Introduction
Ubiquitin is a highly conserved small eukaryotic
protein that is composed of 76 amino acids (1). The process of ubiquitination attaches
ubiquitin to other proteins, and this is achieved by the enzymes
E1, E2 and E3 ligase (1).
Ubiquitination is an important post-translational modification that
regulates protein qualities and quantities via proteasome-mediated
degradation or changes in protein subcellular localization
(2). Protein degradation through
the ubiquitin-proteasome system is the major pathway for
non-lysosomal proteolysis, and ubiquitin E3 ligases are responsible
for tagging target proteins for degradation (2). There are hundreds of E3 ligases
encoded in the human genome, and large studies regarding interplay
between specific E3 ligases and their target proteins have been
widely conducted. These investigations have demonstrated that
dysregulation of the ubiquitin-proteasome system with regard to E3
ligases and their target proteins contributed to several diseases,
such as cancer and neurodegenerative disorders (3). Therefore, understanding protein
degradation and specific E3 ligases that control the process
provides important insight into therapeutic approaches.
Two RING fingers and DRIL1 (TRIAD1) was originally
found to play roles in differentiation and apoptosis (4–6).
Previously, this protein was reported to be highly expressed during
monocyte differentiation (4) and
induced apoptosis in myeloid and cancer cell lines, including MCF7,
A549, U2OS, H1299 and HCT116 (5,6).
Moreover, the anticancer effect of TRIAD1 was mediated by p53
activation. Thus, it does not have any effect on p53-null cancer
cells, while TRIAD1 ablation attenuated p53 transactivation
(6). TRIAD1 was transcribed by
HoxA10 transcription factor, which had an anticancer effect in
acute myeloid leukemia (7).
Notably, HoxA10 upregulated p53 expression and decreased breast
cancer cell invasiveness (8).
Collectively, this evidence suggested that additional
investigations regarding TRIAD1 regulation would stimulate novel
cancer therapeutic research, the mechanism, however, remains to be
fully elucidated. In the present study, we demonstrated that murine
double minute 2 (MDM2), a well-known E3 ligase for p53, also
functioned as an ubiquitin E3 ligase for TRIAD1.
Materials and methods
Plasmids
Human TRIAD1 cDNA was cloned into pcDNA3.1-Myc/His
(Invitrogen, Carlsbad, CA, USA), pEGFP-C1 (Clontech, Mountain View,
CA, USA), and pGEX6p (Promega, Madison, WI, USA) vectors. Human
MDM2 cDNA was cloned into the pCMV-Tag2-FLAG (Stratagene, La Jolla,
CA, USA) vector. The RING domain-mutant MDM2 (MDM2-C438A) was
created using the QuikChange site-directed mutagenesis kit
(Stratagene), according to the manufacturer’s instructions. The
HA-ubiquitin (HA-Ub) plasmid pMT123 was kindly provided by Dr Dirk
Bohmann (University of Rochester, Rochester, NY, USA). MDM2 small
interfering RNA (siRNA) was purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA).
Antibodies and reagents
Antibodies against MDM2 (SMP14), TRIAD1 (F-23) and
ubiquitin (P4D1) were from Santa Cruz Biotechnology. Anti-β-actin
and anti-FLAG antibodies were from Sigma-Aldrich (St. Louis, MO,
USA), anti-green fluorescent protein (GFP) antibody was from Cell
Signaling Technology (Danvers, MA, USA) and GFP antibody for
immunoprecipitation was from Anaspec (Fremont, CA, USA). CPT,
etoposide, crystal violet, propidium iodine and MG132 were obtained
from Sigma-Aldrich. Cycloheximide (CHX) was purchased from Biopure
(Burlington, Ontario, Canada).
Cell culture and transfection
U2OS (KCLB no. 30096) cells were purchased from the
Korean Cell Line Bank (Seoul, Korea). The
p53−/−mdm2−/− MEF
cell line was a kind gift from Dr Wei Gu (Columbia University, New
York, NY, USA). p53−/−
mdm2−/− MEF cells were maintained in
Dulbecco’s minimum essential medium with 10% fetal bovine serum
(FBS) and 1% penicillin/streptomycin (all from Invitrogen). U2OS
cells were maintained in RPMI-1640 media (Invitrogen) with 10% FBS
and 1% penicillin/streptomycin. Transient transfections were
carried out using HilyMax (Dojindo Laboratories, Kumanoto,
Japan).
Ubiquitination assays.
p53−/−mdm2−/−
MEF cells were cotransfected with Myc-TRIAD1,
HA-ubiquitin, FLAG-MDM2 or the FLAG-MDM2 RING mutant for 48 h and
treated with 1 μM MG132 for 12 h. Cells were lysed with lysis
buffer (6 M guanidine-HCl, 0.1 M
Na2HPO4/NaH2PO4 and 10
mM imidazole), sonicated and incubated with nickel-nitrilotriacetic
acid (Ni-NTA, Qiagen) resin for 4 h. Bound pellets were washed 6
times, using washing buffer (25 mM Tris-HCl, pH 6.8 and 20 mM
imidazole). The precipitates were then analyzed by immunoblotting,
using an anti-HA (ubiquitin) antibody.
Immunoprecipitation and western
blotting
For the immunoprecipitation assay, cell lysates in
CHAPS buffer (0.5% CHAPS; 10 mM Tris-HCl, pH 7.5; 1 mM
MgCl2; 1 mM EGTA; 5 mM β-mercaptoethanol; 10% glycerol)
were incubated overnight with the indicated antibody with constant
rotation at 4˚C. Following the addition of protein A/G PLUS-agarose
beads (Santa Cruz Biotechnology), the cell lysates were incubated
for an additional 2 h followed by extensive washing. For the
immunoblotting assay, cells were lysed in RIPA buffer (10 mM
Tris-HCl, pH 7.4; 150 mM NaCl2; 1% NP-40; 1%
deoxycholate; and 0.1% SDS) containing a protease inhibitor
cocktail (Roche Applied Sciences, Indianapolis, IN, USA). Proteins
were electrophoresed and transferred onto a nitrocellulose
membrane, which was incubated overnight with each indicated
antibody at 4˚C. For protein detection, horseradish
peroxidase-conjugated anti-mouse or anti-rabbit antibodies (Cell
Signaling Technology) were used, followed by enhanced
chemiluminescence (ECL; Pierce, Thermo Fisher Scientific, Rockford,
IL, USA) and autoradiography.
Cell viability assays
The cell proliferation ratio was measured using the
3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium
inner salt (MTS) assay kit (Promega), according to the
manufacturer’s instructions. Briefly, cells were transfected with
GFP, GFP-TRIAD1 or FLAG-MDM2, plated onto 96-well culture plates at
a density of 1,000 cells/well and incubated for 48 h. The MTS/PMS
solution was added to each well and incubated at 37˚C for 1–2 h.
The absorbance was measured at 490 nm using a microplate reader.
The cell proliferation ratio relative to that of the control cells
was graphically represented.
Colony formation assays
Cells seeded on 6-well plates (at 4×104
cells/well) were transfected with GFP, GFP-TRIAD1, or FLAG-MDM2
plasmids. Subsequent to a 2-week incubation, colonies were fixed
and stained with 0.1% crystal violet.
Statistical analysis
Statistical analysis was performed using the
two-tailed Student’s t-test. p<0.05 was considered to indicate a
statistically significant difference.
Results
TRIAD1 ubiquitination and degradation is
induced by MDM2 E3 ligase
TRIAD1-mediated apoptosis has been previously
reported to be induced by p53 activation (6). Occasionally, proteins responsible for
p53 activation, such as RUNX3, ribosome protein L11 and p300, might
be targets for MDM2 E3 ligase (9–11).
Therefore, TRIAD1 was examined as a potential novel ubiquitination
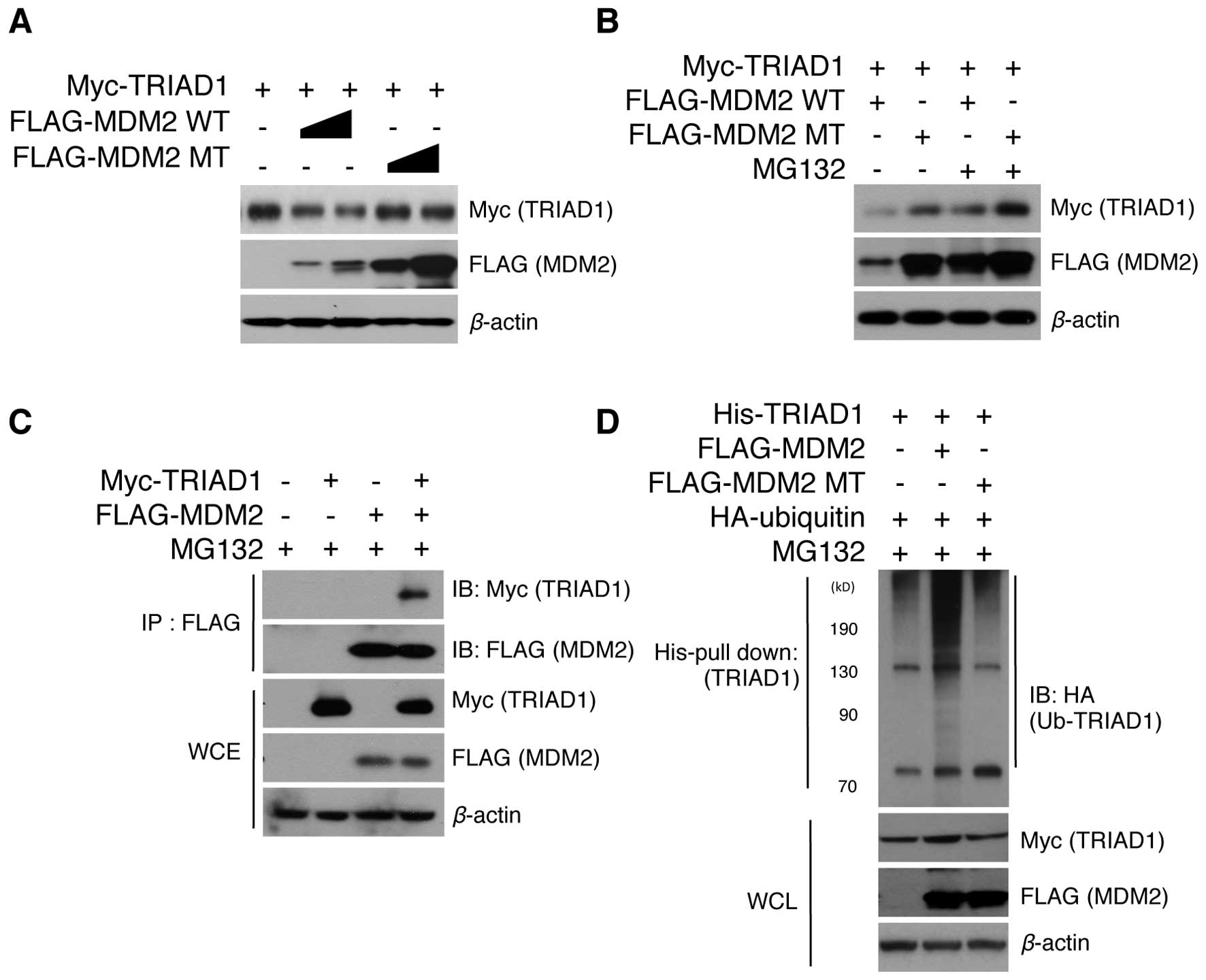
target for MDM2. To test this hypothesis, the protein level of
TRIAD1 upon MDM2 overexpression was initially examined. Notably,
MDM2 greatly decreased TRIAD1 protein levels in a MDM2
concentration-dependent manner (Fig.
1A). MDM2 lacking RING ligase activity was also found not to
have induced TRIAD1 degradation (Fig.
1A), indicating that this process was MDM2 E3 ligase
activity-dependent. These results led to our examining whether
MDM2-mediated TRIAD1 degradation was handled by the proteasomal
degradation system. The MDM2-mediated TRIAD1 decrease was found to
have markedly recovered when proteasomal function was inhibited
with MG132 (Fig. 1B), indicating
that MDM2 induces the proteasomal degradation of TRIAD1.
Furthermore, immunoprecipitation assays demonstrated an interaction
between TRIAD1 and MDM2 in cells (Fig.
1C), suggesting that TRIAD1 is a direct ubiquitination target
for MDM2 E3 ligase. We also assessed whether TRIAD1 is
ubiquitinated by MDM2. As expected, TRIAD1 was determined to have
been ubiquitinated by MDM2 via its RING ligase activity (Fig. 1D). These results indicated that
TRIAD1 was a novel target for MDM2 E3 ligase and was subsequently
degraded via ubiquitin-mediated proteolysis.
TRIAD1 protein stability is regulated by
MDM2 E3 ligase
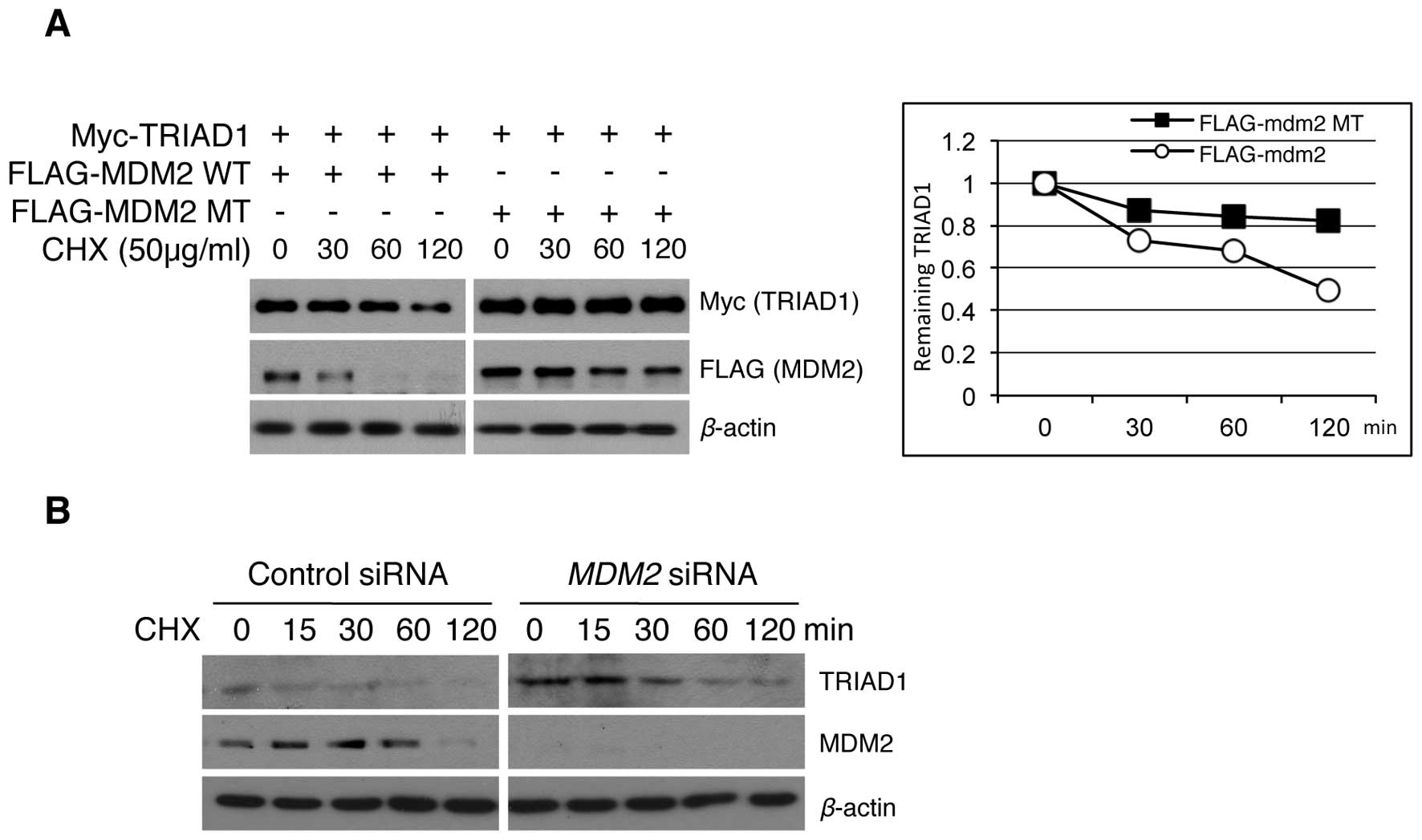
TRIAD1 cellular stability has not been evaluated
previously. The above results of MDM2-induced TRIAD1 degradation
led to our examining whether the regulation of exogenous and
endogenous TRIAD1 protein stability was a result of MDM2 E3 ligase
activity. The exogenous TRIAD1 stability in an MDM2 overexpression
system was examined and results showed that the half-life of the
TRIAD1 protein was decreased in an MDM2 E3 ligase
activity-dependent manner (Fig.
2A). Conversely, the stability of endogenous TRIAD1 was
increased following MDM2 knockdown (Fig. 2B). Collectively, these results
suggest that TRIAD1 was regulated by MDM2 E3 ligase in
vitro.
MDM2 attenuates TRIAD1-mediated cell
growth inhibition
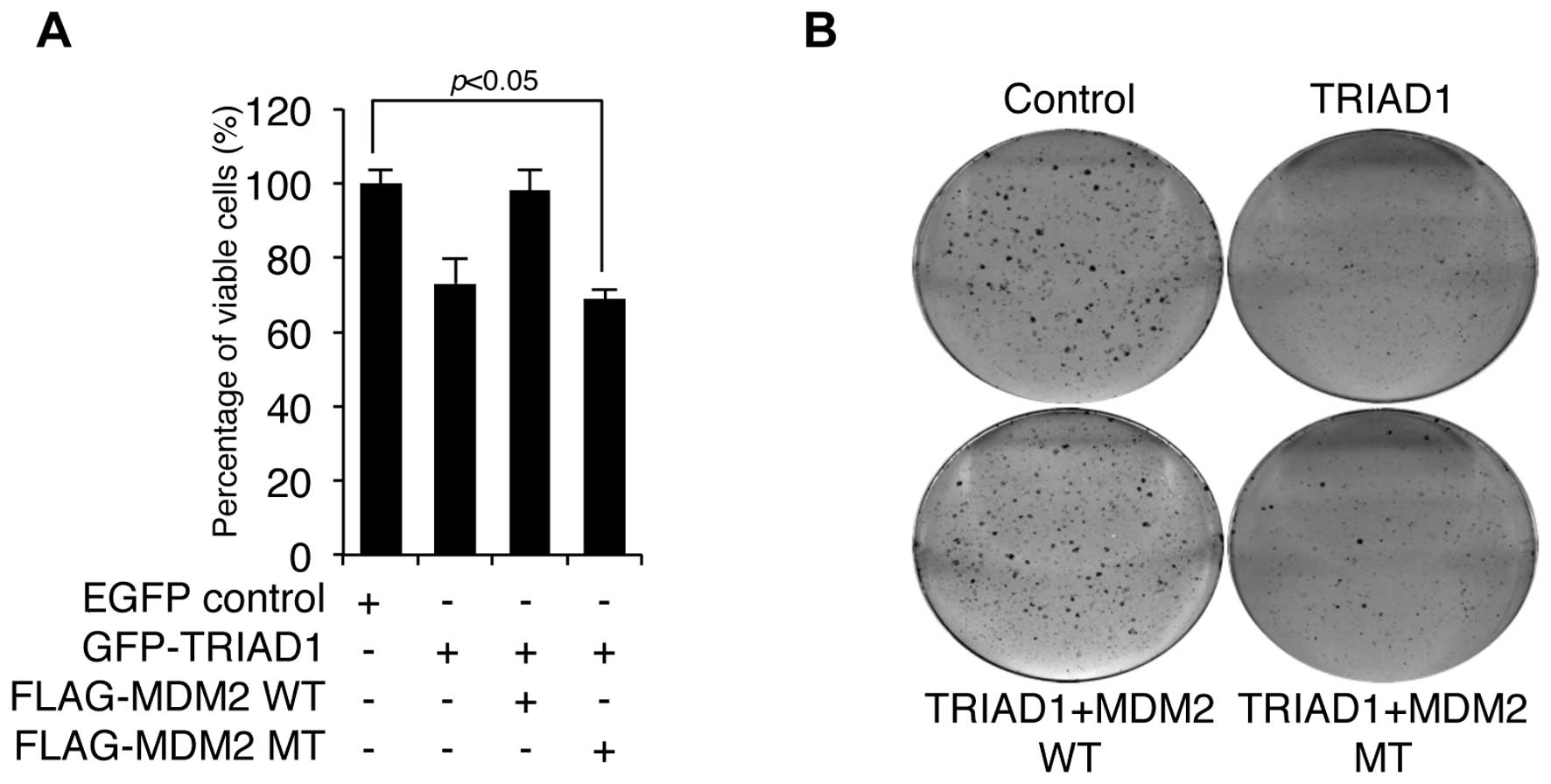
Since E3 ligase usually negatively regulates the
activity of its substrates (12),
we measured the cell growth following TRIAD1 transfection in the
presence or absence of wild-type (WT) or mutant (MT) MDM2 to test
whether MDM2 negatively regulated TRIAD1-mediated cell growth
inhibition. As shown in Fig. 3A,
TRIAD1-mediated cell growth inhibition was completely blocked by
WT, although not MT MDM2, which lacks RING ligase activity.
Furthermore, TRIAD1-mediated clonogenic growth inhibition was
reduced by MDM2 (Fig. 3B). These
results indicate that MDM2 negatively regulated TRIAD1 function via
its RING ligase activity.
Discussion
Results of the present study have demonstrated the
novel regulatory mechanism of the TRIAD1 protein by MDM2 E3 ligase.
TRIAD1 was found to have interacted with MDM2 in vivo.
Although the precise region responsible for the interaction was not
identified in this study, the results suggest that TRIAD1 is
potentially an ubiquitination substrate for MDM2. TRIAD1 was
ubiquitinated and targeted for proteasomal degradation in an
MDM2-dependent manner. Conversely, MDM2 siRNA increased the
stability of endogenous and exogenous TRIAD1. Cell proliferation
and colony-forming assays demonstrated that the antiproliferative
function of TRIAD1 was inhibited by MDM2. Overall, the above data
indicate that TRIAD1 function is regulated by an
ubiquitin-dependent proteasomal pathway through MDM2 E3 ligase.
Although TRIAD1 has been previously reported to have
induced apoptosis via p53 activation (6), the molecular interplay between TRIAD1
and MDM2 has been determined as non-p53 status-dependent, since, in
the present study, TRIAD1 was ubiquitinated and degraded by MDM2 in
p53-null MEF cells. However, the antiproliferative function of
TRIAD1 depends on the p53 status, since TRIAD1 overexpression in
p53-null MEF or p53-null HCT116 cells did not induce apoptosis
[data not shown; (6)]. Therefore,
although MDM2-induced TRIAD1 degradation was independent of p53,
the physiological role of TRIAD1-MDM2 is mediated by p53.
Furthermore, although MDM2 directly inhibits p53 stability and
activity (13), MDM2 is also
believed to negatively regulate p53 activation by inhibiting TRIAD1
for the following reasons: i) TRIAD1 induces apoptosis via p53
activation (6); ii) the ablation of
TRIAD1 attenuates p53 transactivation (6) and iii) TRIAD1 degradation by MDM2 is
independent of p53, while negative MDM2-induced TRIAD1 cellular
function regulation is p53-dependent. Thus, a balance between
TRIAD1 and the MDM2 expression may be the primary factor involved
in p53 regulation.
ARF is a tumor suppressor frequently deleted in
several tumors (14–16). Both ARF and TRIAD1 activate p53
(17). ARF induces MDM2 degradation
(18), whereas TRIAD1-mediated MDM2
regulation is not clearly understood. TRIAD1 and ARF may
synergistically mediate p53 activation, although no such mechanism
has been elucidated. However, if ARF were not required for
TRIAD1-mediated p53 activation, TRIAD1 would be a novel tumor
therapeutic target given that ARF is deleted in several types of
cancer (14–16).
Based on these results, a novel regulatory mechanism
of TRIAD1-MDM2 is proposed, whereby TRIAD1 is regulated by MDM2 via
ubiquitin-mediated proteolysis. The finding that TRIAD1 induces p53
activation emphasizes the importance of the development of
p53-mediated tumor therapeutic targets.
Acknowledgements
The authors would like to thank Dr B.A. van der
Reijden (Radboud University Nijmegen Medical Centre, The
Netherlands), for providing the GFP-TRIAD1 plasmids. The authors
are also grateful to the members of the research group, for their
support and suggestions regarding this study. The present study was
financed by the 2011 Konkuk University research support
program.
References
|
1
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar
|
|
2
|
Mani A and Gelmann EP: The
ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol.
23:4776–4789. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ardley HC and Robinson PA: E3 ubiquitin
ligases. Essays Biochem. 41:15–30. 2005. View Article : Google Scholar
|
|
4
|
van der Reijden BA, Erpelinck-Verschueren
CA, Lowenberg B and Jansen JH: TRIADs: a new class of proteins with
a novel cysteine-rich signature. Protein Sci. 8:1557–1561.
1999.PubMed/NCBI
|
|
5
|
Marteijn JA, van Emst L,
Erpelinck-Verschueren CA, et al: The E3 ubiquitin-protein ligase
Triad1 inhibits clonogenic growth of primary myeloid progenitor
cells. Blood. 106:4114–4123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung JH, Lee SM, Bae S, et al: Triad 1
induces apoptosis by p53 activation. FEBS lett. 584:1565–1570.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang H, Bei L, Shah CA, Horvath E and
Eklund EA: HoxA10 influences protein ubiquitination by activating
transcription of ARIH2, the gene encoding Triad1. J Biol Chem.
286:16832–16845. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chu MC, Selam FB and Taylor HS: HOXA10
regulates p53 expression and matrigel invasion in human breast
cancer cells. Cancer Biol Ther. 3:568–572. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chi XZ, Kim J, Lee YH, et al: Runt-related
transcription factor RUNX3 is a target of MDM2-mediated
ubiquitination. Cancer Res. 69:8111–8119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai MS, Shi D, Jin Y, et al: Regulation of
the MDM2-p53 pathway by ribosomal protein L11 involves a
post-ubiquitination mechanism. J Biol Chem. 281:24304–24313. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin Y, Zeng SX, Lee H and Lu H: MDM2
mediates p300/CREB-binding protein-associated factor ubiquitination
and degradation. J Biol Chem. 279:20035–20043. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leng RP, Lin Y, Ma W, et al: Pirh2, a
p53-induced ubiquitin-protein ligase, promotes p53 degradation.
Cell. 112:779–791. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Momand J, Zambetti GP, Olson DC, George D
and Levine AJ: The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell.
69:1237–1245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Faderl S, Kantarjian HM, Estey E, et al:
The prognostic significance of p16(INK4a)/p14(ARF) locus deletion
and MDM-2 protein expression in adult acute myelogenous leukemia.
Cancer. 89:1976–1982. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fulci G, Labuhn M, Maier D, et al: p53
gene mutation and ink4a-arf deletion appear to be two mutually
exclusive events in human glioblastoma. Oncogene. 19:3816–3822.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pasmant E, Laurendeau I, Heron D, Vidaud
M, Vidaud D and Bieche I: Characterization of a germ-line deletion,
including the entire INK4/ARF locus, in a melanoma-neural system
tumor family: identification of ANRIL, an antisense noncoding RNA
whose expression coclusters with ARF. Cancer Res. 67:3963–3969.
2007. View Article : Google Scholar
|
|
17
|
Midgley CA, Desterro JM, Saville MK, et
al: An N-terminal p14ARF peptide blocks Mdm2-dependent
ubiquitination in vitro and can activate p53 in vivo. Oncogene.
19:2312–2323. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Xiong Y and Yarbrough WG: ARF
promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus
deletion impairs both the Rb and p53 tumor suppression pathways.
Cell. 92:725–734. 1998. View Article : Google Scholar : PubMed/NCBI
|