Introduction
Acetylation is a pivotal post-transcriptional
modification, which strongly influences chromatin structure and
function (1). Due to its wide
variety of targets, it is not only implicated in the regulation of
gene expression via chromatin structure modifications but also in
protein-protein interactions, protein stability, DNA binding, and
subcellular localization (2).
Histone acetylation is mediated by histone acetyltransferases
(HATs). The resulting structural modification of chromatin leads to
nucleosomal relaxation and altered transcriptional activation. The
reverse reaction is mediated by histone deacetylases (HDACs), which
induce deacetylation, chromatin condensation, and transcriptional
repression (3). Alterations
mediated by HAT/HDAC activities are not solely reduced to
chromatin. Mounting evidence accentuates their involvement in
lysine acetylation/deacetylation of non-histone substrates
including transcription factors (such as NF-κB, p53, GATA2, MEF2)
and chromatin-associated co-repressor proteins (4,5). A
balanced histone acetylation status is essential for the proper
progress of cell proliferation, apoptosis and differentiation. An
improper HDAC recruitment or activity, often leads to abnormal gene
expression that is associated with cancer development (6), hence rendering HDAC as an excellent
target in current cancer research. Two HDAC inhibitors,
suberoylanilide hydroxamic acid (SAHA; vorinostat) and the natural
product romidepsin (FK-228) are currently approved for cancer
chemotherapy and many other inhibitors are in clinical trials
(7). So far, eighteen human HDAC
enzymes have been identified and grouped into four classes based on
the structure of their accessory domains. Classes I, II, and IV,
but not III, require a zinc molecule as an essential cofactor in
their active site and are inhibited by Zn2+-binding HDAC
inhibitors such as SAHA and the natural product, trichostatin A.
Class I HDACs comprise HDACs 1, 2, 3 and 8, whereas class II HDACs
include HDACs 4, 5, 6, 7 and 9 that are larger in size than the
other classes (4,5). Recent publication of the X-ray crystal
structure of HDAC8 (8) followed by
several other HDACs (7) fuelled the
research activity in the discovery and development of novel HDAC
inhibitors.
The transcription factor NF-κB is a dimer of
proteins belonging to the Rel family. It is an ubiquitous
transcription factor present in all cell types. The most common
form of NF-κB is the p65/p50 heterodimer. In most cells, NF-κB
complexes are localized to the cytosol as inactive forms with the
inhibitor of κB protein (IκB). Phosphorylation of IκB results in
its ubiquitination and subsequent proteasome-mediated degradation.
Activated NF-κB then translocates to the nucleus where it
transactivates more than 500 target genes (9). Many factors are known to activate
NF-κB, including inflammatory cytokines such as tumor necrosis
factor alpha (TNFα) and interleukin (IL)-1, carcinogens (cigarette
smoke), UV radiation, hyperglycemia and tumor promoters. Over the
last decade, NF-κB became a major target in drug discovery, due to
its key role in cancer development, inflammation, cell
proliferation and death (10).
Recent evidence indicates that the activation of
various transcription factors, including NF-κB, is regulated
through the interaction with HDAC proteins (11–13).
Previous studies with various cell types showed that HDAC1 and
HDAC2 negatively regulate NF-κB activity through direct interaction
with the p65 (RelA) subunit of NF-κB (14,15).
Other studies suggest a physical interaction between p65 (RelA) and
class I type HDACs (HDAC1, 2 and 3), where high expression of class
I HDACs has been linked to increased nuclear translocation of p65
(RelA) (16). Remarkably, Class I
HDAC isoforms are often overexpressed in various types of cancers
where they are usually associated with a poor prognosis (5,17,18).
In addition, HDAC inhibitors are acknowledged as effective
anti-inflammatory agents, some inhibiting NF-κB (19–21)
and may therefore play an important role in the prevention of
cancers that develop as a result of chronic inflammation. The
combined antiproliferative as well as anti-inflammatory potency
represents a highly attractive combination for the treatment of
numerous chronic inflammatory conditions which are frequently
associated with an increased risk of developing cancer.
Chalcones (1,3-diphenyl-2-propenones) are a group of
aromatic compounds that represent a large class of natural products
found in many medicinal plants, fruits, vegetables, spices and
nuts. They are the natural precursors of flavonoids and display a
variety of biological activities. Although the modes of action of
this class of compounds are not fully understood, great efforts are
devoted to elucidate the mechanisms underlying their promising
anti-inflammatory and anticancer activities. Hence several natural
chalcones have been reported to inhibit the NF-κB signaling, and
numerous synthetic derivatives have been evaluated in
structure-activity relationship (SAR) studies (22,23).
Besides the NF-κB inhibition, interference in microtubule formation
is generally thought to be responsible for their anticancer
activities (24,25). Despite the cross-talk and modulation
effects between NF-κB and HDACs, and structural similarity of
chalcones to broad-spectrum HDAC inhibitors SAHA and trichostatin
A, HDACs have not been investigated as potential targets for
natural chalcones. In this study, we aimed to test twenty-one
commercially available chalcones (Table
I) for dual HDACs and NF-κB inhibitory activities in
vitro. Viability assays were also carried out to elucidate the
cytotoxic potential of the chalcones against leukemia cells. We
also aimed to explore SAR to determine the essential
functionalities on the chalcone core for biological activity. We
also performed molecular modeling and docking studies in an attempt
to understand the potential mode and mode/site of binding of
natural chalcones to NF-κB and class I type HDACs.
 | Table IStructural features of natural
chalcones. |
Table I
Structural features of natural
chalcones.
 |
|---|
|
|---|
| Substitution |
|---|
|
|
|---|
| Chalcone | 2′ | 3′ | 4′ | 6′ | 2 | 3 | 4 | Δα,β |
|---|
| Chalcone, no.1 | H | H | H | H | H | H | H | Unsaturated |
| 2′-Hydroxychalcone,
no 2 | OH | H | H | H | H | H | H | Unsaturated |
| 2-Hydroxychalcone,
no 3 | H | H | H | H | OH | H | H | Unsaturated |
| 4-Hydroxychalcone,
no 4 | H | H | H | H | H | H | OH | Unsaturated |
| 4-Methoxychalcone,
no 5 | H | H | H | H | H | H | OCH3 | Unsaturated |
|
3,4-Dimethoxychalcone, no 6 | H | H | H | H | H | OCH3 | OCH3 | Unsaturated |
| 4′-Hydroxychalcone,
no. 7 | H | H | OH | H | H | H | H | Unsaturated |
| 4′-Methoxychalcone,
no 8 | H | H | OCH3 | H | H | H | H | Unsaturated |
|
4,4′-Dimethoxychalcone, no 9 | H | H | OCH3 | H | H | H | OCH3 | Unsaturated |
| Isoliquiritigenin
(2′,4,4′-trihydroxychalcone), no 10 | OH | H | OH | H | H | H | OH | Unsaturated |
| Calomelanone
(2′,6′-dihydroxy-4,4′-dimetoxydihydrochalcone), no 11 | OH | H | OCH3 | OH | H | H | OCH3 | Saturated |
| Butein
(2′,3–4,4′-tetrahydroxychalcone), no 12 | OH | H | OH | H | H | OH | OH | Unsaturated |
| Flavokawain C
(2′,4-dihydroxy-4′,6′-dimethoxychalcone), no 13 | OH | H | OCH3 | OCH3 | H | H | OH | Unsaturated |
| Gymnogrammene
(2′,6′-dihydroxy-4,4′-dimethoxychalcone), no 14 | OH | H | OCH3 | OH | H | H | OCH3 | Unsaturated |
| Homobutein
(2′,4,4′-trihydroxy-3-methoxychalcone), no 15 | OH | H | OH | H | H | OCH3 | OH | Unsaturated |
|
2,3-Dimethoxy-2′-hydroxychalcone, no
16 | OH | H | H | H | OCH3 | OCH3 | H | Unsaturated |
| Flavokawain A
(2′-hydroxy-4,4′,6′-trimethoxychalcone), no 17 | OH | H | OCH3 | OCH3 | H | H | OCH3 | Unsaturated |
| Eriodictyolchalcone
(2′,4′,6′,3,4-pentahydroxychalcone), no 18 | OH | H | OH | OH | H | OH | OH | Unsaturated |
| Phloretin
(2′,4,4′,6′-tetrahydroxydihydrochalcone), no 19 | OH | H | OH | OH | H | H | OH | Saturated |
| Phloridzin
(phloretin-2′-O-glucoside), no 20 | O-Glu | H | OH | OH | H | H | OH | Saturated |
| Marein
(2′,3,3′,4,4′-pentahydroxy-4′-glucosylchalcone), no 21 | OH | OH | O-Glu | OH | H | OH | OH | Unsaturated |
|
| Glu, glucose. | | | | | | | | |
Materials and methods
Chalcones
Twenty-one natural chalcones, namely chalcone,
2′-hydroxychalcone, 2-hydroxychalcone, 4-hydroxychalcone,
4-methoxychalcone, 3,4-dimethoxychalcone, 4′-hydroxychalcone,
4′-methoxychalcone, 4,4′-dimethoxychalcone, isoliquiritigenin
(2′,4,4′-trihydroxychalcone), calomelanone
(2′,6′-dihydroxy-4,4′-dimethoxydihydrochalcone), butein
(2′,3–4,4′-tetrahydroxychalcone), flavokawain C
(2′,4-dihydroxy-4′,6′-dimethoxychalcone), gymnogrammene
(2′,6′-dihydroxy-4,4′-dimethoxychalcone), homobutein
(2′,4,4′-trihydroxy-3-methoxychalcone),
2,3-dimethoxy-2′-hydroxychalcone, flavokawain A
(2′-hydroxy-4,4′,6′-trimethoxychalcone), eriodictyolchalcone
(2′,4′,6′,3,4-pentahydroxychalcone) phloretin
(2′,4,4′,6′-tetrahydroxydihydrochalcone), phloridzin
(phloretin-2′-O-glucoside) and marein
(2′,3,3′,4,4′-pentahydroxy-4′-glucosylchalcone) were studied.
Table 1 shows their chemical
features and substitution patterns. Chalcone was purchased from
Fluka (Steinheim, Germany), whereas all remaining chalcones were
obtained from Extrasynthese (Genay Cedex, France) (purity
>97%).
Cell culture and reagents
Human Philadelphia chromosome-positive chronic
myelogenous leukemia cell line K562 was purchased from Deutsche
Sammlung für Mikroorganismen und Zellkulturen (DSMZ, Braunschweig,
Germany) and cultured in RPMI-1640 medium (Lonza, Verviers,
Belgium) supplemented with 10% fetal calf serum (Hyclone, Perbio,
Erembodegem, Belgium) and 1% (v/v) antibiotic-antimycotic (Lonza,
BioWhittaker™), at 37°C, in a 5% CO2, humidified
atmosphere. Human recombinant TNFα (PeproTech, Rocky Hill, NJ, USA)
was resuspended in 1× phosphate-buffered saline (PBS) sterile
solution containing 0.5% bovine serum albumin (MP Biomedicals,
Asse-Relegem, Belgium) to reach a final concentration of 10
μg/ml.
HDAC activity/inhibition measurement
Direct HDAC inhibition by chalcone derivatives was
estimated using K562 total extracts as an HDAC source and the
enzymatic HDAC activity measurement was performed using a
fluorometric HDAC assay kit (Active Motif, Rixensart, Belgium)
according to the manufacturer’s instructions. Briefly, after being
washed twice with ice-cold 1× PBS, cells were pelleted by
centrifugation, and lysed in M-PER® mammalian protein
extraction reagent (Pierce, Erembodegem, Belgium) and supplemented
with 1× protease inhibitor cocktail (Roche, Prophac, Luxembourg).
The cell suspension was gently mixed on an orbital shaker for 15
min and centrifuged at 14,000 × g at 4°C for 15 min. Protein
content was assessed using the Bradford assay (Bio-Rad, Nazareth,
Belgium), and 10 μg of proteins were incubated with vehicle or
various concentrations of the different chalcones for 1 h at 37°C
in the presence of an HDAC fluorometric substrate. Subsequently,
the HDAC assay developing solution was added and after 15 min of
incubation at room temperature, the fluorescence was measured using
a Gemini EM microplate spectrofluorometer (Molecular Devices,
Belgium) with excitation at 360 nm and emission at 460 nm. The
measured activities were normalized by the vehicle-treated control
enzyme activities and IC50 values were calculated.
Transient transfections
Transient transfections of K562 cells were performed
as previously described (26).
In vitro cytotoxicity assays (viability
assay)
The in vitro growth inhibitory values of
chalcone derivatives on the K562 cell line were determined as
detailed previously (26).
Molecular modeling and docking
studies
The 2D structures of chalcone molecules were drawn
using SketchEI and transferred into the VEGA ZZ molecular modeling
software (27,28) to generate 3D structures. All
molecules were saved into a single mol file, that was used as input
for the OMEGA, OpenEye Scientific Software (Omega version 2.3.2;
http://www.openeye.com) to generate a maximum of
2 low energy conformers with default values. These conformations
were stored as OEB file extension format and their 3D similarity
was compared using the Rocs, OpenEye Scientific Software (Rocs
version 2.3.1; http://www.openeye.com). E-Dragon
Software (29) was utilized to
calculate constitutional and molecular property descriptors. The
descriptors selected to describe the SAR were selected using
Partial Least Squares regression as implemented in the PLSR module
of Virtual Computational Chemistry Laboratory (29) and Gretl software was used to
calculate the correlation between the logarithm of the activity and
predicted molecular properties.
The molecular docking was carried out using Glide
software (Grid-Based Ligand Docking With Energetics) (Schrodinger
Inc., Portland, OR, USA) (30,31)
after the docking targets were prepared using Protein Preparation
Wizard workflow in Maestro (Schrodinger Inc.) by removing water
molecules, adding the hydrogen atoms and assigning all atom force
field (OPSL-2005) charges and atom types. The position of all atoms
was adjusted by minimizations until the average root mean square
deviation reached 0.3 Å. The crystal structures of HDAC8 wild-type
and variant D101 complexed with ligands [Protein Data Bank (pdb)
entries 1T69 and 3EZT] were used for molecular docking of chalcones
into the protein active site. The box encompassing the active site
was selected based on the position of co-crystalized ligands
complex as described in a previous study (32). The crystal structure of NF-κB
complexed to DNA was chosen as a target system to elucidate binding
modes of chalcones (pdb entry 1NFK). Prior to docking the DNA
molecule was removed and the coordinates of the enclosing box of 30
Å (center at × = −1,1958 Å; y = 9.0149 Å; z = 19,7598 Å) were
encompassing the active site residues involved in hydrogen bonds
with the NF-κB recognition site of DNA (Arg54, Arg56, Tyr57, Cys59,
Lys241, Gln306 and Thr143) (35).
Flexible ligand docking simulations were carried out with Glide
using the default settings. The ten best poses obtained using the
Extra-Precision Glide (Glide XP) mode were selected for analysis.
The most favorable poses of molecules showing activity were
subjected to further energy minimization using Macromodel 9.1 and
OPLS2005 force field.
Results
Inhibition of HDAC activity by chalcone
derivatives
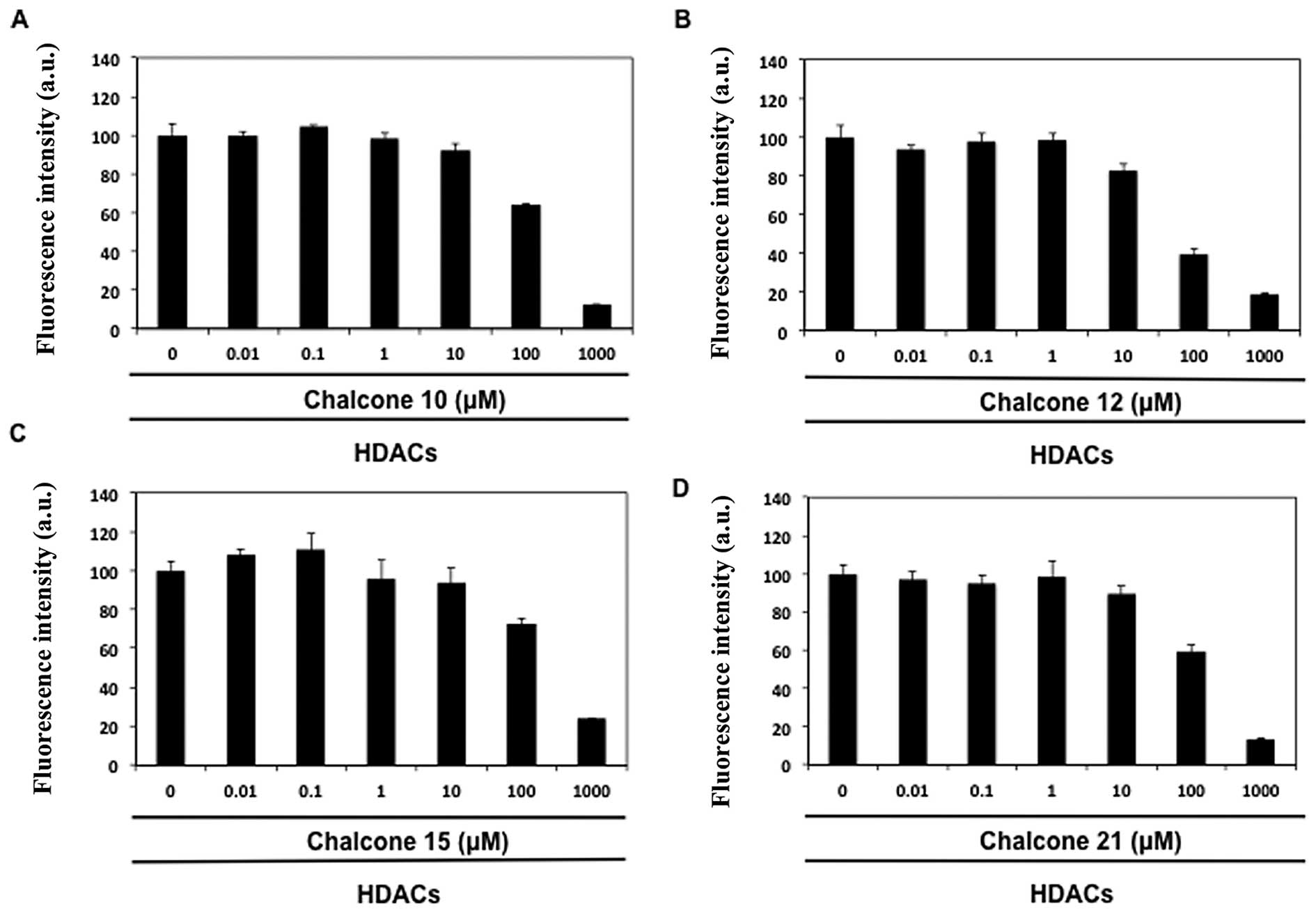
The effect of chalcone derivatives (nos. 1–21) was
examined on total HDAC activity using a fluorescence HDAC assay. As
shown in Table II, four chalcone
aglycones, namely isoliquiritigenin (no. 10), butein (no. 12),
homobutein (no. 15) and the glycoside marein (no. 21), reduced HDAC
activity in a concentration-dependent manner (IC50
values 60–190 μM, Fig. 1). Butein
(no. 12) appeared to be the best inhibitor of HDAC activity. Other
chalcone derivatives were assumed as inactive, because they were
unable to provide distinct inhibitory effect even at the highest
test concentration (1000 μM).
| Table IIBiological activity of natural
chalcones. |
Table II
Biological activity of natural
chalcones.
| Chalcone | HDAC inhibition,
IC50 (μM) | NF-κB inhibition,
IC50 (μM) | Viability |
|---|
| 1 | >1000 | n.m. | 14 |
| 2 | >1000 | n.m. | 28 |
| 3 | >1000 | n.m. | 2 |
| 4 | >1000 | 24 | 31 |
| 5 | >1000 | 29 | 35 |
| 6 | >1000 | n.m. | 38 |
| 7 | >1000 | 28 | 16 |
| 8 | >1000 | n.m. | 38 |
| 9 | >1000 | >200 | n.t. |
| 10 | 110 | 32 | 44 |
| 11 | >1000 | 11 | 31 |
| 12 | 60 | 38 | 13 |
| 13 | >1000 | 8 | 13 |
| 14 | >1000 | >200 | n.t. |
| 15 | 190 | 38 | 29 |
| 16 | >1000 | n.m. | 12 |
| 17 | >1000 | >200 | n.t. |
| 18 | >1000 | >200 | n.t. |
| 19 | >1000 | 41 | 59 |
| 20 | >1000 | >200 | n.t. |
| 21 | 100 | >200 | n.t. |
| Standards |
0.14b |
6.0a |
1.4a |
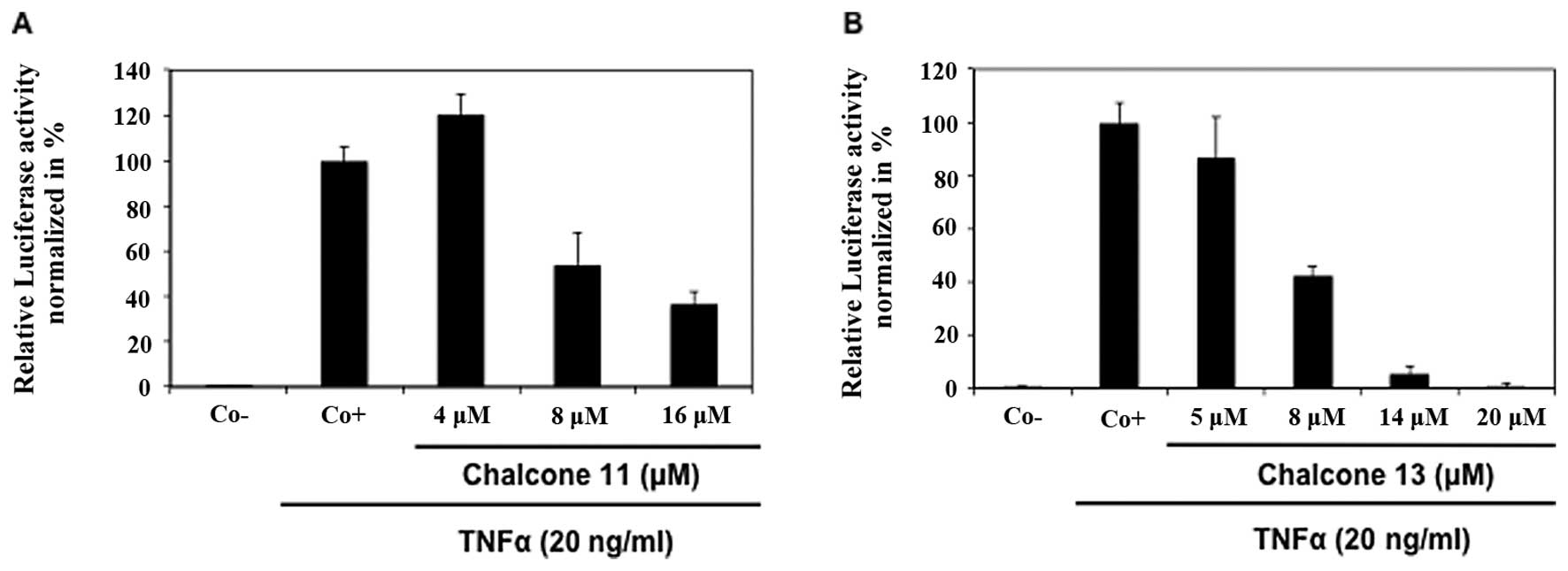
Inhibition of TNFα-induced NF-κB
activation by chalcones
By using a luciferase-based in cellulo NF-κB
reporter assay, chalcones were evaluated for TNFα-induced NF-κB
transcription inhibition activity (Table II). Flavokawain C (no. 13) was the
most potent NF-κB inhibitor, followed by the dihydrochalcone
calomelanone (no. 11) with IC50 values of 8 and 11 μM,
respectively (Fig. 2). Chalcones
4-hydroxychalcone (no. 4), 4-methoxychalcone (no. 5),
4′-hydroxychalcone (no. 7), isoliquiritigenin (no. 10), butein (no.
12), homobutein (no. 15) and phloretin (no. 19) also demonstrated
good potential with IC50 values ranging between 24–41
μM. Three polymethoxychalcones, i.e. 4,4′-dimethoxychalcone (no.
9), gymnogrammene (no. 14), flavokawain A (no. 17), as well as cone
eriodictyolchalcone (no. 18) and two chalcone glycosides,
phloridzin (no. 20) and marein (no. 21), which showed no or limited
inhibitory activity at 200 μM concentration, were considered as
inactive. The remaining compounds, chalcone (no. 1),
2′-hydroxychalcone (no. 2), 2-hydroxychalcone (no. 3),
3,4-dimethoxychalcone (no. 6), 4′-methoxychalcone (no. 8) and
2,3-dimethoxy-2′-hydroxychalcone (16) were cytotoxic at concentrations,
which inhibited NF-κB activation.
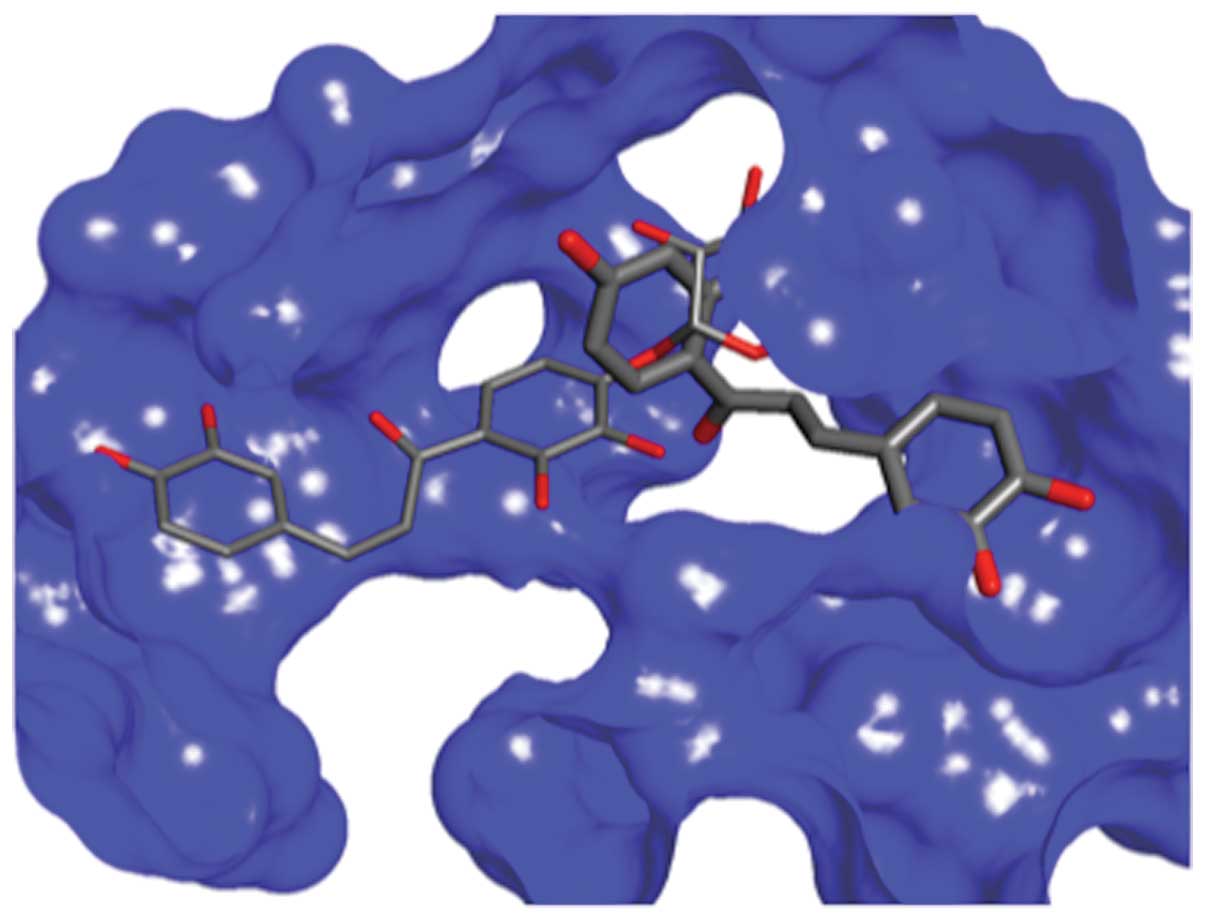
Docking studies of chalcones 12 and
chalcone 21 within the active site of HDAC8
In order to shed light on the potential mode of
action of chalcones, and to understand why some chalcones inhibit
either NF-κB or HDACs and some inhibit both, we have carried out
molecular modeling, molecular similarity and docking studies. The
compounds were docked into the binding sites of HDAC8, and the best
studied HDAC enzyme was selected based on the position of the
co-crystalized ligand in the crystal structure of the complex (pdb
entries 1T69 and 3ETZ) (32). The
GlideScore values were compared to the activities that were
experimentally obtained (Table
III). The results of the docking indicated that all chalcones
could favorably bind in the active site, although not all molecules
showed activity. The most active molecule 12 had a less favorable
GlideScore than chalcone 21 that exhibited the most favorable
binding. The binding mode of these two molecules is different
(Fig. 3) which may be the result of
the large active site of HDAC8 which accommodates two
(4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl]benzamide)
moieties in the interior pocket of the protein. Furthermore, the
docking could not distinguish the third active molecule 10 from the
rest of the group. There is a clear difference between the
GlideScore of the binding of 21 to 20. However, 20 binds almost as
good as 12 and better than 10, resulting in the absence of the
correlation between activity and binding affinity determined by
Glide. We hypothesize that molecules that are not active could
possibly bind preferably elsewhere on the protein surface rather
than on the active site.
| Table IIIGlideScore values obtained for HDAC
and NF-κB proteins. |
Table III
GlideScore values obtained for HDAC
and NF-κB proteins.
| Chalcone | Docking scores
against 1T69 | Docking scores
against 3ETZ | Docking scores
against 1NFK |
|---|
| 1 | −6.83 | −5.27 | −2.39 |
| 2 | −5.90 | −6.08 | −4.05 |
| 3 | −5.40 | −6.35 | −3.06 |
| 4 | −4.71 | −6.27 | −2.38 |
| 5 | −5.52 | −5.34 | −3.22 |
| 6 | −4.92 | −5.87 | −2.76 |
| 7 | −4.88 | −5.96 | −2.88 |
| 8 | −5.78 | −5.86 | −2.48 |
| 9 | −5.88 | −5.70 | −2.6 |
| 10 | −6.03 | −7.03 | −2.84 |
| 11 | −6.75 | −7.21 | −3.15 |
| 12 | −7.00 | −8.00 | −4.71 |
| 13 | −5.32 | −7.45 | −3.48 |
| 14 | −5.63 | −6.52 | −4.29 |
| 15 | −6.44 | −7.06 | −0.71 |
| 16 | −5.31 | −6.57 | −3.1 |
| 17 | −4.47 | −6.79 | −1.75 |
| 18 | −5.69 | −7.24 | −4.06 |
| 19 | −5.15 | −6.47 | 0.12 |
| 20 | −6.85 | −7.57 | −5.53 |
| 21 | −8.08 | −10.51 | −6.03 |
The docking studies of chalcones were also carried
out using a crystal structure of NF-κB dimer in complex with duplex
DNA. We followed a procedure reported by Piccagli et
al(33) and have chosen the DNA
recognition surface to define the docking target. The binding site
included residues Arg54, Arg56, Tyr57, Cys59, Lys241, Gln306 and
Thr143 to gain information on the interaction of our compounds with
NF-κB. As shown in Table III,
there is a lack of correlation between the activity and the
GlideScore results. This can be due to non-specific binding.
Moreover, chalcones could act on a different active site of NF-κB.
To further rationalize the activity of this class of compounds we
have elucidated the SARs and predicted more than 1600 molecular
properties for all molecules using EDragon software (29). This analysis was not aimed to lead
to the development of predictive models since the data set is small
(fifteen molecules with measured NF-κB activity) and thus we did
not create training and test sets. Two different sets of molecules
were subjected to partial least square regression using PLSR module
of the Virtual Computational Chemistry Laboratory to determine
which constitutional and molecular properties correlated with the
negative logarithm of activity (pNF-κB). The first group consisted
of all fifteen tested molecules and the observed correlation was
not satisfactory (r2=0.53), leading us to examine the
second smaller group, consisting of only nine molecules that
displayed activity. The PLS results indicated that a combination of
nineteen descriptors correlated with activity (r2=0.99).
There were some highly correlated and irrelevant descriptors
selected to describe correlation. The selection of descriptors was
optimized by developing least square methods using the Gretl
software and removing descriptors until a minimal number of
descriptors was obtained with a good correlation (r2=
0.914, s=0.250, n=9, F=3.48). The formula used was the following:
pNF-κB = −79.78(±48.55) − 0.425(±.0.344)*SS +
31.582(±24.)*Mp + 43.847(±25.978)*ARR + 1.868
(±1.619)*Hy + 0.265(±.468)*MLOGP + 2.793
(±1.682)*nBO.
The list and values of descriptors, observed and
calculated are shown in Table IV.
The statistics indicate reasonable descriptive value of the model
that shows which molecular properties influence activity of the
molecules in the NF-κB assay. Since we could not develop a
satisfactory model that would differentiate the active and inactive
molecules, we examined the molecular similarity and differences
between most active molecules and the inactive ones. Conformational
search carried out by Omega software and the Merck Molecular Force
Field force revealed that all molecules can exist in several
different conformations due to the free rotation around bonds
between carbonyl carbon and neighboring groups. ROCS search and
comparison of electrostatic forces showed that unsurprisingly
molecules are similar (Shape Tanimoto coefficients are between 0.94
and 0.0.69 and Tverstsky coefficients are between 0.91 and 0.71).
The highest similarity was observed between the most active
chalcone molecule 13 and inactive 9, indicating that shape and
electrostatic properties are not sufficient to explain the
different activities of the group.
| Table IVPredicted molecular properties
correlating to the activity of potent chalcones in the NF-κB
assay. |
Table IV
Predicted molecular properties
correlating to the activity of potent chalcones in the NF-κB
assay.
| Chalcone | Ss | Mp | nBO | Hy | MLOGP | ARR | Observed
pNF-κB | Calculated
pNF-κB |
|---|
| 4 | 41.67 | 0.71 | 18 | −0.371 | 3.148 | 0.667 | 4.62 | 4.59 |
| 5 | 41.17 | 0.69 | 19 | −0.873 | 3.398 | 0.632 | 4.54 | 4.56 |
| 7 | 41.67 | 0.71 | 18 | −0.371 | 3.148 | 0.667 | 4.55 | 4.59 |
| 10 | 53.00 | 0.69 | 20 | 1.096 | 2.552 | 0.600 | 4.49 | 4.37 |
| 11 | 56.67 | 0.65 | 23 | 0.304 | 2.334 | 0.522 | 4.96 | 4.97 |
| 12 | 58.67 | 0.68 | 21 | 1.928 | 1.764 | 0.571 | 4.42 | 4.51 |
| 13 | 57.67 | 0.67 | 23 | 0.304 | 1.746 | 0.522 | 5.10 | 5.02 |
| 15 | 58.17 | 0.67 | 22 | 0.545 | 1.062 | 2.013 | 4.42 | 4.51 |
| 19 | 58.67 | 0.68 | 21 | 1.928 | 1.764 | 0.571 | 4.39 | 4.40 |
Discussion
Chronic inflammation has been linked to most
incurable illnesses, including cancer, cardiovascular and
neurodegenerative diseases. Cancer is regulated by a large number
of genes that are modulated by transcription factors, such as
NF-κB, which controls genes involved in inflammation,
proliferation, angiogenesis and metastasis (23). Any disturbance in the corresponding
pathways leads to the activation of NF-κB and release of cytokines,
thus contributing to the initiation and progression of
tumorigenesis. On the other hand, acetylation and deacetylation act
as regulating mechanisms for activation or inactivation of various
transcription factors, including NF-κB. This process is mediated by
HDAC and can consequently be modulated by HDAC inhibitors (34). Protein complexes involved in the
regulation of cell-cycle progression and apoptosis are also
controlled by this mechanism (2).
The reversible acetylation appears to regulate the interaction
between p65 and IκB, and controls the duration of the NF-κB
response. NF-κB activation leads to apoptosis resistance. Several
NF-κB inhibitors showed potential to overcome this resistance and
induce apoptosis (35,36). Thus, inhibition of NF-κB may
sensitize cancer cells and eventually lead to the induction of
apoptosis. Interestingly, in our study, chalcones 4, 5, 7, 10, 11,
12, 13, 15 and 19 inhibited both NF-κB and the viability of K562
cells.
Interestingly, three chalcones (nos. 10, 12, and 15)
inhibited both NF-κB and HDAC activity. Nevertheless, underlying
mechanisms in the action of chalcones as dual inhibitors remain to
be elucidated in the future. To our knowledge, the mechanisms that
link both inhibitory activities were first reported herein. As an
example, upon interaction with histone deacetylase 3 (HDAC3), p65
is deacetylated leading to efficient interaction with IκB and
subsequent activation via the canonical pathway (37). Compounds that efficiently inhibit
HDAC3 and other HDAC isoforms are considered interesting NF-κB
inhibitors. Moreover, transcription factor STAT1 is physiologically
acetylated and binds p65, thus inhibiting NF-κB. STAT1-associated
HDAC were described to deacetylate STAT1 leading to the liberation
of p65 and subsequent activation of the canonical NF-κB pathway.
Inhibitors of HDAC activity contribute to a shift towards
acetylated STAT1 actively interacting with NF-κB and thus
inhibiting activation of p65 required for inflammatory cell
signaling (38). Even though the
inhibitory activity of selected chalcones appears weak when only
the total HDAC activity is assessed, inhibitory effect against
specific HDAC isoforms are generally stronger as shown for
tubastatin A (39).
Attempts to generate HDAC inhibitors generally focus
on varying the cap group to exploit variability in the HDAC surface
surrounding the active site. However, although efforts are being
made to identify truly class- or isoform-selective HDAC inhibitors
with anticancer and anti-inflammatory properties, the current list
of such compounds remains relatively poor. One of the main reasons
is the lack of structural determinants of selective HDAC inhibition
due to the challenge of studying the interaction of small
inhibitory molecules with multiple large protein complexes that
encompass HDAC activities, which are often dependent on or
regulated by these complexes. Accordingly, the selectivity of small
inhibitory molecules may depend on the context of HDAC complexes
and requires investigation (40).
The small size of the library limits the ability to obtain common
structural features necessary for individual or dual target
activity.
A number of SAR studies have been performed on
synthetic chalcones and their biological effects, including NF-κB
inhibition (22,23). To our knowledge this is the first
study looking at potential SARs among natural chalcones. In the
present study, some chalcones showed interesting NF-κB inhibitory
potential, and some empirical SARs have been obtained. Our results
show that chalcone, the parent compound, or its 2′-hydroxy- or
2-hydroxy- derivatives (nos. 1–3) did not express any NF-κB
inhibition potential. We were able to draw some SARs originating
from the substitutions with an electron donating functional group
such as methoxy or hydroxy, at positions 4, 4′ and 6′. Chalcone
glycosides (nos. 20 and 21) were not active, which might be due to
the reduced permeability through cell membranes. In addition, all
chalcones with reported NF-κB activity contain a highly
electrophilic α,β-unsaturated carbonyl moiety and calomelanone and
phloretin are the first examples of dihydrochalcones with such
activity. For HDAC activity clearer trends were observed, i.e.
hydroxy groups at C-2′, C-4′, C-3 and C-4 were essential. This
substitution pattern also appears to be important for dual
activity. The empirical SARs prompted us to perform some molecular
modeling-docking studies on both targets. The molecular modeling
investigations could not provide a definite rationale for dual
activity of chalcones and failed to provide criteria for
distinguishing the active molecules from inactive. There could be
many reasons underlying this observation. Most of the molecules
have low molecular weight (between 200 to 275 Da) and as such can
be considered as fragments. This could lead to non-specific binding
and lack of correlation between molecular properties and
activity.
Some of the compounds tested herein have previously
been reported as NF-κB inhibitors including isoliquiritigenin (no.
10) (41) and butein (no. 12)
(42). We have previously
demonstrated that 4′-hydroxychalcone showed 26S protease inhibition
activity on three different proteolytic activities (chymotrypsin,
trypsin- and caspase-like) in a dose-dependent manner (26). The involvement of natural chalcones
in cancer tumorigenesis has been reviewed (43). To the best of our knowledge this is
the first study aiming to screen a natural chalcone library and
attempting to draw SARs among them. Several natural chalcones
emerged as relatively good inhibitors of NF-κB. Additionally, HDAC
was identified as a novel potential target for the chalcones
described.
Acknowledgements
During this project B.O. and M.S. were supported by
Télévie grants. In addition, M.S. was supported by a ‘Waxweiler
grant for cancer prevention research’ from the Action Lions
‘Vaincre le Cancer’. This work was supported by the ‘Recherche
Cancer et Sang’ foundation, ‘Recherches Scientifiques Luxembourg’
association and the Télévie Luxembourg. The authors thank ‘Een
Häerz fir Kriibskrank Kanner’ association and the Action Lions
‘Vaincre le Cancer’ for generous support. Publication costs were
covered by the Fonds National de la Recherche Luxembourg.
References
|
1
|
Grunstein M: Histone acetylation in
chromatin structure and transcription. Nature. 389:349–352. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
ten Holte P, Van Emelen K, Janicot M, Fong
PC, de Bono JS and Arts J: HDAC inhibition in cancer therapy: an
increasingly intriguing tale of chemistry, biology and clinical
benefit. Cancer. Topics in Medicinal Chemistry. Bradbury RH:
Springer Verlag; Berlin: pp. 293–332. 2007
|
|
4
|
Folmer F, Orlikova B, Schneckenburger M,
Dicato M and Diederich M: Naturally occurring regulators of histone
acetylation/deacetylation. Curr Nutr Food Sci. 6:78–99. 2010.
View Article : Google Scholar
|
|
5
|
Seidel C, Schnekenburger M, Dicato M and
Diederich M: Histone deacetylase modulators provided by Mother
Nature. Genes Nutr. Feb 12–2012.(Epub ahead of print).
|
|
6
|
Gibbons RJ: Histone modifying and
chromatin remodelling enzymes in cancer and dysplastic syndromes.
Hum Mol Genet. 14(Suppl 1): R85–R92. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lombardi PM, Cole KE, Dowling DP and
Christianson DW: Structure, mechanism, and inhibition of histone
deacetylases and related metalloenzymes. Curr Opin Struct Biol.
21:735–743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vannini A, Volpari C, Filocamo G, et al:
Crystal structure of a eukaryotic zinc-dependent histone
deacetylase, human HDAC8, complexed with a hydroxamic acid
inhibitor. Proc Natl Acad Sci USA. 101:15064–15069. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gupta SC, Sundaram C, Reuter S and
Aggarwal BB: Inhibiting NF-kappaB activation by small molecules as
a therapeutic strategy. Biochim Biophys Acta. 1799:775–787. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vallabhapurapu S and Karin M: Regulation
and function of NF-kappaB transcription factors in the immune
system. Annu Rev Immunol. 27:693–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Batra S, Sahu RP, Kandala PK and
Srivastava SK: Benzyl isothiocyanate-mediated inhibition of histone
deacetylase leads to NF-kappaB turnoff in human pancreatic
carcinoma cells. Mol Cancer Ther. 9:1596–1608. 2010. View Article : Google Scholar
|
|
12
|
Jung ID, Lee JS, Jeong YI, et al:
Apicidin, the histone deacetylase inhibitor, suppresses Th1
polarization of murine bone marrow-derived dendritic cells. Int J
Immunopathol Pharmacol. 22:501–515. 2009.PubMed/NCBI
|
|
13
|
Roth SY, Denu JM and Allis CD: Histone
acetyltransferases. Annu Rev Biochem. 70:81–120. 2001. View Article : Google Scholar
|
|
14
|
Kiernan R, Bres V, Ng RW, et al:
Post-activation turn-off of NF-kappa B-dependent transcription is
regulated by acetylation of p65. J Biol Chem. 278:2758–2766. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu J and Colburn NH: Histone deacetylase
inhibition down-regulates cyclin D1 transcription by inhibiting
nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 3:100–109.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lehmann A, Denkert C, Budczies J, et al:
High class I HDAC activity and expression are associated with
RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC
Cancer. 9:3952009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Florean C, Schnekenburger M, Grandjenette
C, Dicato M and Diederich M: Epigenomics of leukemia: from
mechanisms to therapeutic applications. Epigenomics. 3:581–609.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schnekenburger M and Diederich M:
Epigenetics offer new horizons for colorectal cancer prevention.
Curr Colorectal Cancer Rep. 8:66–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Papeleu P, Wullaert A, Elaut G, et al:
Inhibition of NF-kappaB activation by the histone deacetylase
inhibitor 4-Me2N-BAVAH induces an early G1 cell cycle arrest in
primary hepatocytes. Cell Prolif. 40:640–655. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fabre C, Grosjean J, Tailler M, et al: A
novel effect of DNA methyltransferase and histone deacetylase
inhibitors: NFkappaB inhibition in malignant myeloblasts. Cell
Cycle. 7:2139–2145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Halili MA, Andrews MR, Sweet MJ and
Fairlie DP: Histone deacetylase inhibitors in inflammatory disease.
Curr Top Med Chem. 9:309–319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Srinivasan B, Johnson TE, Lad R and Xing
C: Structure-activity relationship studies of chalcone leading to
3-hydroxy-4,3′,4′,5′-tetramethoxychalcone and its analogues as
potent nuclear factor kappaB inhibitors and their anticancer
activities. J Med Chem. 52:7228–7235. 2009.PubMed/NCBI
|
|
23
|
Yadav VR, Prasad S, Sung B and Aggarwal
BB: The role of chalcones in suppression of NF-kappaB-mediated
inflammation and cancer. Int Immunopharmacol. 11:295–309. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Edwards ML, Stemerick DM and Sunkara PS:
Chalcones: a new class of antimitotic agents. J Med Chem.
33:1948–1954. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lawrence NJ, McGown AT, Ducki S and
Hadfield JA: The interaction of chalcones with tubulin. Anticancer
Drug Des. 15:135–141. 2000.PubMed/NCBI
|
|
26
|
Orlikova B, Tasdemir D, Golais F, Dicato M
and Diederich M: The aromatic ketone 4′-hydroxychalcone inhibits
TNFalpha-induced NF-kappaB activation via proteasome inhibition.
Biochem Pharmacol. 82:620–631. 2011.
|
|
27
|
Pedretti A, Villa L and Vistoli G: VEGA -
an open platform to develop chemo-bio-informatics applications,
using plug-in architecture and script programming. J Comput Aided
Mol Des. 18:167–173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pedretti A, Villa L and Vistoli G:
Atom-type description language: a universal language to recognize
atom types implemented in the VEGA program. Theor Chem Acc.
109:229–232. 2003. View Article : Google Scholar
|
|
29
|
Tetko IV, Gasteiger J, Todeschini R, et
al: Virtual computational chemistry laboratory - design and
description. J Comput Aided Mol Des. 19:453–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Friesner RA, Banks JL, Murphy RB, et al:
Glide: a new approach for rapid, accurate docking and scoring. 1.
Method and assessment of docking accuracy. J Med Chem.
47:1739–1749. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Halgren TA, Murphy RB, Friesner RA, et al:
Glide: a new approach for rapid, accurate docking and scoring. 2.
Enrichment factors in database screening. J Med Chem. 47:1750–1759.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ortore G, Di Colo F and Martinelli A:
Docking of hydroxamic acids into HDAC1 and HDAC8: a rationalization
of activity trends and selectivities. J Chem Inf Model.
49:2774–2785. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Piccagli L, Fabbri E, Borgatti M, et al:
Docking of molecules identified in bioactive medicinal plants
extracts into the p50 NF-kappaB transcription factor: correlation
with inhibition of NF-kappaB/DNA interactions and inhibitory
effects on IL-8 gene expression. BMC Struct Biol. 8:382008.
View Article : Google Scholar
|
|
34
|
Di Gennaro E, Bruzzese F, Caraglia M,
Abruzzese A and Budillon A: Acetylation of proteins as novel target
for antitumor therapy: review article. Amino Acids. 26:435–441.
2004.PubMed/NCBI
|
|
35
|
Shen KH, Chang JK, Hsu YL and Kuo PL:
Chalcone arrests cell cycle progression and induces apoptosis
through induction of mitochondrial pathway and inhibition of
nuclear factor kappa B signalling in human bladder cancer cells.
Basic Clin Pharmacol Toxicol. 101:254–261. 2007. View Article : Google Scholar
|
|
36
|
Lee ST, Wong PF, Cheah SC and Mustafa MR:
Alpha-tomatine induces apoptosis and inhibits nuclear factor-kappa
B activation on human prostatic adenocarcinoma PC-3 cells. PLoS
One. 6:e189152011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen L, Fischle W, Verdin E and Greene WC:
Duration of nuclear NF-kappaB action regulated by reversible
acetylation. Science. 293:1653–1657. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Krämer OH, Baus D, Knauer SK, et al:
Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev.
20:473–485. 2006.PubMed/NCBI
|
|
39
|
Butler KV, Kalin J, Brochier C, Vistoli G,
Langley B and Kozikowski AP: Rational design and simple chemistry
yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J
Am Chem Soc. 132:10842–10846. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tang W, Luo T, Greenberg EF, Bradner JE
and Schreiber SL: Discovery of histone deacetylase 8 selective
inhibitors. Bioorg Med Chem Lett. 21:2601–2605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JY, Park SJ, Yun KJ, Cho YW, Park HJ
and Lee KT: Isoliquiritigenin isolated from the roots of
Glycyrrhiza uralensis inhibits LPS-induced iNOS and COX-2
expression via the attenuation of NF-kappaB in RAW 264.7
macrophages. Eur J Pharmacol. 584:175–184. 2008.PubMed/NCBI
|
|
42
|
Pandey MK, Sandur SK, Sung B, Sethi G,
Kunnumakkara AB and Aggarwal BB: Butein, a tetrahydroxychalcone,
inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene
expression through direct inhibition of IkappaBalpha kinase beta on
cysteine 179 residue. J Biol Chem. 282:17340–17350. 2007.
View Article : Google Scholar
|
|
43
|
Orlikova B, Tasdemir D, Golais F, Dicato M
and Diederich M: Dietary chalcones with chemopreventive and
chemotherapeutic potential. Genes Nutr. 6:125–147. 2011. View Article : Google Scholar : PubMed/NCBI
|