Introduction
Statins are widely used drugs in therapy of
hypercholesterinemia. They competitively inhibit HMG-CoA-reductase,
a key enzyme in cholesterol biosynthesis, and additionally reduce
blood LDL levels by inducing expression of LDL receptors (LDLR).
Statins have been described to support anti-viral therapy (1–3) and to
interfere with tumor growth in patients (4–6).
Statistical correlation of statin use and cancer incidence revealed
protection at least from stomach and liver cancer, as well as from
lymphoma (7).
Although the multi-kinase-inhibitor Sorafenib has
been shown to exert anti-tumor activity and to prolong patient
survival (8,9), hepatocellular carcinoma (HCC) still is
among the most prevalent malignancies worldwide (10). Statins might be able to broaden the
spectrum of drugs useful in therapy of HCC. In fact, various
statins have been tested for their potential to interfere with
hepatoma cell growth alone (11–14) or
in combination (15,16). On the other hand, when applied at
high doses or to patients with impaired liver functions statins
have severe side effects, e.g., myopathy or liver toxicity
(17,18). Therefore, it would be favourable to
use statins in HCC therapy which do not further affect functional
primary hepatocytes. We investigated selectivity of anti-tumor
effects for 5 commonly prescribed statins using human and mouse
hepatoma cell lines in comparison to primary hepatocytes. Our
results in the human and in the mouse system show a more selective
effect of fluva-, simva- and lovastatin on hepatoma cells compared
to primary hepatocytes. Sensitivity of hepatoma cell lines towards
statin incubation was dependent on cellular proliferation and
correlated to the expression of p53. The p53 over-expressing human
hepatoma cell line Huh7 could be sensitized towards statin-induced
apoptosis by a knockdown of p53. Apoptosis induction by statins in
tumor cells was found to be based on inhibition of prenylation,
since restoration of geranyl-geranylation reverted statin
cytotoxicity in vitro. These results further point to
beneficial effects of statin treatment of HCC patients, but also
implicate the use of selected statins and that anti-tumor effects
might depend on individual patient tumor conditions, e.g., p53
expression status of the tumor.
Materials and methods
Reagents
HMG-CoA-reductase inhibitors fluva- and simvastatin
(Cayman Chemical, Ann Arbor, MI, USA), atorvastatin (Sortis, Pfizer
Pharma GmbH), rosuvastatin (Crestor, Astra Zeneca) and lovastatin
(Tocris Bioscience; Bristol, UK) were dissolved in DMSO. As a
vehicle control, DMSO was dissolved to the concentrations used on
statin incubated cells and measured in parallel. Mevalonate (MVLT),
geranyl-geranyl-pyrophosphate (GGPP), and cholesterol were
purchased from Sigma-Aldrich Chemie GmbH; Steinheim, Germany. Final
concentrations (as indicated in the figures and figure legends)
were obtained by dilution in medium.
Cell culture
The human hepatoma cell lines Huh7 (19) and HepG2 (20) were cultured in DMEM containing 10%
fetal calf serum (FCS) (both from Invitrogen GmbH, Karlsruhe,
Germany) and 1% penicillin/streptomycin (Biochrom AG Seromed,
Berlin, Germany). The mouse hepatoma cell line Hepa1-6 (21) was maintained in RPMI-1640 medium
(10% FCS; 1% penicillin/streptomycin). For experimental procedures
cells were seeded into 24- or 96-well plates and allowed to adhere
overnight.
Isolation of primary hepatocytes
Mouse primary hepatocytes (C57Bl/6) were isolated by
a modification of the two-step collagenase perfusion method of
Seglen (22) and cultured in
William’s E+GlutaMAXTM-I medium, supplemented with 10% FCS, 1%
L-glutamine, 2% 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES), 1% sodium pyruvate (all from Invitrogen GmbH) and 1%
penicillin/streptomycin (Biochrom AG Seromed). Primary human
hepatocytes were isolated as described previously (23).
Stable knockdown
ShRNA expressing vectors were based on the
lentiviral pLKO.1 construct (RNAi Consortium vector collection
(24) and purchased from Sigma
Aldrich GmbH. Target sequences for shRNA were TGG GTC CTT ACA CTC
AGC TTT CT for p53 (shp53) and TTA TCG CGC ATA TCA CGC G for E.
coli DNA polymerase as a control gene (shneg). Transduced cells
were selected using 2 μg/ml puromycin (Carl Roth GmbH + Co. KG;
Karlsruhe, Germany).
Analysis of cell viability, proliferation
and apoptosis
Cell viability was measured by using (3–4,
5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma
Aldrich GmbH) according to the manufacturer’s instructions.
Cellular proliferation and viability was measured by the
xCELLigence real-time cell analyzing system (Roche Molecular
Diagnostics, Mannheim, Germany). Briefly, 10,000 cells were seeded
per well of a 96-well E-plate and viability was measured
continuously as impedance and expressed as an arbitrary unit named
cell index. Data were normalized to the time-point of seeding and
represent means of triplicates as described previously (25). Dead cells were visualized by trypan
blue staining and cell counting using a Neubauer chamber (Carl Roth
GmbH + Co. KG). Total cell numbers as well as the percentages of
dead cells were determined at 72-h incubation. To quantify
apoptosis, activation of caspase 3 was measured using the
colorimetric assay (Sigma Aldrich GmbH) according to the
manufacturer’s instructions.
Western blot analyses
Protein (25 μg) was fractionated by 12%
SDS-polyacrylamide gel electrophoresis and blotted onto
nitrocellulose membranes. Western blot analyses were developed
using an enhanced chemiluminescence system (Amersham, GE Healthcare
Europe GmbH, Munich, Germany) according to the manufacturer’s
instructions. Semi-quantitative evaluation was performed using the
VersaDoc Imaging System (Bio-Rad Laboratories GmbH, Munich,
Germany). Antibodies: rabbit anti-p53 (1:1000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and mouse anti-GAPDH
(1:5000; HyTest Ltd., Turku, Finland).
Detection of mRNA by RT-PCR
To verify altered gene expression RNA was
transcribed into cDNA using the Verso™ cDNA kit (Thermo Fisher
Scientific, Waltham, MA, USA). Oligonucleotides for subsequent
PCR-reactions were obtained from Metabion International AG
(Martinsried, Germany) and are summarized in Table I. Real-time RT-PCR was performed
using the CFXTM real-time system (Bio-Rad Laboratories GmbH) and
reagents from ABgene® (Epsom, UK). To confirm
amplification specificity, PCR products were subjected to melting
curve analysis and gel electrophoresis.
 | Table IOligonucleotide sequences for
real-time RT-PCR. |
Table I
Oligonucleotide sequences for
real-time RT-PCR.
| Oligonucleotide | Sequence 5′-3′ |
|---|
| 5′GAPDHhum | 5′-TGA TGA CAT CAA
GAA GGT GG-3′ |
| 3′GAPDHhum | 5′-CGA CCA CTT TGT
CAA GCT C-3′ |
| 5′mATPsynthase | 5′-GCC CAC TTC TTA
CCA CAA GG-3′ |
| 3′mATPsynthase | 5′-GCG ACA GCG ATT
TCT AGG AT-3′ |
| 5′PCNAhum | 5′-GGC GTG AAC CTC
ACC AGT AT-3′ |
| 3′PCNAhum | 5′-TCT CGG CAT ATA
CGT GCA AA-3′ |
Statistical analysis
The results were analyzed using Student’s t-test, if
two groups were compared and Dunnett’s test if more groups were
tested against a control group. If variances were inhomogeneous in
Student’s t-test, the results were analyzed using the Welsh test.
The data in this study are expressed as a mean ± SEM. P≤0.05 was
considered significant.
Results
Fluvastatin, simvastatin and lovastatin
dose-dependently and selectively reduce viability of mouse hepatoma
cells
The use of statins has been shown to affect hepatic
tumor growth in patients (6), but
also bears the risk of severe side effects when over-dosed or
applied to patients with e.g., impaired liver function (17,18).
To investigate which statin might have the highest efficacy in HCC
therapy and to predict the probability of side effects we first
compared viability of freshly isolated primary mouse hepatocytes
(PH) in the presence of increasing concentrations of statins to
mouse hepatoma cells (Hepa1-6) in vitro. Using fluva- (FLV),
simva- (SMV), rosuva- (ROV), atorva- (ATV), as well as lovastatin
(LOV) we found that within 72 h of incubation, FLV (Fig. 1A), SMV (Fig. 1B) and LOV (Fig. 1E) significantly reduced viability of
hepatoma cells in comparison to PH, while ROV (Fig. 1C), and ATV (Fig. 1D) did not. At a concentration of 10
μM FLV, SMV and LOV effects on mouse hepatoma cells were
significantly more pronounced than effects on primary mouse
hepatocytes. We also chose this concentration to compare the impact
of all 5 statins on viability of primary human hepatocytes (PHhum).
Our results show that PHhum in general were much more resistant
towards statin incubation than mouse hepatocytes, with only mild
toxic effects being observed for FLV and LOV (Fig. 1F).
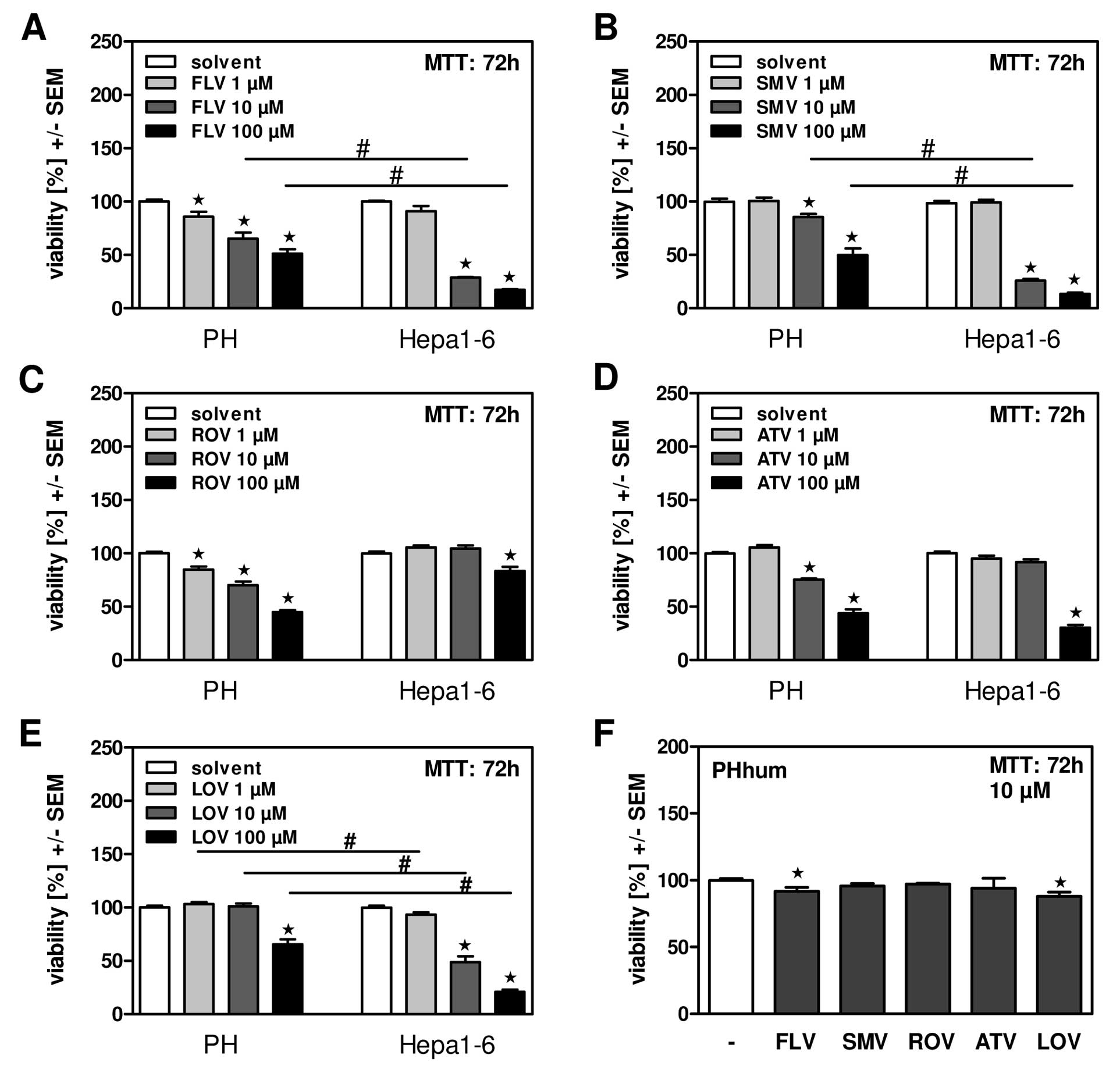 | Figure 1FLV, SMV and LOV dose-dependently and
selectively reduce viability of mouse hepatoma cells. Primary mouse
hepatocytes (PH) or mouse hepatoma cells (Hepa1-6) were incubated
in the presence of fluvastatin (FLV; A), simvastatin (SMV; B),
rosuvastatin (ROV; C), atorvastatin (ATV; D), or lovastatin (LOV;
E) at 1, 10 or 100 μM for 72 h. Cell viability was measured by MTT
assay. *P≤0.05 for statin vs. solvent incubated cells;
#P≤0.05 for statin incubated PH vs. Hepa1-6 cells. (F)
Primary human hepatocytes (PHhum) were incubated in the presence of
FLV, SMV, ROV, ATV, or LOV at 10 μM for 72 h. Cell viability was
measured by MTT assay. *P≤0.05 for statin vs. solvent
incubated cells. |
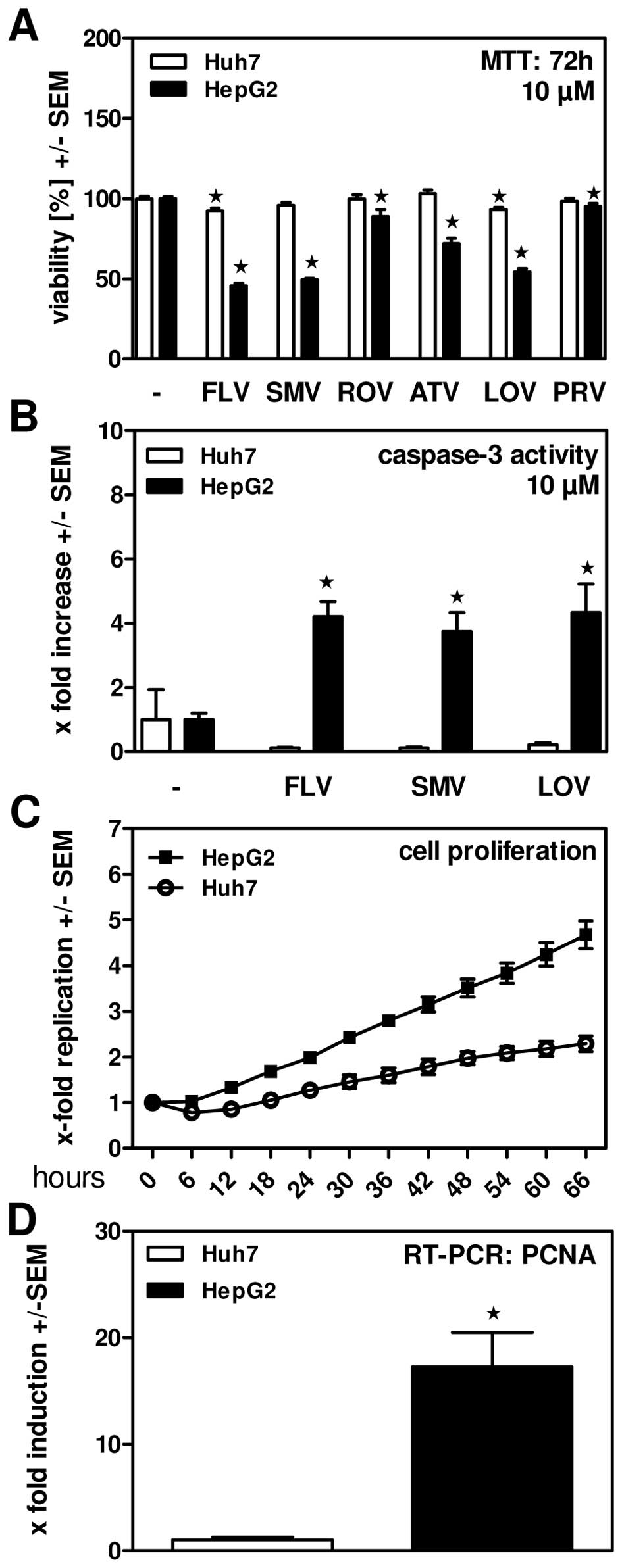
FLV, SMV and LOV efficiently reduce
viability of HepG2 but not Huh7 human hepatoma cells
Incubating the human hepatoma cell lines Huh7 and
HepG2 in the presence of 10 μM of statins we found that FLV and LOV
showed mild toxic effect on Huh7 cells, while none of the other
statins reduced viability of this cell line (Fig. 2A). In contrast, all statins
significantly interfered with viability of HepG2 cells (Fig. 2A). Again, most pronounced effects
were observed for FLV, SMV and LOV, while effects of ATV were
intermediate and ROV had only moderate effects on viability of
HepG2 cells (Fig. 2A). We also
measured apoptosis induction by FLV, SMV and LOV in Huh7 and HepG2
cells and found that these statins induced apoptosis in HepG2, but
not in Huh7 human hepatoma cells (Fig.
2B). These results indicate that statins in principle are able
to induce apoptosis in tumor cells. Measuring real-time
proliferation of HepG2 and Huh7 cells we found that HepG2 cells
were proliferating much faster than Huh7 cells (Fig. 2C). This finding was in line with
results from real-time RT-PCR, where expression of proliferating
cell nuclear antigen (PCNA) was found to be significantly higher in
HepG2 cells (Fig. 2D).
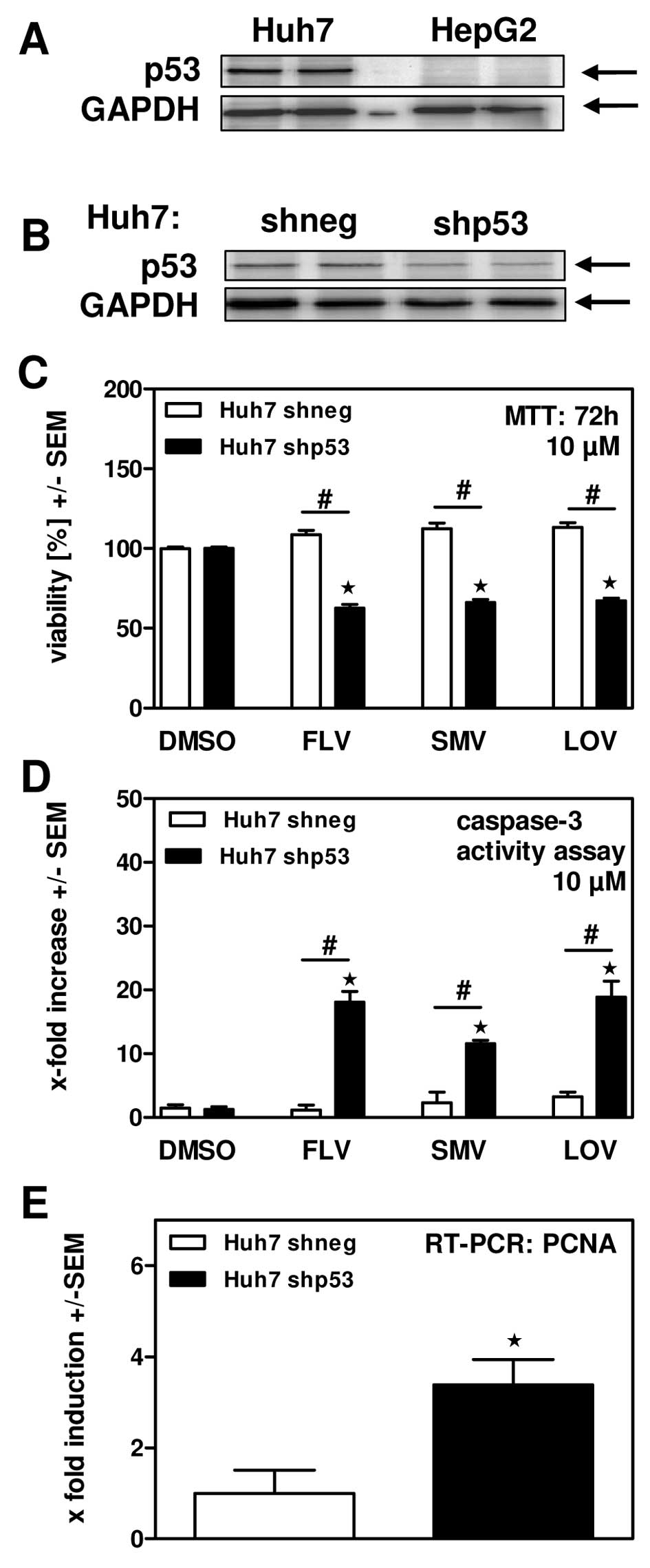
Protection of Huh7 cells against statin
induced cytotoxicity seems to depend on over-expression of p53
As shown in Fig. 2A and
B Huh7 human hepatoma cells were much more resistant against
statin incubation than HepG2 cells. One fundamental difference
between these cell lines is their content of p53 tumor suppressor
protein. While HepG2 cells express normal amounts of p53, Huh7
cells accumulate high amounts of p53 (26). We confirmed this observation in our
cell lines by Western blot analysis (Fig. 3A). Based on the hypothesis that high
amounts of p53 would interfere with cell proliferation, which might
be the reason for decreased sensitivity of Huh7 cells towards
statin-induced cytotoxicity, we generated Huh7 cells with a stable
knockdown of p53 (shp53). Western blot analysis (Fig. 3B) revealed a p53 knockdown of ~50%
on protein level compared to cells expressing shRNA directed
against E. coli polymerase (shneg) as a control for
unspecific knockdown. Incubation of both cell lines in the presence
of FLV, SMV or LOV revealed significantly reduced cellular
viability (Fig. 3C) and increased
apoptosis induction (Fig. 3D) by
statins in the presence of the p53 knockdown, providing evidence
that p53 might contribute to resistance against anti-tumor effects
of statins. Our hypothesis was further supported by the finding
that p53 knockdown increased expression of PCNA as a marker for
proliferation activity (Fig.
3E).
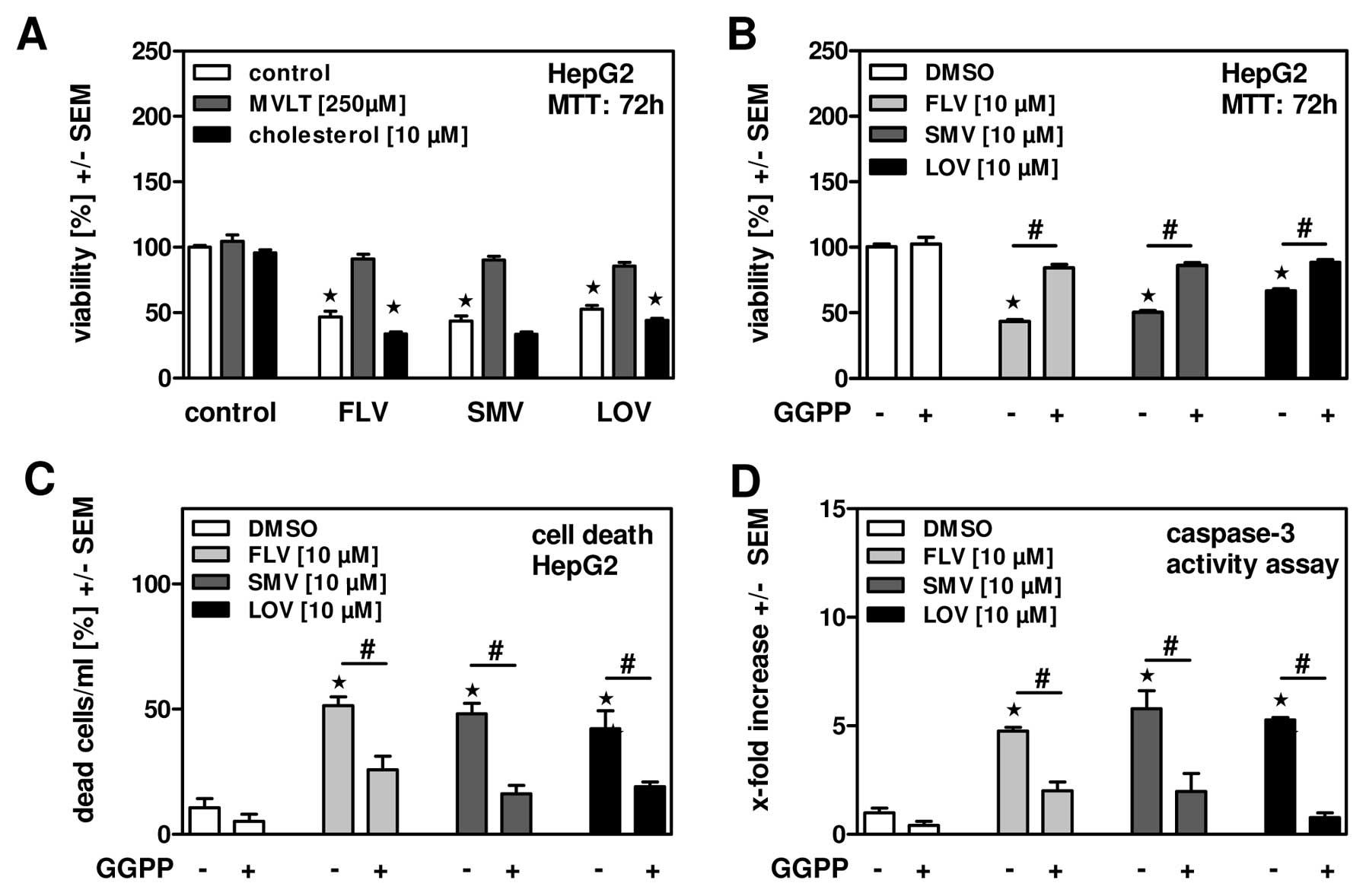
Statins induce tumor cell apoptosis by
interfering with geranyl-geranylation
To further characterize apoptosis induction by
statins in hepatoma cells we investigated if reduced biosynthesis
of cholesterol itself or an intermediate step in the cholesterol
biosynthesis pathway might be responsible. Therefore, HepG2 cells
were incubated in the presence of FLV, SMV or LOV with or without
cholesterol or mevalonate (MVLT) supplementation. Our results show
that cholesterol was not able to restore cell viability (Fig. 4A), while MVLT did, indicating that
inhibition of the cholesterol synthesis pathway rather than a lack
of cholesterol seems to be responsible for induction of hepatoma
cell death by statins. By interfering with the cholesterol
biosynthesis pathway statins deprive cells of signaling molecules,
e.g., geranyl-geranyl-pyrophosphate (GGPP), necessary for
prenylation of small G-proteins and subsequent maintenance of
cellular integrity. This might be especially important for fast
proliferating tumor cells. To investigate this hypothesis we
incubated HepG2 cells in parallel with statins and GGPP and found a
partially restoration of cell viability (Fig. 4B) and inhibition of cell death, as
measured by counting dead cells as a percentage of whole cell
numbers (Fig. 4C) or apoptosis
induction (Fig. 4D). The same
effects were observed when incubating mouse hepatoma cells
(Hepa1-6) in the presence of statins and GGPP (data not shown),
indicating a generalized mechanism. In conclusion, our results
implicate that FLV, SMV and most prominently LOV induce apoptosis
in hepatoma cell lines by interfering with cellular integrity.
Discussion
Besides their role in regulation of endogenous
cholesterol synthesis and lipoprotein metabolism statins have been
shown to exert pleiotopic effects, e.g., on pancreatic or prostate
cancer (4,5). Moreover, it has been observed that
diabetes patients with additional statin therapy have a lower risk
to develop HCC (6). It has been
described that e.g., FLV and SMV induce apoptosis and cell cycle
arrest in hepatoma cell lines (11,12),
ATV blocks both Myc phosphorylation and activation, suppressing
tumor initiation and growth in vivo(13) and that SMV modifies the expression
of cell adhesion molecules leading to reduced tumor cell growth and
invasion (14). Moreover, statins
showed synergistic anti-tumor effects with e.g., the COX-2
inhibitor celecoxib (15) or the
PKC beta inhibitor enzastaurin (16).
In the present study, we compared anti-tumor effects
of FLV, SMV, ATV, ROV and LOV on human and mouse hepatoma cell
lines to effects on primary human or mouse hepatocytes. Our general
observation was that anti-tumor effects of FLV, SMV and LOV on
hepatoma cell lines were significantly more pronounced compared to
effects on primary hepatocytes, which implicates that these statins
might be especially useful and better tolerated in therapy of HCC
patients.
Our results also show that there are profound
differences in the susceptibility of human hepatoma cell lines
towards statin treatment, pointing to the fact that not every HCC
patient might benefit from statin therapy. It seems that anti-tumor
effects of statins are tightly linked to the proliferative capacity
of cells. We found that slow proliferating Huh7 cells, like the
in vitro almost quiescent primary human hepatocytes, were
significantly less affected by statins than e.g., the fast
proliferating human hepatoma cell line HepG2. Along with inhibition
of cholesterol biosynthesis statins also deprive hepatocytes of
intermediate products, e.g., mevalonate or the isoprenoids
geranyl-geranyl-pyrophosphate (GGPP) and farnesyl-pyrophosphate
(FPP), which are necessary for post-translational modifications and
activation of cellular proteins, e.g., small G-proteins of the Ras
superfamily (27). These proteins
are regulators of important biological processes, including cell
survival and cell growth, organization of the cytoskeleton and cell
motility, intracellular vesicle formation and trafficking as well
as nucleo-cytoplasmic transport (28). Fast growing tumor cells have a
higher consumption of those intermediate products for reproduction
of the cytoskeleton due to increased cell cycle activity and
permanent cell division. Inline with our observations, statins have
also been shown to affect prostate cancer cells by inhibition of
prenylation (29). While these
observations indicate that cancer therapy with certain statins
might preferentially target fast proliferating tumor cells, these
findings also implicate that necessary regenerative processes
within the liver might be subject to statin toxicity. Therefore,
only statins with high selectivity for tumor cells, i.e., FLV, SMV
or LOV might be useful in supplementation of HCC treatment.
A reason for reduced proliferation and
susceptibility of Huh7 cells towards statins might be their
enhanced content of the tumor suppressor protein p53 compared to
e.g., HepG2 cells. Huh7 hepatoma cells accumulate high amounts of
p53 (26) and show reduced cellular
proliferation. In fact we found that the ability of statins to
induce apoptosis in hepatoma cell lines directly correlated with
the levels of p53. Huh7 cells did not respond to statin incubation
at concentrations achievable in patients (the maximum daily dose of
FLV for a patient would correspond to ~30 μM in vitro),
while a knockdown of p53 by ~50% increased statin effects
significantly. This implicates that only HCC with a particularly
high expression or availability of p53 might be protected against
statin toxicity, which is a rare event and supports the potential
use of statins in HCC therapy. Nevertheless, the p53 pathway seems
to play a role in the mechanism of apoptosis induction by
statins.
G1/S cell cycle arrest in human HCC cell lines has
been proposed as a mechanism for statins-induced apoptosis
(30). Regarding the balance
between tolerable statin concentrations for primary cells and a
certain necessity for cell proliferation to achieve statin toxicity
it has to be evaluated if repeated cycles rather than high doses of
statin treatment might have a more beneficial impact on tumor
regression rates. It also has to be evaluated if the combination of
statins with anti-proliferative tumor therapy is recommendable.
Acknowledgements
This work was supported by the Deutsche
Forschungs-gemeinschaft (DFG): grant SA 1378/3-1 to G.S. and M.D.;
SFB 841 graduate school ‘Inflammation & Regeneration’ to G.T.
The expert technical assistance of Elena Tasika is gratefully
acknowledged.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
LDLR
|
LDL receptor
|
|
GGPP
|
geranyl-geranyl-pyrophosphate
|
|
FLV
|
fluvastatin
|
|
SMV
|
simvastatin
|
|
ROV
|
rosuvastatin
|
|
ATV
|
atorvastatin
|
|
LOV
|
lovastatin
|
|
MVLT
|
mevalonate
|
|
PHhum
|
primary human hepatocytes
|
|
PCNA
|
proliferating cell nuclear antigen
|
References
|
1
|
Bader T and Korba B: Simvastatin
potentiates the anti-hepatitis B virus activity of FDA-approved
nucleoside analogue inhibitors in vitro. Antiviral Res. 86:241–245.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Delang L, Paeshuyse J, Vliegen I, Leyssen
P, Obeid S, Durantel D, Zoulim F, Op de Beeck A and Neyts J:
Statins potentiate the in vitro anti-hepatitis C virus activity of
selective hepatitis C virus inhibitors and delay or prevent
resistance development. Hepatology. 50:6–16. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ikeda M, Abe K, Yamada M, Dansako H, Naka
K and Kato N: Different anti-HCV profiles of statins and their
potential for combination therapy with interferon. Hepatology.
44:117–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murtola TJ, Tammela TL, Määttänen L,
Huhtala H, Platz EA, Ala-Opas M, Stenman UH and Auvinen A: Prostate
cancer and PSA among statin users in the finnish prostate cancer
screening trial. Int J Cancer. 127:1650–1659. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ishikawa S, Nagai Y, Masuda T, Koga Y,
Nakamura T, Imamura Y, Takamori H, Hirota M, Funakosi A, Fukushima
M and Baba H: The role of oxysterol binding protein-related protein
5 in pancreatic cancer. Cancer Sci. 101:898–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
El-Serag HB, Johnson ML, Hachem C and
Morgana RO: Statins are associated with a reduced risk of
hepatocellular carcinoma in a large cohort of patients with
diabetes. Gastroenterology. 136:1601–1608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuoppala J, Lamminpää A and Pukkala E:
Statins and cancer: a systematic review and meta-analysis (review).
Eur J Cancer. 44:2122–2132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR,
Seitz JF, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici
M, Voliotis D and Bruix J; SHARP Investigators Study Group.
Sorafenib in advanced hepatocellular carcinoma. N Engl J Med.
359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J,
Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D and Guan Z:
Efficacy and safety of sorafenib in patients in the Asia-Pacific
region with advanced hepatocellular carcinoma: a phase III
randomised, doubleblind, placebo-controlled trial. Lancet Oncol.
10:25–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang W, Wu J, Zhou L, Xie HY and Zheng
SS: Fluvastatin, a lipophilic statin, induces apoptosis in human
hepatocellular carcinoma cells through mitochondria-operated
pathway. Indian J Exp Biol. 48:1167–1174. 2010.
|
|
13
|
Cao Z, Fan-Minogue H, Bellovin DI,
Yevtodiyenko A, Arzeno J, Yang Q, Gambhir SS and Felsher DW: MYC
phosphorylation, activation, and tumorigenic potential in
hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer
Res. 71:2286–2297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Relja B, Meder F, Wang M, Blaheta R,
Henrich D, Marzi I and Lehnert M: Simvastatin modulates the
adhesion and growth of hepatocellular carcinoma cells via decrease
of integrin expression and ROCK. Int J Oncol. 38:879–885. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao J, Jia WD, Li JS, Wang W, Xu GL, Ma
JL, Ge YS, Yu JH, Ren WH, Liu WB and Zhang CH: Combined inhibitory
effects of celecoxib and fluvastatin on the growth of human
hepatocellular carcinoma xenografts in nude mice. J Int Med Res.
38:1413–1427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim W, Yoon JH, Kim JR, Jang IJ, Bang YJ,
Kim YJ and Lee HS: Synergistic anti-tumor efficacy of lovastatin
and protein kinase C-beta inhibitor in hepatocellular carcinoma.
Cancer Chemother Pharmacol. 64:497–507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neuvonen PJ: Drug interactions with
HMG-CoA reductase inhibitors (statins): the importance of CYP
enzymes, transporters and pharmacogenetics (review). Curr Opin
Investig Drugs. 11:323–332. 2010.PubMed/NCBI
|
|
18
|
Brown WV: Safety of statins. Curr Opin
Lipidol. 19:558–562. 2008. View Article : Google Scholar
|
|
19
|
Nakabayashi H, Taketa K, Yamane T,
Miyazaki M, Miyano K and Sato J: Phenotypical stability of a human
hepatoma cell line, HuH-7, in long-term culture with chemically
defined medium. Gann. 75:151–158. 1984.PubMed/NCBI
|
|
20
|
Aden DP, Fogel A, Plotkin S, Damjanov I
and Knowles BB: Controlled synthesis of HBsAg in a differentiated
human liver carcinoma-derived cell line. Nature. 282:615–616. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Darlington GJ, Bernhard HP, Miller RA and
Ruddle J: Expression of liver phenotypes in cultured mouse hepatoma
cells. J Natl Cancer Inst. 64:809–819. 1980.PubMed/NCBI
|
|
22
|
Seglen PO: Preparation of rat liver cells.
3 Enzymatic requirements for tissue dispersion. Exp Cell Res.
82:391–398. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dandri M, Burda MR, Török E, Pollok JM,
Iwanska A, Sommer G, Rogiers X, Rogler CE, Gupta S, Will H, Greten
H and Petersen J: Repopulation of mouse liver with human
hepatocytes and in vivo infection with hepatitis B virus.
Hepatology. 33:981–988. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moffat J, Grueneberg DA, Yang X, Kim SY,
Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK,
Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC,
Lander ES, Sabatini DM and Root DE: A lentiviral RNAi library for
human and mouse genes applied to an arrayed viral high-content
screen. Cell. 124:1283–1298. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gloesenkamp CR, Nitzsche B, Ocker M, Di
Fazio P, Quint K, Hoffmann B, Scherübl H and Höpfner M: AKT
inhibition by triciribine alone or as combination therapy for
growth control of gastroenteropancreatic neuroendocrine tumors. Int
J Oncol. 40:876–888. 2012.PubMed/NCBI
|
|
26
|
Bressac B, Galvin KM, Liang TJ,
Isselbacher KJ, Wands JR and Ozturk M: Abnormal structure and
expression of p53 gene in human hepatocellular carcinoma. Proc Natl
Acad Sci USA. 87:1973–1977. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chan KKW, Oza AM and Siu LL: The statins
as anticancer agents. Clin Cancer Res. 9:10–19. 2003.
|
|
28
|
Konstantinopoulos PA, Karamouzis MV and
Papavassiliou AG: Post-translational modification and regulation of
the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug
Discov. 6:541–555. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roy M, Kung HJ and Ghosh PM: Statins and
prostate cancer: role of cholesterol inhibition vs. prevention of
small GTP-binding proteins. Am J Cancer Res. 1:542–561.
2011.PubMed/NCBI
|
|
30
|
Sutter AP, Maaser K, Höpfner M, Huether A,
Schuppan D and Scherübl H: Cell cycle arrest and apoptosis
induction in hepatocellular carcinoma cells by HMG-CoA reductase
inhibitors. Synergistic antiproliferative action with ligands of
the peripheral benzodiazepine receptor. J Hepatol. 43:808–816.
2005. View Article : Google Scholar
|