Introduction
The incidence of malignant melanoma is increasing
worldwide. Stage IV melanoma is still a devastating disease with a
median survival time of 6–9 months depending on the bulk and
localisation of disease. Despite first successful immunotherapies
as with ipililumab, a CTLA4 antibody in first and second line
treatment of metastatic melanoma (1,2) and
targeted therapy for patients with BRAF mutations with
PLX4032 (3,4), there is still a need to better define
patients responding to alkylating substances such as temozolomide
especially in BRAF wild-type patients. Single agent dacarbazine has
been the standard of care for many years. Response rates (RRs) of
5.5–13% (5) have been reported in
recent large phase III trials with a further 15–28% of patients
having stable disease (SD), however, few responses are long-lasting
(6).
Temozolomide is an oral alkylating agent with a
mechanism of action similar to dacarbazine (7), that is spontaneously converted to its
active metabolites (8). This oral
drug has been used successfully in the treatment of primary brain
tumors. It was shown that the gene promoter methylation status of
the O6-methylguanine-DNA-methyltransferase gene
(MGMT) is of critical importance for survival in
glioblastoma patients treated with temozolomide (9). MGMT silencing by promoter
methylation impairs the ability of the MGMT protein to remove alkyl
groups from the O6-position of guanine, thereby
increasing the mutation rate in rapidly dividing tumor cells and
improving patient outcome. Recently, we reported that about
one-third of brain metastases from various origins including
malignant melanoma revealed a methylated MGMT promoter
(10). Moreover, homogeneous MGMT
immunoreactivity correlates with an unmethylated MGMT
promoter status suggesting that brain metastases may be a potential
target for therapy with alkylating substances.
Temozolomide demonstrated clinical activity in a
phase I trial with 4 out of 23 patients responding to treatment
(11). This was supported by a
phase II trial where 21% of the patients responded to treatment. A
9 months longer survival was seen in responders (12). Two large phase III randomized
controlled trials of intravenous dacarbazine versus oral
temozolomide every 4 weeks or dose dense every second week as
first-line treatment in patients with melanoma demonstrated
toxicity and response rate (13,14)
with a trend for superiority in progression-free survival (PFS) and
some quality of life domains in one trial in favour of temozolomide
(13). A quite recently published
phase III trial indicates that extended schedule escalated dose
temozolomide has an acceptable safety profile, but does not improve
overall survival (OS) and PFS in stage IV melanoma when compared to
standard dose dacarbazine (14).
Blocking the vascular endothelial cell growth factor
(VEGF) by bevacizumab showed significant survival advantage when
combined with chemotherapy in advanced colorectal (15) and non-small cell lung cancer
(16). The combination of
bevacizumab and temozolomide was chosen in this trial because of
the lack of overlapping toxicities. Furthermore, the combination of
VEGF antibodies and chemotherapy improved survival in colon and
lung cancer (15).
The optimal use of temozolomide in the treatment of
metastatic melanoma is under discussion. The combined therapy of
temozolomide and bevacizumab used in this phase II trial was an
attempt to improve the response rate (17). A disease stabilization rate
confirmed by independent external review of 52% at 12 weeks of the
trial treatment is considered promising for further investigations.
The response rate (16.1%) as well as the PFS (4.2 months) obtained
in our trial was slightly higher than reported for single agent
temozolomide (9.8–12%) and 2.1 months, respectively, despite having
included also patients at higher risk (LDH >2 UNL; ECOG PS 2).
Overall survival (9.3 months) in our trial was comparable to single
agent temozolomide (12,13,18).
The investigation of the methylation status of the
MGMT promoter was one of the prospectively defined
translational research projects in this SAKK trial. Therefore, we
tested MGMT promoter methylation and MGMT protein expression
in our patient set of SAKK 50/07 with metastatic melanoma to
elucidate the molecular basis of tumor response and outcome in
patients treated with temozolomide and bevacizumab.
Material and methods
Patients and tumors
Formalin-fixed, paraffin-embedded tissue samples of
62 patients previously untreated for metastatic melanoma with ECOG
performance status ≤2 were selected for MGMT promoter
methylation analysis and immunohistochemical analysis. The patients
were treated with temozolomide at 150 mg/m2 days 1–7
orally and bevacizumab at 10 mg/kg body weight day 1 intravenous
every 2 weeks until disease progression or unacceptable toxicity.
Response was assessed every 6 weeks by computed tomography
according to the RECIST (response evaluation criteria in solid
tumors) criteria.
The protocol was approved by local ethics review
boards and all patients gave written informed consent. The trial
was registered at the National Institute of Health (www.clinicaltrial.gov; identifier number:
NCT00568048).
DNA extraction and methylation-specific
PCR
All tumors were histologically reviewed by one
pathologist (H.M.) on the basis of hematoxylin and eosin-stained
tissue sections. One to three biopsy cylinders (0.6-mm diameter)
were punched from tumor areas that contained at least 70% tumor
cells. Genomic DNA extraction, sodium bisulfite modification of
isolated DNA and the analysis of the methylation status of
MGMT was done using a one-step PCR approach with 40 cycles
and the nested primer pair described by Palmisano et
al(19). DNA of normal
lymphocytes and human colorectal adenocarcinoma cell-line SW620 was
used as a negative and positive control for methylated alleles of
MGMT, respectively.
Immunohistochemistry
Immunohistochemistry of melanoma sections was
performed as described (10).
Sections were incubated with clone MT3.1 (dilution 1:160;
NeoMarkers, Newmarket, UK). MGMT-immunopositive cells revealed a
strong nuclear staining. Lymphocytes and endothelial cells served
as an internal positive control. The immunoreactivity was scored
semi-quantitatively as follows: 0: <5% positive tumor cells, 1+:
5–75% positive tumor cells, 2+: >75–95% positive tumor cells,
3+: >95% positive tumor cells.
Statistical analyses
The endpoints of interest defined in the trial
protocol and considered for this project include disease
stabilization rate at week 12 (DSR12) consisting of either a
complete response (CR), partial response (PR) or SD, best overall
response, PFS and OS. PFS was defined as the time from trial
registration until either a disease progression or death, with
patients censored at the time of starting a second line therapy or
the last time they were known to be alive without progression. For
survival-type endpoints subgroups were compared using the log-rank
test. Associations between categorical variables were assessed by
either the Chi-squared or Fisher’s exact test as well as the
Mantel-Haenszel Chi-square test of trend if a variable was ordinal.
The data were analyzed in SAS (Statistical Analysis Systems,
version 9.2).
Results
Patients
Between January 2008 and April 2009, 62 patients (40
male) were enrolled in the trial SAKK 50/07. None of the patients
were ineligible or withdrew participation before treatment start.
The median age at enrollment was 59 years (range: 29–82) and the
median follow-up time was 20.1 (range: 1.7–32.6) months. All
patients were administered at least one cycle of therapy. The
independently reviewed DSR12 was 52% including 10 patients with a
PR and 22 patients with SD.
MGMT promoter methylation and protein
expression
Tumor tissue was not available for 9 of the 62
patients enrolled in the trial. MGMT methylation status was
reliably determined in 42 of 53 (79%) formalin-fixed and
paraffin-embedded samples. Overall, a methylated MGMT
promoter was detectable in 11 of 42 (26%) of the metastatic
melanomas by MS-PCR.
MGMT immunoreactivity was assessed in 49 of 53 (92%)
tumors. Of 53 tumors, 4 were excluded from further calculations
because endothelial cells and/or leukocytes used as internal
control for MGMT positivity were MGMT negative possibly indicating
tissue fixation problems. Seventeen (35%) tumor samples were 3+, 11
(22%) were 2+, whereas 4 (8%) were 1+ and 17 (35%) lacked
immunoreactivity for MGMT.
Both the methylation status of the MGMT
promoter as well as the MGMT immunoprofile were available in 39
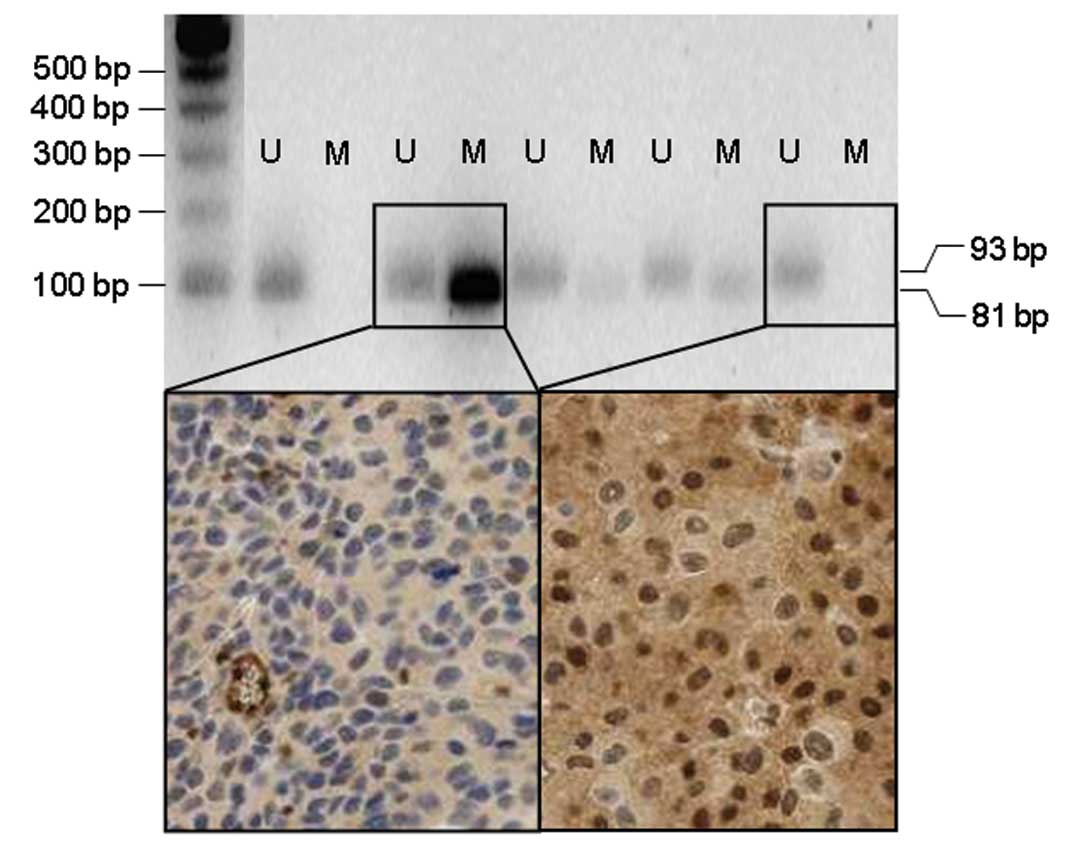
patients. Examples of melanomas with unmethylated and methylated
MGMT promoter as well as with strongly positive and negative
MGMT protein expression are shown in Fig. 1. No significant association was
observed when the MGMT expression patterns in the methylated and in
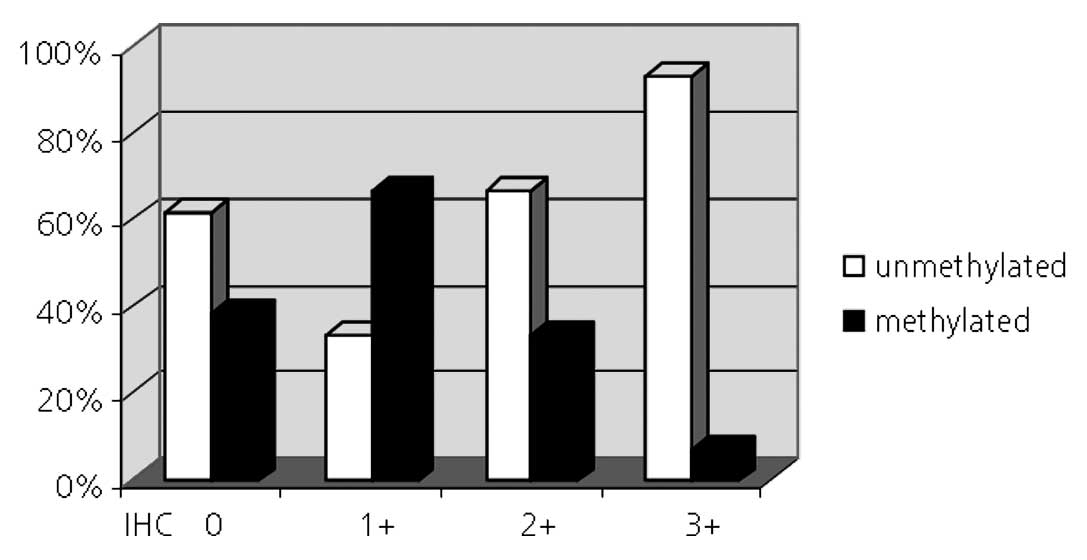
the unmethylated patient subgroups were compared. Of 28 (46%)
melanomas with an unmethylated MGMT promoter, 13 displayed a
homogeneous 3+ MGMT immunoreactivity, 6 (21%) tumors had a 2+
score, one (4%) had a 1+ score and 8 (29%) were MGMT negative.
Within the subgroup of melanomas which had a methylated MGMT
promoter 5 (45%) were also MGMT negative, two (18%), three (27%)
and one (9%) were scored 1+, 2+ and 3+, respectively (Fig. 2).
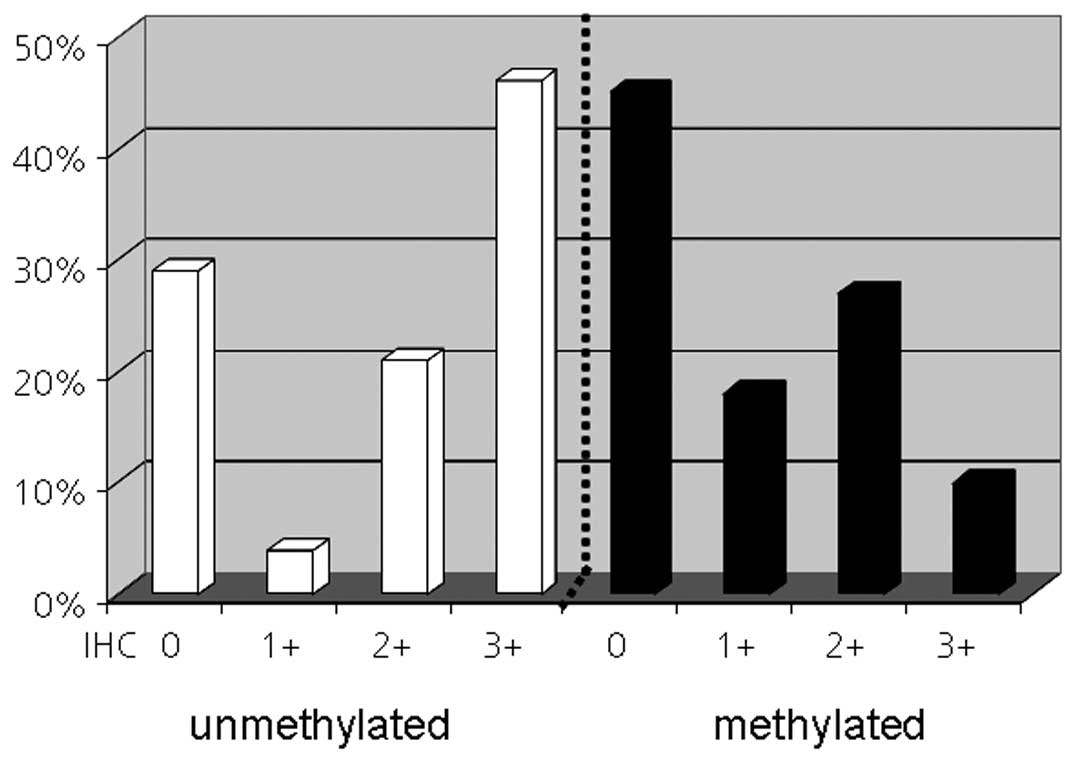
When we looked at the methylation status in the four
MGMT expression subgroups we noted that in the patient subgroup
with a 3+ score 13 of 14 (93%) tumors were also unmethylated. In
contrast, no association was observed between MGMT negativity and
promoter methylation. Only 5 of 13 (38%) MGMT negative tumors had a
methylated promoter (Fig. 3).
However, no statistical correlation existed between expression and
methylation status.
MGMT promoter methylation status and
patient outcome
Patients with methylated MGMT promoter had
higher response rates according to RECIST, when CR + PR versus SD +
PD were compared (p=0.0426, Table
I). No significant differences were obtained for other response
groupings.
 | Table IAssociation between response and
MGMT promoter methylation status. |
Table I
Association between response and
MGMT promoter methylation status.
| Methylation
status | Response | Mantel-Haenszel
Chi-square test of trend p-value |
|---|
| Methylated vs. | CR, PR, SD, PD | 0.0803 |
| Unmethylated | CR + PR + SD, PD | 0.5573 |
| CR + PR, SD + PD | 0.0426 |
PFS and OS were stratified by MGMT
methylation status. Median PFS time in the methylated subgroup was
8.1 months [95% confidence interval (CI): 1.7, 8.4] versus 3.4
months (95% CI: 1.4, 5.4) in the unmethylated subgroup. Median OS
time in the methylated subgroup was 9.9 months (95% CI: 8.4, 15.9)
versus 9.1 months (95% CI: 6.6, 11.9) in the unmethylated subgroup.
Although, in the methylated subgroup median PFS was exceeded by
more than 4 months compared to the unmethylated subgroup the
results did not reach statistical significance.
MGMT protein expression and patient
outcome
The different groupings for the MGMT protein
expression (0–5, 5–75, 75–95 and 95–100% nuclear positive tumor
cells) were compared in various groupings with regard to best
overall response. The best response outcome was obtained from
patients that were MGMT negative (<5%) versus otherwise when CR
+ PR + SD versus PD were compared (p=0.05).
PFS and OS were stratified by the MGMT expression
groups. In general, patients with negative or heterogeneous MGMT
expression had longer PFS and OS rates than patients with high and
homogeneous MGMT expression. However, these differences did not
reach statistical significance (data not shown). Best overall
response by MGMT protein level and promoter methylation status is
summarized in Table II.
| Table IIBest overall response by MGMT protein
expression and promoter methylation status. |
Table II
Best overall response by MGMT protein
expression and promoter methylation status.
| | MGMT expression (%
positive tumor cells) |
|---|
| |
|
|---|
| Methylation
status | Best response | Unknown | 0–5% | 5–75% | 75–95% | 95–100% |
|---|
| Methylated | Complete
response | - | - | - | - | - |
| Partial
response | - | 5 | - | - | 1 |
| Stable disease | - | - | 1 | 2 | - |
| Disease
progression | - | - | 1 | 1 | - |
| Unkown | - | - | - | - | - |
| Unmethylated | Complete
response | - | - | - | - | 1 |
| Partial
response | - | 1 | - | 2 | 1 |
| Stable disease | 2 | 4 | - | 3 | 6 |
| Disease
progression | 1 | 2 | 1 | 1 | 5 |
| Unknown | - | 1 | - | - | - |
| Unknown | Complete
response | | | | | |
| Partial
response | 2 | 1 | - | - | 1 |
| Stable disease | 3 | 3 | - | - | 2 |
| Disease
progression | 1 | 1 | 2 | 2 | 2 |
| Unknown | - | - | - | - | - |
Discussion
We investigated the epigenetic silencing of
MGMT by both MGMT promoter methylation and the MGMT
protein expression status in metastatic melanomas to assess the
predictive value for benefit from temozolomide in combination with
bevacizumab. In our study, 26% of metastatic melanomas revealed a
methylated MGMT promoter. This result is in line with
previous studies on similar patient cohorts resulting in
MGMT promoter methylation between 20 and 31% (10,20,21).
Of note, a methylated MGMT promoter correlated significantly
with complete and partial responses. Compared to patients with
unmethylated MGMT promoter this patient group had also a
trend to prolonged median PFS and OS of more than 4 months.
Our combined analysis of MGMT promoter
methylation status and protein expression is unique and revealed a
strong association between homogeneous MGMT expression and
unmethylated MGMT promoter. Ninety-three percent of the tumors with
>95% MGMT-positive tumor cells had an unmethylated MGMT
promoter. In contrast, only 44% of the tumors with no or
heterogeneous MGMT positivity had a methylated MGMT
promoter. Similar results were observed in patients with brain
metastases of various solid tumors, as previously published
(10).
There are several possible explanations for this
discrepancy: i) gene mutations may lead to loss of functional MGMT
expression independent of the MGMT promoter methylation
status; ii) in a certain set of unmethylated and MGMT negative
melanomas, MGMT may be induced after addition of alkylating,
DNA damaging agents (22,23); iii) both discordance between
MGMT promoter methylation and individual metastases from the
same patient (20) and/or
intra-tumoral heterogeneity of promoter hypermethylation (21) may significantly weaken MGMT
methylation as a reliable predictor of chemosensitivity; iv) some
tumors may be interpreted unmethylated because the corresponding
PCR product is not detectable or not reproducible. In contrast to
MS-PCR, pyrosequencing enables to identify promoter regions
methylated from 0 to 100%. A previous melanoma study showed that in
50% of the tumors, MGMT promoter methylation was 10% or less
(18). It is therefore tempting to
speculate that tumors with low MGMT methylation are
considered unmethylated when MS-PCR is used.
In a recently published study methylation of the
MGMT promoter was analyzed in metastatic melanoma with the
combined bisulfite restriction analysis technique (24). In contrast to our findings, those
patients with a methylated MGMT promoter neither had a
survival advantage nor a better response rate but suffered from
more severe adverse events compared to those with an unmethylated
promoter. One possible explanation for the discrepant outcomes may
be the difference in treatment. The lack of MGMT in
temozolomide-treated melanoma cells causes an increase of toxicity,
which may even be enhanced in the presence of bevacizumab by
attenuating vascularization and therefore oxygen and energy supply
in the tumor.
We also believe that the use of different methods
and the analysis of different CpG regions within the MGMT
promoter may lead to contradictory results. Consequently, a
standardized protocol with a still to be defined threshold that
more reliably separates MGMT methylated (inactive) from
unmethylated (active) melanoma patients would be highly desirable.
Here we selected to use a protocol which was previously described
for investigating the MGMT methylation status in glioma
patients (9) and which allowed us
to directly compare the results obtained from melanoma and glioma
patients.
A reproducible and reliable promoter methylation
analysis was only possible in 42 of 53 cases. DNA treated with
formalin and bisulfite is highly modified and degraded and thus of
limited quality to perform reliable PCR. To minimize the
amplification of artificial PCR products we used a one-step PCR
approach. The success rate for DNA amplification was much higher
compared to that obtained from a recent study in which nested PCR
with 80 cycles was used (10). All
53 melanoma DNA samples were amplifiable, the results of 11 (19%)
cases, however, were not reproducible. Similar error rates were
reported in the literature (10,25,26).
The reasons why some of the DNA samples extracted from formalin
fixed tissue are not suitable for MS-PCR may be a too long tissue
fixation time, the use of unbuffered formalin or insufficient
dehydration of the tissue.
Based on our data we suggest that both molecular and
immunohistochemical approaches may be helpful to identify
responders (methylated) as well as non-responders (unmethylated and
homogeneous expression). For routine diagnosis further optimized
standard protocols with preferably unfixed, snap-frozen biopsies
are necessary to improve specificity and sensitivity.
Acknowledgements
We thank Sonja Schmid, Martina Storz and Silvia
Behnke for their excellent technical assistance. This work was
supported by Roche Pharma (Schweiz) AG, Schering Plough, Essex, UK
and the Swiss State Secretariat for Education and Research.
References
|
1
|
Hodi FS, O’Day SJ, McDermott DF, et al:
Improved survival with ipilimumab in patients with metastatic
melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar
|
|
2
|
Robert C, Thomas L, Bondarenko I, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chapman PB, Hauschild A, Robert C, et al:
Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Flaherty KT, Puzanov I, Kim KB, et al:
Inhibition of mutated, activated BRAF in metastatic melanoma. N
Engl J Med. 363:809–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bedikian AY, Millward M, Pehamberger H, et
al: Bcl-2 antisense (oblimersen sodium) plus dacarbazine in
patients with advanced melanoma: the Oblimersen Melanoma Study
Group. J Clin Oncol. 24:4738–4745. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chapman PB, Einhorn LH, Meyers ML, et al:
Phase III multicenter randomized trial of the Dartmouth regimen
versus dacarbazine in patients with metastatic melanoma. J Clin
Oncol. 17:2745–2751. 1999.PubMed/NCBI
|
|
7
|
Stevens MF, Hickman JA, Langdon SP, et al:
Antitumor activity and pharmacokinetics in mice of
8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one
(CCRG 81045; M&B 39831), a novel drug with potential as an
alternative to dacarbazine. Cancer Res. 47:5846–5852.
1987.PubMed/NCBI
|
|
8
|
Tsang LL, Quarterman CP, Gescher A and
Slack JA: Comparison of the cytotoxicity in vitro of temozolomide
and dacarbazine, prodrugs of
3-methyl-(triazen-1-yl)imidazole-4-carboxamide. Cancer Chemother
Pharmacol. 27:342–346. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hegi ME, Diserens AC, Gorlia T, et al:
MGMT gene silencing and benefit from temozolomide in glioblastoma.
N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ingold B, Schraml P, Heppner FL and Moch
H: Homogeneous MGMT immunoreactivity correlates with an
unmethylated MGMT promoter status in brain metastases of various
solid tumors. PLoS One. 4:e47752009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Newlands ES, Blackledge GR, Slack JA, et
al: Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC
362856). Br J Cancer. 65:287–291. 1992. View Article : Google Scholar
|
|
12
|
Bleehen NM, Newlands ES, Lee SM, et al:
Cancer Research Campaign phase II trial of temozolomide in
metastatic melanoma. J Clin Oncol. 13:910–913. 1995.PubMed/NCBI
|
|
13
|
Middleton MR, Grob JJ, Aaronson N, et al:
Randomized phase III study of temozolomide versus dacarbazine in
the treatment of patients with advanced metastatic malignant
melanoma. J Clin Oncol. 18:158–166. 2000.PubMed/NCBI
|
|
14
|
Patel PM, Suciu S, Mortier L, et al:
Extended schedule, escalated dose temozolomide versus dacarbazine
in stage IV melanoma: Final results of a randomised phase III study
(EORTC 18032). Eur J Cancer. 47:1476–1483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kabbinavar F, Hurwitz HI, Fehrenbacher L,
et al: Phase II, randomized trial comparing bevacizumab plus
fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with
metastatic colorectal cancer. J Clin Oncol. 21:60–65. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johnson DH, Fehrenbacher L, Novotny WF, et
al: Randomized phase II trial comparing bevacizumab plus
carboplatin and paclitaxel with carboplatin and paclitaxel alone in
previously untreated locally advanced or metastatic non-small-cell
lung cancer. J Clin Oncol. 22:2184–2191. 2004. View Article : Google Scholar
|
|
17
|
von Moos R, Seifert B, Simcock M, et al:
First-line temozolomide combined with bevacizumab in metastatic
melanoma: a multicentre phase II trial (SAKK 50/07). Ann Oncol.
23:531–536. 2011.PubMed/NCBI
|
|
18
|
Rietschel P, Wolchok JD, Krown S, et al:
Phase II study of extended-dose temozolomide in patients with
melanoma. J Clin Oncol. 26:2299–2304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Palmisano WA, Divine KK, Saccomanno G, et
al: Predicting lung cancer by detecting aberrant promoter
methylation in sputum. Cancer Res. 60:5954–5958. 2000.PubMed/NCBI
|
|
20
|
Kohonen-Corish MR, Cooper WA, Saab J,
Thompson JF, Tren RJ and Millward MJ: Promoter hypermethylation of
the O(6)-methylguanine DNA methyltransferase gene and
microsatellite instability in metastatic melanoma. J Invest
Dermatol. 126:167–171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rastetter M, Schagdarsurengin U, Lahtz C,
et al: Frequent intra-tumoural heterogeneity of promoter
hypermethylation in malignant melanoma. Histol Histopathol.
22:1005–1015. 2007.PubMed/NCBI
|
|
22
|
Fritz G, Tano K, Mitra S and Kaina B:
Inducibility of the DNA repair gene encoding
O6-methylguanine-DNA methyltransferase in mammalian
cells by DNA-damaging treatments. Mol Cell Biol. 11:4660–4668.
1991.PubMed/NCBI
|
|
23
|
Grombacher T, Mitra S and Kaina B:
Induction of the alkyltransferase (MGMT) gene by DNA damaging
agents and the glucocorticoid dexamethasone and comparison with the
response of base excision repair genes. Carcinogenesis.
17:2329–2336. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hassel JC, Sucker A, Edler L, et al: MGMT
gene promoter methylation correlates with tolerance of temozolomide
treatment in melanoma but not with clinical outcome. Br J Cancer.
103:820–826. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hamilton MG, Roldan G, Magliocco A,
McIntyre JB, Parney I and Easaw JC: Determination of the
methylation status of MGMT in different regions within glioblastoma
multiforme. J Neurooncol. 102:255–260. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Preusser M, Elezi L and Hainfellner JA:
Reliability and reproducibility of PCR-based testing of
O6-methylguanine-DNA methyltransferase gene (MGMT)
promoter methylation status in formalin-fixed and paraffin-embedded
neurosurgical biopsy specimens. Clin Neuropathol. 27:388–390.
2008.PubMed/NCBI
|