They reported an
11-fold induction of total FDXR mRNA after 5-FU administration for
18 h. We confirmed their findings and extended them to human
tissue. Besides interindividual variations, also different time
points, 18 h vs. 14 days in this report, may have accounted for the
observed discrepancies. Comparing uninduced levels in tumor and
normal tissue, we found that the level of the splice variants was
approximately 2-fold higher in tumor than in normal tissue
(FDXR-18: 2-fold, FDXR+18: 1.85-fold). Since FR is involved in the
energy supply of cells, the observed higher level in tumor tissue
may reflect a higher demand for FDXR enzyme activities of tumors,
either based on increased transcription and/or stabilized mRNA in
tumor tissue.
Looking at the ratios between the splice variants,
we observed an overrepresentation of ratios between 10:1 and 1000:1
in contrast to the published fixed relation of 100:1 in favor of
the −18-variant. We speculate that these differences could be based
on the different methods used. Our wide range is most likely a
result of the very sensitive assay (quantitative RT-PCR) used in
this study. On the other hand, we cannot formally rule out that
levels may be different in intestinal tissue since no results have
been published to date for this tissue and tumor tissue of cancer
patients. Indeed, we found an even more unbalanced ratio in tumor
tissue, although it was reported that they are generated by a
non-regulated mechanism (9). After
treatment, the functional −18-variant was slightly underrepresented
in the tumors (0.94-fold), and somewhat increased values were found
in normal tissue (1.28-fold). In contrast, the level of the
inactive splice variant (+18-variant) increased drastically in
tumor (17-fold) and normal tissue (31-fold). Overall, constant
predominance of the −18-variant (100:1) was only found in 4 normal
tissues (n=37) and in 6 tumors (n=38). Thus, the −18/+18 ratio
shifted extremely to the +18-variant. Some cases showed even a
predominance of the +18-variant after treatment. This was found in
normal (n=10) and tumor tissue (n=2). We explored several
explanations for these results. Although not reported for the
splice process of FDXR, differential regulation of splicing may
account for our findings. Then, 5-FU interfered with the splicing
process causing the observed increase in the +18-variant. On the
other hand, the results could have been due to the increased
stability of the +18-variant. It has previously been reported that
binding elements in introns or alternative splicing at the
beginning of an exon such as in FDXR pre-mRNA may change the
stability of the +18 mRNA. In this context, the induced apoptosis
and cell death actively reduce levels of the FDXR-18 mRNA (20).
To this end, we speculated that induction of the
nonfunctional +18-variant might be associated with nonsuccessful
treatment. We found that high +18-variant levels were inversely
associated with successful treatment based on histological
evaluation of specimens (r=−0.62); while no or a minor induction
(<1.3) was associated with successful treatment (8 of 14
patients). From these results, it can be speculated that the
association of the +18-form with ineffective treatment may depend
on the inability to generate oxidative stress. The alternative
splicing at the end of exon 7 directly adjacent to the
NADPH-binding site of FR would probably disrupt the second β-sheet
of the ferredoxin reductase βαβ-structure of the NADPH-binding site
(8). Consequently, binding of NADPH
and the substrate hydroxylation and otherwise an induction of
apoptosis via the leak of electrons for generation of O2
radicals (18) would not be
possible for the +18-form. In addition, it has been shown that the
+18-form did not exhibit reductase activity (15). Further efforts may clarify whether
the +18-form is really unable to produce oxidative stress and to
induce apoptosis and may also elucidate the process of 5-FU-induced
+18-variant expression.
Abbreviations:
|
RT
|
reverse transcriptase
|
|
PCR
|
polymerase chain reaction
|
|
q-RT-PCR
|
quantitative reverse transcriptase
polymerase chain reaction
|
|
5-FU
|
5-fluorouracil
|
|
FDXR
|
ferredoxin reductase gene
|
|
FR
|
ferredoxin reductase protein
|
|
ROS
|
reactive oxygen species
|
|
5′DFUR
|
5-FU prodrug doxifluridine
|
References
|
1
|
Heidelberger C, Chaudhuri NK, Danneberg P,
et al: Fluorinated pyrimidines, a new class of tumour-inhibitory
compounds. Nature. 179:663–666. 1957. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kelder W, Hospers GA and Plukker JT:
Effects of 5-fluorouracil adjuvant treatment of colon cancer.
Expert Rev Anticancer Ther. 6:785–794. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Navolanic PM and McCubrey JA:
Pharmacological breast cancer therapy (Review). Int J Oncol.
27:1341–1344. 2005.PubMed/NCBI
|
|
4
|
Patt YZ, Hassan MM, Lozano RD, Brown TD,
Vauthey JN, Curley SA and Ellis LM: Phase II trial of systemic
continuous fluorouracil and subcutaneous recombinant interferon
alpha-2b for treatment of hepatocellular carcinoma. J Clin Oncol.
21:421–427. 2003. View Article : Google Scholar
|
|
5
|
Santi DV, McHenry CS and Sommer H:
Mechanism of interaction of thymidylate synthetase with
5-fluorodeoxyuridylate. Biochemistry. 13:471–481. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parker WB and Cheng YC: Metabolism and
mechanism of action of 5-fluorouracil. Pharmacol Ther. 48:381–395.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hwang PM, Bunz F, Yu J, et al: Ferredoxin
reductase affects p53-dependent, 5-fluorouracil-induced apoptosis
in colorectal cancer cells. Nat Med. 7:1111–1117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu G and Chen X: The ferredoxin reductase
gene is regulated by the p53 family and sensitizes cells to
oxidative stress-induced apoptosis. Oncogene. 21:7195–7204. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brentano ST, Black SM, Lin D and Miller
WL: cAMP post-transcriptionally diminishes the abundance of
adrenodoxin reductase mRNA. Proc Natl Acad Sci USA. 89:4099–4103.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miller WL: Molecular biology of steroid
hormone synthesis. Endocr Rev. 9:295–318. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jefcoate CR, McNamara BC and DiBartolomeis
MJ: Control of steroid synthesis in adrenal fasciculata cells.
Endocr Res. 12:315–350. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Solish SB, Picado-Leonard J, Morel Y, Kuhn
RW, Mohandas TK, Hanukoglu I and Miller WL: Human adrenodoxin
reductase: two mRNAs encoded by a single gene on chromosome
17cen----q25 are expressed in steroidogenic tissues. Proc Natl Acad
Sci USA. 85:7104–7108. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chandra J, Samali A and Orrenius S:
Triggering and modulation of apoptosis by oxidative stress. Free
Radic Biol Med. 29:323–333. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin D, Shi YF and Miller WL: Cloning and
sequence of the human adrenodoxin reductase gene. Proc Natl Acad
Sci USA. 87:8516–8520. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brandt ME and Vickery LE: Expression and
characterization of human mitochondrial ferredoxin reductase in
Escherichia coli. Arch Biochem Biophys. 294:735–740. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tanaka-Nozaki M, Tajiri T, Tanaka N, et
al: Intratumoral induction of thymidylate synthase mRNA by 5-FU in
colorectal cancer patients: association with survival. Oncol Rep.
10:1425–1429. 2003.PubMed/NCBI
|
|
17
|
Chomczynski P and Sacchi N: Single-step
method of RNA isolation by acid guanidinium
thiocyanate-phenol-chloroform extraction. Anal Biochem.
162:156–159. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanukoglu I, Rapoport R, Weiner L and
Sklan D: Electron leakage from the mitochondrial NADPH-adrenodoxin
reductase-adrenodoxin-P450scc (cholesterol side chain cleavage)
system. Arch Biochem Biophys. 305:489–498. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bedi A, Pasricha PJ, Akhtar AJ, et al:
Inhibition of apoptosis during development of colorectal cancer.
Cancer Res. 55:1811–1816. 1995.PubMed/NCBI
|
|
20
|
Nott A, Meislin SH and Moore MJ: A
quantitative analysis of intron effects on mammalian gene
expression. RNA. 9:607–617. 2003. View Article : Google Scholar : PubMed/NCBI
|