Introduction
The epidermal growth factor receptor (EGFR) is a
transmembrane glycoprotein with an extracellular EGF-binding domain
and an intracellular domain possessing intrinsic tyrosine kinase
activity (1,2). Ligand binding activates the receptor's
tyrosine kinase, initiating cascades of intracellular signaling
such as those via the Ras protein (3). High levels of EGFR expression have
been reported in a wide range of human malignancies (4–6) and
enhanced expression of EGFR has previously been shown in non-small
cell lung cancer (NSCLC) (7).
Since it was reported that EGFR overexpression is a
factor of poor prognosis (8,9),
treatments targeting EGFR would be expected to show survival
benefits. Erlotinib (Tarceva®) is an oral, small
molecule tyrosine kinase inhibitor that reversibly binds to the
intracellular domain of EGFR. This blocks autophosphorylation of
EGFR with subsequent inhibition of the downstream signaling
pathways which promote cell proliferation. Erlotinib is used for
metastatic NSCLC and pancreatic cancer in many countries. Clinical
results have demonstrated that erlotinib monotherapy or combination
therapy with gemcitabine showed a survival benefit for NSCLC or
pancreatic cancer, respectively (10,11).
However, most of these patients developed progressive disease (PD)
during such therapies and it is usually considered best to switch
to chemomonotherapy after developing PD. It is reported that the
major mechanisms of erlotinib resistance are gatekeeper mutation
(T790M) of EGFR and c-Met amplification (12,13) in
tumor cells. On the other hand, it is reported that the tumor cells
express active EGFR even after acquiring resistance to erlotinib
(13,14). Considering that EGFR overexpression
is a factor of poor prognosis, discontinuing erlotinib treatment
after PD has developed may be an inappropriate option and combining
erlotinib with the next stage of chemotherapy may be an appropriate
therapy. We have previously reported that the combination of
docetaxel with erlotinib showed a synergistic effect in NSCLC cell
lines in vivo irrespective of EGFR or K-RAS mutation status
(15).
Therefore, we investigated the antitumor effect of
combination therapies of erlotinib with various chemotherapeutic
agents docetaxel, irinotecan and gemcitabine, using
erlotinib-resistant tumor cell xenografts as well as an in
vivo erlotinib PD xenograft model, to show the clinical
relevance of continuing erlotinib treatment after development of
PD.
Materials and methods
Chemicals
Erlotinib was provided by F. Hoffman-La Roche
(Basel, Switzerland) as a fine powder and was dissolved in
distilled water containing 6% (w/v) Captisol (CyDex
Pharmaceuticals, KS, USA) and diluted with saline for in
vivo experiments. Erlotinib was dissolved in DMSO for in
vitro experiments. Docetaxel was synthesized by Kanto Chemical
Co., Inc. (Tokyo, Japan) as a fine powder and was dissolved in
saline containing 2.5% (v/v) polysorbate 80 (Sigma-Aldrich Co.,
USA) and 2.5% (v/v) ethanol for in vivo experiments.
Irinotecan was purchased from Daiichi Sankyo Pharmaceutical Co.,
Ltd. (Tokyo, Japan) as an aqueous solution and diluted with
saline.
Animals
Male 5-week-old BALB-nu/nu mice
(CAnN.Cg-Foxn1<nu>/CrlCrlj nu/nu) were obtained from Charles
River Japan (Kanagawa, Japan). All animals were allowed to
acclimatize and recover from shipping-related stress for 1 week
prior to the study. The health of the mice was monitored by daily
observation. Chlorinated water and irradiated food were provided
ad libitum, and the animals were kept in a controlled
light-dark cycle (12 h-12 h). The protocol was reviewed by the
Institutional Animal Care and Use Committee of Chugai
Pharmaceutical Co., Ltd., and all mouse experiments were performed
in accordance with the Guidelines for the Accommodation and Care of
Laboratory Animals promulgated in Chugai Pharmaceutical Co.,
Ltd.
Tumor cells
Human non-small cell lung cancer (NSCLC) cell lines,
HCC827 (exon 19 deletion EGFR) and H1975 (T790M mutation in EGFR),
and human pancreatic cancer cell line, HPAC (wild-type EGFR), were
obtained from the American Type Culture Collection. Human NSCLC
cell line, EBC-1 (c-Met-amplification) was obtained from the
RIKEN BRC (Ibaraki, Japan). Erlotinib-resistant cell line HCC827TR3
was established in-house by exposing HCC827 cells to increasing
concentrations of erlotinib in vitro. The HCC827, HCC827TR3
and H1975 cells were maintained at 37°C under 5% CO2 in
RPMI-1640 medium (Sigma-Aldrich Co.) containing 10% FBS, 10 mM
HEPES, 1 mM sodium pyruvate, and 4.5 g/l glucose. The HPAC cell
line was maintained in DMEM: Ham's F12 combined medium (1:1)
(Invitrogen, USA) containing 5% FBS, 2 μg/ml insulin, 5 μg/ml
transferrin, 40 ng/ml hydrocortisone, and 10 ng/ml EGF. The EBC-1
was maintained in EMEM (Sigma-Aldrich Co.) containing 10% FBS.
Evaluation of antitumor activity
Study 1
HCC827TR3, EBC-1, H1975 xenograft models and
treatment. A suspension of tumor cells (5×106
cells/mouse) was inoculated subcutaneously into the right flank of
mice. Tumors were allowed to reach 0.1–0.3 cm3 in size,
mice were randomly allocated to the control group, erlotinib group,
chemotherapy group and combination of erlotinib with chemotherapy
group and these were treated with vehicle of erlotinib and vehicle
of chemotherapy, erlotinib and vehicle of chemotherapy, vehicle of
erlotinib and chemotherapy, or erlotinib and chemotherapy,
respectively. Erlotinib was administered orally (p.o.) once a day
from Day 2. Docetaxel was administered intravenously (i.v.) once in
3 weeks (Day 1). Irinotecan was administered intravenously (i.v.)
once in 2 weeks (Day 1). To evaluate the antitumor effect and
tolerability, tumor volume and body weight were measured twice a
week. The tumor volume (V) was estimated from the equation V =
ab2/2, where a and b were tumor length and width,
respectively.
Study 2
Establishment of in vivo erlotinib PD model
and treatment. To establish an in vivo erlotinib PD model, a
suspension of HPAC cells (5×106 cells/mouse) was
inoculated subcutaneously into the right flank of the mice. Tumors
were allowed to reach 0.1–0.3 cm3 in size, mice were
randomly allocated to control and erlotinib groups. Erlotinib was
administered orally (p.o.) once a day starting from Day 1 to Day
18.
After establishment of PD during erlotinib treatment
was confirmed, mice were re-randomized and allocated to the control
group, erlotinib group, gemcitabine group, and combination of
gemcitabine with erlotinib group and these were treated with
vehicle of erlotinib and vehicle of gemcitabine, erlotinib and
vehicle of gemcitabine, vehicle of erlotinib and gemcitabine, or
erlotinib and gemcitabine, respectively. Erlotinib was administered
orally (p.o.) on Days 21–25, 28–32, 35–40. Gemcitabine was
administered i.v. once a week (on Days 20, 27 and 34). To evaluate
the antitumor effect and tolerability, tumor volume and body weight
were measured twice a week. The tumor volume (V) was estimated from
the equation V = ab2/2, where a and b were tumor length
and width, respectively.
Western blotting
Cells (HCC827, HCC827TR3, EBC-1 and H1975) were
seeded into 6-well plates at a concentration of 5×105
cells per well and were preincubated overnight. Then, erlotinib was
added and incubation continued for 2 h. Cells were stimulated with
100 ng/ml of EGF (Invitrogen) for the last 15 min of the
incubation. HPAC tumor tissues of the in vivo PD model were
pulverized in liquid nitrogen. Cellular total protein was prepared
from cell lysates and the pulverized frozen tumors. Proteins (100
μg each of HPAC, EBC-1 and H1975; 5 μg each of HCC827 and
HCC827TR3) were electrophoresed on SDS-PAGE with 7.5% gel and
transferred onto PVDF membranes (GE Healthcare Japan, Tokyo,
Japan). The membranes were blocked with a blocking buffer (Thermo
Fisher Scientific, Kanagawa, Japan), immunoblotted with primary
antibody against EGFR (Santa Cruz Biotechnology Inc., CA, USA),
pY1068 pEGFR (Cell Signaling Technology Inc.) and GAPDH (Santa Cruz
Biotechnology Inc.). The protein-antibody complex was detected by
chemiluminescence (GE Healthcare Japan).
Cell proliferation assay
Cells were seeded at a density of 1000 or 3000
cells/well in 96-well plates and were preincubated overnight. The
cells were then treated with erlotinib for 96 h. Cell proliferation
was evaluated by Cell Counting Kit-8 (Dojindo, Kumamoto,
Japan).
Statistical analysis
Statistical analysis to evaluate the antitumor
activity was performed using the Mann-Whitney U test. For in
vitro experiments, Student's t-test was used. Differences were
considered to be significant at P≤0.05. Statistical analysis was
carried out using the SAS preclinical package (SAS Institute, Inc.,
Tokyo, Japan).
Results
Erlotinib sensitivity, EGFR expression
and effect of erlotinib on phosphorylation of EGFR and downstream
signaling molecules in erlotinib-resistant NSCLC cells
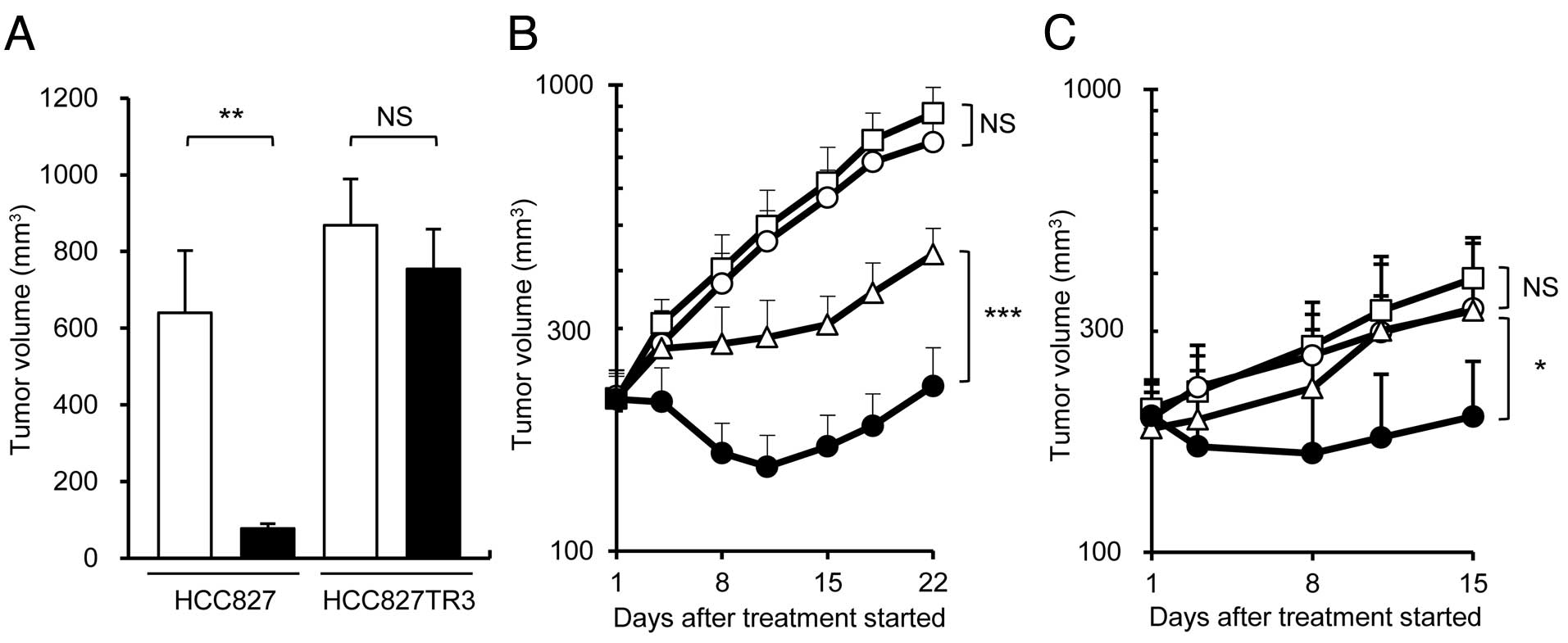
First, we examined the growth inhibition of tumor
cells, namely HCC827, HCC827TR3, EBC-1 and H1975. HCC827TR3 was
1000 times more resistant to erlotinib than parental HCC827
(Fig. 1A) in vitro. We found
that the mechanism of erlotinib resistance of HCC827TR3 was neither
c-Met amplification nor T790M mutation in EGFR (data not
shown). Almost no growth inhibition was observed in EBC-1 and H1975
cells up to 3 μmol/l of erlotinib (Fig.
1B). Next, we examined EGFR expression in the tumor cells and
the effect of erlotinib on the phosphorylation of EGFR, as well as
its major downstream signal molecules such as Akt, ERK, Stat3, by
Western blotting. All of the cell lines expressed EGFR and
phosphorylated EGFR (Fig. 1C). The
EGFR phosphorylation was completely suppressed by erlotinib in
HCC827, HCC827TR3 and EBC-1, although erlotinib did not inhibit the
proliferation of HCC827TR3 and EBC-1. On the other hand, erlotinib
did not suppress the phosphorylation of EGFR in H1975 cells
(Fig. 1C). Erlotinib suppressed the
phosphorylation of Akt and ERK in HCC827 cells. However, out of the
three erlotinib-resistant cell lines, only a slight inhibition of
ERK phosphorylation in HCC827TR3 was observed (Fig. 1C).
Antitumor effect of combination therapy
of chemotherapeutic agents with erlotinib in erlotinib-resistant
tumor xenografts
Because EGFR phosphorylation was suppressed by
erlotinib in the erlotinib-resistant cells (EBC-1, HCC827TR3), it
may be of value to administer erlotinib concurrently with a
chemotherapeutic agent when treating erlotinib-resistant tumors.
Therefore, we next examined the antitumor activity of combination
therapy of a chemotherapeutic agent with erlotinib against these
erlotinib-resistant cell lines in xenografts.
First, we examined the antitumor effect of docetaxel
monotherapy and docetaxel + erlotinib therapy using the HCC827TR3
xenograft model. In this model, erlotinib monotherapy did not show
any antitumor effect even at a dose of 25 mg/kg, which was higher
than the effective dose for parental HCC827 xenograft model
(Fig. 2A). However, docetaxel in
combination with erlotinib showed a significantly higher antitumor
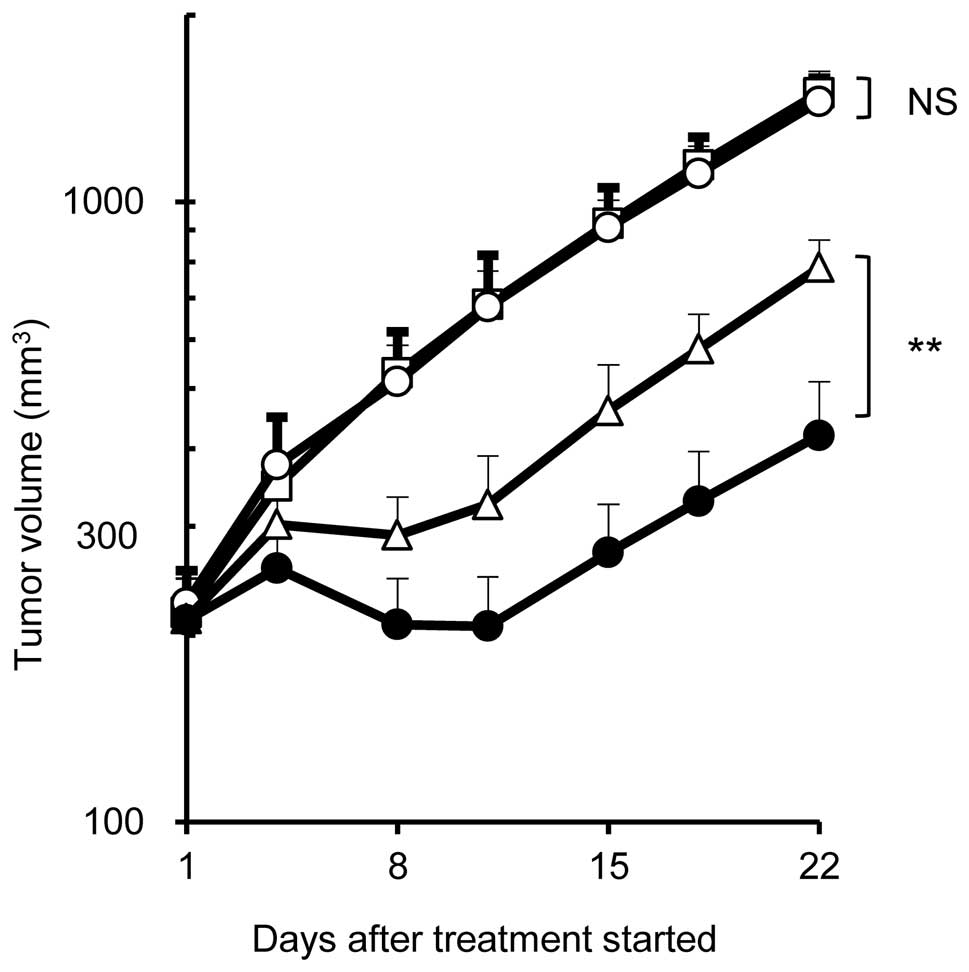
activity compared with docetaxel monotherapy (Fig. 2B). A similar result was obtained in
the combination therapy of irinotecan with erlotinib in the same
xenograft model (Fig. 2C). In the
EBC-1 xenograft model, similarly, significantly higher antitumor
effect was obtained in the combination therapy of docetaxel (5
mg/kg) with erlotinib (75 mg/kg) compared to docetaxel monotherapy
whereas erlotinib did not show any antitumor effect at the same
dose (Fig. 3). Namely, the
combination therapy of chemotherapeutic agent with erlotinib showed
a significantly higher antitumor effect compared with
chemomonotherapy while erlotinib monotherapy showed no effect in
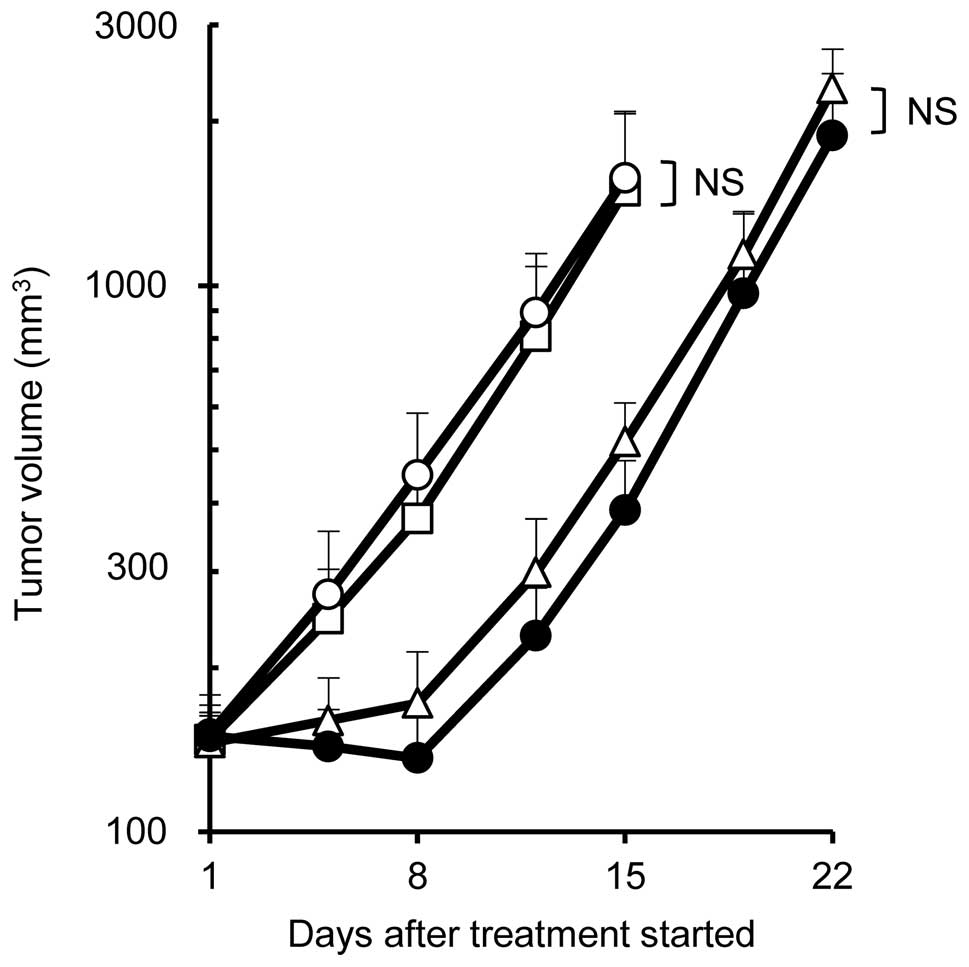
HCC827TR3 or EBC-1 xenografts. On the other hand, no significant
effect was seen between docetaxel monotherapy (5 mg/kg) and
combination of docetaxel (5 mg/kg) with erlotinib (75 mg/kg) in the
H1975 xenograft model (Fig. 4).
 | Figure 2Antitumor effect in parental HCC827
and resistant HCC827TR3 xenograft models. (A) Erlotinib monotherapy
at Day 22. □, control; ■, erlotinib 15 mg/kg (HCC827), 25 mg/kg
(HCC827TR3), n=5/group. (B) Combination therapy of docetaxel with
erlotinib in HCC827TR3 xenograft model. □, control; ○, erlotinib 25
mg/kg; ▵, docetaxel 20 mg/kg; ●, combination, n=7/group. (C)
Combination therapy of irinotecan with erlotinib in HCC827TR3
xenograft model. □, control; ○, erlotinib 25 mg/kg; ▵, irinotecan
60 mg/kg; ●, combination, n=5/group. Statistically significant
differences are shown. NS, not significant; *P≤0.05,
**P≤0.01, ***P≤0.001 by Wilcoxon test. |
Establishment of in vivo
erlotinib-resistant model and antitumor activity of gemcitabine in
combination with erlotinib
To mimic the clinical PD phenomenon and examine the
effect of combination therapy of docetaxel with erlotinib, we
established an in vivo erlotinib-resistant model using
EGFR-positive pancreatic cancer cell line HPAC. The HPAC cells were
subcutaneously inoculated into BALB/c-nu/nu mice, and erlotinib (75
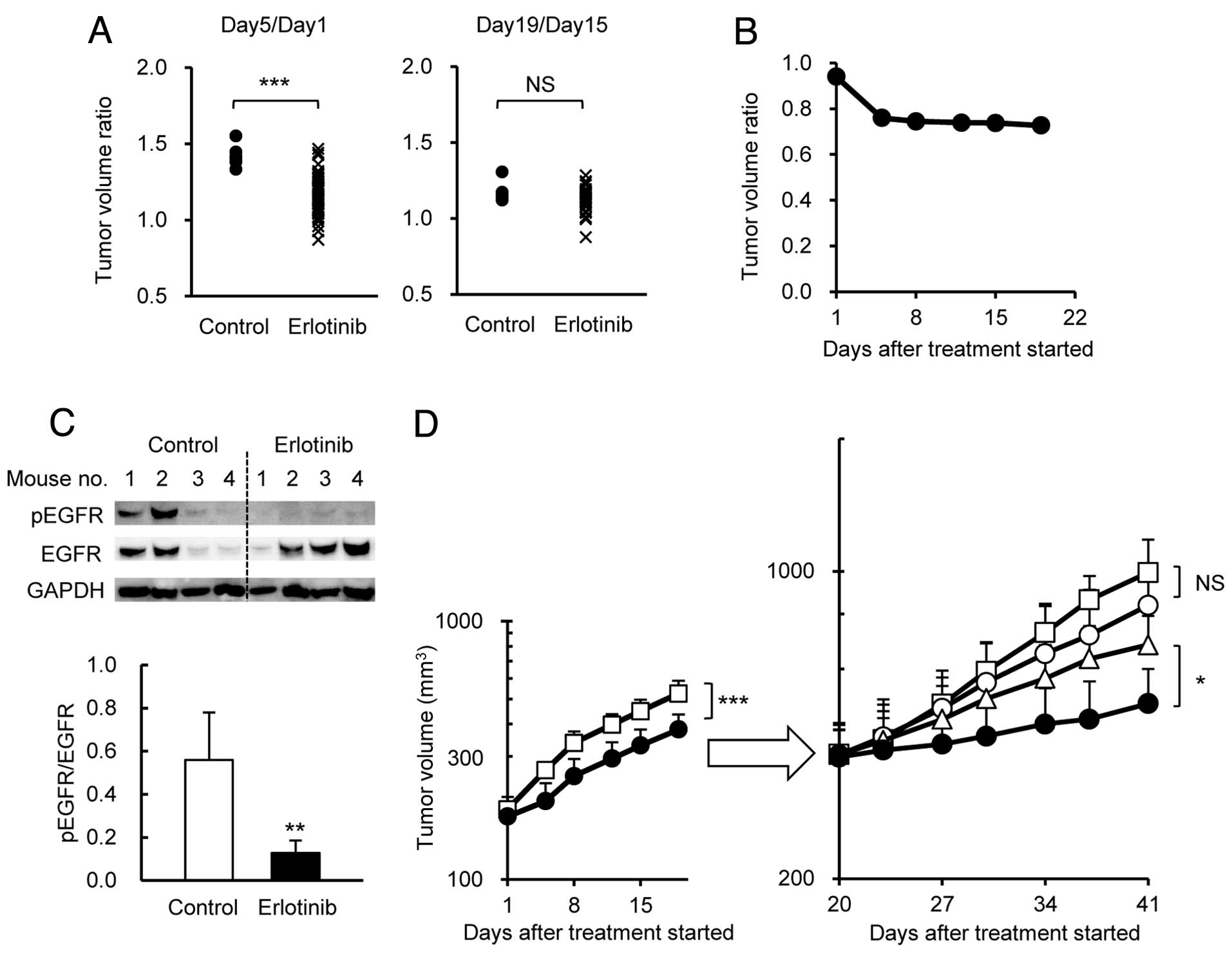
mg/kg) was administered p.o. once a day for 18 days. In this model,
erlotinib significantly inhibited tumor growth up to 5 days after
the start of administration (Fig.
5A). Subsequently, however, the tumor growth inhibition effect
by erlotinib disappeared, even though erlotinib was continuously
administered (Fig. 5A). Fig. 5B shows the constant tumor volume
ratio of erlotinib group to vehicle group after around Day 8. On
Day 20, the mice in the erlotinib group were randomly allocated to
4 groups, namely, vehicle group, erlotinib group, gemcitabine
group, and gemcitabine + erlotinib group. Although EGFR protein
remained positive and its phosphorylation had been substantially
reduced by erlotinib by Day 21 (Fig.
5C), the erlotinib group did not show significant tumor growth
inhibition compared with the vehicle group (Fig. 5D). This indicated that the HPAC
tumors had become resistant to erlotinib.
Using this model, we examined the antitumor activity
of combination therapy of gemcitabine (25 mg/kg) with erlotinib (75
mg/kg). The results indicated that the combination therapy showed a
significant antitumor effect compared with gemcitabine monotherapy
(Fig. 5D) even though erlotinib
monotherapy showed no tumor inhibitory effect.
Discussion
By using two types of tumor models, we were able to
investigate the mechanism by which NSCLC and pancreatic cancer
become resistant to erlotinib. Although EBC-1 and H1975 show
amplification of c-Met and mutation of T790M, respectively,
HCC827TR3, which was established in-house, has neither. In the
HCC827TR3 cells, neither EGFR down-regulation nor reduction of EGFR
phosphorylation was observed (Fig.
1C). The fact that EGFR phosphorylation was inhibited by
erlotinib in HCC827TR3 cells but the PI3K pathway was not inhibited
and the Ras-ERK/MAPK pathway only partially inhibited (Fig. 1C) indicates that the resistance
mechanism may be the activation of these pathways by protein
kinase(s) other than c-MET.
Erlotinib completely inhibited EGFR phosphorylation
in EBC-1 and HCC827TR3 cells but not in H1975 cells (Fig. 1C). This coincides well with the
previous reports (12,13,14)
which state that, in cells with c-Met amplification,
erlotinib resistance is activated in the cell growth signaling
pathway through heterodimer formation of MET and HER3 molecules.
Thus, EGFR remains intact in c-Met amplification cells such
as EBC-1, and erlotinib is able to inhibit EGFR phosphorylation. In
the case of HCC827TR3, although the precise mechanism of resistance
is not yet clear, it would seem that EGFR phosphorylation was
inhibited by a similar mechanism. On the other hand, erlotinib
could not inhibit EGFR phosphorylation in H1975 cells because the
T790M mutation in EGFR causes a conformation change at the ATP
binding pocket, thus decreasing the affinity between erlotinib and
EGFR.
Since all of the erlotinib-resistant cell lines
express EGFR, we examined the antitumor effect of combination
therapy of docetaxel with erlotinib or irinotecan. In these models,
erlotinib monotherapy did not show significant antitumor effect
compared with the control group (Figs.
2A, 3 and 4). Interestingly, however, combination
therapy of docetaxel with erlotinib showed a synergistic effect in
HCC827TR3 (Fig. 2B) and EBC-1
(Fig. 3) xenografts. A similar
result was obtained in HCC827TR3 xenografts using irinotecan as a
chemotherapeutic agent (Fig. 2C).
These results may indicate that the chemotherapeutic agent used in
the combination therapy need not be restricted to a specific drug.
On the other hand, no significant increase of antitumor effect of
combination therapy compared with docetaxel monotherapy was
observed in H1975 xenografts (Fig.
4). These results coincide well with the report of Okabe et
al in which gefitinib and S-1 were used in combination in H1975
and HCC827GR5 xenografts (16).
Since EGFR phosphorylation was completely inhibited by erlotinib in
HCC827TR3 cells and EBC-1 cells but not in H1975 cells (Fig. 1C), it is possible that inhibition of
EGFR phosphorylation is prerequisite for the combination therapy to
be effective. EGFR phosphorylation activates signal transduction
pathways, such as PI3K and Ras-ERK/MAPK, and erlotinib inhibits
these pathways. However, the role of erlotinib in combination
therapy in erlotinib-resistant xenograft models may be inhibition
of signal pathway(s) other than the PI3K or Ras-ERK/MAPK pathways,
because erlotinib monotherapy did not show any antitumor effect in
HCC827TR3 and EBC-1 xenografts. In the H1975 xenograft model,
erlotinib failed to inhibit EGFR phosphorylation (Fig. 1C) hence the antitumor effect of
combination therapy was not enhanced. Okabe et al reported
that the combination effect of S-1 with gefitinib was attributed to
the down-regulation of thymidylate synthase (TS) by gefitinib and
the mechanism could work even after the tumor cells became
resistant to gefitinib (16). We
consider that similar mechanisms are involved in our system,
although the target molecules have so far not been specified. In
the case of EBC-1, combination therapy using docetaxel was expected
to reduce c-MET in cells, but this was not observed (data not
shown). It was reported that erlotinib restores the effect of
chemotherapeutic agents through direct inhibition of PgP or BCRP
(17,18). However, this is unlikely because
verapamil, a PgP or BCRP inhibitor, did not restore the sensitivity
to docetaxel in HCC827TR3 cells (data not shown).
In our HPAC in vivo model which mimics PD in
clinical therapy, the combination therapy of gemcitabine with
erlotinib showed significantly strong antitumor effect compared
with gemcitabine monotherapy (Fig.
5D). EGFR expression and phosphorylated EGFR were detected in
the tumors of the control group after PD had developed.
Surprisingly, phosphorylation of EGFR was completely inhibited in
the tumors of the erlotinib group (Fig.
5C). These results indicate the usefulness of the combination
therapy of a chemotherapeutic agent with erlotinib against in
vivo-induced erlotinib-resistant tumors.
Erlotinib is currently approved for the treatment of
NSCLC and pancreatic cancer. In the present study, we showed that
combination therapy of a chemotherapeutic agent with erlotinib is
efficacious against two erlotinib-resistant NSCLC cell lines
(EBC-1, HCCC827TR3) and one pancreatic cancer cell line (HPAC)
which had become erlotinib resistant, suggesting that this form of
treatment would be useful against NSCLC and pancreatic cancer which
developed PD. Erlotinib has been reported to have an excellent
benefit for patients with NSCLC harboring mtEGFR and to prolong the
overall survival of patients with NSCLC harboring wtEGFR (1,10). The
combination therapy may be effective regardless of the EGFR
mutation status because it was effective on both HCC827TR3 (mtEGFR)
and HPAC (wtEGFR) cells.
The results suggest that combination therapy of a
chemotherapeutic agent with erlotinib showed stronger antitumor
effect compared with chemomonotherapy against erlotinib-resistant
tumors in that erlotinib inhibited the phosphorylation of EGFR in
the tumor. It may be possible to obtain evidence for the
suitability of the combination therapy by monitoring the EGFR
phosphorylation level in tumors after PD has developed following
erlotinib treatment. However, this test cannot distinguish tumors
which had intrinsically low EGFR phosphorylation and, to solve the
problem, it may be necessary to test the EGFR phosphorylation level
before the start of erlotinib therapy. In the present study,
docetaxel, irinotecan and gemcitabine were used as chemotherapeutic
agents. Whether or not similar results can be obtained with other
chemotherapeutic agents is an issue for future research. If a
patient goes into PD during combination therapy, a possible
treatment modality may be to change the chemotherapeutic agent
while continuing erlotinib.
References
|
1
|
Cohen S, Ushiro H, Stoscheck C and
Chinkers M: A native 170,000 epidermal growth factor
receptor-kinase complex from shed plasma membrane vesicles. J Biol
Chem. 257:1523–1531. 1982.PubMed/NCBI
|
|
2
|
Carpenter G and Cohen S: Epidermal growth
factor. J Biol Chem. 265:7709–7712. 1990.
|
|
3
|
Pawson T: Protein modules and signalling
networks. Nature. 373:573–580. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yasui W, Hata J, Yokozaki H, et al:
Interaction between epidermal growth factor and its receptor in
progression of human gastric carcinoma. Int J Cancer. 41:211–217.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ozanne B, Richards CS, Hendler F, Burns D
and Gusterson B: Over-expression of the EGF receptor is a hallmark
of squamous cell carcinomas. J Pathol. 149:9–14. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hollstein MC, Smits AM, Galiana C, et al:
Amplification of epidermal growthfactor receptor gene but no
evidence of ras mutations in primary human esophageal cancers.
Cancer Res. 48:5119–5123. 1988.PubMed/NCBI
|
|
7
|
Haeder M, Rotsch M, Bepler G, et al:
Epidermal growth factor receptor expression in human lung cancer
cell lines. Cancer Res. 48:1132–1136. 1988.PubMed/NCBI
|
|
8
|
Ritter CA and Arteaga CL: The epidermal
growth factor receptor-tyrosine kinase: a promising therapeutic
target in solid tumors. Semin Oncol. 30:3–11. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Selvaggi G, Novello S, Torri V, et al:
Epidermal growth factor receptor overexpression correlates with a
poor prognosis in completely resected non-small cell lung cancer.
Ann Oncol. 15:28–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shepherd FA, Pereira JR, Ciuleanu T, et
al: Erlotinib in previously treated non-small cell lung cancer. N
Engl J Med. 353:123–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moore MJ, Goldstein D, Hamm J, et al:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: a phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar
|
|
12
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang Z, Du R, Jiang S, et al: Dual
MET-EGFR combinatorial inhibition against T790M-EGFR-mediated
erlotinib-resistant lung cancer. Br J Cancer. 99:911–922. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furugaki K, Iwai T, Shirane M, Kondoh K,
Moriya Y and Mori K: Schedule-dependent antitumor activity of the
combination with erlotinib and docetaxel in human non-small cell
lung cancer cells with EGFR mutation, KRAS mutation or both
wild-type EGFR and KRAS. Oncol Rep. 24:1141–1146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okabe T, Okamoto I, Tsukioka S, et al:
Addition of S-1 to the epidermal growth factor receptor inhibitor
gefitinib overcomes gefitinib resistance in non-small cell lung
cancer cell lines with MET amplification. Clin Cancer Res.
15:907–913. 2009. View Article : Google Scholar
|
|
17
|
Noguchi K, Kawahara H, Kaji A, Katayama K,
Mitsuhashi J and Sugimoto Y: Substrate-dependent bidirectional
modulation of P-glycoprotein-mediated drug resistance by erlotinib.
Cancer Sci. 100:1701–1707. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Z, Peng XX, Kim IW, et al: Erlotinib
(Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B
member 1 and ATP-binding cassette subfamily G member 2-mediated
drug resistance. Cancer Res. 67:11012–11020. 2007. View Article : Google Scholar : PubMed/NCBI
|