Introduction
Bile acids are amphipathic molecules that are
synthesized from cholesterol in the liver. They are essential to
the digestion and absorption of lipids, but high concentration of
bile acids exert pathological activities in hepatic and colorectal
tissues (1). The hydrophobicity of
bile acids seems to be closely linked to their pathological
activities, and highly hydrophobic bile acids, such as deoxycholic
acid (DCA), are potent apoptotic inducers and have been identified
as tumor promoters (2). Several
studies have demonstrated that bile acid-mediated hepatic injury is
mainly due to hepatocellular apoptosis and colonic carcinogenesis
caused by alterations in cell signaling and gene expression. In
contrast, less hydrophobic (hydrophilic) bile acids such as
ursodeoxycholic acid (UDCA) possess an opposite activity against
hydrophobic bile acids. UDCA relieves cholestatic liver diseases by
exerting cytoprotective and anti-apoptotic activities in
hepatocytes, and it is implicated in the prevention of colonic
cancer through cell cycle arrest and suppression of oncogenic
factors including Ras and COX-2 (3,4).
Bile acids are known to induce both apoptotic and
survival mechanisms in parallel (5,6), and
the regulatory mechanism components governing cell death and
survival include death receptor signaling, epidermal growth factor
receptors (EGFR) and mitogen-activated protein kinases (MAPKs)
(2,7). Death receptor-mediated apoptosis is
controlled by membrane translocation of Fas/CD95 and overexpression
of TNF-related apoptosis-inducing ligand receptor 2 (TRAIL-R2/DR5)
(2,7–9).
Several lines of research have reported the importance of
TRAIL-R2/DR5 induction in bile acid-triggered apoptosis (7,10).
MAPKs are well understood enzymes that play critical roles in
various cellular responses including cell growth, differentiation
and apoptosis.
MAPKs belong to an evolutionarily conserved family
of enzymes that includes three subfamilies: extracellular
signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK1/2) and
p38 MAPK. In hepatocytes, prolonged activation of JNK1/2 or p38
MAPK promotes bile acid-induced apoptosis, whereas ERK1/2 are
mainly involved in the cell survival pathway and their inhibition
enhances bile acid-induced apoptosis (5,11,12).
Hydrophobic bile acid, DCA, and hydrophilic bile acid, UDCA, have
both been shown to activate the ERK pathway, and bile acid-mediated
ERK activation seems to be essential in the cytoprotective pathway
that prevents liver damage. However, when it comes to cancer cells,
hydrophobic bile acid-induced ERK activation seems to be oncogenic
because the ERK pathway has been observed to be involved in COX-2
expression in esophageal cancer cells, it increases the
invasiveness of colon cancer cells and chemoresistance in
hepatocellular carcinoma cells, and it suppresses apoptosis in
colon cancer cells (13–15). Indeed, deregulation of the ERK
pathway has often been correlated with the malignant progression of
human cancers (16,17).
In our previous study, we observed that UDCA
performs a tumor preventing role in gastric carcinoma cells
(18). However, the role of bile
acids in the ERK pathway of gastric cancer cells remains unclear.
In this study, we explored the effect of UDCA on the ERK pathway
and found that the pro-apoptotic ERK pathway is activated in SNU601
gastric cancer cells. However, DCA-mediated ERK activation exerted
an anti-apoptotic activity in this cell line, and this finding may
point to one of the possible mechanisms of the anti-tumor effect of
UDCA in gastric cancer cells.
Materials and methods
Cell culture and dosing
The SNU601 human gastric cancer cell line was
obtained from the Korea Cell Line Bank and grown in RPMI-1640
medium (Invitrogen) supplemented with 10% (v/v) fetal bovine serum
and 1% antibiotics at 37°C in a 5% CO2 atmosphere.
Dosing of the cells was performed by adding 600–1000 μM UDCA (ICN
Biomedicals) or 300 μM DCA (Calbiochem) to the culture medium and
incubation for 48 h, unless otherwise specified. Cells were
pretreated for 1 h with 30 μM of a MEK1 inhibitor (PD98059), 10 μM
MEK1/2 inhibitor (U0126), 10 μM EGFR inhibitor (AG1478), 5 mM
N-acetyl cysteine, 100 μM BHA (butylated hydroxyanisole) or 1 mM
methyl-β-cyclodextrin (MBCD).
Apoptosis measurement
Treated cells were stained with 1 μg/ml Hoechst
33342 (HO) for 15 min at room temperature in the dark. Then, both
the floating and attached cells were collected and centrifuged. The
pooled cell pellets were washed with ice-cold phosphate-buffered
saline (PBS), fixed in 3.7% formaldehyde on ice, washed again with
PBS, resuspended, and then a fraction of the suspension was
centrifuged in a cytospinner (Thermo Shandon). Slides were
prepared, air-dried, mounted in anti-fade solution and observed
under a fluorescence microscope (DM5000, Leica) as described
elsewhere (19). Any
condensed/fragmented nuclei were assessed as apoptotic cells. A
total of 500 cells from randomly chosen microscope viewing fields
were counted and the number of apoptotic cells was expressed as a
percentage of the total number of cells counted.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
viability assays
For performance of the MTT assay, cells were plated
in the wells of a 96-well plate at a density of 1×104
cells/well, incubated for 24 h and then treated with drugs for 48
h. The MTT solution (0.5 mg/ml) was added to the wells and
incubated for 4 h. The plates were centrifuged at 600 g for 10 min,
and then the culture medium was removed. The cells were solubilized
using dimethyl sulfoxide (DMSO) and the solubilized formazan
product was quantified using an enzyme-linked immunosorbent assay
(ELISA) plate reader at 595 nm. The absorbance of the untreated
cells was designated as 100% and the cell survival was expressed as
a percentage of this value.
Immunoblotting
Using a standard technique, equal amounts of protein
were electrophoretically separated using SDS-PAGE and then
transferred to a nitrocellulose membrane. Antibodies were used to
probe for active caspase-6, −3, phospho-p38, p38, phosphor-MEK1/2,
MEK1/2 (Cell Signaling Technology), PARP, phospho-ERK1/2, ERK2,
phosphor-JNK1/2, JNK1/2, α-tubulin (Santa Cruz) and DR5 (Pro Sci).
An image analyzer (Image Station 4000MM, Kodak) was used for
acquisition of the probe signals.
Caspase-8 activity assay
According to the manufacturer’s protocol, a
FADD-like IL-1β-converting enzyme (FLICE) colorimetric assay kit
(BioVision) was used to perform the caspase-8 activity assay.
Briefly, 200 μg of protein lysates in a 50-μl volume was mixed with
reaction buffer, mixed with IETD-pNA substrate, and then incubated
for 90 min. The resulting absorbance was measured at a wavelength
of 405 nm. Fold increase in FLICE activity was determined by
comparison of the results of the treated samples with the level of
the untreated control.
RNA interference (RNAi)
For the RNAi experiment, siRNA of ERK1, 5′-CUC UCU
AAC CGG CCC AUC U(dTdT)-3′ (S) and 5′-AGA UGG GCC GGU UAG AGA
G(dTdT)-3′ (AS), ERK2, 5′-CAC CAU UCA AGU UCG ACA U(dTdT)-3′ (S)
and 5′-AUG UCG AAC UUG AAU GGU G(dTdT)-3′ (AS), EGFR, 5′-GAU CCA
CAG GAA CUG GAU A(dTdT)-3′ (S) and 5′-UAU CCA GUU CCU GUG GAU
C(dTdT)-3′ (AS), and control siRNA, 5′-CCUACGCCACCAAUUUCGU(dTdT)-3′
(S) and 5′-ACGAAAUUGGUGGCGUAGG(dTdT)-3′ (AS) were purchased from
Bioneer (Daejeon, Korea). Using an Amaxa transfection kit, cells
(106) were transfected with 5~8 μg siRNA, and the
transfected cells were then stabilized for 24 h prior to
dosing.
Results and Discussion
UDCA induces pro-apoptotic ERK1/2
activation in SNU601 cells
Generally, the bile acid-induced ERK pathway in
gastrointestinal cancer stimulates cell proliferation, inhibits
apoptosis and causes chemoresistance, as observed in other human
carcinomas (5,14,15).
However, the precise role of hydrophilic bile acid in the ERK
pathway in gastric carcinoma cells remains unclear. In this study,
we examined the role of the ERK pathway in UDCA-induced apoptosis
of the SNU601 gastric carcinoma cell line. First, we examined the
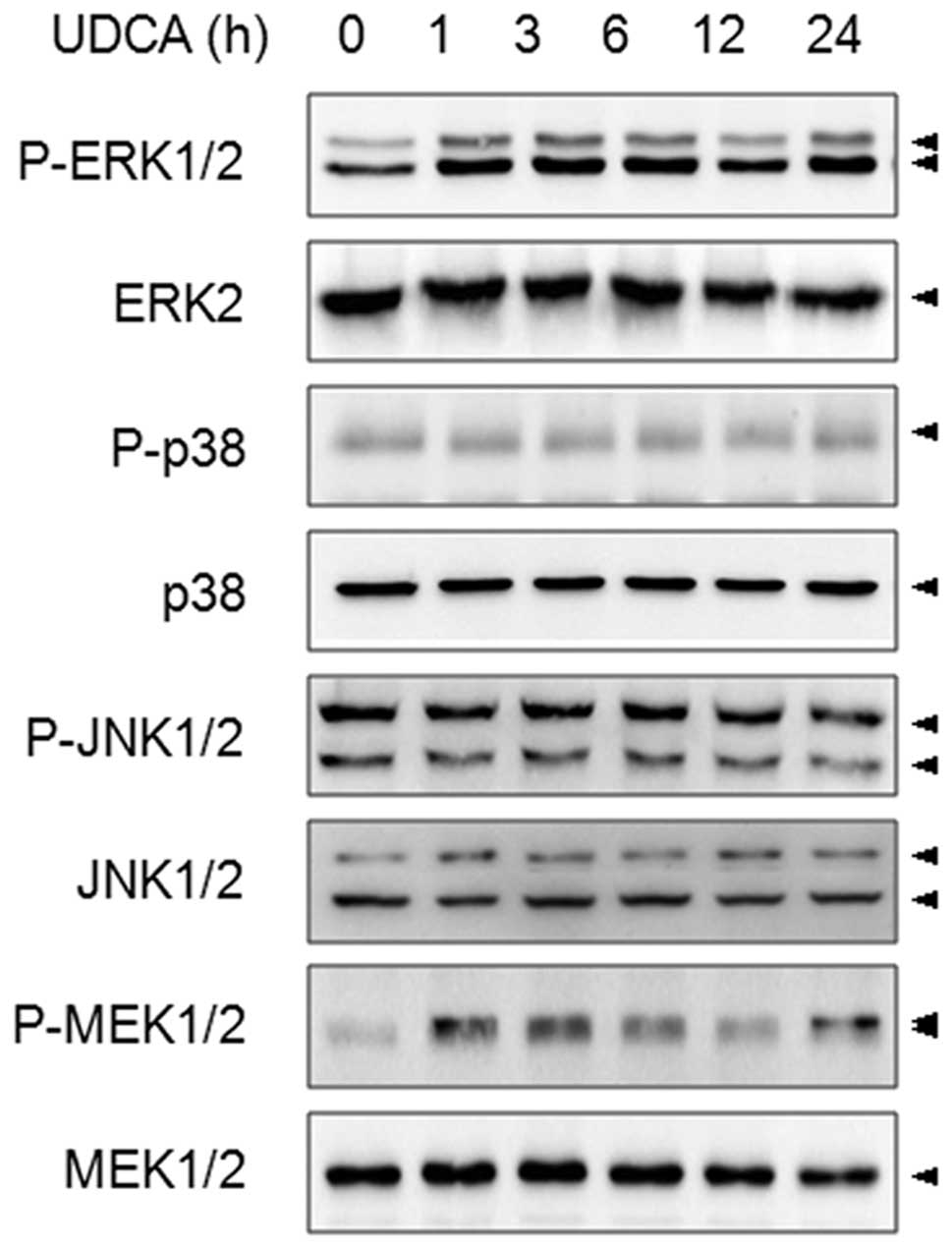
effect of UDCA on the MAPK family members. SNU601 cells were
exposed to 600 μM UDCA for various time intervals, and as active
MAPKs can be estimated by measuring the appearance of
phosphorylated forms of MAPKs, the phosphorylation patterns of
ERK1/2, p38, JNK1/2 and MEK1/2 were determined. As shown in
Fig. 1, treatment by UDCA increased
the phosphorylation levels of ERK1/2 and MEK1/2, but had no effect
on p38 and JNK1/2 in SNU601 cells. In order to elucidate the effect
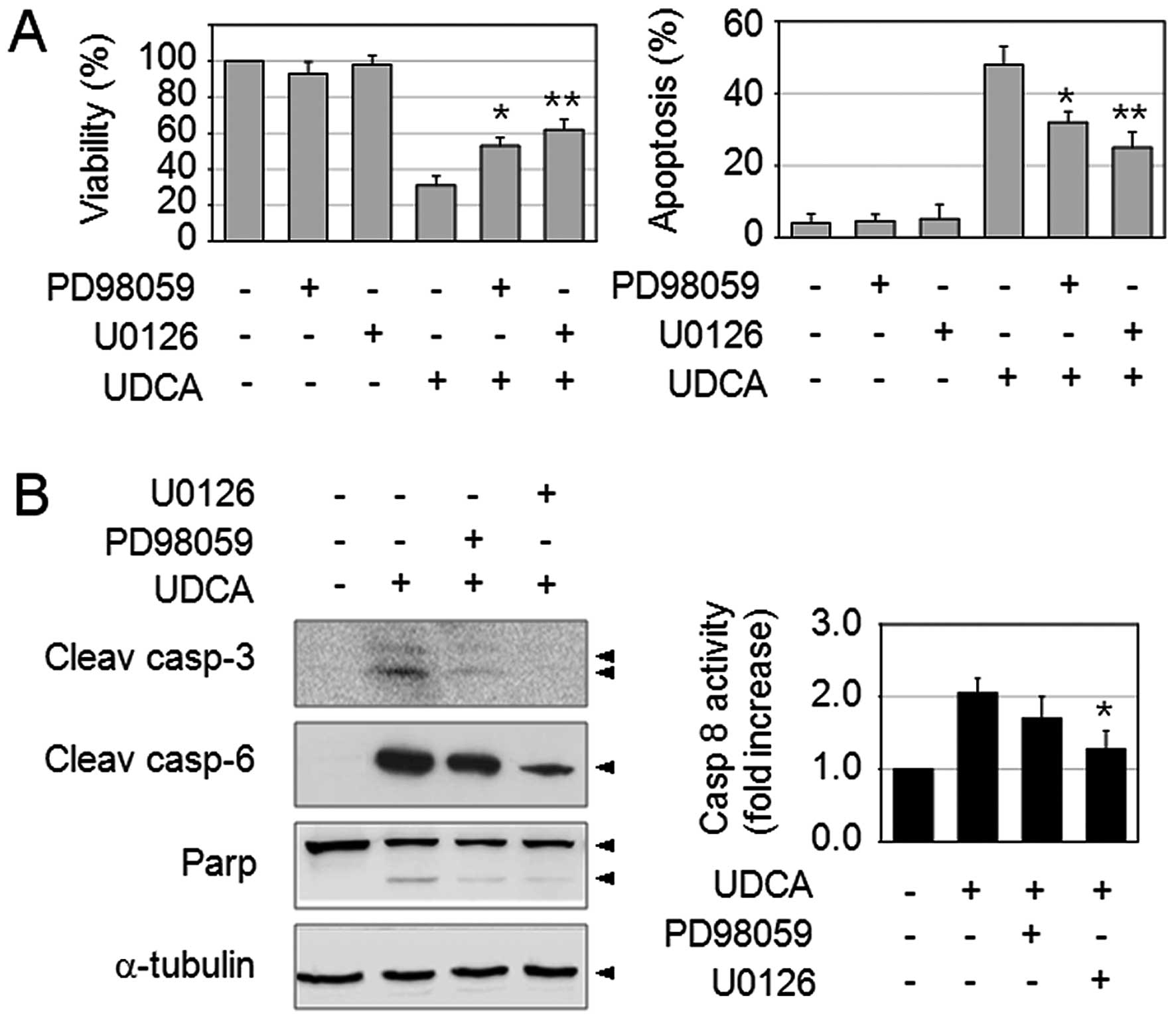
of activation of the ERK pathway upon exposure to UDCA, the SNU601
cells were pre-incubated with 30 μM PD98059 (MEK1 inhibitor) or 10
μM U0126 (MEK1/2 inhibitor) for 1 h, and then further exposed to
UDCA for 48 h. As reported above, UDCA significantly reduced cell
viability and increased apoptosis in SNU601 cells. Interestingly,
combined treatment of UDCA with MEK inhibitors partially enabled
the recovery of cell viability and reduced the amount of apoptosis
(Fig. 2A). In addition, as detected
by immunoblotting and FLICE-like enzyme activity assay, MEK
inhibitors also reduced the quantity of the active form of
caspase-3, −6 and resulting PARP cleavage, as well as caspase-8
activity (Fig. 2B). These results
indicated that UDCA-induced ERK activation plays a pro-apoptotic
role in SNU601 cells. Although it plays an anti-apoptotic role in
general, recent studies have shown several exceptional roles for
ERK. α-Tocopheryl succinate-induced apoptosis has been reported to
be modulated by ERK1/2 in gastric cancer cells (20). In addition, ERK has been shown to
play a pro-apoptotic function upon exposure to cisplatin in
multiple cancer cells including cervical carcinoma, osteosarcoma,
neuroblastoma and myeloid leukemia (21–25).
Hence, various cancer cells may be able to use the ERK pathway to
mediate a pro-apoptotic signal under certain conditions.
The ERK pathway is involved in
UDCA-induced DR5 overexpression
In our previous study, we found that DR5
overexpression was largely responsible for the UDCA-induced
apoptosis in gastric cancer cells (18). Although DR5 induction was shown to
be regulated by PKCδ activation (18), we questioned whether or not the ERK
pathway is also connected to UDCA-induced DR5 expression signaling.
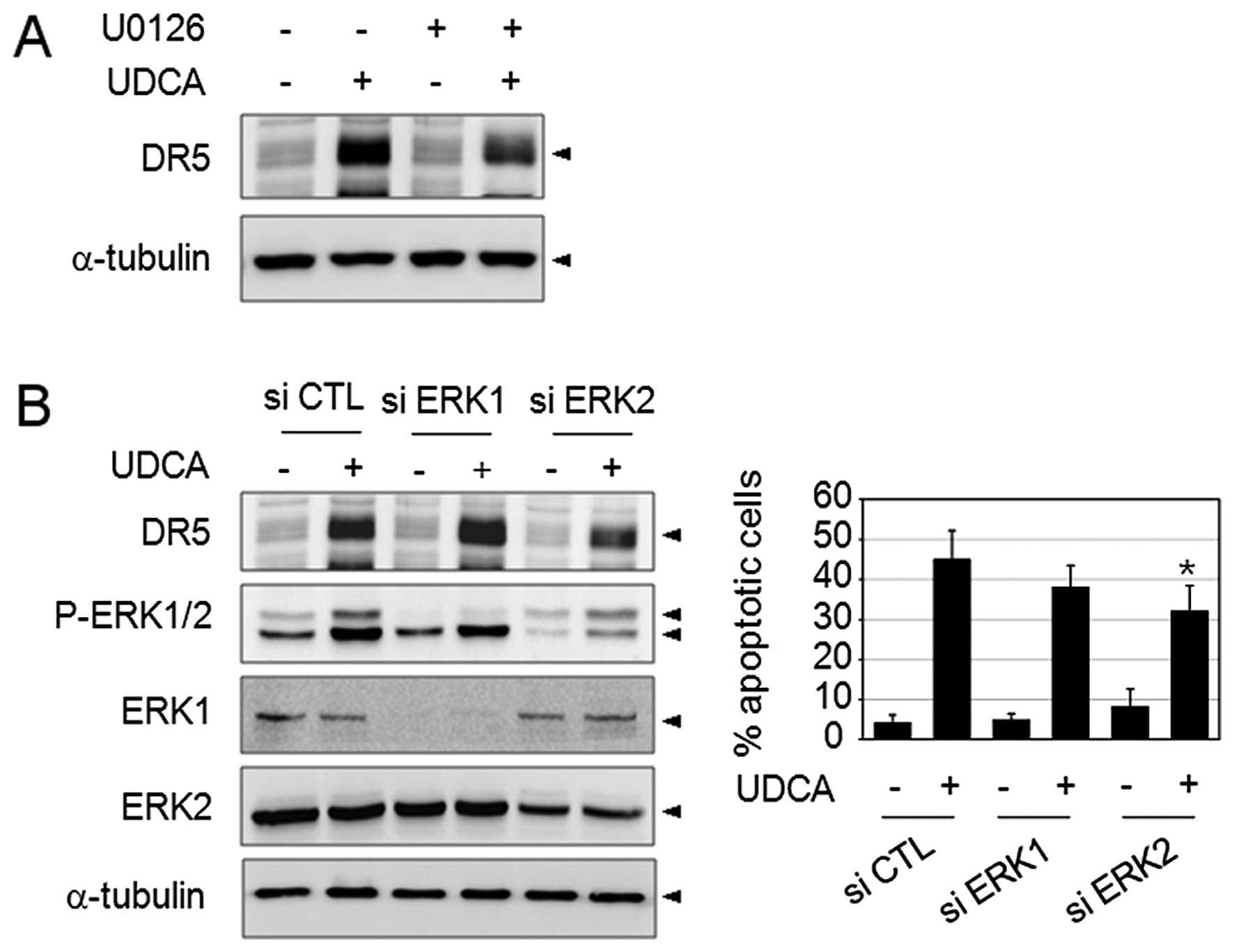
To this end, we assessed the DR5 expression level under suppression
of ERK activation. SNU601 cells were treated with UDCA in the
absence or presence of 10 μM U0126 (MEK1/2 inhibitor) for 24 h, and
then analyzed by immunoblotting using anti-DR5 antibody. As shown
in Fig. 3A, treatment by UDCA
highly increased the expression of DR5, and treatment in
combination with U0126 partially decreased the DR5 expression level
as compared to UDCA-only treated samples. This result suggested the
partial involvement of the ERK pathway in the UDCA-induced DR5
expression pathway. Then, in order to confirm the role of ERK in
UDCA-induced apoptosis, we silenced ERK expression using siRNA
specific to ERK1 and ERK2, and examined its effects on UDCA-induced
DR5 expression and apoptosis. The result of the silencing effect in
the reduction of ERK1 and ERK2 protein levels was confirmed by
immunoblotting. Transfection with siRNA targeting ERK1 appeared to
slightly reduce the number of apoptotic cells, but the results were
statistically insignificant and did not affect the DR5 expression
level as compared with the control siRNA. However, siRNA targeting
ERK2 reduced the UDCA-induced DR5 expression level and
significantly decreased the apoptotic cell rate. These results
suggested that ERK2 activity may be linked to the pro-apoptotic
signaling that contributes to DR5 upregulation in response to UDCA
exposure.
ERK phosphorylation is lipid
raft-dependently controlled
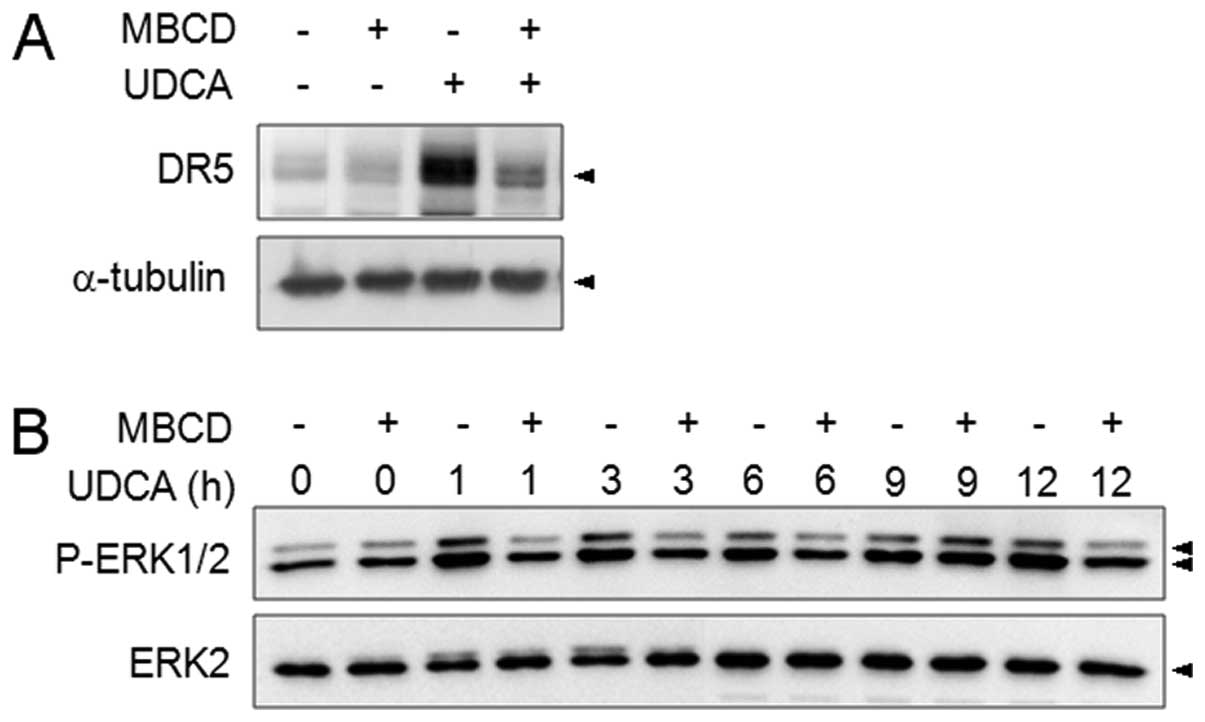
In order to examine the upstream regulation of the
MEK/ERK pathway, we assessed the role of lipid rafts in ERK
phosphorylation. Previously, PKCδ-mediated DR5 expression was shown
to be controlled by lipid rafts upon exposure to UDCA (18). The role of lipid rafts in DR5
induction was reconfirmed using lipid raft disrupting agent, MBCD
(Fig. 4A) and MBCD clearly reduced
UDCA-induced ERK phosphorylation at all measured time-points.
However, the suppression of PKCδ did not affect ERK activation and
vice versa (data not shown). These results indicated that the ERK
pathway is also lipid raft-regulated but that it is a
PKCδ-independently activated pathway. We further explored whether
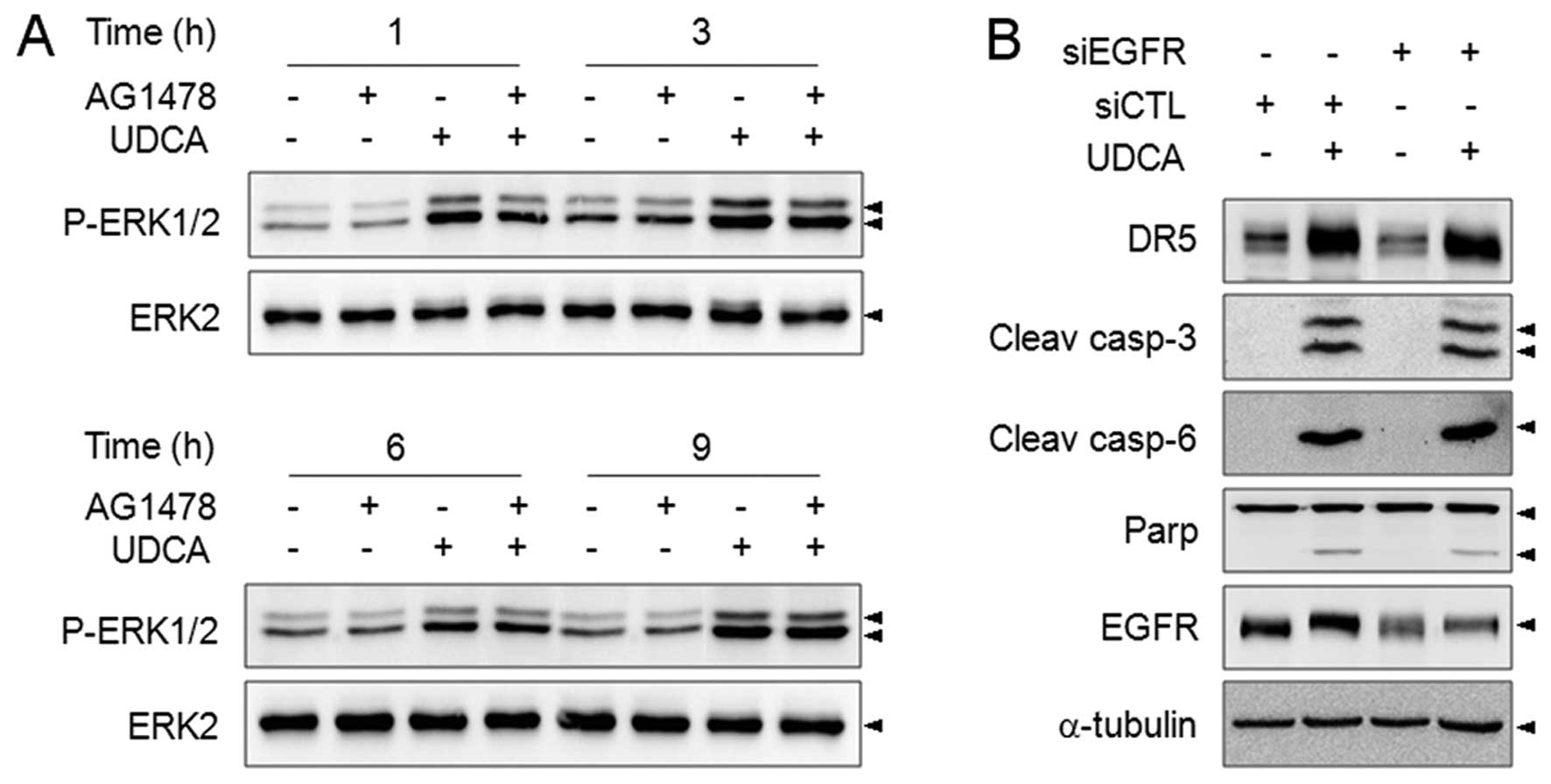
or not EGFR is involved in UDCA-induced ERK activation. The role of
EGFR signaling has been implicated in bile acid-induced ERK
activation in hepatocytes, but UDCA did not induce EGFR activation
in colon cancer cells (26). A
specific inhibitor of EGFR, AG1478 scarcely affected UDCA-induced
ERK phosphorylation (Fig. 5A).
Furthermore, although the interference of EGFR expression was
confirmed, silencing of the expression of EGFR by specific siRNA
did not alter the DR5 protein level, or cleavage of caspase-3, −6
and PARP in response to UDCA (Fig.
5B). Therefore, UDCA-triggered ERK activation may not be
regulated by the EGFR pathway in SNU601 cells.
DCA-induced ERK activation plays an
anti-apoptotic role
Tumor promoting hydrophobic bile acids such as DCA
have also been reported to activate the ERK pathway in hepatocytes
and colon cancer cells. The hydrophobic bile acid-induced ERK
pathway has been suggested to be associated with various tumor
promoting properties in cancer cells. Therefore, we questioned
whether or not hydrophobic bile acid DCA can also trigger ERK
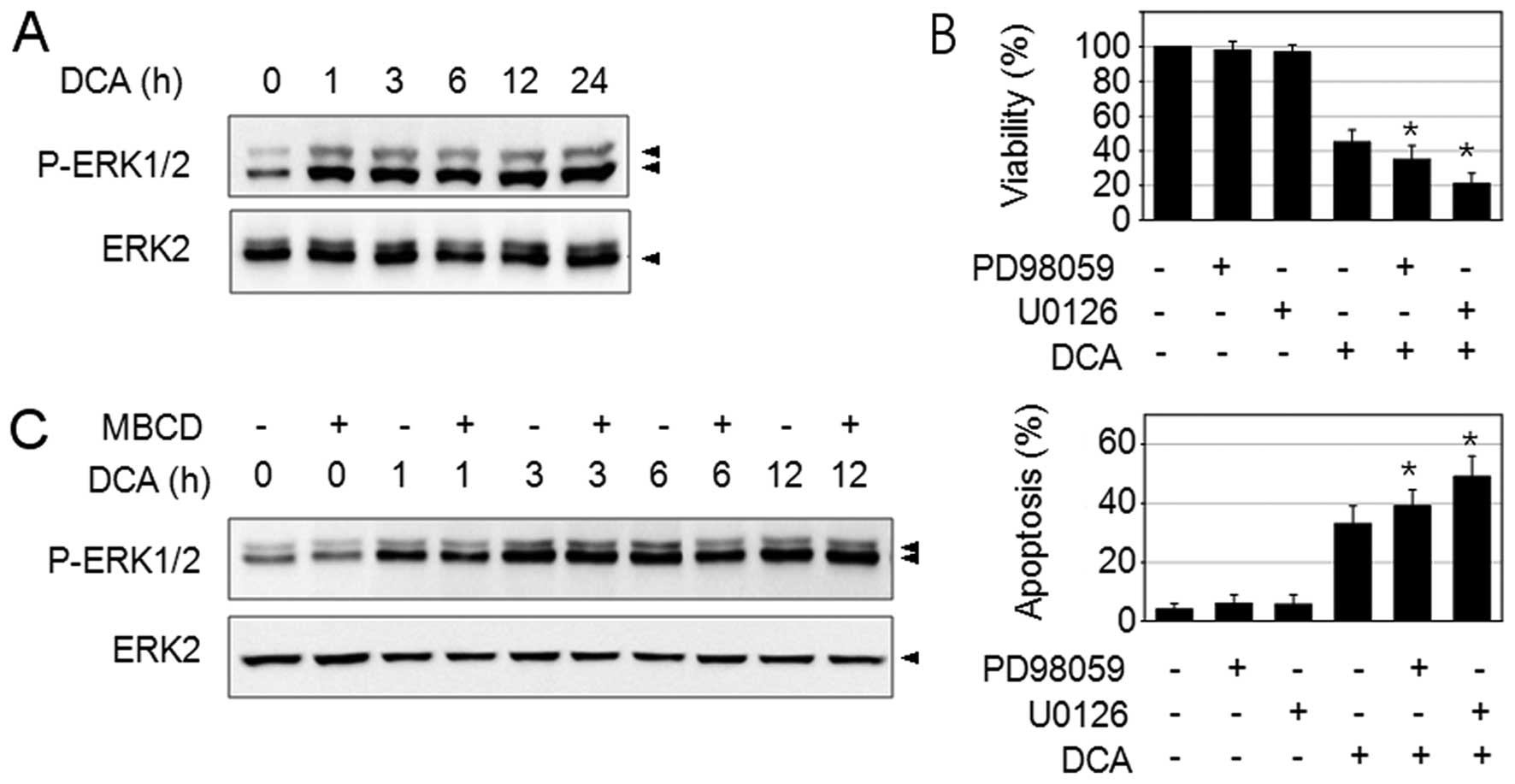
activation in SNU601 cells. DCA treatment strongly increased ERK1/2
phosphorylation as shown in Fig.
6A. Then, we examined the role of DCA-induced ERK activation in
SNU601 cells. When cells were treated with DCA their viability was
reduced and apoptosis was observed. The combination of DCA with MEK
inhibitors, 30 μM of PD98059 or 10 μM of U0126, further reduced
cell viability and increased apoptosis (Fig. 6B). These results agreed with
previous reports in which ERK was observed to play an
anti-apoptotic role in DCA-induced apoptosis (5). Therefore, the results obtained above
indicated that the DCA-induced ERK pathway exerts an opposite
activity from which it was induced by UDCA in SNU601 gastric cancer
cells. Next, we examined if DCA-induced ERK activation requires
lipid rafts or not. We found that combined treatment of MBCD with
DCA did not alter the ERK phosphorylation level as compared to the
DCA treated samples (Fig. 6C),
indicating that DCA-induced ERK activation is not mediated by lipid
rafts. Therefore, UDCA and DCA seem to regulate ERK activation via
a differential signaling mechanism and lead to opposite responses
through the ERK signaling molecule.
Previously, we hypothesized UDCA anti-tumor activity
by demonstrating that UDCA induces apoptosis of gastric cancer
cells. We found that UDCA triggered DR5 overexpression through
lipid rafts, ROS, and in a PKCδ-dependent manner, and the
overexpressed DR5 was translocated to the lipid raft region and
recruited by DISC proteins to initiate caspase-8 activation
(18). However, tumor-promoting
hydrophobic bile acids are also strong apoptotic inducers and it
was observed that DCA induced apoptosis as well as necrosis in
gastric cancer cells (18). In
hepatocytes, the cytoprotective and tumor preventing features of
UDCA that distinguishes it from other hydrophobic bile acids is
often assumed to be the result of its mild hydrophobicity, because
strong hydrophobicity will destroy membrane structures by detergent
effects. Nevertheless, we found that the ERK pathway triggered by
UDCA and DCA played an opposite role in the viability of SNU601
cells. This result indicated that, more than simply having a milder
effect as compared to DCA, UDCA may possess tumor-preventing
activities by inducing differential responses. Indeed, strong
hydrophobic bile acid-induced ERK activity has been reported to be
linked with various tumor-promoting functions. Lithocholic acid
(LCA) has been shown to induce expression of urokinase-type
plasminogen activator receptor (uPAR) and enhances cell
invasiveness in colon cancer cells (14). Furthermore, the ERK pathway is
involved in DCA-upregulated mucin gene transcription, which is
often implicated in colon neoplasia (27). Our finding suggested that UDCA and
DCA trigger differential responses in cancer cells using ERK. Thus,
UDCA exposure can produce an anti-tumor effect.
Acknowledgements
This study was supported by a National Research
Foundation of Korea (NRF) grant funded by the Korean government
(MEST) (2009-0075493) and through the Research Center for Resistant
Cells (R13-2003-009).
References
|
1
|
Greim H, Trulzsch D, Czygan P, Rudick J,
Hutterer F, Schaffner F and Popper H: Mechanism of cholestasis. 6.
Bile acids in human livers with or without biliary obstruction.
Gastroenterology. 63:846–850. 1972.PubMed/NCBI
|
|
2
|
Qiao L, Studer E, Leach K, McKinstry R,
Gupta S, Decker R, Kukreja R, Valerie K, Nagarkatti P, El Deiry W,
et al: Deoxycholic acid (DCA) causes ligand-independent activation
of epidermal growth factor receptor (EGFR) and FAS receptor in
primary hepatocytes: inhibition of EGFR/mitogen-activated protein
kinase-signaling module enhances DCA-induced apoptosis. Mol Biol
Cell. 12:2629–2645. 2001. View Article : Google Scholar
|
|
3
|
Loddenkemper C, Keller S, Hanski ML, Cao
M, Jahreis G, Stein H, Zeitz M and Hanski C: Prevention of
colitis-associated carcinogenesis in a mouse model by diet
supplementation with ursodeoxycholic acid. Int J Cancer.
118:2750–2757. 2006. View Article : Google Scholar
|
|
4
|
Alberts DS, Martinez ME, Hess LM, Einspahr
JG, Green SB, Bhattacharyya AK, Guillen J, Krutzsch M, Batta AK,
Salen G, et al: Phase III trial of ursodeoxycholic acid to prevent
colorectal adenoma recurrence. J Natl Cancer Inst. 97:846–853.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qiao D, Stratagouleas ED and Martinez JD:
Activation and role of mitogen-activated protein kinases in
deoxycholic acid-induced apoptosis. Carcinogenesis. 22:35–41. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rust C, Karnitz LM, Paya CV, Moscat J,
Simari RD and Gores GJ: The bile acid taurochenodeoxycholate
activates a phosphatidylinositol 3-kinase-dependent survival
signaling cascade. J Biol Chem. 275:20210–20216. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Higuchi H and Gores GJ: Bile acid
regulation of hepatic physiology: IV. Bile acids and death
receptors. Am J Physiol Gastrointest Liver Physiol. 284:G734–G738.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Higuchi H, Grambihler A, Canbay A, Bronk
SF and Gores GJ: Bile acids up-regulate death receptor
5/TRAIL-receptor 2 expression via a c-Jun N-terminal
kinase-dependent pathway involving Sp1. J Biol Chem. 279:51–60.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Higuchi H, Bronk SF, Taniai M, Canbay A
and Gores GJ: Cholestasis increases tumor necrosis factor-related
apoptotis-inducing ligand (TRAIL)-R2/DR5 expression and sensitizes
the liver to TRAIL-mediated cytotoxicity. J Pharmacol Exp Ther.
303:461–467. 2002. View Article : Google Scholar
|
|
10
|
Higuchi H, Bronk SF, Takikawa Y, Werneburg
N, Takimoto R, El-Deiry W and Gores GJ: The bile acid
glycochenodeoxycholate induces trail-receptor 2/DR5 expression and
apoptosis. J Biol Chem. 276:38610–38618. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qiao L, Han SI, Fang Y, Park JS, Gupta S,
Gilfor D, Amorino G, Valerie K, Sealy L, Engelhardt JF, et al: Bile
acid regulation of C/EBPbeta, CREB, and c-Jun function, via the
extracellular signal-regulated kinase and c-Jun NH2-terminal kinase
pathways, modulates the apoptotic response of hepatocytes. Mol Cell
Biol. 23:3052–3066. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Looby E, Abdel-Latif MM, Athie-Morales V,
Duggan S, Long A and Kelleher D: Deoxycholate induces COX-2
expression via Erk1/2-, p38-MAPK and AP-1-dependent mechanisms in
esophageal cancer cells. BMC Cancer. 9:1902009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baek MK, Park JS, Park JH, Kim MH, Kim HD,
Bae WK, Chung IJ, Shin BA and Jung YD: Lithocholic acid upregulates
uPAR and cell invasiveness via MAPK and AP-1 signaling in colon
cancer cells. Cancer Lett. 290:123–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liao M, Zhao J, Wang T, Duan J, Zhang Y
and Deng X: Role of bile salt in regulating Mcl-1 phosphorylation
and chemoresistance in hepatocellular carcinoma cells. Mol Cancer.
10:442011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oka H, Chatani Y, Hoshino R, Ogawa O,
Kakehi Y, Terachi T, Okada Y, Kawaichi M, Kohno M and Yoshida O:
Constitutive activation of mitogen-activated protein (MAP) kinases
in human renal cell carcinoma. Cancer Res. 55:4182–4187.
1995.PubMed/NCBI
|
|
17
|
Sivaraman VS, Wang H, Nuovo GJ and Malbon
CC: Hyperexpression of mitogen-activated protein kinase in human
breast cancer. J Clin Invest. 99:1478–1483. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lim SC, Duong HQ, Choi JE, Lee TB, Kang
JH, Oh SH and Han SI: Lipid raft-dependent death receptor 5 (DR5)
expression and activation are critical for ursodeoxycholic
acid-induced apoptosis in gastric cancer cells. Carcinogenesis.
32:723–731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lim SC, Choi JE, Kang HS and Si H:
Ursodeoxycholic acid switches oxaliplatin-induced necrosis to
apoptosis by inhibiting reactive oxygen species production and
activating p53-caspase 8 pathway in HepG2 hepatocellular carcinoma.
Int J Cancer. 126:1582–1595. 2010.
|
|
20
|
Zhao Y, Zhao X, Yang B, Neuzil J and Wu K:
alpha-Tocopheryl succinate-induced apoptosis in human gastric
cancer cells is modulated by ERK1/2 and c-Jun N-terminal kinase in
a biphasic manner. Cancer Lett. 247:345–352. 2007. View Article : Google Scholar
|
|
21
|
Schweyer S, Soruri A, Meschter O, Heintze
A, Zschunke F, Miosge N, Thelen P, Schlott T, Radzun HJ and Fayyazi
A: Cisplatin-induced apoptosis in human malignant testicular germ
cell lines depends on MEK/ERK activation. Br J Cancer. 91:589–598.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh S, Upadhyay AK, Ajay AK and Bhat MK:
p53 regulates ERK activation in carboplatin induced apoptosis in
cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett.
581:289–295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar
|
|
24
|
Woessmann W, Chen X and Borkhardt A:
Ras-mediated activation of ERK by cisplatin induces cell death
independently of p53 in osteosarcoma and neuroblastoma cell lines.
Cancer Chemother Pharmacol. 50:397–404. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Amran D, Sancho P, Fernandez C, Esteban D,
Ramos AM, de Blas E, Gomez M, Palacios MA and Aller P:
Pharmacological inhibitors of extracellular signal-regulated
protein kinases attenuate the apoptotic action of cisplatin in
human myeloid leukemia cells via glutathione-independent reduction
in intracellular drug accumulation. Biochim Biophys Acta.
1743:269–279. 2005. View Article : Google Scholar
|
|
26
|
Im E and Martinez JD: Ursodeoxycholic acid
(UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via
modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells.
J Nutr. 134:483–486. 2004.PubMed/NCBI
|
|
27
|
Lee HY, Crawley S, Hokari R, Kwon S and
Kim YS: Bile acid regulates MUC2 transcription in colon cancer
cells via positive EGFR/PKC/Ras/ERK/CREB,
PI3K/Akt/IkappaB/NF-kappaB and p38/MSK1/CREB pathways and negative
JNK/c-Jun/AP-1 pathway. Int J Oncol. 36:941–953. 2010.PubMed/NCBI
|