Introduction
Glioma is the most common tumor of the brain and
accounts for 45–55% of all brain tumors. Based on histological
features (nuclear atypia, mitosis, microvascular enrichment and
necrosis) and malignancy, glioma is classified into four grades
(1). Grade 1 is innocent and
curable with surgical resection; grade 2 is a low grade diffuse
glioma and can progress into higher grades; grade 3 and 4 gliomas
are malignant and have increased capability to proliferate and
invade. The grade 4 glioma, glioblastoma multiforme (GBM), is the
most aggressive.
Surgical resection is the primary therapy to treat
glioma (2). However, surgical
removal is very restrictive, thus it is difficult to completely
remove diffusive tumor tissues, especially higher grade gliomas.
These glioma tissues invade into the white matter or diffuse along
the ventricle surface, so other strategies such as chemotherapy are
very important to remove cancer cells as completely as possible
(3–5). Chemotherapy increases the life of GBM
patients. Clinical statistics reveals that, chemotherapy treatment
leads to a 15% reduction of mortality, and a 6% increase of 1-more
year survival. However, the effect of chemotherapy on glioma is not
satisfactory, largely due to three problems. The first problem is
the diagnosis of diffusive glioma tissues, which are tiny and grow
deep in the brain tissue and are hard to access. The second problem
is the blockade of chemotherapy drug by the blood-brain barrier.
The third problem is that glioma is highly tolerant under
chemotherapy treatment. Understanding the fundamentals, especially
the molecular mechanisms, is critical to increase the
susceptibility of glioma to chemotherapy.
Tumor cells originated from cells with abnormal
proliferation and dysregulation of the cell cycle (6,7).
Molecular profiling studies showed that the genes controlling cell
growth and restricting cell division are silenced, and genes
promoting cell proliferation and facilitating cell cycle are
overexpressed. Many genes that promote cell cycle are found to be
increased in transformed tumor cells (8). In cell cycle, CDK4 and 6 phosphorylate
tumor suppressor Rb and promote G1 progression and G1/S transition
(9). CDK6 is arrested and inhibited
by INK4 and cyclin D (10). The
CDK4/6 signaling was found to be highly activated in tumor cells,
including sarcoma and leukemia (11,12).
Lam et al reported that 12 in 14 tumor samples showed
increased expression of CDK6 (13).
Moreover, mir-125b blocked G1/S transition and increased apoptosis
by inhibiting CDK6 expression (14). Another microRNA mir-34a
down-regulated CDK6 expression and significantly inhibited the
proliferation of glioma cells (15). However, whether CDK6 mediates the
chemotherapy resistance of glioma remains unknown.
In this study, we tested the hypothesis that
increased expression and activation of CDK6 in glioma contribute to
its resistance to chemotherapy treatment. Using
immunohistochemistry, we found that the expression level of CDK6
correlated well with the tumor grade of gliomas. We then found that
knocking down of CDK6 led to significant reduction on the cell
cycle progression and proliferation of glioma cells. Finally, we
found that glioma cells transfected with CDK6 shRNA showed
increased apoptosis when exposed to temozolomide (TMZ) compared to
cells transfected with control scRNA. Moreover, CDK6 knockdown
resulted in down-regulated expression of several genes relating to
drug resistance. Our findings suggested that CDK6 is an important
molecular determinant contributing to chemotherapy resistance.
Materials and methods
Cell culture
The widely used glioma cell line U251 (ATCC,
Manassas, VA) was cultured for our study. Cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad,
CA), supplemented with 10% fetal bovine serum (Thermo Fisher
Scientific, Inc., Waltham, MA) and antibiotics (penicillin and
streptomycin, 50 U/ml each) (Invitrogen). Cells were grown in 60 mm
dish and incubated in a humidified incubator with 5% CO2
at 37°C. Cells were inoculated at 1×104 cells/dish, and
were used at a confluent of ~75%. For chemotherapy treatment, cells
were treated with TMZ (10 nM) for 24, 48 or 72 h.
Collection of human glioma and normal
tissue samples
Surgically removed human glioma tissue samples and
normal brain tissues were obtained frozen or paraffin-embedded from
Changhai Hospital (China Secondary Military Medical University,
Shanghai, China). Gliomas were graded by the Pathology Department
of Changhai Hospital according to the World Health Organization
grading system, presence and absence of nuclear atypia, mitosis,
microvascular enrichment, and necrosis. Human normal brain tissues
(mostly from the cortex) were obtained from patients with physical
injuries to the brain. These specimens were collected from the
patients registered at the above-mentioned hospitals, and written
informed consent was obtained from the patients. The use of human
tissues was approved by the ethics committees of the hospitals.
RT-PCR
Total RNA from mouse brain tissue or culture was
extracted using TRIzol reagent (Invitrogen). The RNA was treated
with DNase I to remove genomic DNA contamination and then reverse
transcribed on a 96-well temperature cycler (ABI) using an M-MLV RT
kit (Invitrogen). For PCR, a SYBR Premix Ex Taq kit (Takara) was
used to prepare PCR solution and programs were run on Mastercycler
Gradient PCR machine (Eppendorf) or Rotor-Gene 3000 Realtime Cycler
(Corbett Research, Inc.). The primers used are: cdk6
forward, GCGCCTATGGGAAGGTGTTC; cdk6 reverse,
TTGGGGTGCTCGAAGGTCT. Mdr-1 forward,
5-TGGTTCAGGTGGCTCTGGAT-3, mdr-1 reverse,
5-CTGTAGACAAACGATGAGCTATCACA-3; mrp forward,
5-GGCAAAGAAATAAAGCGACTGAA-3, mrp reverse,
5-GGCTGTTGTCTCCATAGGCAAT-3.
Western blotting and immunohistology
For western blotting, total proteins were extracted
from tissues and U251 culture using the RIPA buffer [10 mM Tris-HCl
(pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium
deoxycholate, 1 mM Na3VO4, 1 mM PMSF, and
protease inhibitor cocktail]. Protein concentration was measured
using BCA reagent (Pierce), diluted in sample loading buffer and
subjected to SDS-PAGE gel electrophoresis, then transferred to PVDF
membrane (0.45 μm, PALL), and blocked by 0.05% TBST with 5% milk
and then blotted with primary antibodies at 4°C overnight and
secondary antibodies at room temperature for 1 h. Bound antibodies
were detected by the ECL immunoblotting detection reagent (GE
Healthcare) and exposed to X-ray films. Band densities were
quantified by Gel-Pro Analyzer, and the densitometric results are
shown. The relative amount of proteins was determined by
normalizing the density value of target protein to internal control
and the external control.
For immunohistochemistry, 4 μm thickness
paraffin-processed sections were mounted on poly-D-lysine-coated
glass slides. Each slide was dewaxed in 100% xylene and rehydrated
by incubation in decreasing concentrations (100, 95 and 75%) of
alcohol. Sections were incubated in 3% hydrogen peroxide to quench
endogenous peroxidase. Antigen retrieval was performed in a
microwave oven (95°C for 15 min). The immunoreactions were
performed with ABC kit (Maixin Biotech, China). Briefly, sections
were blocked with horse serum and incubated overnight with diluted
mouse anti-human CDK6 antibody (Santa Cruz) at 4°C. Following
washes with TBST (3×5 min), the slides were incubated with the
appropriate anti-mouse IgG (Dako Envision) at room temperature for
30 min, and then washed with TBST (3×5 min). Sections were
developed using DAB reagent and counterstained with hematoxylin to
stain the nucleus. Sections were dehydrated by incubating in
increasing concentrations (75, 95 and 100%) of alcohol and in 100%
xylene before coverslips were mounted onto the sections.
RNAi and transfection
To knockdown cdk6, three siRNA targeting
sequences were designed using online siRNA Target Finder (Ambion).
The three sets of siRNA was synthesized and annealed to form double
strand siRNA and transfected into U251 cells by Lipofectamine 2000
(Invitrogen). The knockdown efficiency was validated by RT-PCR and
Western blotting, and finally the most specific and efficient
targeting sequence was used for functional assay. The sequence is:
5′-CATGTCGATCAAGACTTGA-3′. For vector based shRNA construct, an
shRNA with the selected targeting sequence was cloned into
pSuper-GFP or pLVTHM. For transfection of U251 cells, 4 μg of shRNA
or control plasmid was dissolved in Opti-MEM and coated with
Lipofectamine 2000 reagent for a 60-mm dish. Six hours later, the
culture medium was replaced, and 48 h later, the cells were
subjected for analysis.
MTT assay
Cells were cultured in a 96-well plate at a density
of 5×103. Two hours before examination, cell-counting
kit-8 reagent (10 μl/well) was added into the wells and incubated
for 2 h. The reaction was terminated by adding SDS solution (0.1%
final concentration). Before reading, the plate was shaked by the
supplied plate shaker and then read at 450 nm.
Flow cytometry to examine cell cycle
progression
Flow cytometry assays were used to study the effect
of knocking down CDK6 cell cycle progression. U251 cells were
harvested, treated with 0.25% trypsin, washed in PBS, centrifuged
(1500 rpm, 5 min) and resuspended with binding buffer. The cell
density was adjusted to 3×105 cells/ml. Cell suspension
(195 μl) and 5 μl Annexin V/FITC were combined and incubated at RT
for 10 min. Next, propidium iodide was added to a final
concentration of 1 μg/ml and incubated at 37°C for 30 min. In each
sample, 6×104 cells were assayed on FACSCalibur
(Becton-Dickinson, Franklin Lakes, NJ) and the cell cycle phase
distribution was analyzed by CellQuest Pro software
(Becton-Dickinson).
Statistical analysis
The data are presented as mean ± SD. All statistical
analysis were done using One-way ANOVA and SPSS software.
Results
Significant upregulation of CDK6 in
glioma
Although CDK6 expression was reported to be
increased in gliomas, it remains unclear whether the malignant
grade correlates with the upregulation of CDK6 expression. To
examine this, we collected 34 glioma samples at different grades
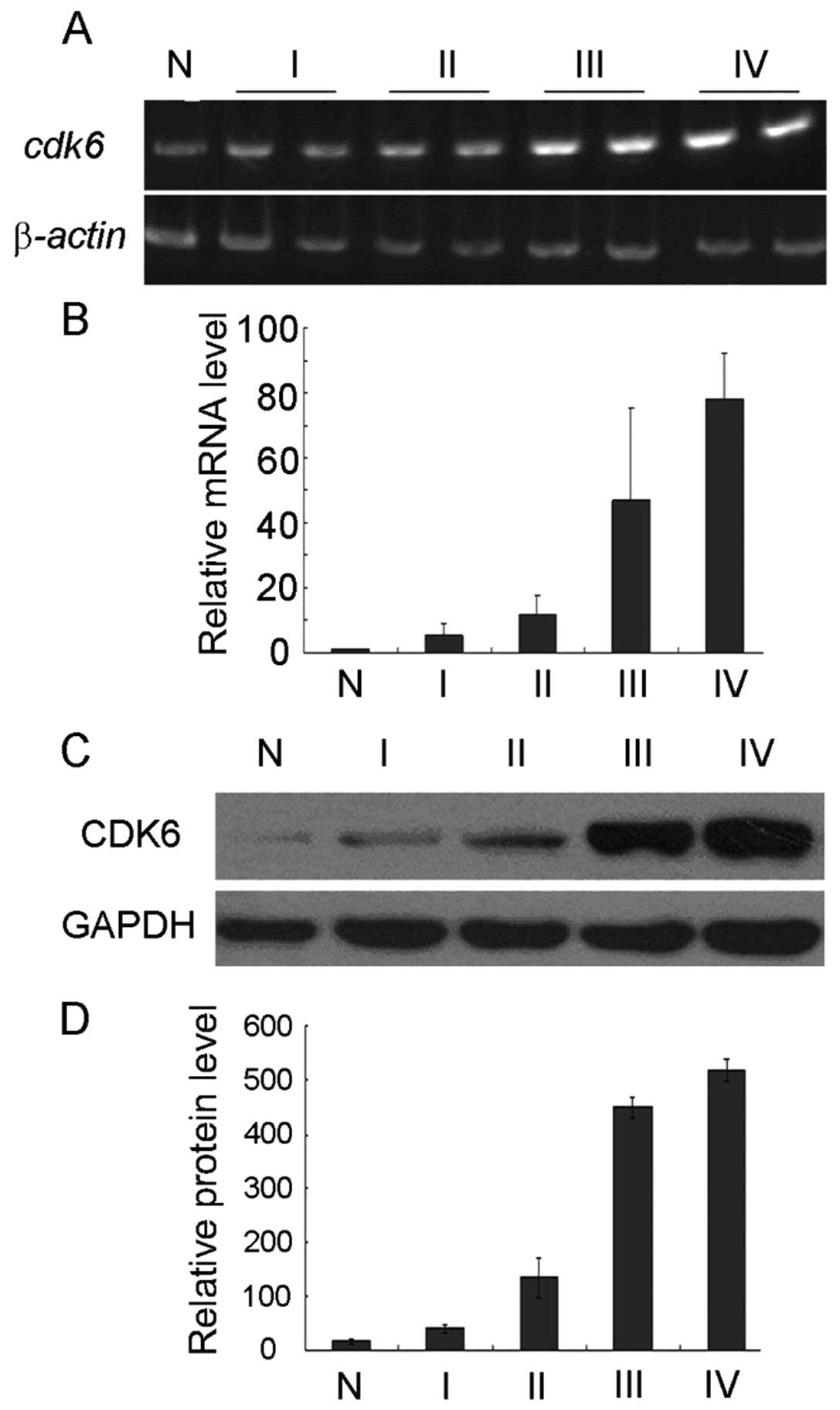
and 4 samples of normal brain tissue. The mRNA of CDK6 showed good
correlation with different grades of gliomas (normal tissue,
1.02±0.25; grade 1, 5.50±0.25; grade 2, 11.78±5.90; grade 3,
46.74±28.81; grade 4, 78.11±14.04, Fig.
1A and B). The expression change of CDK6 protein was also
studied using Western blotting, and similar upregulation pattern
was found in glioma samples (Fig. 1C
and D). In normal tissue, the protein level of CDK6 was
16.81±3.25, whereas in grade 1 glioma, CDK6 expression was
increased to 41.33±7.38, and in grade 2 glioma to 35.11±37.54. In
highly malignant glioma, significant elevation of CDK6 protein was
observed (grade 3, 449.45±20.17; grade 4, 518.61±19.61).
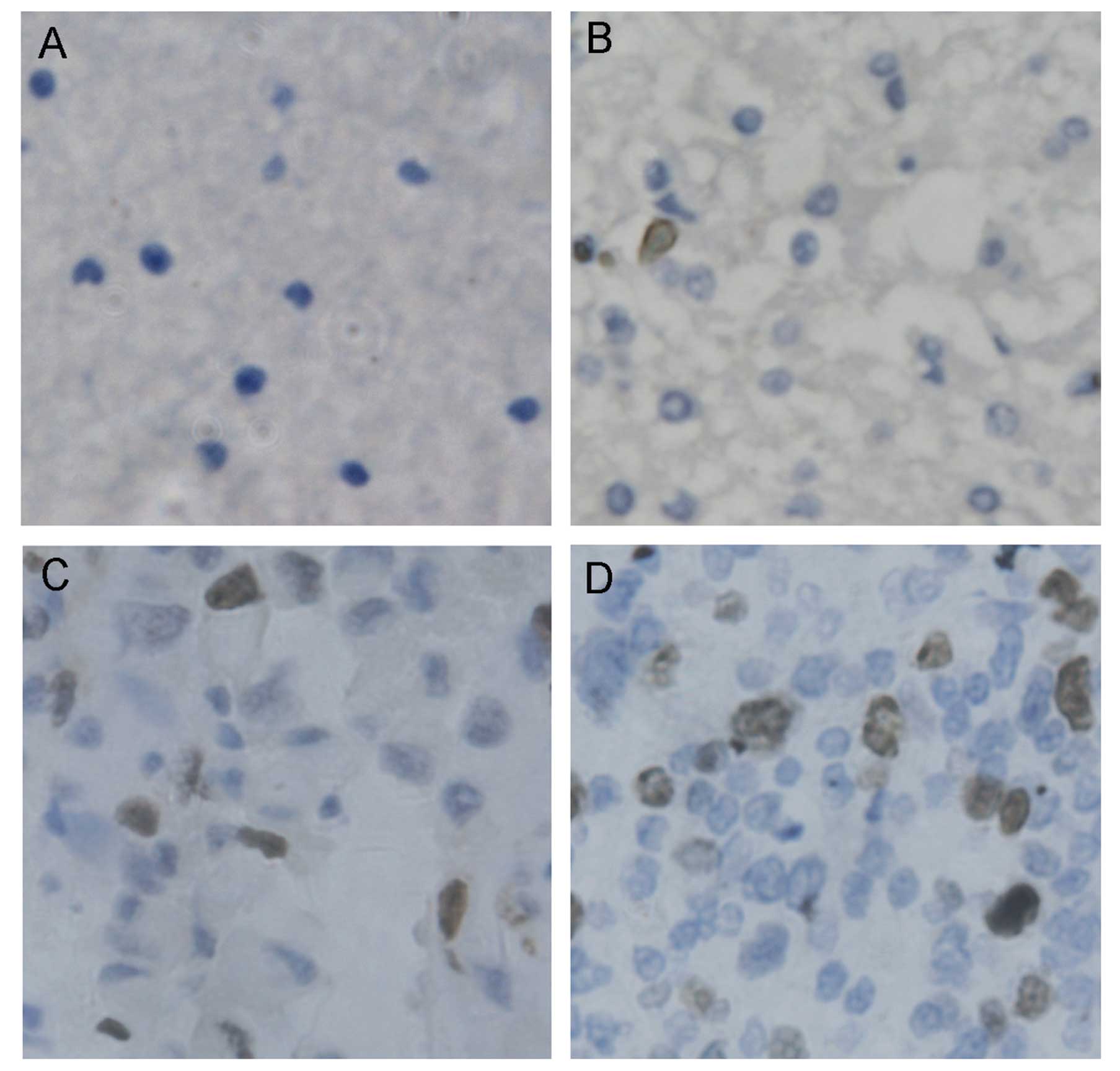
Furthermore, immunostaining showed that, CDK6 was kept at much low
level in normal brain tissue (Fig.
2A). In grade 1 glioma tissue, CDK6 was increased moderately
(Fig. 2B). In grade 2 glioma, CDK6
was highly upregulated (Fig. 2C),
whereas in more malignant grade 3 or 4 glioma, higher upregulation
of CDK6 expression was observed (Fig.
2D).
We also employed microarray chips to do whole-genome
comparison of gene expression profiles between glioma and normal
tissues. Among those highly upregulated genes in glioma tissue,
CDK6 also showed good correlation with glioma grades and survival
time according to follow-up visit data of patients (data not
shown).
Knockdown of CDK6 mRNA in glioma inhibits
glioma growth
Increased high level of CDK6 may promote the G1
progression and G1/S transition, thus leads to uncontrolled cell
division and proliferation, and finally tumorigenesis and
malignancy (13,16,17).
Although upregulation of CDK6 expression correlated with glioma
grades, whether CDK6 contributed to the higher proliferation and
invasion capability requires further determination.
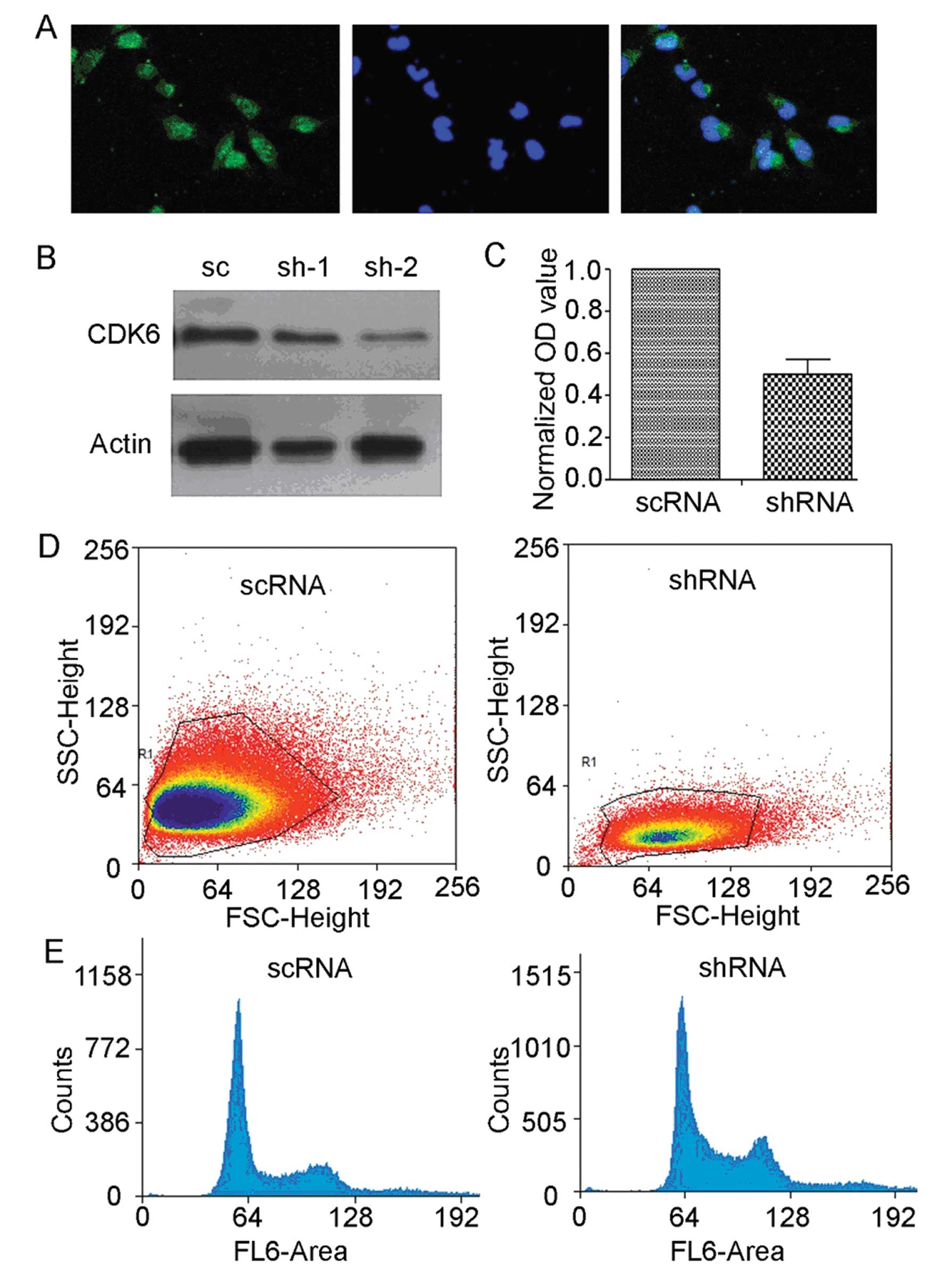
We used U251 cell line to test whether CDK6 played a
role in regulating glioma proliferation (Fig. 3A). To knock down CDK6, an shRNA was
cloned into pSuper-GFP construct, and transfected into U251 cells
(Fig. 3B), 72 h later, cell cycle
distribution of U251 cells was analyzed by flow cytometry. Compared
to scrambled shRNA (scRNA), cdk6-targeting shRNA (shRNA)
resulted in significantly increased retention of U251 cells in G1
phase (scRNA, 51.07±1.33; cdk6 shRNA, 59.77±1.85; p<0.05,
Fig. 3D and E), thereby leading to
a great reduction of the ratio of cells in G2/M phase (scRNA,
25.93±0.50;cdk6 shRNA, 21.85±1.32; p<0.05, Fig. 3D and E).
Knocking down cdk6 inhibited cell cycle
progression, thus blocking glioma proliferation and tumor growth.
To examine the function of cdk6 in cell growth, we used MTT
assay to analyze U251 cells after shRNA transfection (Fig. 3C). Compared to control scRNA,
cdk6 shRNA expression greatly reduced the MTT absorption
value (scRNA, 0.613±0.075; cdk6 shRNA, 0.417±0.065;
p<0.05), indicating significant inhibition of U251 cell growth
by RNAi of CDK6.
Enhanced chemotherapy effect of TMZ on
CDK6-knockdown glioma
Chemotherapy is an indispensible treatment for
higher grade gliomas (4,18). However, the low permeability of
drugs across blood-brain barrier reduced drug concentration in
cerebral-spinal fluid. On the other hand, glioma cell are resistant
to chemotherapy treatment, mainly due to high activity of DNA
repair mechanism, tolerance to cell cycle check points, reduced
uptake of drug and quick metabolism of drug molecules (18–20).
In the above studies, we found that CDK6 expression is highly
upregulated in glioma cells. As CDK6 is a transducer of cytoplasm
signal to nuclear transcription activity and DNA repair mechanism,
we speculated that the chemotherapy-resistance of glioma is mainly
mediated by increased level of CDK6.
To test this hypothesis, we first established a
culture model of chemotherapy. Temozolomide (TMZ) is an alkylating
agent and effective for the treatment of higher grade glioma
(5). We thus tested the effect of
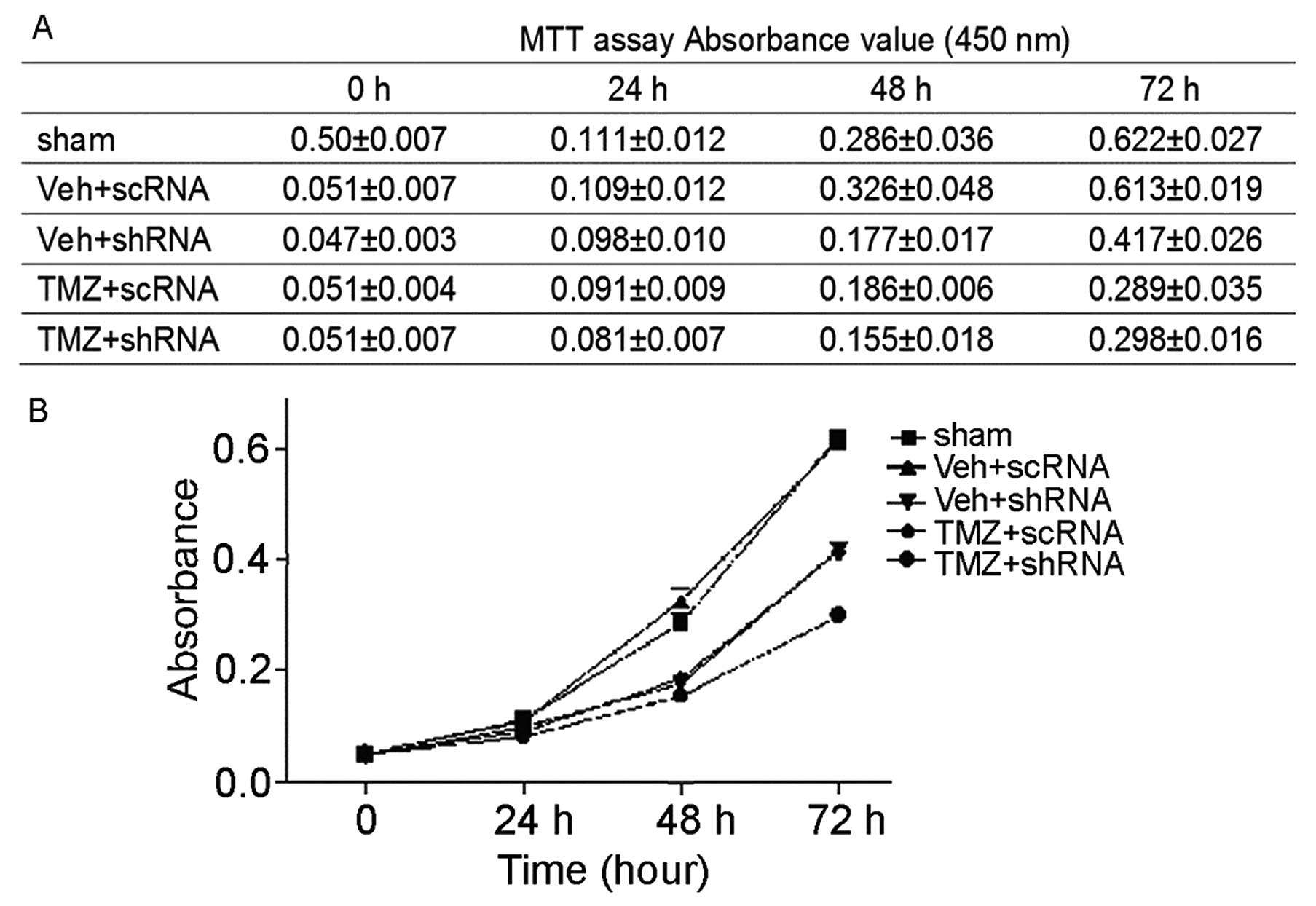
TMZ on glioma in vitro (Fig.
4). Compared to control reagent, TMZ showed inhibition of U251
cell growth even at 24 h after treatment (con, 0.111±0.012; TMZ,
0.098±0.010). At 48 h, significant inhibition of U251 cell growth
by TMZ can be observed (con, 0.286±0.036; TMZ, 0.186±0.006;
P<0.01). Next, we compared the effect of combined treatment of
U251 cells with TMZ and cdk6 knockdown to single treatment
with TMZ. Interestingly, combined treatment of U251 cells with TMZ
and cdk6 shRNA resulted in significant decrease of U251 cell
growth at 48 h after treatment (0.155±0.018, p<0.05), lower than
single treatment with either TMZ (0.186±0.006) or cdk6 shRNA
(0.177±0.017). These results strongly suggested that increased
expression of CDK6 contributed to the resistance of glioma to
chemotherapy.
Reduced drug-resistance to chemotherapy
in CDK6-knockdown glioma
The facilitation of TMZ-mediated chemotherapy
toxicity on U251 glioma cell growth by CDK6 knockdown indicated
that aberrant cell signaling of glioma cells underlies the
resistance of glioma to chemotherapy. To test whether CDK6
knockdown had an effect on drug resistance-related genes, we
analyzed the expression change of several key genes by RT-PCR and
western blotting. MDR-1 is a transmembrane glycoprotein of the
ATP-binding cassette superfamily (21). MDR-1 acts as a bump to transport
drug molecules out of the cells, thereby reducing drug toxicity to
cancer cells. In cdk6-knockdown cells, MDR-1 expression was
greatly down-regulated (shRNA, 0.44±0.18; scRNA, 3.07±0.49; sham,
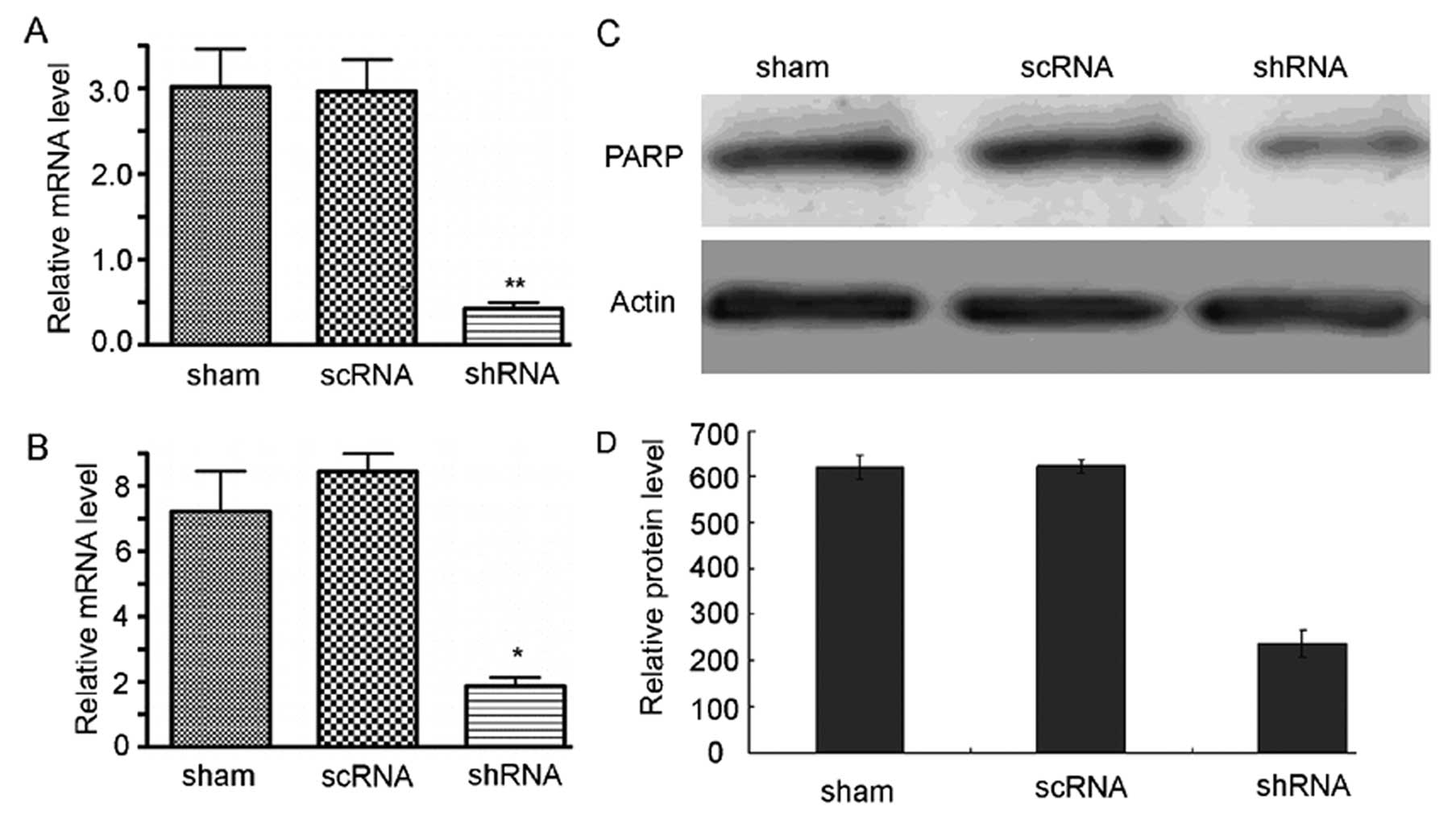
3.09±0.27; p<0.01, Fig. 5A). MRP
is another member of the ABC transporter superfamily (22). We also found that MRP expression in
cdk6-knockdown U251 cells was lower than scRNA transfected
cells (shRNA, 1.93±1.40; scRNA, 7.27±1.49; sham, 8.39±1.17;
p<0.01, Fig. 5B).
Drug resistance can also result from high level of
DNA repair activity. PARP is a DNA polymerase which plays an
important role in DNA repair (23).
PARP can detect DNA single-strand break and bind to the break
sequence and initiate the repair process by the synthesis of a
poly(ADP-ribose) chain and recruiting other DNA repair proteins.
PARP1 is highly expressed in glioma (24), and its expression can be increased
by TMZ treatment. In turn, the upregulated PARP1 protects glioma
cells from DNA-damage induced cell cycle retention and apoptosis.
In cdk6-knockdown cells, PARP1 protein was significantly
down-regulated (scRNA, 623.77±15.36; cdk6 shRNA,
237.23±28.09; p<0.05, Fig. 5C and
D). Conclusively, increased level of CDK6 resulted in enhanced
expression of several key genes responsible for drug resistance,
and CDK6 knockdown or inhibition was effective in reducing their
expression, thereby reducing chemotherapy drug-resistance of glioma
cells.
Discussion
Gene mutation and their aberrant expression are very
common in tumor cells. Microarray studies revealed that in each
type of tumor cells, more than one hundred genes exhibited abnormal
expression or mutation (25). Thus,
finding the key genes that participate in or regulate the
tumorigenesis process has become an important task in cancer
research. CDK6 regulates cell cycle progression, especially in the
transition from G1 to S phase (9).
We first demonstrated that CDK6 upregulation correlated with the
malignant grades of glioma, which implied that the expression level
of CDK6 might determine the proliferation and invasion capability.
As an upstream regulator of cancer repressor Rb, CDK6 expression
level is an indicator of cell division activity (16). CDK6 phosphorylates Rb and activates
transcription factor E2F1/2, thus pushing cells into S phase and
triggering DNA synthesis (6).
The control of G1/S transition is the first gate to
cell division, thus becomes extremely important for cell cycle
control. The highly upregulated CDK6 in glioma cells make the G1/S
checkpoint permissive, thereby enabling cell proliferation. We
found that cdk6 knockdown resulted in significant decrease
of cell growth, indicating that CDK6 is an organizer of cell cycle,
and higher expression of CDK6 may underlie the chemotherapy
resistance of glioma. As a kinase, CDK6 activity can easily be
inhibited by small molecules, which can pass across the blood-brain
barrier. Many drug-resistant genes are upregulated in glioma cells,
some of which may be downstream genes of E2F transcription
activator complex (26,27). Therefore, inhibiting CDK6 may have
considerable effect on enhancing the sensitivity of glioma cells to
chemotherapy drugs. In our study, we observed that combined
treatment of glioma cells with chemotherapy drug TMZ and CDK6 shRNA
significantly inhibited cell growth, which is more effective than
single treatment either with TMZ or CDK6 shRNA.
The mechanism underlying chemotherapy-resistance of
glioma has been intensively studied (18,19,21,22).
Many drug-resistant genes have been found, which mainly belongs to
DNA repair enzymes, small-molecule transmembrane transporters and
drug metabolism proteins. Given their wide divergence and
differential protein structures, it may not be practicable to
interfere with a single drug-resistant gene. Therefore, finding the
upstream regulators maybe more effective to increase drug uptake
and toxicity. CDK6 knockdown resulted in significant
down-regulation of MDR1, MRP and PARP, indicating that CDK6 is an
upstream activator of these drug-resistance related genes, and
should be a promising target to reduce drug-resistance of glioma
cells. Future work will be needed to study how CDK6 overexpression
leads to the upregulation of these genes.
Acknowledgements
This study was supported by grants from the Key
Foundation Project of the National Natural Science Foundation of
China (no. 30930094).
References
|
1
|
Behin A, Hoang-Xuan K, Carpentier AF and
Delattre J-Y: Primary brain tumours in adults. Lancet. 361:323–331.
2003.
|
|
2
|
Lacroix M, Abi-Said D, Fourney DR, et al:
A multivariate analysis of 416 patients with glioblastoma
multiforme: prognosis, extent of resection, and survival. J
Neurosurg. 95:190–198. 2001.
|
|
3
|
Stewart L: Chemotherapy in adult
high-grade glioma: a systematic review and meta-analysis of
individual patient data from 12 randomised trials. Lancet.
359:1011–1018. 2002.
|
|
4
|
Reardon DA, Rich JN, Friedman HS and
Bigner DD: Recent advances in the treatment of malignant
astrocytoma. J Clin Oncol. 24:1253–1265. 2006.
|
|
5
|
Stupp R, Dietrich P-Y, Kraljevic SO, et
al: Promising survival for patients with newly diagnosed
glioblastoma multiforme treated with concomitant radiation plus
temozolomide followed by adjuvant temozolomide. J Clin Oncol.
20:1375–1382. 2002.
|
|
6
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009.
|
|
7
|
Schwartz GK and Shah MA: Targeting the
cell cycle: a new approach to cancer therapy. J Clin Oncol.
23:9408–9421. 2005.
|
|
8
|
Deshpande A, Sicinski P and Hinds PW:
Cyclins and cdks in development and cancer: a perspective.
Oncogene. 24:2909–2915. 2005.
|
|
9
|
Harbour JW, Luo RX, Santi AD, Postigo AA
and Dean DC: Cdk phosphorylation triggers sequential intramolecular
interactions that progressively block Rb functions as cells move
through G1. Cell. 98:859–869. 1999.
|
|
10
|
Ezhevsky SA, Nagahara H, Vocero-Akbani AM,
Gius DR, Wei MC and Dowdy SF: Hypo-phosphorylation of the
retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results
in active pRb. Proc Natl Acad Sci USA. 94:10699–10704. 1997.
|
|
11
|
Hayette S, Tigaud I, Callet-Bauchu E, et
al: In B-cell chronic lymphocytic leukemias, 7q21 translocations
lead to overexpression of the CDK6 gene. Blood. 102:1549–1550.
2003.
|
|
12
|
Van Dross R, Yao S, Asad S, et al:
Constitutively active K-cyclin/cdk6 kinase in Kaposi
sarcoma-associated herpesvirus-infected cells. J Natl Cancer Inst.
97:656–666. 2005.
|
|
13
|
Lam PY, Di Tomaso E, Ng HK, Pang JC,
Roussel MF and Hjelm NM: Expression of p19INK4d, CDK4, CDK6 in
glioblastoma multiforme. Br J Neurosurg. 14:28–32. 2000.
|
|
14
|
Shi L, Zhang J, Pan T, et al: MiR-125b is
critical for the suppression of human U251 glioma stem cell
proliferation. Brain Res. 1312:120–126. 2010.
|
|
15
|
Li Y, Guessous F, Zhang Y, et al:
MicroRNA-34a inhibits glioblastoma growth by targeting multiple
oncogenes. Cancer Res. 69:7569–7576. 2009.
|
|
16
|
Mendrzyk F, Radlwimmer B, Joos S, et al:
Genomic and protein expression profiling identifies CDK6 as novel
independent prognostic marker in medulloblastoma. J Clin Oncol.
23:8853–8862. 2005.
|
|
17
|
Easton J, Wei T, Lahti JM and Kidd VJ:
Disruption of the cyclin D/cyclin-dependent
kinase/INK4/retinoblastoma protein regulatory pathway in human
neuroblastoma. Cancer Res. 58:2624–2632. 1998.
|
|
18
|
Bredel M and Zentner J: Brain-tumour drug
resistance: the bare essentials. Lancet Oncol. 3:397–406. 2002.
|
|
19
|
Feun LG, Savaraj N and Landy HJ: Drug
resistance in brain tumors. J Neurooncol. 20:165–176. 1994.
|
|
20
|
Bredel M: Anticancer drug resistance in
primary human brain tumors. Brain Res Rev. 35:161–204. 2001.
|
|
21
|
Declèves X, Fajac A, Lehmann-Che J, et al:
Molecular and functional MDR1-Pgp and MRPs expression in human
glioblastoma multiforme cell lines. Int J Cancer. 98:173–180.
2002.
|
|
22
|
Haga S, Hinoshita E, Ikezaki K, et al:
Involvement of the multidrug resistance protein 3 in drug
sensitivity and its expression inhuman glioma. Cancer Sci.
92:211–219. 2001.
|
|
23
|
de Murcia JM, Niedergang C, Trucco C, et
al: Requirement of poly(ADP-ribose) polymerase in recovery from DNA
damage in mice and in cells. Proc Natl Acad Sci USA. 94:7303–7307.
1997.
|
|
24
|
Cheng CL, Johnson SP, Keir ST, et al:
Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide
resistance in a DNA mismatch repair-deficient malignant glioma
xenograft. Mol Cancer Ther. 4:1364–1368. 2005.
|
|
25
|
Rhee CH, Hess K, Jabbur J, Ruiz M, Yang Y,
Chen S, Chenchik A, Fuller GN and Zhang W: cDNA expression array
reveals heterogeneous gene expression profiles in three
glioblastoma cell lines. Oncogene. 18:2711–2717. 1999.
|
|
26
|
Johnson DG, Schwarz JK, Cress WD and
Nevins JR: Expression of transcription factor E2F1 induces
quiescent cells to enter S phase. Nature. 365:349–352. 1993.
|
|
27
|
Chen H-Z, Tsai S-Y and Leone G: Emerging
roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009.
|