Introduction
A small population of malignant cells within an
organ can rapidly expand and ultimately disrupt organ function
(1). These cells were previously
referred to as cancer-initiating cells or cancer stem-like cells
(CSCs) based on their high capacity for self-renewal, multilineage
differentiation, and elevated capacity to induce malignancy
(2,3). CSCs, first identified in acute
leukemia, have now been isolated from several human malignancies,
including breast, brain, prostate, ovarian and lung cancers
(4–6).
Lung cancer is the most common malignancy worldwide.
While advancements in early diagnosis and treatment have been made
in the hope of improving survival, recurrence remains a major
obstacle to achieving a cure. After complete surgical resection,
reported recurrence rates range from 30 to 75% (7). Accumulating evidence suggests that
CSCs are responsible for tumor regeneration following chemotherapy
due to their high drug resistance and tumorigenicity (8–10). The
initial isolation of lung cancer stem-like cells (LCSCs) relied on
a basic knowledge of normal stem cells in the bronchioalveolar duct
junction, assays for their functionality, and expression levels of
the specific markers Sca+ and CD34+ on the
cell membranes. Taken together, this knowledge provided robust
means for the identification of the elusive CSCs within an induced
mouse lung cancer model (11). In
an additional study, cells co-expressing
CD133+/CD326+ were isolated from lung cancer
patients and were characterized as human lung cancer stem-like
cells (12). In the absence of such
specific targets for identification purposes, lung cancer stem-like
cells were also identified using flow cytometry-based side
population cell sorting from lung cancer cell lines (13,14).
These methods, however, do not provide a direct model to assess the
mechanism by which cancer stem cells recover from chemotherapy
treatment (15,16). Here, we report another approach to
enriching drug-resistant stem-like cells from the human lung cancer
cell line A549. This model was used to study the role of stem-like
cells in the recurrence of lung cancer in vitro.
The potential for CSCs to proliferate and self-renew
is remarkable. As few as 10 CSCs can initiate tumor formation in a
mouse model (17). There is
evidence to suggest that certain signaling pathways regulating
self-renewal and proliferation are abnormal in CSCs (18,19).
Polycomb group protein B lymphoma Mo-MLV insertion region 1 homolog
(Bmi1) was originally identified as an oncogene that cooperates
with c-Myc in a murine lymphomagenesis model. There is also
evidence indicating that Bmi1 plays an essential role in
maintaining the self-renewal of mouse bronchioalveolar stem cells
in a mouse model (20). However,
the role of Bmi1 in the recurrence of the tumor mass derived from
stem-like cells following chemotherapy was not studied.
In this study, we used cisplatin treatment to enrich
a population of stem-like cells in the human cell line A549 and
elucidated the mechanisms underlying the self-renewal and expansion
of cisplatin-enriched stem-like cells (CESCs). We demonstrated that
Bmi1 is consistently overexpressed in CESCs. Additionally, we
showed that the downregulation of Bmi1 suppressed the self-renewal
of CESCs and the growth of xenografts. Our study demonstrates that
Bmi1 is involved in controlling the self-renewal of stem-like cells
after cisplatin treatment in human A549 cells, providing insight
into the mechanism underlying the recurrence of lung cancer
following chemotherapy.
Materials and methods
Cell culture
The human lung cancer cell line A549 was obtained
from the American Type Culture Collection (ATCC, Rockville, MD) and
was cultured in RPMI-1640 (HyClone, Logan, UT) with 10% fetal
bovine serum (FBS) (Gibco, Grand Island, MI) at 37°C and 5%
CO2.
Cisplatin-enriched stem-like cells
Following cisplatin treatment (5 μg/ml) for 3 days,
A549 cells were cultured for 4 weeks until drug-surviving colonies
were established (21). The
remaining drug-surviving cells (DSCs) were then cultured in
DMEM/F12 serum-free medium containing 50 μg/ml insulin, 100 μg/ml
apo-transferrin, 10 μg/ml putrescine, 0.03 mM sodium selenite, 2 μM
progesterone, 0.6% glucose, 5 mM HEPES, 0.1% sodium bicarbonate,
0.4% BSA, glutamine and antibiotics (Hyclone), as well as 20 ng/ml
EGF, 10 ng/ml bFGF and 10 ng/ml LIF (8). The third-generation of spheres that
developed from DSCs was termed CESCs.
Cell surface marker analysis by flow
cytometry
Cells (2×105) were placed in 100 μl PBS
and incubated with 10 μl anti-human CD133-PE conjugated antibody
(Miltenyi, Auburn, CA). Cells were then washed, resuspended in PBS
with 2 μl of 7-AAD solution (Pharmingen, San Diego, CA), and
analyzed using an Epic XL flow cytometer (Beckman Coulter, Brea,
CA).
Immunofluorescent staining
Tumor spheres were collected and placed on
Matrigel-coated coverslips. After being cultured for 2 h, the
spheres were fixed in 4% paraformaldehyde for 20 min at RT. A549
cells, DSCs, and CESCs were then labeled with anti-human CD133
(1:300, rabbit polyclonal, Abcam, Cambridge, UK) and anti-human
Bmi1 (1:50, rabbit polyclonal, Santa Cruz Biotechnology, Santa
Cruz, CA) antibodies at 4°C overnight. Secondary antibodies,
including FITC-conjugated goat anti-rabbit IgG or Cy3-conjugated
goat anti-rabbit IgG (1:200 dilution, Jackson ImmunoResearch, West
Grove, PA), were incubated with samples for 1 h at 37°C.
To assess the multipotency of the collected tumor
spheres, anti-human CK8 and anti-human CK18 antibodies were used to
stain differentiated spheres. All cells were also counterstained
with 4,6-diamidino-2-phenylindole (DAPI; 100 mg/ml, Sigma) to
detect cell nuclei. Images were collected and analyzed using a
Leica laser scanning confocal microscope and LAS AF software
(Leica, Mannheim, Germany).
Sphere-forming assay
A549 cells, DSCs and tumor spheres were dissociated
with TrypLE™ Express (Invitrogen, Carlsbad, CA) into single-cell
suspensions. The cells were then inoculated into DMEM/F12
serum-free medium containing 50 ng/ml insulin, 100 ng/ml
apo-transferrin, 10 ng/ml putrescine, 0.03 mM sodium selenite, 2 nM
progesterone, 0.6% glucose, 5 mM HEPES, 0.1% sodium bicarbonate,
0.4% BSA, glutamine and antibiotics (Hyclone), as well as 20 ng/ml
EGF, 10 ng/ml bFGF, and 10 ng/ml LIF using serial diluting methods
into 96-well plates (Corning Inc., Corning, NY). Only wells
containing a single cell were counted. Wells containing more than
one cell or no cells were marked and dismissed from statistical
data. After 6–10 days of culture, wells that contained spheres were
counted using inverted phase contrast microscopy (Leica). The
percentage cells with sphere-forming capacity was calculated as the
numbers of wells with sphere/numbers of total wells with single
cell × 100%.
Differentiation
Cells dissociated from T3 spheres were plated at a
density of 1×104 cells/ml on 24-well plates pre-coated
with collagen IV (BD Biosciences, San Jose, CA) in culture media
supplemented with 10% FBS without growth factors. Upon confluence,
cells were stained with anti-human CK8 and anti-human CK18
antibodies as described above.
In vivo analysis of tumor growth
All procedures involving animals were approved by
the Third Military Medical University Animal Care and Use Committee
(Chongqing, China; No.2008-03). A549 cells and T3 spheres (100,
1,000, 10,000 or 100,000 cells) were injected subcutaneously into
the left flank of 5-week-old NOD/SCID mice (6 mice/group). Tumor
size was measured using calipers, and tumor volume was calculated
using the equation V = π/6 (length × width × height).
Generation of Bmi1 RNAi lentiviruses
After testing knockdown efficiencies of several
shRNA constructs, the following shRNA oligonucleotides were
utilized: 5′-AAGGAGGAGGT GAATGATAAA-3′ (to target the open reading
frame of Bmi1), 5′-AGAATTGGTTTCTTGGAAA-3′ and 5′-CGGAAAGA
ATATGCATAGA-3′ (to target two sites in the 3′ untranslated region
of Bmi1), the loop sequence (TTCAAGAGA), and the reverse complement
of each targeting sequence. A non-specific shRNA was also
synthesized as a control, and each shRNA was cloned into a pGCL-GFP
plasmid containing the U6 promoter and green fluorescent protein
(GFP) (Genechem, Shanghai, China). The plasmids pHelper 1.0 and
pHelper 2.0 were also included to provide the necessary packaging
elements needed for lentivirus production. For viral transduction,
shRNA lentiviral vectors at a multiplicity of infection (MOI) of 50
were added to dispersed CESCs just after plating. Bmi1 protein
levels and GFP fluorescence were measured 72 h
post-transduction.
Quantitative real-time RT-PCR
cDNA (2 μl) was subjected to real-time quantitative
RT-PCR using SYBR-Green as a fluorescent reporter and Real Master
Mix (Tiangen, Beijing, China). Specific gene primers for
ABCG2, Bmi1, and the internal control gene, β-actin,
were amplified in separate reaction tubes. Threshold cycle numbers
(CTs) of triplicate reactions were determined using ABI-7500
software and averaged. Levels of specific gene expression were
normalized to β-actin levels using the formula 2−ΔCT,
where ΔCT is the CT of the housekeeping gene (β-actin) subtracted
from the CT of the target gene. The absence of primer dimers was
confirmed, and the specificity of the products was obtained using
melt curve analysis. The primers sequences used included: ABCG2,
5′-C AGGTGGAGGCAAATCTTCGT-3′ (forward) and 5′-ACAC
ACCACGGATAAACTGA-3′ (reverse) (22); Bmi1, 5′-CTGG TTGCCCATTGACAGC-3′
(forward) and 5′-CAGAAAATG AATGCGAGCCA-3′ (reverse) (23); β-actin, 5′-CCTGGCAC CCAGCACAAT-3′
(forward) and 5′-GCCGATCCACACG GAGTACT-3′ (reverse).
Western blot analysis
Cell extracts of CESCs, CESCsNC− and
CESCsBmi1− were collected, and protein concentrations
were determined using a BCA Protein Assay Kit (Hyclone-Pierce).
Protein samples (50 μg) were then electrophoresed on 12%
SDS-polyacrylamide gels and transferred onto polyvinylidene
fluoride (PVDF) membranes. Membranes were blocked in 5% non-fat dry
milk in Tris-buffered saline (TBS) for 1 h at RT and incubated with
anti-Bmi1, anti-p14ARF, anti-MDM2, and anti-p53 antibodies (diluted
1:200, Santa Cruz Biotechnology) overnight at 4°C. After 3 washes
with TBS-Tween-20 (TBST), membranes were incubated with anti-mouse
IgG or anti-rabbit IgG horseradish peroxidase conjugated antibodies
(diluted 1:2000, Zhongshan Goldenbridge Biotechnology, Beijing,
China) for 2 h at RT. Each membrane was also incubated with an
anti-α-tubulin antibody (Santa Cruz Biotechnology) as a loading
control. Membranes were washed three times with TBST, and bound
antibodies were detected using ECL (Beyotime, Jiangsu, China).
Protein levels were quantitated by densitometry using Quantity One
software (Bio-Rad Laboratories, Munich, Germany).
WST-1 assay
CESC, CESCNC− and CESCBmi1−
spheres (1×103 cells each) were plated on 24-well plates
and cultured for 6 days. WST-1
((4-[3-(4-Iodophenyl)-2H-5-tetrazolio)]-1,3-benzene-disulfonate) is
cleaved to formazan, which is soluble in culture medium and can be
correlated with the number of metabolically active cells present in
the culture. The proliferation index for these assays was
determined each day for 6 days after seeding using the Cell
Proliferation Reagent WST-1 (Beyotime, Jiangsu, China).
Statistical analyses
All experiments were performed at least three times
and representative results are presented as the mean values ±
standard deviation (SD). The data were analyzed using SPSS v13.0
software (SPSS Inc., Chicago, IL, USA). Statistical analysis was
performed by one-way analysis of variance (ANOVA), and comparisons
among groups were achieved using independent sample t-tests.
Statistical significance was established at values of
P<0.05.
Results
In vitro cisplatin treatment combined
with serum-free culture selective enrichment for self-renewing
tumor spheres
Tumors that recur after an initial response to
chemotherapy are resistant to multiple drugs (24). Cancer cells are thought to acquire
resistance to chemotherapy through two mechanistic categories,
innate drug resistance and acquired drug resistance (25). Similar to normal stem cells, cancer
stem-like cells are capable of innate drug resistance due to their
ability to pump out toxic drugs (26). We treated the lung cancer cell line
A549 with the conventional chemotherapy drug cisplatin for 3 days.
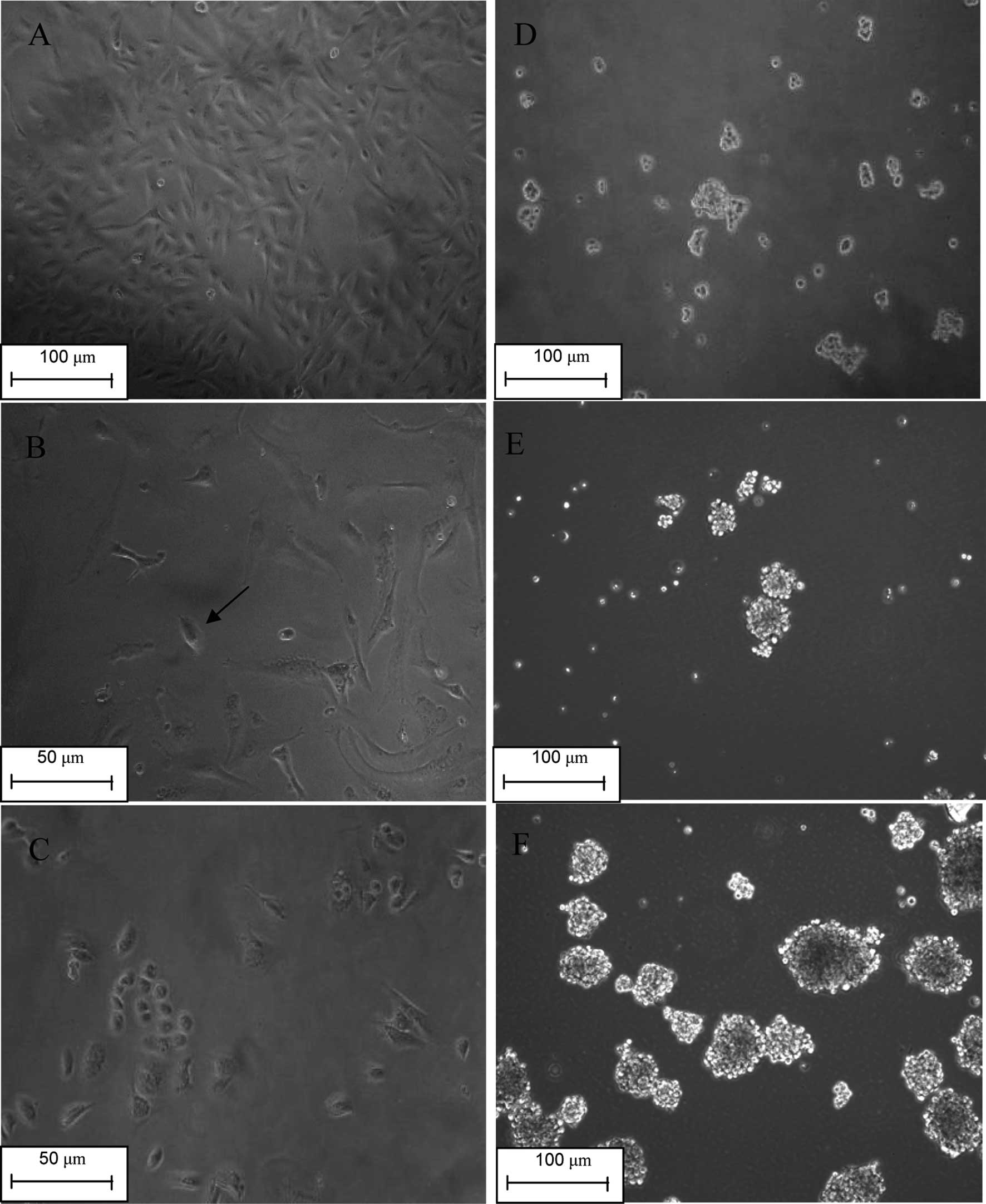
As described by Levina et al(21), a vast majority of the cells died. A
portion of the surviving cells resembled senescent cells with
enlarged and flattened morphology. These cells grew larger in size
and disaggregated during the 10–20 days following drug treatment.
Small, round, or spindle-shaped cells with lower adherence appeared
during the first week after drug treatment, and their growing
colonies gradually replaced the ‘senescent’ cells (Fig. 1A-C). These cells were termed drug
surviving cells (DSCs).
DSCs were then transferred into a low-adherence
plate and cultured at clonal density. A complete serum-free medium
was used to selectively ‘starve’ the cells, which depend on serum
and plate attachment to expand, with acquired drug resistance. As
expected, a portion of the cells did not form tumor spheres and
gradually died, while some tumor spheres were observed floating in
the medium (Fig. 1D). A second
generation of tumor spheres (T2) was produced by dissociating the
first generation (T1) into single cells and culturing accordingly
(Fig. 1E). We observed fewer dead
cells and more spheres, indicating that the population of acquired
drug-resistant cells gradually diminished. In the medium of the
third generation of tumor spheres (T3), very few dead cells were
detected (Fig. 1F). Based on our
observations, we propose that the self-renewing tumor spheres were
selectively enriched in the T3 tumor spheres.
Identifying stem-like cell properties of
T3 tumor spheres
To verify that T3 tumor spheres possess the
characteristics of cancer-initiating cells, the T3 tumor spheres
were analyzed for expression of the stem cell markers CD133 and
ABCG2, sphere-forming capacity, differentiation ability, and
tumorigenic potential.
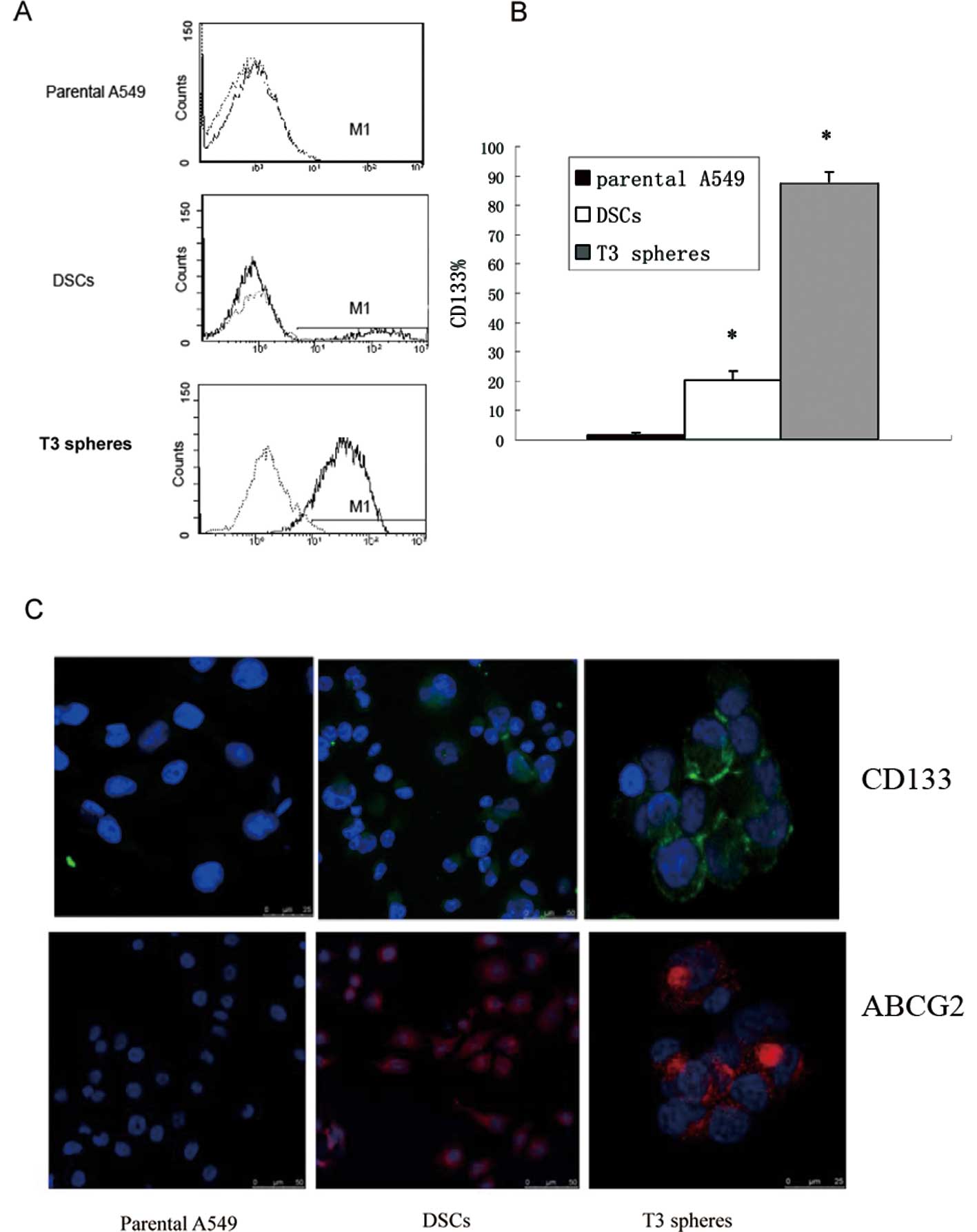
CD133 is a widely accepted marker for stem cells,
and expression of CD133 was detected on parental A549, DSCs and
isolated T3 spheres by flow cytometry. Based on the flow cytometry
assays, ~0.5–1.3% of A549 cells were CD133+, ~20% of
DSCs were CD133+, and >85% of T3 spheres were
CD133+ (Fig. 2A).
Additionally, extensive expression of CD133 was detected in T3
sphere samples, while lower levels of CD133 expression were
detected for the parental A549 and DSCs based on immunostaining
assays (Fig. 2B). Another stem cell
marker, which is also responsible for the efflux of Hoechst 33342
by the ‘side population’, was detected. There was little ABCG2
expression in parental A549, while higher levels of ABCG2 were
present in DSCs and T3 spheres (Fig.
2C).
The sphere-forming cells among the parental A549
cells, DSCs, and T3 spheres were examined for their ability to form
new spheres after being initially cultured as a single cell. After
2 weeks, 85.34% of wells with a single cell derived from T3 spheres
formed a new set of spheres, while only 23.81% of wells with a
single cell derived from DSCs and 1.63% of parental A549 cells were
able to form spheres (Fig. 3A).
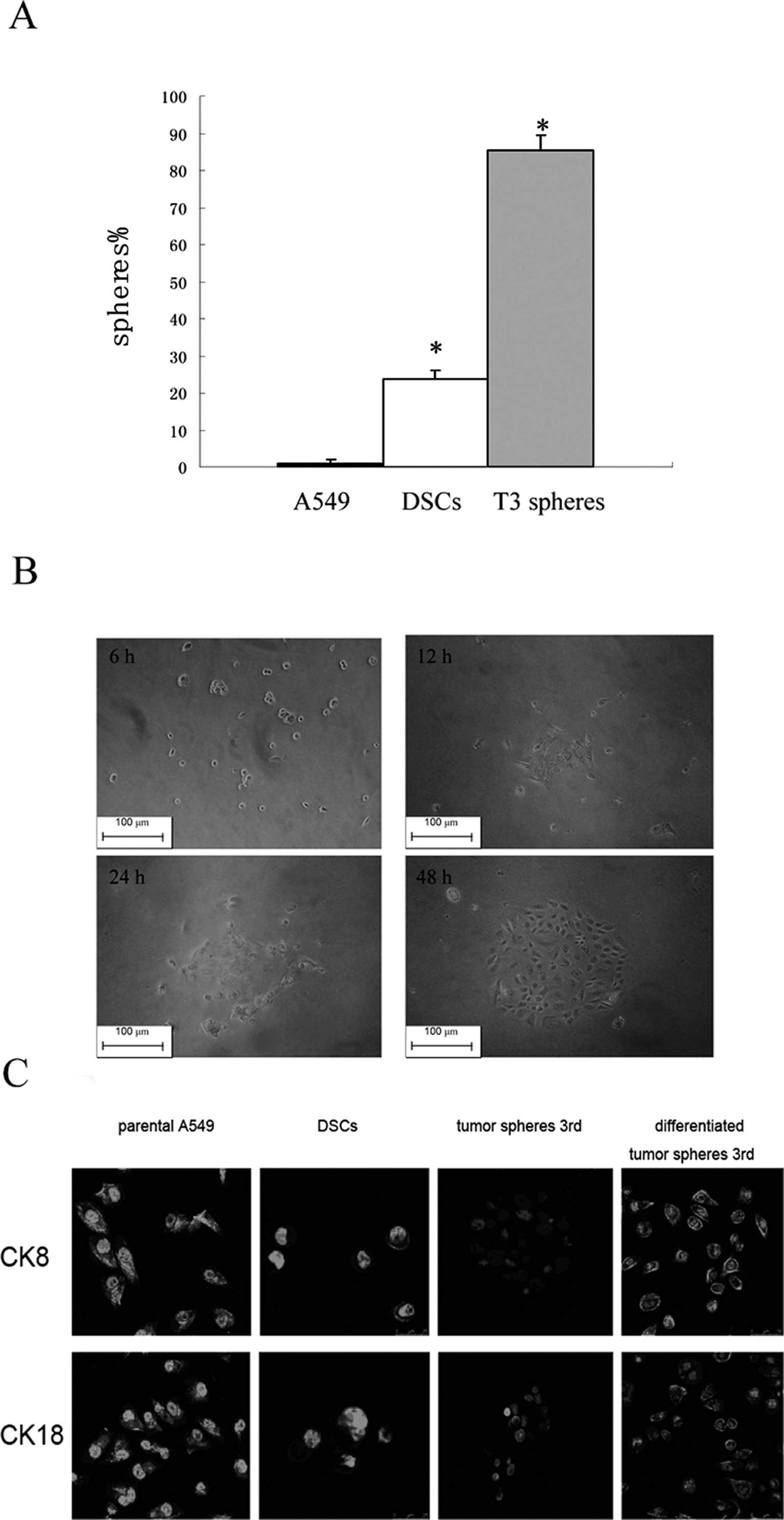 | Figure 3Stem cell properties of the third
generation of tumor spheres. (A) The proportion of wells with
single cells forming new spheres by parental A549 cells, DSCs, and
T3 spheres were quantified (spheres %). Data presented are the
means ± SD. *P<0.001 compared with parental A549. (B)
T3 spheres are dissociated, removed from growth factors, and plated
on collagen for 6, 12, 24 and 48 h. (Original magnification, ×200).
(C) Immunofluoresence staining of differentiated cell markers (CK8
and CK18 as indicated), expressed by parental A549 cells, DSCs, and
undifferentiated and differentiated third-generation tumor spheres.
After further differentiation (48 h), they develop into elongated
cells with subpopulations staining for either differentiated
subtype. |
Type I CK18 cytokeratin and type II CK8 cytokeratin
are markers for the identification of lung cancer cells (27). These were both detected in parental
A549 cells and T3 spheres. Expression of both markers was observed
in parental A549 cells, while the same was not observed in T3
spheres (Fig. 3B). When T3 spheres
were cultured in media supplemented with 10% FBS, however, they
gradually became adherent and exhibited a morphology similar to
that of parental A549 cells, and expression of CK8 and CK18
cytokeratins was restored (Fig.
3C). These results suggest that less differentiated tumor
spheres can propagate into differentiated progenies.
To test the hypothesis that T3 spheres would be more
tumorigenic because of their enhanced stem-like properties, T3
spheres and parental A549 cells were injected subcutaneously into
NOD/SCID mice in a limiting dilution experiment (i.e., 100, 1,000,
10,000 and 100,000 cells). It was observed that as few as 100
sphere-forming tumor cells could produce a tumor, while absence of
tumor formation was observed up to 90 days following the
inoculation of 100 or 1,000 parental A549 cells (Table I).
 | Table IIncidence of tumors of parental A549
cells and third generation tumor spheres serially transplanted on
NOD/SCID mice. |
Table I
Incidence of tumors of parental A549
cells and third generation tumor spheres serially transplanted on
NOD/SCID mice.
| Cell number | 105 | 104 | 103 | 102 |
|---|
| Parental A549
cells | 5 | 2 | 0 | 0 |
| | 104 | 103 | 102 |
| T3 spheres | | 6 | 4 | 2 |
Based on the results described above, the enriched
T3 spheres were defined as cisplatin-enriched stem-like cells
(CESCs).
CESCs have enhanced Bmi1 expression and
are not quiescent
Prior studies demonstrated that cancer stem cells
acquire the self-renewal ability of normal stem cells, yet refuse
to remain quiescent (28). We have
previously demonstrated that inoculating 10,000 CESCs
subcutaneously in NOD/SCID mice can induce the formation of tumor
nodules that are palpable in the 8 days following implantation.
This time span is significantly shorter than the 4 weeks at minimum
required for 10,000 parental A549 cells to form palpable tumor
nodules (data not shown). Although these abilities are supposed to
remain quiescent, these data indicate that the proliferation and
self-renewal of CESCs are accelerated. Elevated levels of Bmi1, a
member of the Polycomb group (PcG) gene family, have been observed
in hematopoietic stem cell populations. Bmi1 is also known to be
required for the self-renewal of mouse bronchioalveolar stem cells
in a mouse model. To evaluate whether Bmi1 regulates the
self-renewing capacity of CESCs, the expression level of Bmi1 in
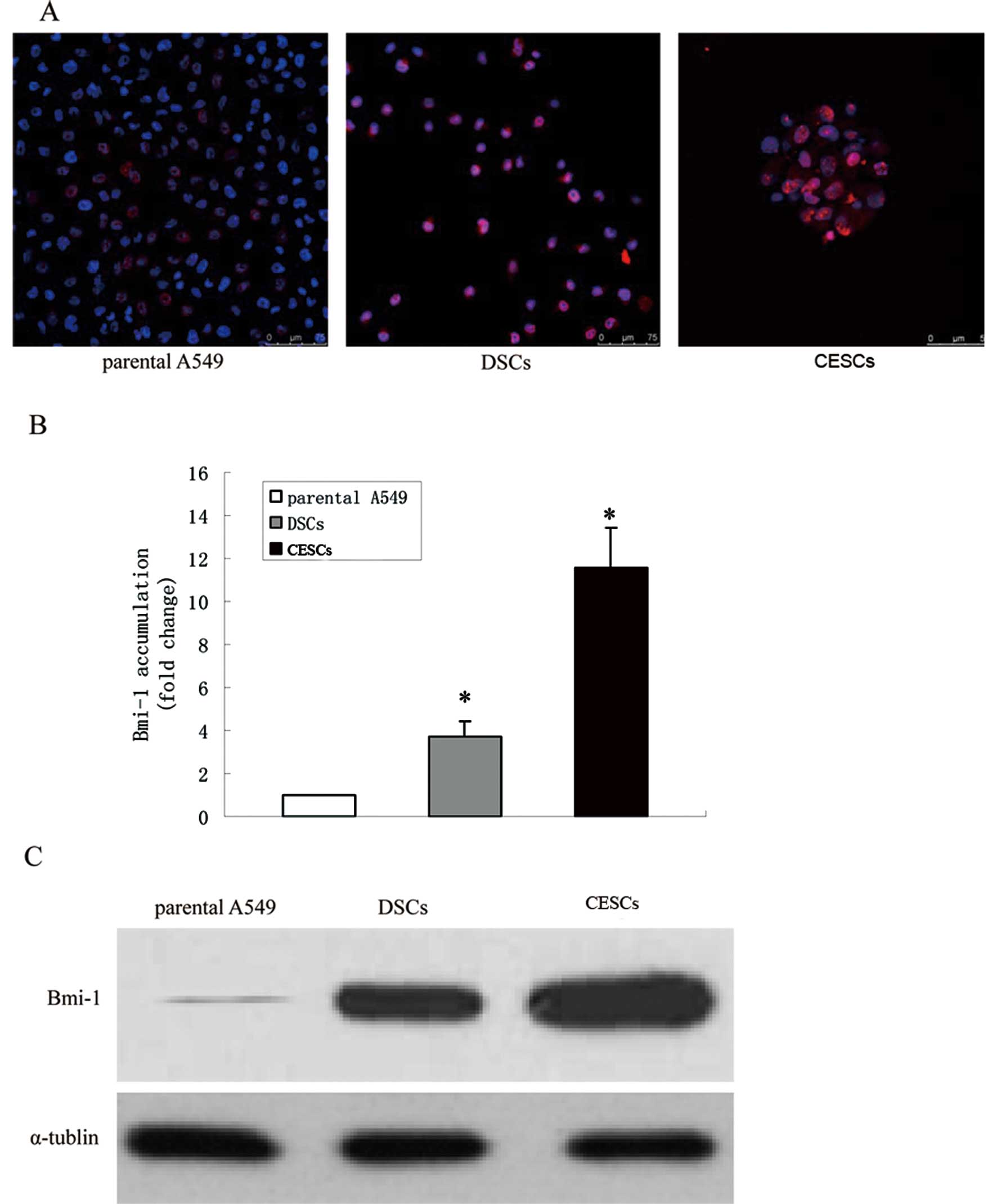
CESCs was initially examined. Bmi1 was expressed in the nuclei of
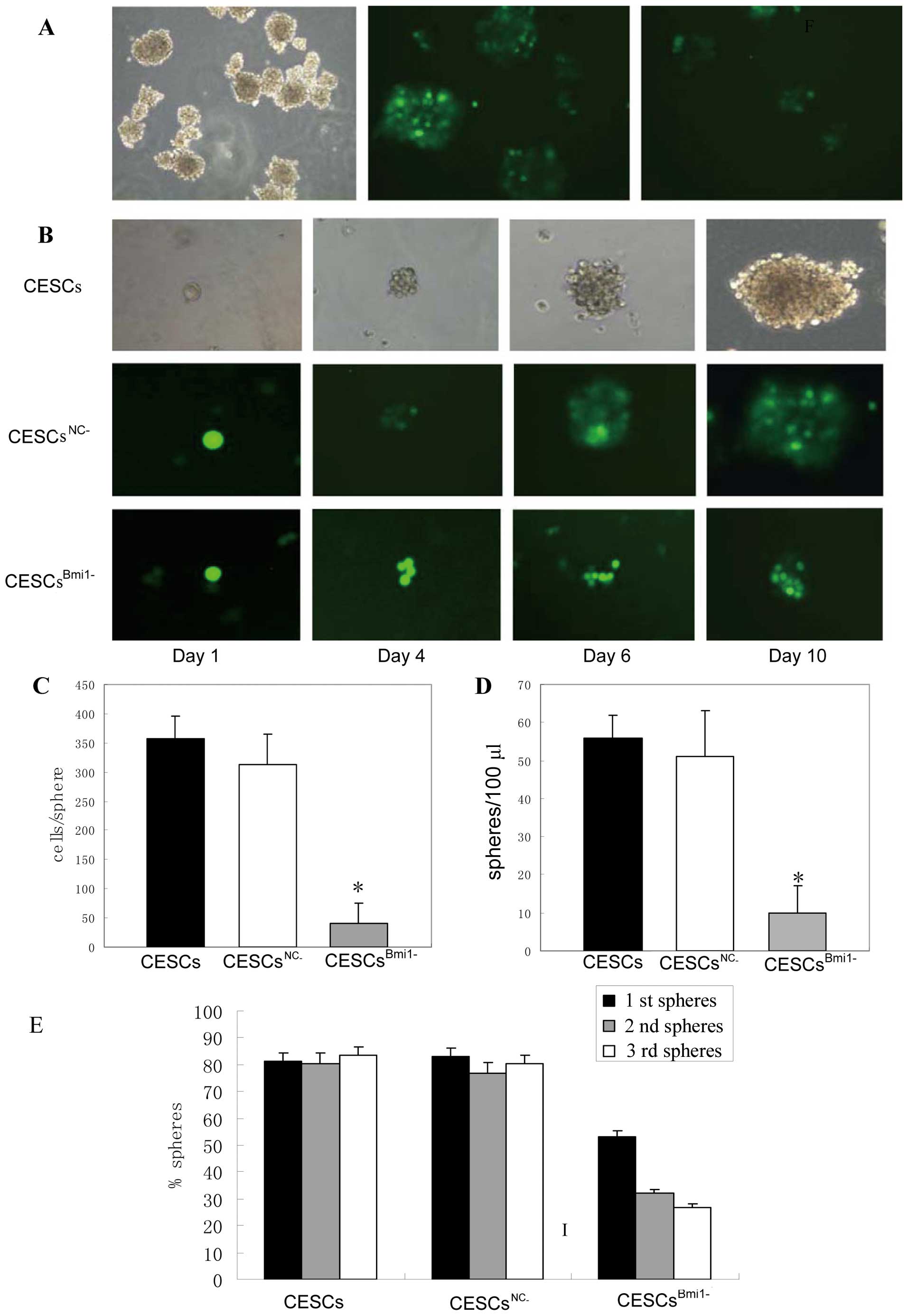
parental A549 cells, DSCs and CESCs (Fig. 4A) but was more widely expressed in
DSCs and CESCs. Additionally, real-time PCR assays were performed
to detect Bmi1 mRNA levels. In these assays, Bmi1
mRNA abundance was 3.73±0.72-fold and 11.55±1.8-fold higher in DSCs
and CESCs, respectively, compared to the parental A549 cells
(Fig. 4B). Similar results from
western blot assays using the same cell types confirmed the finding
(Fig. 4C). Together, these findings
suggest that Bmi1 is overexpressed in CESCs relative to parental
A549 cells.
Bmi1 Knockdown slows down the
proliferation of CESCs
To investigate the role of Bmi1 in CESCs
proliferation, lentiviruses containing a Bmi1-specific shRNA
sequence (CESCsBmi1−) or a scrambled control shRNA
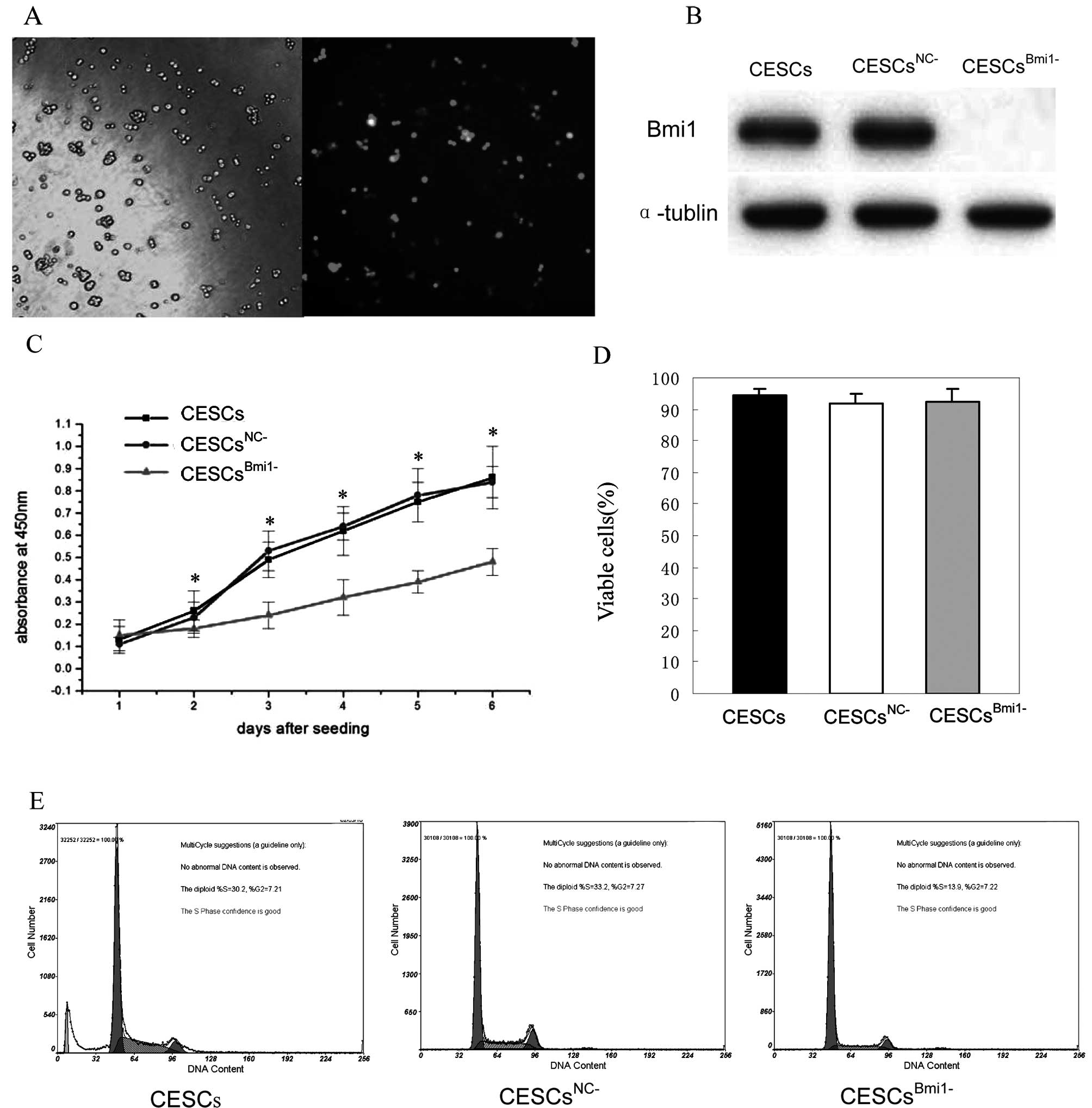
sequence (CESCsNC−) were used to infect CESCs. GFP
fluorescence imaging confirmed the high efficiency of infection
(Fig. 5A), and western blot assays
showed downregulation of Bmi1 expression in CESCsBmi1−
compared to CESCsNC− (Fig.
5B). Using WST-1 assays, proliferation of CESCsBmi1−
was also observed to be significantly slower than that of either
CESCs or CESCsNC− beginning the third day after seeding
(Fig. 5C; P<0.01). To determine
whether the slower growth rate exhibited by CESCsBmi1−
was due to an induction of cell death, a trypan blue exclusion test
was performed to assess cell viability. As shown in Fig. 5D, significant differences in cell
viability were not observed between CESCs, CESCsBmi1−,
and CESCsNC−; therefore, the slower growth rate
associated with the CESCBmi1− cells was not due to an
increase in cell death. Furthermore, the cell cycle analysis of
CESCsBmi1−, CESCs, and CESCsNC− assessed
using flow cytometry revealed an appreciable arrest in the G1 phase
in CESCsBmi1− relative to CESCs and CESCsNC−
(Fig. 5E). These results suggest
that cell proliferation of CESCs slowed following Bmi1
knockdown.
Bmi1 is required for the self-renewal and
tumorigenesis of CESCs
To test whether Bmi1 is important for the
self-renewal of CESCs, the rate of sphere formation was assayed for
CESCsBmi1−, CESCs and CESCsNC−. The size and
rate of sphere formation were both reduced in
CESCsBmi1−, with the spheres being 1/5 to 1/6 the size
of control CESCs (Fig. 6A and B).
This was due to a lower number of cells being contained in
CESCBmi1− spheres, which was determined to have 41
cells/sphere vs. 357 cells/sphere and 313 cells/sphere for CESC and
CESCNC− spheres, respectively (Fig. 6C). In addition, ~100 spheres/ml, 560
spheres/ml, and 510 spheres/ml were detected in
CESCsBmi1−, CESCs and CESCsNC−, respectively
(Fig. 6D). It would be more
convincing to carry out a sphere-forming assay from single cells
and use these cells to determine the role of Bmi1. We measured the
rate of sphere formation using single CESCsBmi1−, CESCs
and CESCsNC− for three passages. The rate of sphere
formation gradually declined in CESCsBmi1− but remained
stable in CESCs and CESCsNC− (Fig. 6E). Taken together, these results
indicate that the downregulation of Bmi1 leads to the loss of
self-renewal in some CESCs.
Tumor formation assays were also performed by
subcutaneously injecting 100, 1,000, 10,000 and 100,000 CESCs,
CESCsBmi1− and CESCsNC− into NOD/SCID mice.
CESCsBmi1− cells were associated with a decreased level
of tumorigenicity relative to CESCs and CESCsNC− because
at least 10,000 CESCsBmi1− were needed to establish a
xenograft tumor (Table II). These
data indicate that Bmi1 is required for CESCs to form
xenografts.
| Table IIIncidence of tumors of CESCs,
CESCsBmi1− and CESCsNC− cells serially
transplanted on NOD/SCID mice. |
Table II
Incidence of tumors of CESCs,
CESCsBmi1− and CESCsNC− cells serially
transplanted on NOD/SCID mice.
| Cell number | 105 | 104 | 103 | 102 |
|---|
| CESCs | 6 | 6 | 5 | 3 |
| 105 | 104 | 103 | 102 |
|
CESCsNC− | 6 | 6 | 4 | 3 |
| 105 | 104 | 103 | 102 |
|
CESCsBmi1− | 4 | 1 | 0 | 0 |
Discussion
Lung cancer is the most common malignancy worldwide.
Despite a variety of traditional therapeutic methods ranging from
surgery to chemotherapy to the use of recently developed
EGFR-targeted drugs, recurrence of lung cancer remains high. A
growing body of evidence suggests that cancer stem-like cells
(CSCs) are chemoresistant and may be responsible for the observed
tumor recurrence following treatment with chemotherapy. Due to the
difficulty in recognizing CSCs, little is known regarding the
regulatory mechanisms responsible for their capacity to self-renew
and initiate tumor formation; therefore, the role of CSCs in tumor
recurrence remains unclear.
Previous study has reported that chemotherapy drugs
selectively kill non-stem-like cells (non-CSCs) lacking ABCG2 or
other drug pumping mechanisms, resulting in an enrichment of
stem-like cells (21). In this
study, A549 lung cancer cells were treated with cisplatin to select
for stem-like cells. To further exclude serum-dependent non-CSCs
that may have been retained among the CSCs, cisplatin-surviving
cells were sequentially passaged in serum-free media to obtain
third-generation tumor spheres. These cells were considered to have
the properties of stem-like cells, including the expression of
CD133 and ABCG2, enhanced sphere formation capacities,
differentiation, as well as the ability to form tumors in
immune-deficient mice. Based on in vitro sphere forming
assays and the proportion of CD133+ cells detected, it
is estimated that >85% of the T3 spheres obtained were stem-like
cells. Additionally, low levels of epithelial differentiation
markers (i.e., cytokeratins 8/18) confirmed the undifferentiated
status of T3 spheres. Based on these results, T3 spheres were
considered to represent cisplatin-enriched stem-like cells (CESCs)
and were further characterized.
Stem cells are traditionally regarded as quiescent
cells and only respond to a proliferative signal when tissue repair
is needed (29). In this study, the
majority of A549 cells were eradicated using cisplatin treatment,
creating a phenomenon resembling tissue repair. We observed that
CESCs were not quiescent and formed tumor nodules within 1 week,
whereas parental A549 cells required 4 weeks. These data indicate
that CESCs had a high potential to proliferate and expand.
Investigating the mechanism regulating CESCs proliferation and
self-renewal is important for understanding how cancer stem-like
cells initiate the tumor mass. The Polycomb group protein Bmi1 has
been implicated in the development and progression of several
malignancies (30,31). With regard to stem cell biology,
Bmi1 is essential for the self-renewal of adult murine
hematopoietic stem cells and neuronal stem cells (32,33).
Recently, it has been reported that Bmi1 is critical for
bronchioalveolar stem cell expansion and lung tumorigenesis in a
mouse model (20). In this study,
we initially observed the expression of Bmi1 in CESCs and also
found that Bmi1 was transcriptionally and translationally
upregulated in CESCs compared with parental A549 cells. This may be
due to the regulation of Bmi1 by certain transcriptional factors
under the pressure of cisplatin or to the cells' need to
proliferate. This mechanism is not of direct relevance to our study
and will not be discussed here.
Because we have confirmed that Bmi1 is highly
expressed in CESCs, it is reasonable to study the function of Bmi1
by decreasing its level of expression. We therefore developed a
Bmi1-targeted shRNA and inserted this construct into the A549 cell
genome using a lentivirus, establishing a stable Bmi1
low-expressing cell line, CESCsBmi1−. Bmi1-targeted
shRNA resulted in a decrease in the proliferative potential of
CESCs as shown on the WST-1 assay that was unrelated to cell death.
Additionally, flow cytometry demonstrated a G1 phase arrest in
CESCsBmi1−, indicating that knocking down Bmi1
influenced the cell cycle and resulted in impaired cell
proliferation. We also tested the function of Bmi1 in the
regulation of CESCs self-renewal using the sphere-forming assay
both from colonies and from single cells. CESCsBmi1−
formed fewer spheres compared to CESCs, and the size of
CESCsBmi1− spheres was smaller due to a lower average
cell count in CESCsBmi1− spheres relative to CESCs.
Self-renewal of CESCsBmi1−, CESCs and
CESCsNC− at the single cell level was also studied.
Knock down of Bmi1 resulted in a decreased ratio of sphere-forming
cells, indicating the Bmi1 is required to maintain stem cell number
by promoting self-renewal. Additionally, in vivo studies
demonstrated that CESCsBmi1− injected subcutaneously
into NOD/SCID mice exhibited a lower tumorigenic potential. Based
on these data, Bmi1 plays a crucial role in maintaining the
self-renewing capacity of CESCs.
Drug-resistance of CSCs represents a significant
obstacle in the treatment of cancers with chemotherapy. In lung
cancer particularly, non-small cell lung cancer (NSCLC),
chemotherapy often results in tumor recurrence within 1–2 years
following treatment, as well as a restoration of pre-treatment
tumor size. In this study, we used the lung cancer A549 cell line
to study the role of stem-like cells in tumor recurrence following
chemotherapy. We enriched CESCs using cisplatin treatment and found
that Bmi1 is a critical gene regulating CESCs proliferation and
self-renewal. Our data provide important insight into the mechanism
of lung cancer recurrence following chemotherapy.
Acknowledgements
This project was supported by a grant from the
National Natural Science Foundation of China (NSFC, no. 30772144).
We thank Jin Li (Respiratory Disease Research Center, The Second
Affiliated Hospital of Third Military Medical University,
Chongqing, China) for her technical assistance in flow cytometry,
and Lixia Guang (Central Laboratory, The Second Affiliated Hospital
of Third Military Medical University, Chongqing, China) for
providing laboratory instruments and analytic software.
References
|
1
|
Shackleton M, Quintana E, Fearon ER and
Morrison SJ: Heterogeneity in cancer: cancer stem cells versus
clonal evolution. Cell. 138:822–829. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vermeulen L, Todaro M, de Sousa Mello F,
et al: Single-cell cloning of colon cancer stem cells reveals a
multi-lineage differentiation capacity. Proc Natl Acad Sci USA.
105:13427–13432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dick JE: Stem cell concepts renew cancer
research. Blood. 112:4793–4807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harrison H, Farnie G, Howell SJ, et al:
Regulation of breast cancer stem cell activity by signaling through
the Notch4 receptor. Cancer Res. 70:709–718. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim CF, Jackson EL, Woolfenden AE, et al:
Identification of bronchioalveolar stem cells in normal lung and
lung cancer. Cell. 121:823–835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eramo A, Lotti F, Sette G, et al:
Identification and expansion of the tumorigenic lung cancer stem
cell population. Cell Death Differ. 15:504–514. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seo DC, Sung JM, Cho HJ, et al: Gene
expression profiling of cancer stem cell in human lung
adenocarcinoma A549 cells. Mol Cancer. 6:752007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ho MM, Ng AV, Lam S and Hung JY: Side
population in human lung cancer cell lines and tumors is enriched
with stem-like cancer cells. Cancer Res. 67:4827–4833. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sullivan JP, Minna JD and Shay JW:
Evidence for self-renewing lung cancer stem cells and their
implications in tumor initiation, progression, and targeted
therapy. Cancer Metastasis Rev. 29:61–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maeda R, Yoshida J, Hishida T, et al: Late
recurrence of non-small cell lung cancer more than 5 years after
complete resection: incidence and clinical implications in patient
follow-up. Chest. 138:145–150. 2010.PubMed/NCBI
|
|
14
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar
|
|
15
|
Dylla SJ, Beviglia L, Park IK, et al:
Colorectal cancer stem cells are enriched in xenogeneic tumors
following chemotherapy. PLoS One. 3:e24282008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eramo A, Ricci-Vitiani L, Zeuner A, et al:
Chemotherapy resistance of glioblastoma stem cells. Cell Death
Differ. 13:1238–1241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tamase A, Muraguchi T, Naka K, et al:
Identification of tumor-initiating cells in a highly aggressive
brain tumor using promoter activity of nucleostemin. Proc Natl Acad
Sci USA. 106:17163–17168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Malanchi I, Peinado H, Kassen D, et al:
Cutaneous cancer stem cell maintenance is dependent on beta-catenin
signalling. Nature. 452:650–653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Korkaya H, Paulson A, Iovino F and Wicha
MS: HER2 regulates the mammary stem/progenitor cell population
driving tumorigenesis and invasion. Oncogene. 27:6120–6130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dovey JS, Zacharek SJ, Kim CF and Lees JA:
Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem
cell expansion. Proc Natl Acad Sci USA. 105:11857–11862. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Levina V, Marrangoni AM, DeMarco R,
Gorelik E and Lokshin AE: Drug-selected human lung cancer stem
cells: cytokine network, tumorigenic and metastatic properties.
PLoS One. 3:e30772008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bea S, Tort F, Pinyol M, et al: BMI-1 gene
amplification and overexpression in hematological malignancies
occur mainly in mantle cell lymphomas. Cancer Res. 61:2409–2412.
2001.
|
|
23
|
Tsunoda S, Okumura T, Ito T, et al: ABCG2
expression is an independent unfavorable prognostic factor in
esophageal squamous cell carcinoma. Oncology. 71:251–258. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Donnenberg VS and Donnenberg AD: Multiple
drug resistance in cancer revisited: the cancer stem cell
hypothesis. J Clin Pharmacol. 45:872–877. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tredan O, Galmarini CM, Patel K and
Tannock IF: Drug resistance and the solid tumor microenvironment. J
Natl Cancer Inst. 99:1441–1454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eyler CE and Rich JN: Survival of the
fittest: cancer stem cells in therapeutic resistance and
angiogenesis. J Clin Oncol. 26:2839–2845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanaji N, Bandoh S, Fujita J, Ishii T,
Ishida T and Kubo A: Compensation of type I and type II cytokeratin
pools in lung cancer. Lung Cancer. 55:295–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li L and Neaves WB: Normal stem cells and
cancer stem cells: the niche matters. Cancer Res. 66:4553–4557.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Beachy PA, Karhadkar SS and Berman DM:
Tissue repair and stem cell renewal in carcinogenesis. Nature.
432:324–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park IK, Morrison SJ and Clarke MF: Bmi1,
stem cells, and senescence regulation. J Clin Invest. 113:175–179.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang HW, Ding J, Jin JL, et al: Defects
in mesenchymal stem cell self-renewal and cell fate determination
lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner
Res. 25:640–652. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shakhova O, Leung C and Marino S: Bmi1 in
development and tumorigenesis of the central nervous system. J Mol
Med. 83:596–600. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van der Lugt NM, Domen J, Linders K, et
al: Posterior transformation, neurological abnormalities, and
severe hematopoietic defects in mice with a targeted deletion of
the bmi-1 proto-oncogene. Genes Dev. 8:757–769. 1994.
|