Introduction
Pancreatic cancer, characterized by aggressive
malignancy and high resistance to chemotherapy agents, is
considered as one of the most lethal tumors with a 5-year mortality
of 97–98% (1,2). Surgery is the only curative treatment
for pancreatic cancer. However, <20% patients have resectable
tumor at the time of diagnosis due to the poor diagnosis (3). Except for the surgical therapy, both
of chemotherapy and radiation regimens hardly ameliorate the
outcome of pancreatic cancer patients, which causes ~90% of cases
death within 1 year after diagnosis (4). Since the end of 20th century,
gemcitabine, a deoxycitidine analogue, was confirmed to improve the
objective response rate in patients with advanced pancreatic
carcinoma, and has been applied as a first-line chemotherapy agent
(5,6). However, only 5.4% of all patients are
sensitive to the gemcitabine-based chemotherapy (7,8). Thus,
novel chemotherapeutic drugs and candidate compounds are urgently
needed to improve the outcome of patients with the lethal
malignancy.
Ursolic acid (UA) is a pentacyclic triterpenoid
compound extracted and purified from many types of medicinal
plants, herbs, and edible vegetables (9). UA contains a variety of
pharmacological activities. The anti-oxidative and
anti-inflammatory actions of UA are widely applied to
hepatoprotection, neuroprotection, cardiovascular protection and so
on (10–14). Besides these effects, UA has been
proved to possess multiple anti-tumor activities including
inhibiting tumorigenesis and progression (9). UA causes cytotoxicity to diverse
malignancies such as fibrosarcoma, prostate cancer, hepatocellular
carcinoma, colorectal cancer, melanoma, and breast cancer (15–20).
However, the cytotoxic role of UA in gemcitabine-resistant human
pancreatic cancer and its potential molecular mechanisms are still
unclear.
In this study, we investigated the effect of UA on
growth restriction and apoptosis in human pancreatic resistant
cancer in vitro and in vivo. Additionally, we
explored possible mechanisms of action, such as PI3K/Akt/NF-κB and
c-Jun N-terminal kinase (JNK) pathways.
Materials and methods
Cell culture and reagents
The human pancreatic resistant cancer cell lines MIA
PaCa-2 and PANC-1 were kindly provided by Shanghai Institute of
Materia Medica. The human pancreatic resistant cancer cell line
Capan-1 was kindly provided by Department of Pharmacology, Sun
Yat-Sen University. The paclitaxel-resistant endometrial cell line
HEC-1A was purchased from Cell Bank of Chinese Academy of Sciences.
The platinum-resistant ovarian cancer cell line OVCAR-3 was
purchased from Experimental Animal Center of Sun Yat-Sen
University. The cells were routinely cultured in DMEM (Invitrogen,
Grand Island, NY) supplemented with 10% fetal bovine serum (FBS)
(Invitrogen), 100 U/ml penicillin, and 100 mg/ml streptomycin in 5%
CO2 at 37°C. Ursolic acid (UA) (National Institutes for
Food and Drug Control, China) was dissolved in DMSO (Sigma-Aldrich,
St. Louis, MO) as a 100 mM stock solution and stored at −20°C.
Further dilution was done in serum-free cell culture medium. PI3K
inhibitor LY294002 (Cell Signaling Technology, Beverly, MA) was
dissolved in DMSO (Sigma) as a 50 mM stock solution and stored at
−20°C. Similarly, JNK inhibitor SP600125 (Sigma) was dissolved and
stored as a 10 mM stock solution by DMSO at −20°C.
Cell viability and proliferation
Cells were seeded into 96-well plates at a density
of 2×103/well and allowed to attach overnight before
being treated by different concentrations of UA for 24 h. Then cell
viability and proliferation were evaluated. Cell viability was
tested by the 3-(4,5-dimethylthiaziazol-2-yl)-2, 5-diphenyl
tetrazolium bromide (MTT, Sigma) assay as described in the
literature (21). Detection of cell
proliferation was through the colorimetric
5′-bromo-2′-deoxy-uridine (BrdU) incorporation assay (Roche
Diagnostic Corp., Indianapolis, IN, USA) according to the
instruction of the manufacturer. Briefly, cells were labeled by
addition of BrdU for 4 h in 5% CO2 at 37°C. Then,
FixDenat solution was used for cells fixation. The anti-BrdU-POD
antibody was added to locate the BrdU label in the DNA. After the
incubation of the peroxidase substrate, the immune complex was
detected by a multi-well spectrophotometer reader using an iMark™
Microplate Reader (Bio-Rad, Richmond, CA).
Hoechst 33258 staining
The three types of cells at the density of
2×106/well in a 6-well plate were fixed by 4% paraform
for 10 min. Then Hoechst 33258 (Sigma) at 5 μg/ml was added to the
cells for 15 min at room temperature in the dark. The nuclei
morphology was observed by fluorescence microscopy (Olympus,
Melvie, NY) with a 340-nm excitation filter.
Caspase-3/7, −8 and −9 activity
measurements
Caspase-3/7, −8 and −9 activity were assessed by
Caspase-Glo luminescent-based assays (Promega, Madison, WI).
Accordance with the instructions, cells (1×104/well)
were seeded into a 96-well plate overnight to ensure adherence.
After various concentrations of UA treatment for 24 h in a 5%
CO2-humidified atmosphere at 37°C, 100 μl of 3/7, −8 and
−9 Caspase-Glo reagents were added to each well, respectively.
Mixed and incubated for 1 h, the mixtures were transferred to a
fluorescence microtiter plate and quantified fluorescence intensity
by a luminometer. The corresponding OD values from MTT assay
described above were considered as the viable cell number to
normalize the fluorescence intensity.
Cytochrome C ELISA assay
Cytoplasmic cytochrome C levels were determined by a
cytochrome C ELISA kit (R&D Systems, Minneapolis, MN). The
cytoplasmic protein was collected by a nuclear and cytoplasmic
extraction reagents (Pierce, Rockford, IL) from adherent cells
(2×106/well) in a 6-well plate after UA incubation for
24 h. The BCA protein assay kit (Pierce) was used to normalize the
cytoplasmic protein. The protein was stored in −80°C until it was
analyzed by cytochrome C ELISA kit according to the manufacturer's
instructions.
Western blotting and immunofluorescent
analysis
Western blotting was performed as described
previously (22). The following
antibodies were used: antibodies against PI3K, Akt, phospho-Akt,
GSK-3β, phospho-GSK-3β, NF-κB phospho-p65, JNK, phospho-SAPK/JNK,
phospho-MDM2, IκB (1:1000, Cell Signaling Technology); NF-κB p65,
phospho-IκB (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA), and
actin (1:1000, Thermo Scientific IHC, Fremont, CA).
The immunofluorescent analysis was performed as
previous protocol (23). After UA
treatments, the MIA PaCa-2 cells were fixed in 4% paraformaldehyde
for 30 min and perforated by PBS containing 0.1% Triton X-100. The
cells were blocked with 5% bovine serum albumin (BSA) for 1 h at
37°C and incubated the NF-κB p65 antibody (1:200) at 4°C overnight.
After washing by PBS, Cy3-conjugated goat anti-rabbit IgG (1:50,
Proteintech, Chicago, IL) was used as the secondary antibody for 1
h at 37°C. Nuclear staining was achieved by incubating the cells in
DAPI for 10 min. The fluorescence was photographed with
fluorescence microscopy (Olympus).
Mouse xenograft model and
immunohistochemical analysis (IHC)
To establish a subcutaneous xenograft model, MIA
PaCa-2 cells (5×106) were inoculated subcutaneously on
the flanks of 6-week-old nude mice (female, purchased from
Experimental Animal Center of Southern Medical University). When
the average tumor size reached ~100 mm3, mice with tumor
xenografts were randomized into three groups with six mice in each
group including: i) vehicle alone (normal saline with 1% DMSO
i.p.), ii) low dose UA (100 mg/kg twice a week i.p.), iii) high
dose UA (200 mg/kg twice a week i.p.). All of the treatments were
administered at a dose of 0.1 ml/10 g bodyweight. The formula,
(length × width2)/2, was used to estimate tumor sizes.
Both the body weight and tumor sizes were recorded twice a week. At
the end of the experiment, the animals were sacrificed and the
tumors were dissected and weighed to calculate the inhibitory rate.
Animal studies were performed according to the institutional
guidelines of Experimental Animal Center in Sun Yat-sen
University.
The paraffin tissue sections were obtained from the
tumor tissues from the nude mice, IHC were performed as described
previously (24). Briefly, the
sections were deparaffinizated with xylene and microwaved for 10
min in the presence of 10 mM citric acid buffer. After quenching
the endogenous peroxidases by hydrogen peroxide, the sections were
blocked by 5% BSA/0.3% Triton X-100 for 20 min. Then the sections
were incubated with cleaved caspase-3 (1:200) antibody overnight at
4°C and secondary antibody, goat anti-rabbit IgG (1:50,
Proteintech) for 30 min at 37°C, and then signal was generated by
using the DAB substrate kit (Boster, Wuhan, China) for
peroxidase.
Statistical analysis
Data are expressed as mean ± standard deviation (SD)
of three separate experiments and analyzed by SPSS. P<0.05 was
considered significant and determined by the two-sample Student's
t-test or one-factor ANOVA analysis.
Results
Ursolic acid (UA) decreases the viability
and proliferation of the human pancreatic resistant cancer cell
lines
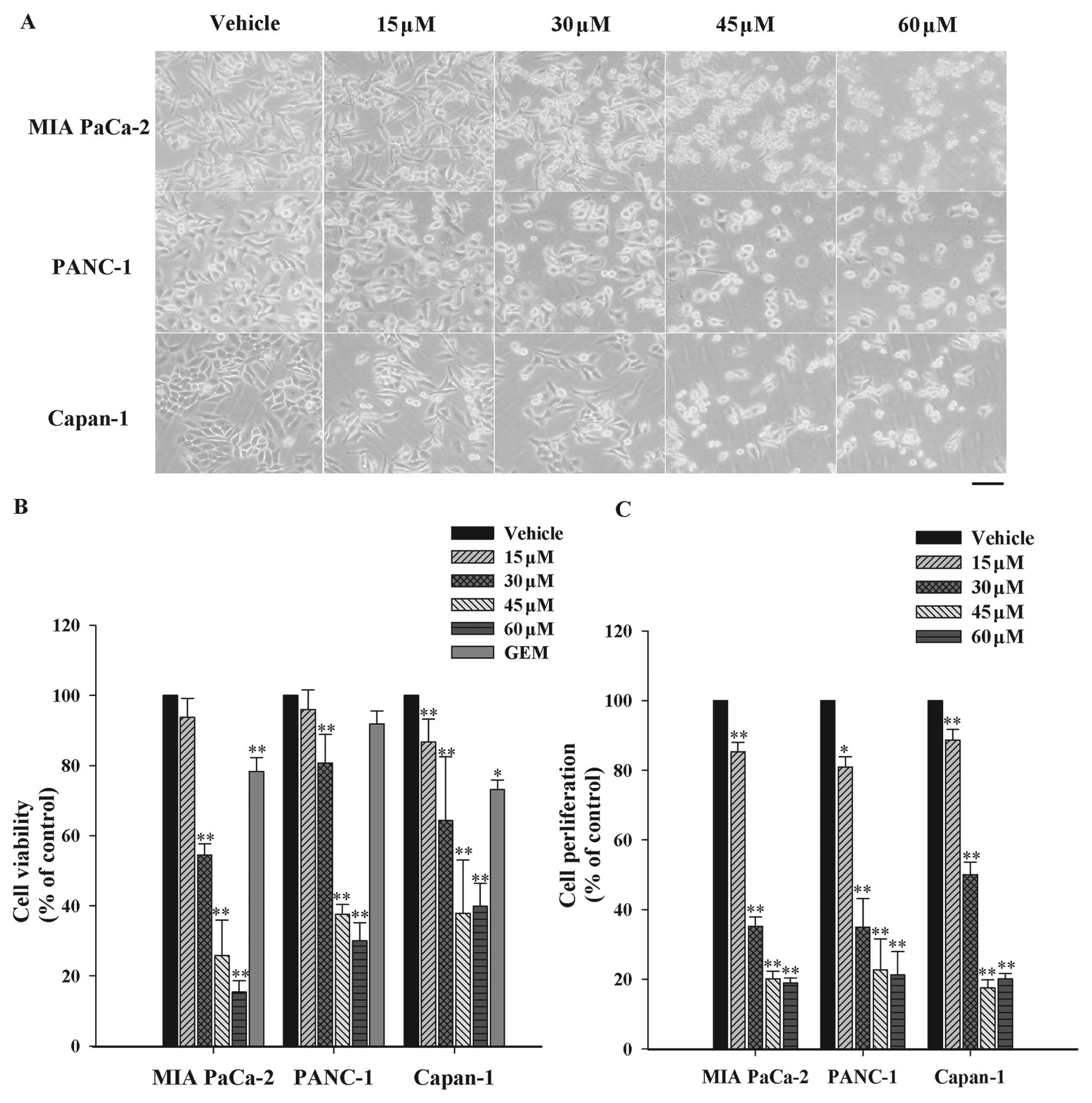
In order to appraise the cytotoxicity of UA on
pancreatic-resistant carcinoma, three different malignancy grades
of pancreatic resistant cancer cell lines including MIA PaCa-2
(grade 3), PANC-1 (grade 2) and Capan-1 (grade 1) were exposed to
increasing concentrations of UA (15, 30, 45 and 60 μM) for 24 h
(6,25–27).
Firstly, the morphology of three cell lines under microscopic
observation revealed the corresponding reduction of surviving cell
numbers and morphological changes after UA treatments (Fig. 1A). In addition, cell viability was
analyzed by MTT assay. As shown in Fig.
1B, UA dose-dependently decreased the viability of three cell
lines with the half maximal inhibitory concentration
(IC50 value) at 40.8, 45.3 and 41.3 μM, respectively.
Gemcitabine as the positive control at the concentration of 45 μM
did not induce significant cytotoxicity to PANC-1 cells comparing
with the vehicle for 24 h. However, UA decreased the viability by
62.5% at the same concentration in the identical cell lines.
Similarly, UA achieved stronger cytotoxic effect than gemcitabine
in MIA PaCa-2 and Capan-1 cells. The proliferation inhibition of UA
was further investigated by 5′-bromo-2′-deoxy-uridine (BrdU)
incorporation assay. As observed in Fig. 1C, growth inhibition effect of UA in
MIA PaCa-2 cells started at 15 μM and increased up to 60 μM. The
results were in agreement with the outcomes of MTT assay. These
data suggested that UA was capable of causing growth inhibition to
pancreatic-resistant carcinoma in vitro.
Both intrinsic and extrinsic pathways are
involved in UA-triggered apoptosis
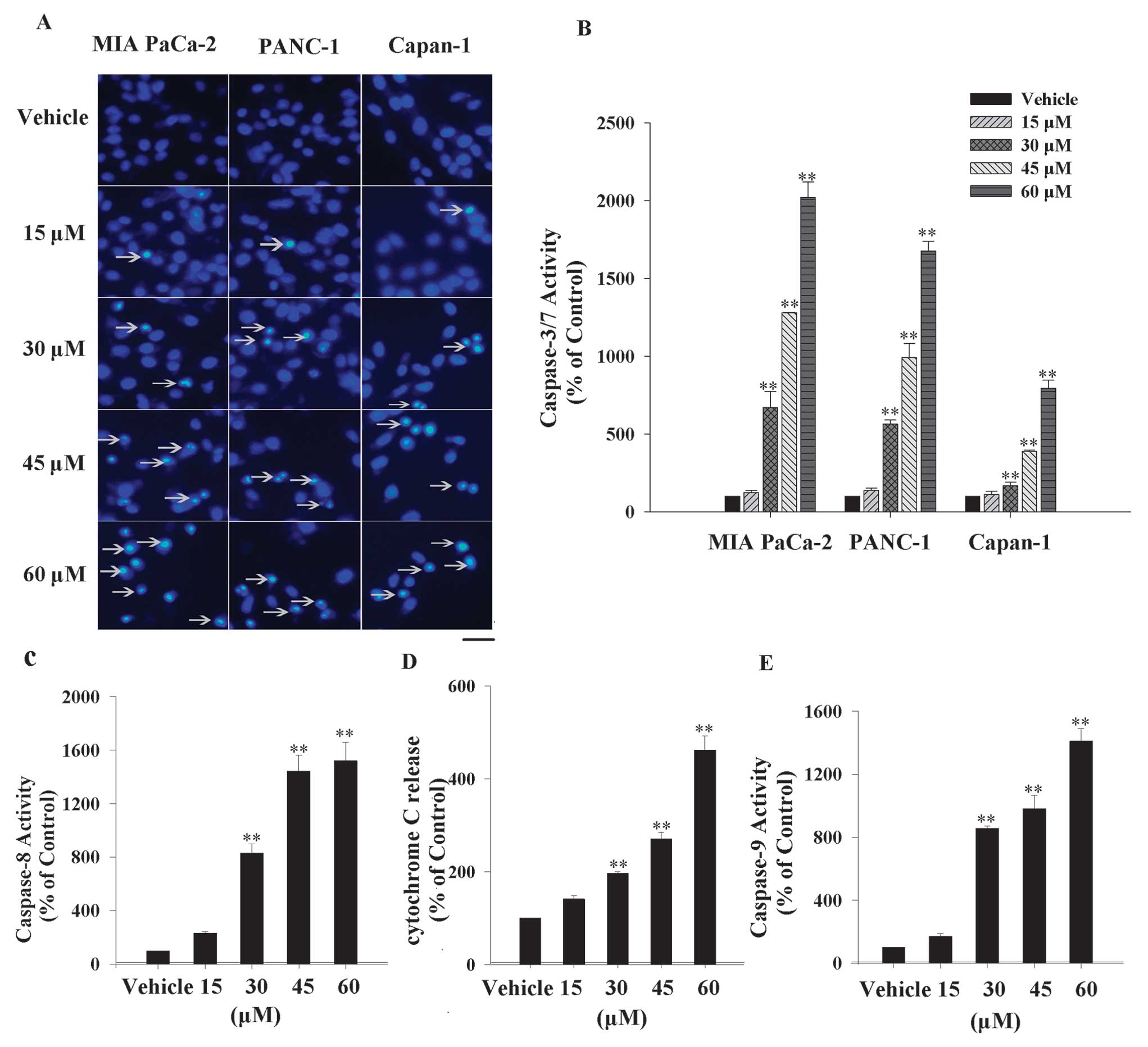
UA has been shown to induce apoptosis in multiple
cancer cell lines (16–20). To identify whether UA has the same
effect in pancreatic-resistant cancer cell lines, the morphologic
analysis was performed by Hoechst 33258 staining. MIA PaCa-2,
PANC-1 and Capan-1 cells were exposed to various concentrations
(15, 30, 45 and 60 μM) of UA for 24 h, the morphological
characteristics of apoptotic cells such as condensed nuclei and
cell shrinkage were discovered and apoptotic proportion grew with
upward dose (Fig. 2A). To confirm
the UA-triggered apoptosis in three cell lines, another apoptotic
marker, caspase-3/7 activity, was examined. Clearly, the
caspase-3/7 activity was promoted gradually along with the rising
concentrations of UA. Fig. 2B shows
that the caspase-3/7 activity was enhanced 20-, 16- and 8-fold,
respectively, in MIA PaCa-2, PANC-1 and Capan-1 cells at 60 μM
UA.
The caspase-dependent cascade is mediated by
endogenous and exogenous signal transductions (22). To study by which UA induces
caspase-3/7 activation, several markers belonging to the two
pathways were inspected. Caspase-8 is a downstream apoptotic
molecule of the death receptor, which mediates the death receptor
signaling pathway (extrinsic pathway). As shown in Fig. 2C, caspase-8 activity increased by
~15-fold with 60 μM UA treatment. Cytochrome C released from
mitochondria into cytoplasm was the initial step in the intrinsic
apoptotic pathway. Fig. 2D exhibits
the cytochrome C relative release rate increase by ~4.5-fold at 60
μM UA. Besides, caspase-9 activity, another marker of the intrinsic
apoptotic pathway, was also increased by ~14-fold after 60 μM UA
intervention (Fig. 2E).
c-Jun-terminal kinase (JNK) pathway is
potentially involved in UA-induced endogenous apoptosis
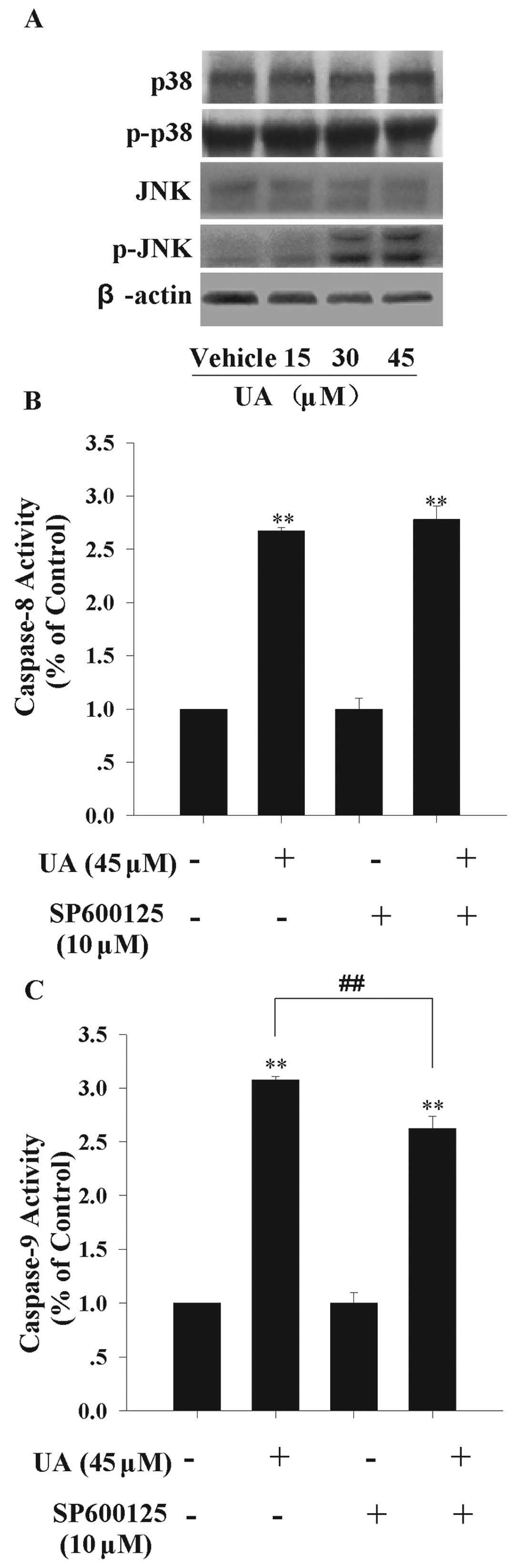
The mitogen-activated protein kinase (MAPK) family
is closely related to cell apoptosis (28). There are some reports revealing that
UA is able to activate JNK, which is one of crucial components in
MAPK signaling pathway (16,29).
Here, we detected JNK and P38 expression levels, both of which were
reported as vital molecules of MAPK family in the programmed cell
death, to explore more potential signal channels (30). We found that JNK activity, measured
by phosphorylated-JNK level, was significantly up-regulated at the
presence of 30 and 45 μM UA. But, UA had no obvious effect on JNK,
P38 and phosphorylated-P38 protein levels (Fig. 3A). This result indicated that JNK
may contribute to UA-induced apoptosis. To further study the
apoptotic pathway in which JNK was involved, we used the JNK
inhibitor SP600125 to block the activity of JNK pathway. MIA PaCa-2
cells were pretreated with 10 μM SP600125 for 1 h, and then treated
with 45 μM UA for 12 h. As shown in Fig. 3B and C, pretreatment by SP600125
partially inhibited the caspase-9 activity by 16%, while caspase-8
activity was not altered significantly. These data indicated that
JNK is at least partly involved in UA-induced endogenous apoptotic
signaling transduction.
UA inhibits the PI3K/Akt pathway and
down-regulates the expression of downstream proteins
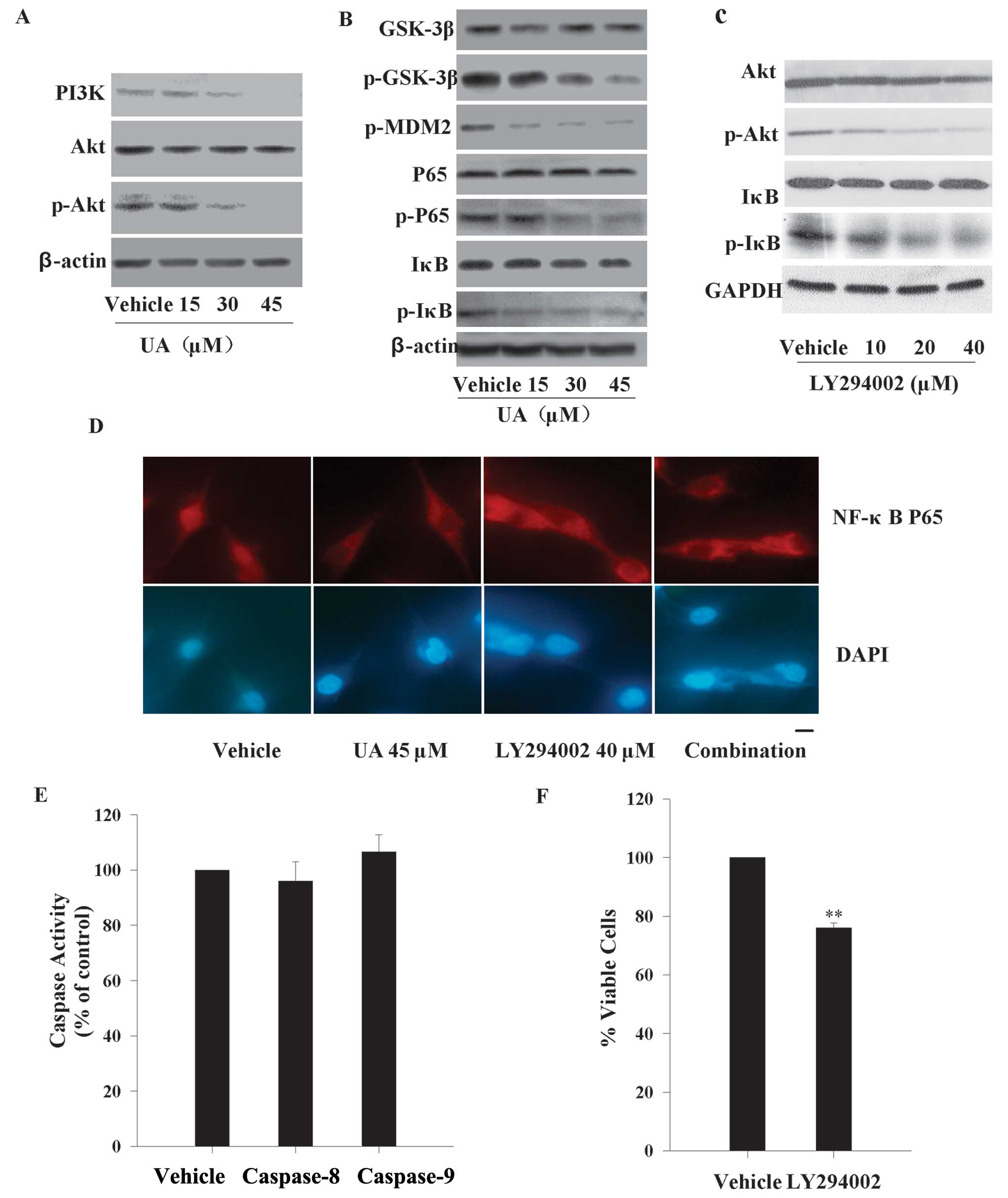
To ascertain the potential mechanism by which UA
caused growth suppression and apoptosis to pancreatic cancer, we
also examined the activity of PI3K/Akt pathway in MIA PaCa-2 cell
line. When cells were in the presence of increasing concentrations
of UA (15, 30 and 45 μM) for 12 h, the results by western blotting
demonstrated the total PI3K and the phosphorylated Akt levels were
attenuated gradually in a dose-dependent manner, while the total
Akt protein expressions did not alter (Fig. 4A). PI3K/Akt pathway is
constitutively activated in most tumor cell lines and regulates a
series of biological characteristics of tumors such as growth and
apoptosis (31). Therefore, we
checked the possible Akt targeting effector molecules downstream.
Similar changes to Akt, were recorded for the phosphorylation level
of GSK-3β, MDM2, P65, IκB that also decreased gradually, whereas
the total proteins of GSK-3β, P65, IκB did not show any
corresponding alteration (Fig.
4B).
PI3K/Akt/NF-κB pathway is involved in
UA-induced cell viability decrease, but not intrinsic and extrinsic
apoptosis
Aberrant activation of NF-κB greatly contributes to
chemoresistance in pancreatic cancer, and suppression of NF-κB
sensitizes the treatment outcome of gemcitabine (32). To assess whether the UA-induced
NF-κB activation is a downstream event of PI3K/Akt signaling
pathway, IκB and its phosphorylated form were further investigated.
We inhibited the phosphorylation of Akt by 10, 20, 40 μM LY294002
for 12 h. As seen in Fig. 4C, 20 μM
LY294002 and above abolished more than half of the phosphorylated
level of Akt protein. Phosphorylated IκB was evidently modulated
downward in a dose-dependent pattern. However, there was no
alteration of total IκB within 12 h. In addition, the nuclear
translocation of the cytoplasmic NF-κB p65 subunit was also checked
by immunofluorescence. UA (45 μM) and/or 40 μM LY294002 for 12 h
repressed the NF-κB p65 nuclear translocation compared with vehicle
(Fig. 4D). These results suggested
there was crosstalk between PI3K/Akt and NF-κB signaling pathways
in MIA PaCa-2 cells.
We next examined the involvement of PI3K/Akt/NF-κB
pathway in the apoptotic cascade. The MIA PaCa-2 cells were
incubated with 40 μM LY294002 for 12 h. As seen in Fig. 4E and F, inhibiting the activation of
Akt can not activate caspase-8 and −9. Hence, PI3K/Akt/NF-κB
pathway is not the main participant in UA-induced intrinsic and
extrinsic apoptotic signaling transductions. However, the cell
viability was decreased by ~25% after exposure to 40 μM LY294002
for 24 h. These findings indicated that PI3K/Akt/NF-κB pathway was
involved in UA-induced cytotoxicity to MIA PaCa-2 cells, but the
mechanism was not caspase-8- and −9-dependent.
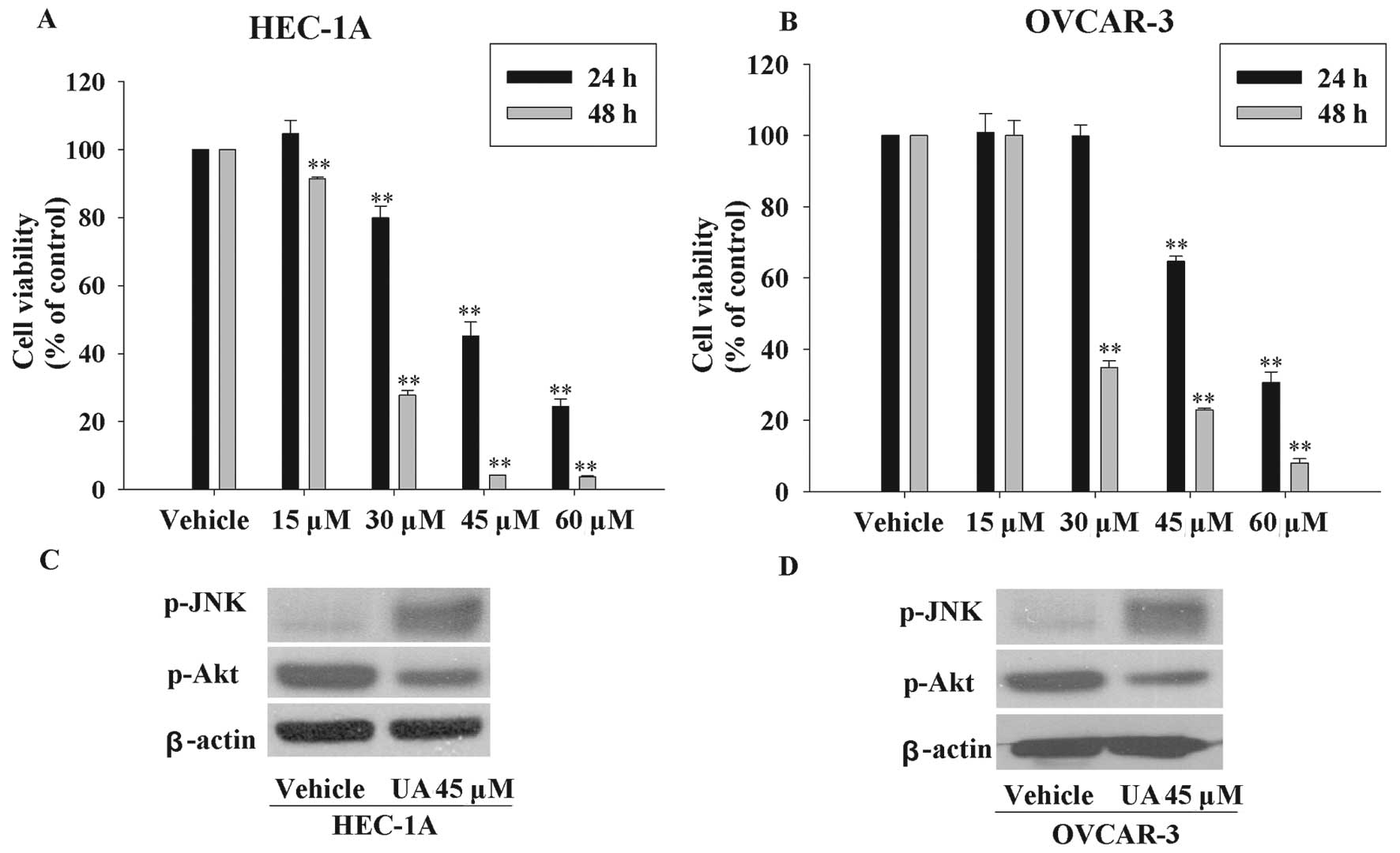
UA overcomes chemoresistance in other
resistant cancer cell lines
We also tested whether UA possessed the potential to
induce other resistant cancer cells death in the similar fashion. A
paclitaxel-resistant endometrial cell line (HEC-1A) and a
platinum-resistant ovarian cancer cell line (OVCAR-3) were used to
examine the cell viability after UA exposure (33,34).
As seen in Fig. 5A and B, UA
inhibited the cell viability in a time and dose-dependent manner.
Moreover, UA also increased the phosphorylated JNK, and decreased
the expression of phosphorylated Akt (Fig. 5C and D). These results further
confirmed that the mechanism of action of UA was associated with
JNK and Akt pathways.
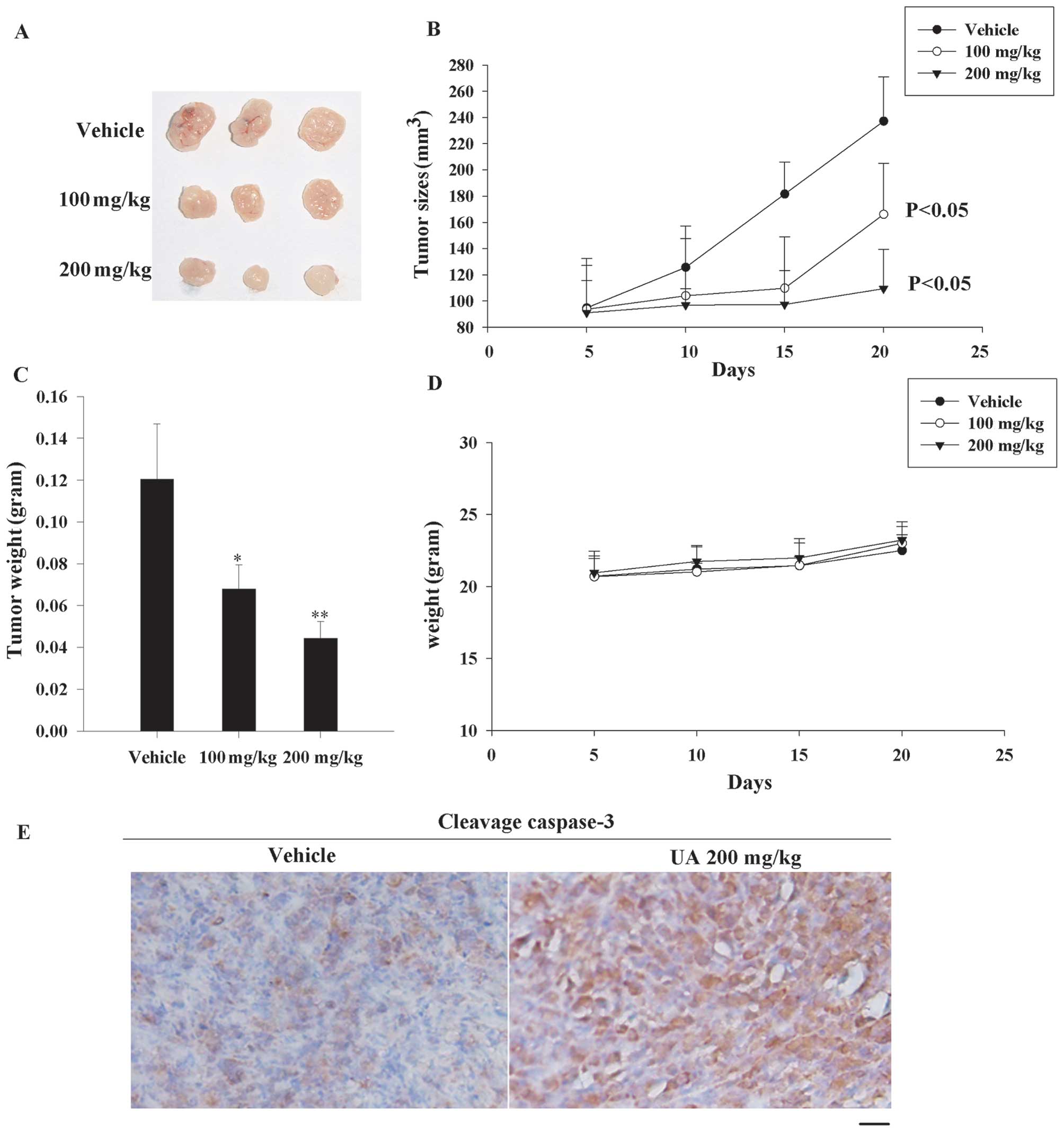
UA causes tumor regression in MIA PaCa-2
xenograft model
The data described above clearly showed that UA was
a potent compound to inhibit proliferation and caused deaths of
various chemotherapy-resistant cancer cell lines. Thus we tried to
apply UA in a nude mouse xenograft model to provide evidence for
clinical application. Until the tumor volume reached ~100
mm3, the mice were treated i.p. with either vehicle or
UA at 100/200 mg/kg twice a week. Significant inhibition of tumor
growth by UA was observed after UA treatment for 20 days (Fig. 6A and B). Tumor weights in UA-treated
groups were also lower than the vehicle group (30% less in 100
mg/kg group and 50% less in 200 mg/kg group) (Fig. 6C). In order to measure the safety of
UA, body weights of mice were regularly detected. As seen in
Fig. 6D, there were no significant
differences compared with control group after the UA treatment.
Furthermore, we checked the cleavage caspase-3 expression by
immunohistochemistry to reveal that the tumor in UA-exposed group
had more visible apoptosis. These data elucidated that UA caused
resistant pancreatic cancer death also in vivo.
Discussion
Due to the poor early diagnosis and serious
chemotherapy resistance, pancreatic carcinoma is highly fatal.
Although gemcitabine is known as the most active chemotherapy agent
for this tumor, it has only a slight survival benefit and shows an
objective tumor response rate of <10% owing to pre-existing or
acquired chemo-resistance (35,36),
thus demanding urgent development of effective therapeutic options.
Here we demonstrate that a small molecule ursolic acid (UA), a well
known anti-oxidative and anti-inflammatory agent, significantly
suppresses the growth and induces apoptosis of resistant pancreatic
cancer through activation of c-Jun-terminal kinase (JNK) pathway
and inhibition of PI3K/Akt/NF-κB pathway.
UA, belonging to pentacyclic triterpenoid, is a
soluble, natural, and small molecule compound, which has displayed
its proliferation-inhibiting and apoptosis-inducing effects in some
chemotherapy-resistant tumors (16,17,37).
Previous study showed that UA only decreased the cell viability by
~40% in a hormone refractory prostate cancer cell line at 72 h
(16). In contrast to the prostate
cancer cells, UA exhibits much more potent cytotoxicity to
pancreatic tumors. The half maximal inhibitory concentration
(IC50 value) of UA was 40.8 μM in highly malignant
pancreatic cancer cells (MIA PaCa-2 cells) within 24 h in
vitro. In our data, both modulation of proliferation and
induction of apoptosis by UA were observed within the time course
of 24 h. However, UA had suppressed the proliferation significantly
at 15 μM rather than any type of caspase activity or cytochrome C
relative release rate. The alterations of proliferation inhibiting
rate and caspase-3/7 activity were detected at 30–45 μM UA.
Furthermore, UA at the higher concentration (60 μM) was only
perceived to be effective in apoptosis induction. Hence, the
results seem to indicate that decreased proliferation and increased
apoptosis by UA together impact cell viability of pancreatic cancer
cells.
The JNK pathway reported as important apoptosis
inducing factor exerts antagonistic effects on apoptosis (30). JNK, a mitogen-activated protein
kinase (MAPK), can be activated primarily by exposure to
environmental stress (38).
Although the extracellular signal-regulated kinase (ERK) pathway is
considered as a great contributor to oncogenesis, previous study
has demonstrated that it plays a lesser role in mitogen-induced
survival of pancreatic cancer (39). However, many studies have reported
that JNK pathway plays a crucial role in UA-induced apoptosis in
many cancers (16,29,40).
In our study, the activation of JNK, but not P38, was induced by UA
in a dose-dependent manner in MIA PaCa-2 cells. Two major apoptotic
signaling transductions, namely the receptor-mediated/exogenous and
mitochondrial/endogenous pathways, are activated in UA-induced
apoptosis in MIA PaCa-2 cells. The trigger of endogenous pathway
depends on the release of cytochrome C from mitochondria to cytosol
and activation of caspase-9 (41).
In the extrinsic pathway, the binding of death ligands to their
homologous receptors drives activity of caspase-8 (42). Here, we showed that SP600125, a
specific JNK inhibitor, could partly protect UA-induced caspase-9
activation rather than caspase-8. Our data are consistent with a
previous report (16). Teraishi
et al(43) have proved that
JNK activation is required for gemcitabine to achieve its
cytotoxicity and the weaker activity of JNK may result in
gemcitabine resistance. Our data displayed that JNK pathway was
activated excessively in the gemcitabine resistant cell line (MIA
PaCa-2). We proved similarly that JNK pathway participated in
UA-induced mitochondrial apoptotic signaling transduction in MIA
PaCa-2 cells. Thus we speculate that UA-induced JNK activation may
contribute to overcome gemcitabine resistance and induces
programmed cell death in gemcitabine resistant pancreatic
cancer.
The current consensus to PI3K/Akt signaling
transduction is that this cascade contributes greatly to tumor
progression and chemotherapy resistance (44). PI3K is a broadly expressed lipid
kinase, the main action of which is to execute the catalyzed
reaction to synthesize the second messenger-PIP3 from
PIP2(44,45). Akt, acting downstream of PI3K, is
usually activated by directly binding to PIP3 through its
pleckstrin-homology (PH) domain and phosphorylation (44). Akt activation is closely relevant to
anti-apoptosis and proliferative signal transduction. Primarily,
Akt stimulates the degradation of IκB which results in the nuclear
translocation of P65 and activation of target genes (46). Akt also catalyzes phosphorylation of
MDM to improve its function to bind to P53 and enhance p53
degradation (47,48). In the aspect of cell proliferative
promotion, Akt inhibits the kinase activity of GSK3β by maintaining
the phosphorylated level to protect cyclin D from degradation
(49). Our results showed that UA
strongly suppressed the activation of Akt and blocked the activity
of its downstream molecules mentioned above.
We proved that suppressing the Akt activity by
LY294002, a selective Akt inhibitor, down-regulated the NF-κB
activation by reducing the p65 nuclear translocation and
phosphorylating IκB in MIA PaCa-2 cells. We propose that PI3K/Akt
pathway seems crosstalk with NF-κB pathway in pancreatic cancer
cells. Arlt et al(32) has
reported previously the evident correlation between basal NF-κB
activity and gemcitabine resistance in pancreatic cancer cells.
There is some evidence showing that PI3K/Akt inhibition promoted
anti-tumor effectiveness of gemcitabine via inhibiting PI3K/Akt
phosphorylation in vitro and in vivo(50,51).
UA may demonstrate its potent anti-cancer effects via
PI3K/Akt/NF-κB pathway in MIA PaCa-2 cells. We observed that
LY294002 did not protect MIA PaCa-2 cells from caspase-8/9
activations. Therefore, exogenous and endogenous apoptosis are not
affected equally by UA-triggered Akt activation. However, LY294002
decreased the viability of MIA PaCa-2 cells to some extent, and it
is well-known that cell viability is a combination of cell
proliferation and death. We argue that the UA-induced
PI3K/Akt/NF-κB pathway suppression may only affect the pancreatic
cancer cell proliferative rate. Besides, recent study also
demonstrated that UA was able to induce tumor cells apoptosis
through a caspase-independent pathway (17). That phenomenon means UA has the
chance to apply for combined treatment to overcome chemotherapy
resistance. In our study, we can not exclude that the
PI3K/Akt/NF-κB pathway also triggers caspase-independent apoptosis.
But further studies are needed to prove the hypothesis.
To examine whether the effect of UA on JNK and Akt
pathway is confined to resistant pancreatic cancer cells solely or
is a general phenomenon which is applicable to other resistant
cancer cells, we examined the effect of UA on an endometrial cell
line (HEC-1A) and an ovarian cancer cell line (OVCAR-3) because
both of them are highly resistant to chemotherapy. According to our
data, the JNK activity was inhibited, but Akt was excessively
activated in these cells. UA significantly decreased the cell
viability of HEC-1A cells and OVCAR-3 cells, which was associated
with an increase of JNK activation and a decrease of Akt
activation. For future clinical application, the safety and
efficacy of UA were further detected in vivo. We presented
evidence that UA suppressed tumor growth without any obvious side
effects. As a natural compound extracted from edible foods and
herbs, UA is relatively low-toxic with LD50 value
>637 mg/kg intraperitoneally in mice. Our studies provide
further evidence that UA is a potent compound to apply to overcome
chemoresistance in oncological therapy.
In conclusion, UA is a small, safe, potent and
multifuntional molecule which is easily produced and relatively
safe. UA has been applied in clinical trial in some areas to treat
cancer (52). Consequently, UA is
comparable or even superior to conventional anti-tumor drugs,
encouraging further the use of this small molecular compound in
gemcitabine resistant pancreatic cancer and other chemo-insensitive
cancers.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (no. 30973202), Doctoral Fund of Ministry of
Education of China (no. 20090171110059), Natural Science Foundation
of Guangdong Province (no. S2011010004621), and Guangdong
Provincial Fund of Industry, Education and Academy (no.
2008B090500194).
References
|
1
|
Stathis A and Moore MJ: Advanced
pancreatic carcinoma: current treatment and future challenges. Nat
Rev Clin Oncol. 7:163–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Campbell PJ, Yachida S, Mudie LJ, et al:
The patterns and dynamics of genomic instability in metastatic
pancreatic cancer. Nature. 467:1109–1113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herrmann R and Jelic S: Pancreatic cancer:
ESMO clinical recommendations for diagnosis, treatment and
follow-up. Ann Oncol. 19(Suppl 2): I25–I26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosewicz S and Wiedenmann B: Pancreatic
carcinoma. Lancet. 349:485–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burris HR, Moore MJ, Andersen J, et al:
Improvements in survival and clinical benefit with gemcitabine as
first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
6
|
Ou YQ, Zhu W, Li Y, et al: Aspirin
inhibits proliferation of gemcitabine-resistant human pancreatic
cancer cells and augments gemcitabine-induced cytotoxicity. Acta
Pharmacol Sin. 31:73–80. 2010. View Article : Google Scholar
|
|
7
|
Ina S, Hirono S, Noda T and Yamaue H:
Identifying molecular markers for chemosensitivity to gemcitabine
in pancreatic cancer: increased expression of interferon-stimulated
gene 15 kDa is associated with intrinsic chemoresistance. Pancreas.
39:473–485. 2010. View Article : Google Scholar
|
|
8
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar
|
|
9
|
Liu J: Oleanolic acid and ursolic acid:
research perspectives. J Ethnopharmacol. 100:92–94. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu J: Pharmacology of oleanolic acid and
ursolic acid. J Ethnopharmacol. 49:57–68. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shih YH, Chein YC, Wang JY and Fu YS:
Ursolic acid protects hippocampal neurons against kainate-induced
excitotoxicity in rats. Neurosci Lett. 362:136–140. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chung YK, Heo HJ, Kim EK, et al:
Inhibitory effect of ursolic acid purified from Origanum majorana L
on the acetylcholinesterase. Mol Cells. 11:137–143. 2001.PubMed/NCBI
|
|
13
|
Tsai SJ and Yin MC: Antioxidative and
anti-inflammatory protection of oleanolic acid and ursolic acid in
PC12 cells. J Food Sci. 73:H174–H178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Somova LO, Nadar A, Rammanan P and Shode
FO: Cardiovascular, antihyperlipidemic and antioxidant effects of
oleanolic and ursolic acids in experimental hypertension.
Phytomedicine. 10:115–121. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cha HJ, Bae SK, Lee HY, et al:
Anti-invasive activity of ursolic acid correlates with the reduced
expression of matrix metalloproteinase-9 (MMP-9) in HT1080 human
fibrosarcoma cells. Cancer Res. 56:2281–2284. 1996.PubMed/NCBI
|
|
16
|
Zhang Y, Kong C, Zeng Y, et al: Ursolic
acid induces PC-3 cell apoptosis via activation of JNK and
inhibition of Akt pathways in vitro. Mol Carcinog. 49:374–385.
2010.PubMed/NCBI
|
|
17
|
Yang L, Liu X, Lu Z, et al: Ursolic acid
induces doxorubicin-resistant HepG2 cell death via the release of
apoptosis-inducing factor. Cancer Lett. 298:128–138. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xavier CP, Lima CF, Preto A, Seruca R,
Fernandes-Ferreira M and Pereira-Wilson C: Luteolin, quercetin and
ursolic acid are potent inhibitors of proliferation and inducers of
apoptosis in both KRAS and BRAF mutated human colorectal cancer
cells. Cancer Lett. 281:162–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Manu KA and Kuttan G: Ursolic acid induces
apoptosis by activating p53 and caspase-3 gene expressions and
suppressing NF-kappaB mediated activation of bcl-2 in B16F-10
melanoma cells. Int Immunopharmacol. 8:974–981. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang JS, Ren TN and Xi T: Ursolic acid
induces apoptosis by suppressing the expression of FoxM1 in MCF-7
human breast cancer cells. Med Oncol. 29:10–15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu W, Ou Y, Li Y, et al: A small-molecule
triptolide suppresses angiogenesis and invasion of human anaplastic
thyroid carcinoma cells via down-regulation of the nuclear
factor-kappa B pathway. Mol Pharmacol. 75:812–819. 2009. View Article : Google Scholar
|
|
22
|
Li J, Zhu W, Leng T, et al:
Triptolide-induced cell cycle arrest and apoptosis in human renal
cell carcinoma cells. Oncol Rep. 25:979–987. 2011.PubMed/NCBI
|
|
23
|
He D, Xu Q, Yan M, et al: The NF-kappaB
inhibitor, celastrol, could enhance the anti-cancer effect of
gambogic acid on oral squamous cell carcinoma. BMC Cancer.
9:3432009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stairs DB, Nakagawa H, Klein-Szanto A, et
al: Cdx1 and c-Myc foster the initiation of transdifferentiation of
the normal esophageal squamous epithelium toward Barrett's
esophagus. PLoS One. 3:e35342008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia Y, Liu Y, Wan J, et al: Novel triazole
ribonucleoside down-regulates heat shock protein 27 and induces
potent anticancer activity on drug-resistant pancreatic cancer. J
Med Chem. 52:6083–6096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jonckheere N, Fauquette V, Stechly L, et
al: Tumour growth and resistance to gemcitabine of pancreatic
cancer cells are decreased by AP-2alpha overexpression. Br J
Cancer. 101:637–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyamoto H, Murakami T, Tsuchida K, Sugino
H, Miyake H and Tashiro S: Tumor-stroma interaction of human
pancreatic cancer: acquired resistance to anticancer drugs and
proliferation regulation is dependent on extracellular matrix
proteins. Pancreas. 28:38–44. 2004. View Article : Google Scholar
|
|
28
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang YX, Kong CZ, Wang LH, et al: Ursolic
acid overcomes Bcl-2-mediated resistance to apoptosis in prostate
cancer cells involving activation of JNK-induced Bcl-2
phosphorylation and degradation. J Cell Biochem. 109:764–773.
2010.PubMed/NCBI
|
|
30
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Falasca M: PI3K/Akt signalling pathway
specific inhibitors: a novel strategy to sensitize cancer cells to
anti-cancer drugs. Curr Pharm Des. 16:1410–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arlt A, Gehrz A, Muerkoster S, et al: Role
of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma
cell lines against gemcitabine-induced cell death. Oncogene.
22:3243–3251. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X, Yang Y, Xu C, et al: CHFR
suppression by hypermethylation sensitizes endometrial cancer cells
to paclitaxel. Int J Gynecol Cancer. 21:996–1003. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lange TS, Kim KK, Singh RK, Strongin RM,
McCourt CK and Brard L: Iron(III)-salophene: an organometallic
compound with selective cytotoxic and anti-proliferative properties
in platinum-resistant ovarian cancer cells. PLoS One. 3:e23032008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Welch SA and Moore MJ: Combination
chemotherapy in advanced pancreatic cancer: time to raise the white
flag? J Clin Oncol. 25:2159–2161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mamaghani S, Patel S and Hedley DW:
Glycogen synthase kinase-3 inhibition disrupts nuclear
factor-kappaB activity in pancreatic cancer, but fails to sensitize
to gemcitabine chemotherapy. BMC Cancer. 9:1322009. View Article : Google Scholar
|
|
37
|
Shan JZ, Xuan YY, Ruan SQ and Sun M:
Proliferation-inhibiting and apoptosis-inducing effects of ursolic
acid and oleanolic acid on multi-drug resistance cancer cells in
vitro. Chin J Integr Med. 17:607–611. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Perugini RA, McDade TP, Vittimberga FJ and
Callery MP: Pancreatic cancer cell proliferation is
phosphatidylinositol 3-kinase dependent. J Surg Res. 90:39–44.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang YX, Kong CZ, Wang HQ, Wang LH, Xu CL
and Sun YH: Phosphorylation of Bcl-2 and activation of caspase-3
via the c-Jun N-terminal kinase pathway in ursolic acid-induced
DU145 cells apoptosis. Biochimie. 91:1173–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar
|
|
42
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Teraishi F, Zhang L, Guo W, et al:
Activation of c-Jun NH2-terminal kinase is required for
gemcitabine's cytotoxic effect in human lung cancer H1299 cells.
FEBS Lett. 579:6681–6687. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kane LP, Shapiro VS, Stokoe D and Weiss A:
Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 9:601–604.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mayo LD and Donner DB: A
phosphatidylinositol 3-kinase/Akt pathway promotes translocation of
Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA.
98:11598–11603. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B and
Hung MC: HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2
phosphorylation. Nat Cell Biol. 3:973–982. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pham NA, Tsao MS, Cao P and Hedley DW:
Dissociation of gemcitabine sensitivity and protein kinase B
signaling in pancreatic ductal adenocarcinoma models. Pancreas.
35:E16–E26. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ng SS, Tsao MS, Nicklee T and Hedley DW:
Wortmannin inhibits pkb/akt phosphorylation and promotes
gemcitabine antitumor activity in orthotopic human pancreatic
cancer xenografts in immunodeficient mice. Clin Cancer Res.
7:3269–3275. 2001.PubMed/NCBI
|
|
52
|
Sultana N: Clinically useful anticancer,
antitumor, and antiwrinkle agent, ursolic acid and related
derivatives as medicinally important natural product. J Enzyme
Inhib Med Chem. 26:616–642. 2011. View Article : Google Scholar
|