Introduction
Nickel (II) has been considered a hazardous heavy
metal due to its ability to induce cytotoxicity and carcinogenicity
as the result of environmentally persistent exposure. In 1990, the
International Agency for Research on Cancer (IARC) classified
nickel compounds as group 1 carcinogens (confirmed carcinogen) in
humans on the basis of experiments conducted in animals which
showed several types of tumors, such as lung and nasal cancers
(1). Although the molecular
mechanisms of toxicity and carcinogenicity of nickel remain poorly
understood, one commonly suggested mechanism for nickel toxicity is
the induction of oxidative DNA damage (2). Nickel compounds generate reactive
oxygen species (ROS) via a Fenton-type reaction, or via the
inactivation of enzyme activities involved in cellular defenses
against reactive oxygen systems (3), leading to single and double-strand DNA
breaks (4,5).
Intracellular redox homeostasis is maintained by
balancing ROS production with ROS removal via cellular antioxidant
defense systems (6). The nuclear
factor erythroid-2 related factor 2 (Nrf2) is one of the major
factors inherent to cellular defenses against oxidative stress
(7,8). Particulary, Nrf2-antioxidant response
element (ARE) signaling is regarded as a promising strategy in
cancer prevention due to its pivotal role in the transcriptional
activation of cytoprotective genes facilitating the detoxification
of carcinogens (9). Ramos-Gomez
et al(10) have reported
that increased sensitivity to carcinogen and abrogated
chemoprotective efficacy of enzyme inducers were found in
Nrf2-deficient mice. The Nrf2 cytoprotective adaptive response has
evolved as a component of the molecular protection strategies in
organisms against exposure to environmental toxicants (11). In our previous study, we have
consistently demonstrated the protective effects of Nrf2 on
genotoxicity status induced by cadmium treatment in Nrf2-deficient
cells (12).
Recently, it has been emphasized the significance of
gene-environment interactions in lethal diseases including cancer.
A possible definition of gene-environment interaction is that it
occurs when a genetic factor and environmental exposure work
together to cause a disease outcome in some or all cases. In
particular, genetically based variability (silencing, polymorphism)
of the carcinogen metabolizing proteins may influence
susceptibility to environmental carcinogens (13). Masuko et al(14) have reported that the incompetence of
Nrf2 might accelerate the development of inflammatory obstructive
lung diseases, such as asthma and chronic obstructive pulmonary
disease (COPD) after smoking a cigarette containing various heavy
metals.
Microarray techniques allow for the simultaneous
quantitative analysis of more than one thousand genes, and may
prove to be a powerful tool in toxicological studies as well
(15,16). In particular, this tool has been
applied to evaluate the putative toxicity of environmental
pollutants, and established several databases of gene expression
patterns induced by toxic chemicals (17). Determining the meaning of the
observed biological changes in transcript levels requires the clear
elucidation of biological interdependencies. Therefore, it is
necessary to analyze the relevant pathways or networks, including
significantly regulated genes, in order to explain whole chains of
events observed in microarray experiments (18). Several reports have demonstrated
that heavy metals as environmental pollutant exhibited a variety of
potential toxicity mechanisms, including oxidative stress,
disturbances in the homeostasis of essential metals, and
interference of interactions with cellular macromolecules (19,20).
In this study, we investigated the protective role
of Nrf2 against nickel-induced toxicity in terms of the inhibition
of oxidative stress and DNA damage. Additionally, we carried out
DNA microarray analyses to profile the gene expression pattern and
pathway analysis to identify the specific networks among these
genes in environmental pollutant nickel- and/or a Nrf2
gene-specific siRNA-treated human cell line.
Materials and methods
Cell culture
RKO (ATCC CRL-2577), human colon cancer cells were
cultured in RPMI-1640 (Gibco, Carlsbad, CA, USA), supplemented with
10% fetal bovine serum (FBS) (Gibco) and 1% antibiotics (Gibco) at
37°C in a 5% CO2 incubator.
Transfection of Nrf2-siRNA
RKO cells were plated in 6-well plates at a density
of 1×105 cells/well. The next day, the cells were
transfected with Nrf2 siRNA using Oligofectamine (Invitrogen GmbH,
Karlsruhe, Germany) in accordance with the manufacturer’s
recommendation. Specific silencing was confirmed using western blot
analysis (data not shown). The Nrf2-targeted siRNA sequence (sense,
5′-GAGUAUGAGCU GGAAAAACdTdT-3′ and antisense: 5′-GUUUUUCCAGC
UCAUACUCdTdT-3′) were synthesized by Dharmacon (Thermo Scientific,
USA).
Nickel treatment
Nickel (II) acetate was purchased from Sigma Co.
(St. Louis, MO, USA). We determined the sublethal concentration of
nickel for our experimental design using the MTT
(3-[4,5-dimethyl-2-thiazol-2-yl]-2,5-diphenyltetrazolium bromide)
assay and FACS analysis (data not shown). After 24 h of siRNA
transfection, cells were treated with low concentration of 20 μM
nickel acetate for 12 or 24 h.
Detection of oxidative stress
In order to assess changes in the intracellular ROS
levels as the result of nickel treatment and the absence of Nrf2,
we employed an oxidant-sensitive fluorescent probe,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA)
(Sigma). Non-fluorescent H2DCFDA turned into the
florescent H2DCF as the result of a reaction with
intracellular ROS. After heavy metal treatment in either Nrf2
wild-type or Nrf2 siRNA-treated cells, H2DCFDA was added
at 0.5 μM diluted in PBS and incubated for 30 min at 37°C. The
cells were observed in PBS under a Leica DM IRB fluorescence
microscope (Wetzlar, Germany).
Comet assay
The comet assay (single-cell gel electrophoresis)
was conducted as described with minor modifications (21). In brief, cells were suspended in
0.5% (w/v) low-melting point agarose, and spread onto microscope
slides pre-coated with 1% (w/v) normal-melting point agarose, then
covered with a further layer of low-melting agarose. The slides
were subsequently dipped into a lysis solution (2.5 M NaCl, 100 mM
Na2EDTA and 10 mM Tris, pH 10.0) and placed overnight at
4°C in darkness, in order to solublize the cell membranes and
cytoplasm. Upon completion of lysis, the slides were placed in a
horizontal gel electrophoresis tank with alkaline electrophoresis
buffer (300 mM NaOH and 1 mM Na2EDTA, pH 13.0) and left
in the solution for 20 min at 4°C to allow the DNA to unwind and
the alkali-labile sites to express. Electrophoresis was conducted
for 15 min at 25 V and 250 mA. After being run, slides were
neutralized with neutralization buffer and washed in PBS. The
slides were air-dried at room temperature. Then, the slides were
stained with SYBR Gold solution and observed under a fluorescence
microscope (Nikon Eclipse 50i, Nikon, Japan). One hundred cells
were scored for DNA damage intensity at each experiment point from
two different slides and the means were calculated.
γ-H2AX immunofluorescence staining
The formation of nuclear foci containing
phosphorylated histone H2AX was assessed by immunofluorescence.
Briefly, either RKO or Nrf2 knockdown cells grown on glass
coverslip were treated without and with nickel for 24 h, followed
by incubation in selective fresh media for additional 12 h. The
cells were fixed with iced methanol for 30 min at −20°C and rinsed
with iced acetone for a few seconds. After washing with PBS, the
cells were incubated with an anti-γ-H2AX primary antibody (Active
Motif, Inc., Carlsbad, CA, USA) overnight at 4°C and an
anti-rabbit-Cy3 secondary antibody (Jackson ImmunoResearch, USA)
for 1 h at room temperature. After PBS washing, the cells were
mounted with mounting solution containing DAPI nuclear stain. The
coverslips were placed onto slides and the foci were visualized
under fluorescence microscope (Nikon, Japan).
Total RNA preparation for microarray
experiment
To isolate total RNA, frozen cell pellets were
homogenized in lysis buffer containing 2-mercaptoethanol. Total RNA
was subsequently purified using RNeasy kits (Qiagen, Valencia, CA,
USA) in accordance with the manufacturer’s recommendations, prior
to the microarray experiments.
Focused DNA microarray and data
analysis
The genes involved in DNA damage, DNA repair,
apoptosis, oxidative stress, and cell cycle were selected to design
the focused microarray chip. The information of all genes was found
in the UniGene Build 189 and Agilent databases (http://earray.chem.agilent.com). Transcriptomic
studies were conducted using an 8*15K Oligo chip (Agilent) and a
Quick Amp Labeling kit in accordance with the manufacturer’s
protocols. Subio platform ver. 1.6 was used for the initial
analysis of expression data. Expression changes are described as
fold-changes (expression ratio between control- and
treated-signals) (22).
Real-time quantitative RT-PCR
(qRT-PCR)
Real-time qRT-PCR was performed using Power
SYBR-Green PCR kit (Applied Biosystems, Carlsbad, CA, USA)
according to the manufacturer’s instructions. The primers of 10
genes analyzed from microarray data were designed for qRT-PCR
(Table III). Thermal cycling
conditions were undertaken as follows: 50°C for 2 min and 95°C for
10 min followed by 40 cycles of 95°C for 30 sec and 60°C for 30
sec, 72°C for 30 sec. The real-time PCR analysis was performed on
an Applied Biosystems Prism 7900HT Sequence Detection System (PE
Applied Biosystems).
 | Table IIIList of gene-specific primer
sequences used in the qRT-PCR validation study. |
Table III
List of gene-specific primer
sequences used in the qRT-PCR validation study.
| Gene name | Forward | Reverse |
|---|
| CAV1 | TCT CTA CAC CGT TCC
CAT CC | ACT TGC TTC TCG CTC
AGC TC |
| FOSL2 | AGC CTT GGA GAA CTC
GGT TT | AAC AAA GGG ACA GGA
ATG GTC |
| MICA | TTC CAT GTT TCT GCT
GTT GC | ACT GGG TGT TGA TCC
AGG AC |
| PIM2 | GGA ATG GAA GAT GGA
CAC CA | AAA CAG CAA GCC TTA
TTT CCC |
| RUNX1 | GGA TCT CGC TGT AGG
TCA GG | CTC CGG GAA TCT TCC
TGT TT |
| SLC7A6 | TGC TCT GGC CTA ATG
GAT CT | GAC TGC CTC ACT GTT
GAC CA |
| APLP1 | GCC TGC CTG GTG AAT
TTG TG | GCC TCC GGG TTG AAC
TCT C |
| CLSPN | ATG ATT CCC AGA TGG
ACT TG | AGC CAC TGC TCT CGT
TCA AT |
| PCAF | AAA GAT GGC CGT GTT
ATT GG | CCA TAG CCC TTG ACT
TGC TC |
| PRAME | ATG GAA CGA AGG CGT
TTG TG | GTG TCT CCC GTC AAA
GGC T |
Pathway analysis
We used Pathway Studio 7.1 software (Ariadne
Genomics, Rockville, MD, USA) to define the cellular networks and
interactions among genes expressed in our microarray experiment.
This software contains >100,000 regulations, interactions,
modifications and cell process events between proteins and small
molecules. The database has been compiled by the application of the
text-mining tool MedScan to the entirety of PubMed (23). Additionally, Pathway Studio enables
the visualization of gene expression values and status in the
context of protein interaction networks and pathways.
Results
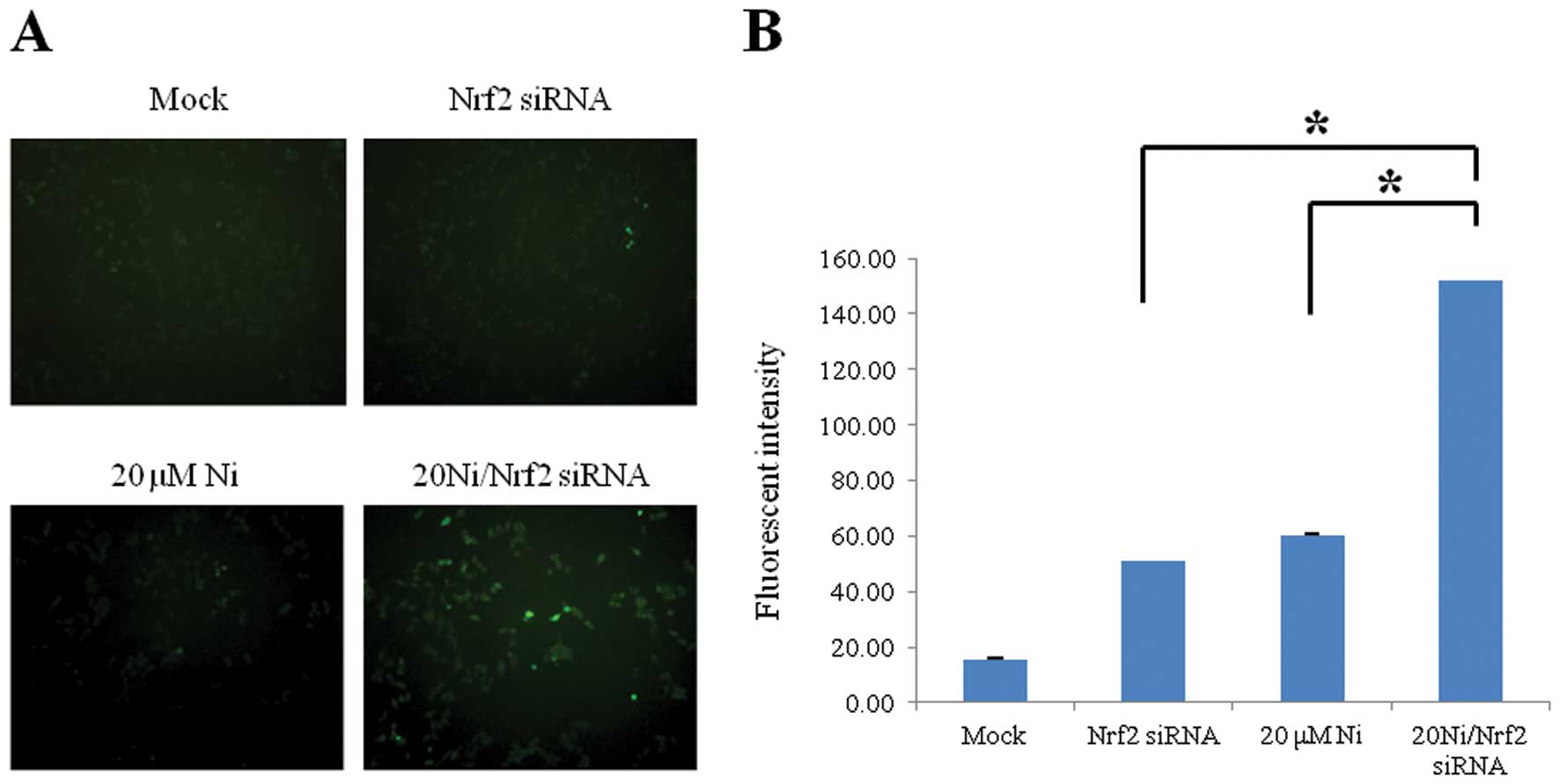
ROS level was increased in Nrf2 knockdown
cells in response to nickel
Nrf2 as a redox factor has a vital role in
protection by regulating the cellular oxidation/reduction status.
To evaluate the protective roles of Nrf2 against oxidative stress
induced by a sub-lethal 20 μM nickel exposure (data not shown), we
constructed a transient Nrf2 knockdown system in human RKO cells
via siRNA transfection (data not shown). The level of oxidative
stress was assessed via immunofluorescence detection using
ROS-sensitive H2DCHDA. We observed significantly increased
intracellular ROS levels in the nickel-treated Nrf2 lacking cells
than that in the nickel-treated wild-type cells (Fig. 1). This result indicates that Nrf2
might be an important redox modulator to suppress nickel-induced
oxidative stress.
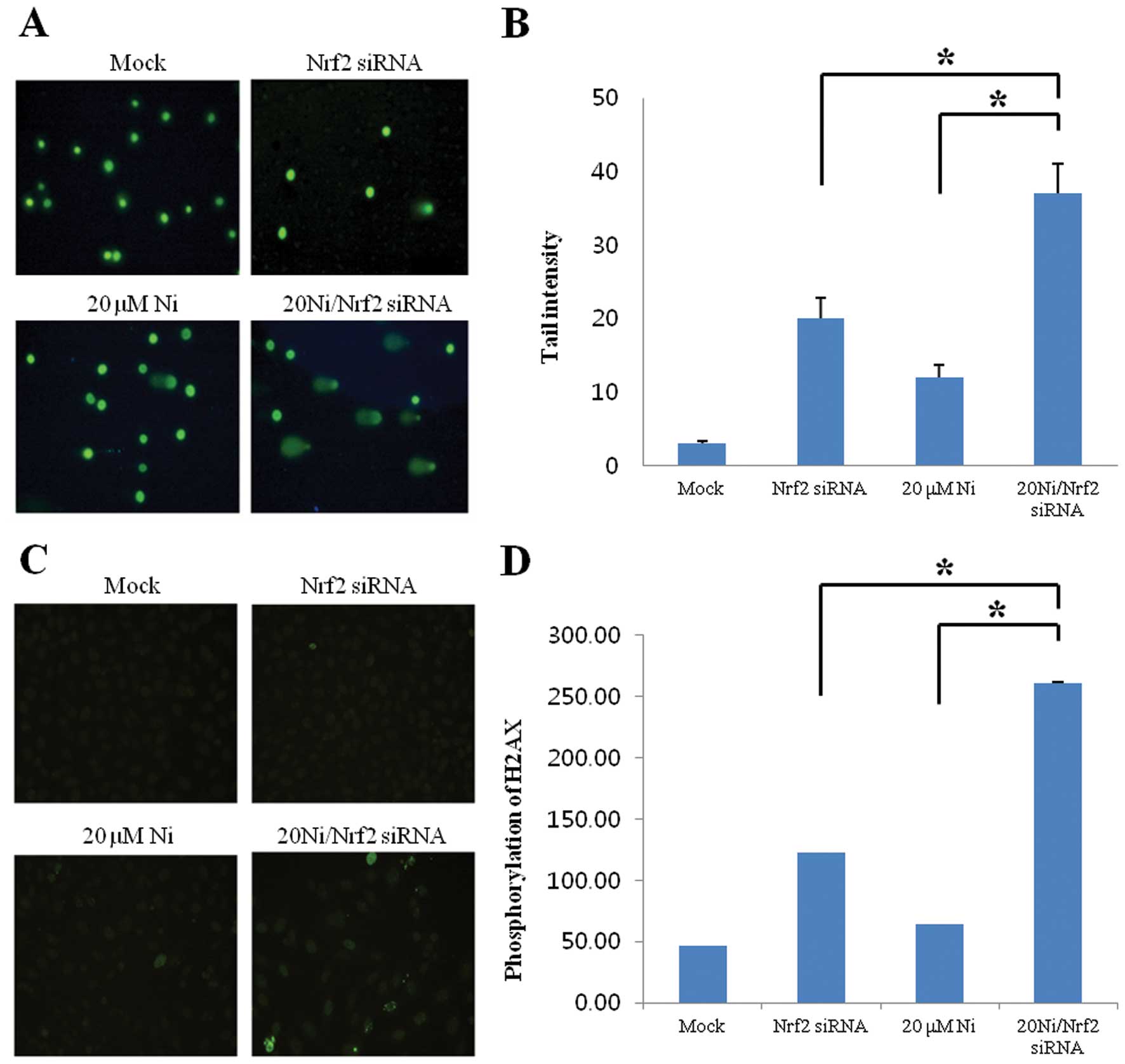
The amount of DNA strand breaks were
significantly augmented in nickel-treated Nrf2-silencing cells
Nickel exposure induces ROS with the subsequent
generation of oxidative DNA damage (2). To evaluate the functions of Nrf2 in
inhibition of nickel-induced DNA damage, we employed the comet
assay and γ-H2AX immunofluorescence staining to detect DNA strand
breaks as indicator of oxidative DNA damage. In Fig. 2, we demonstrated a significant
increase in damaged DNA in Nrf2-lacking RKO compared to wild-type
cells under nickel treatment, thereby indicating the critical
protective role of Nrf2 factor against nickel-caused DNA
damage.
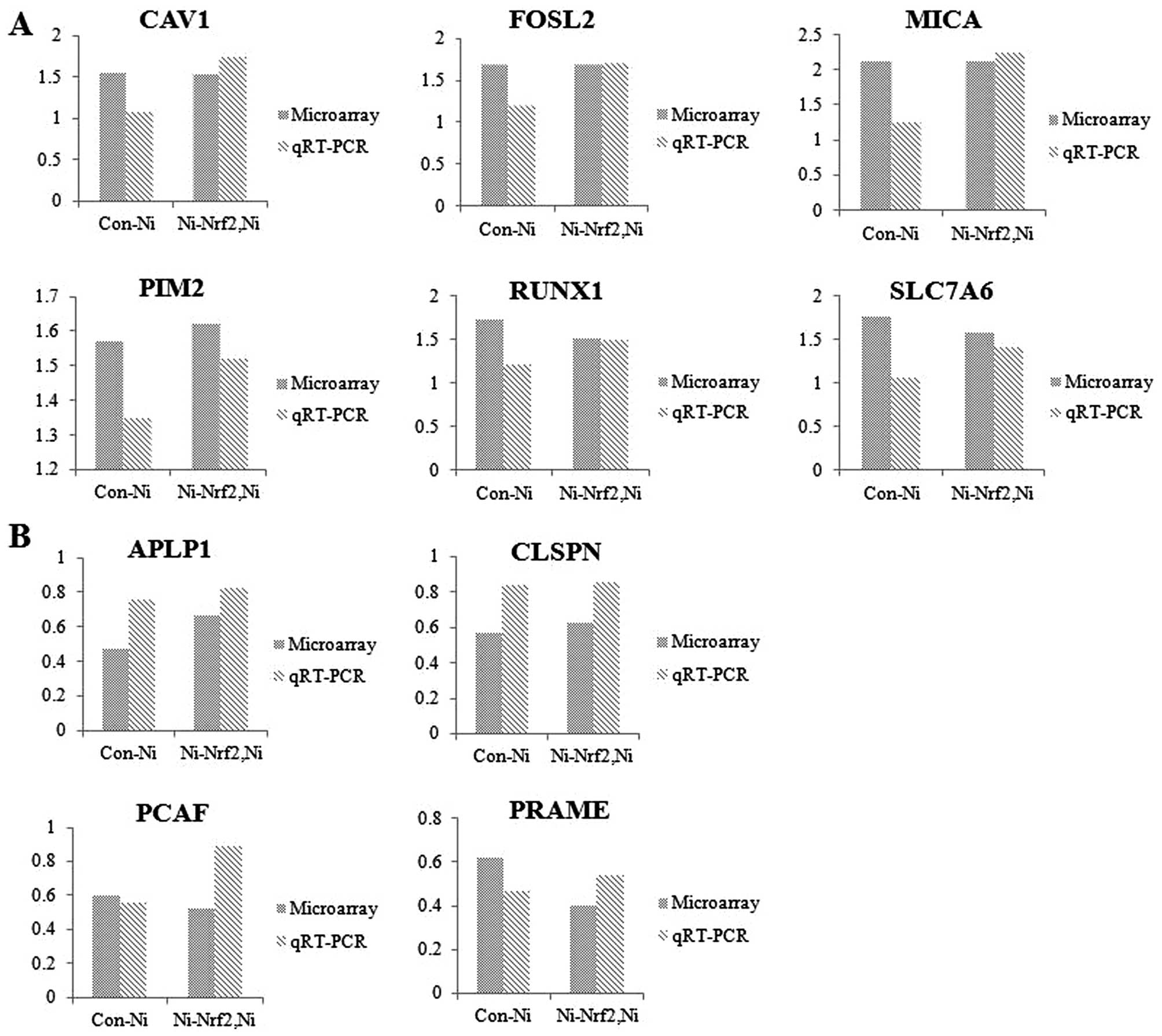
The potential nickel- and Nrf2-target
genes were discovered using toxicogenomic tool
The alteration of genomic expression patterns
against nickel depending on functional Nrf2 status was analyzed
using focused DNA microarray experiments. Microarray experiments
were conducted after 24 h of nickel treatment at sub-lethal
concentration in Nrf2 knockdown cells compared to wild-type cells.
As a results of the toxicogenomics assays, we detected a total of
17 genes that were significantly altered by Nrf2 knockdown under
nickel exposure condition (>1.5-fold and P<0.05). Six notable
genes were upregulated (Table I)
and 11 genes were downregulated (Table
II). Additionally, we demonstrated that the expression levels
from the microarray results of 10 genes (CAV1, FOSL2, MICA, PIM2,
RUNX1, SLC7A6, APLP1, CLSPN, PCAF and PRAME) were consistent with
those shown in the qRT-PCR experiment (Fig. 3). These data implied that these
validated genes might be regarded as potential markers for
understanding the interaction of Nrf2-oriented genes in response to
nickel-induced toxicity.
| Table IList of 1.5-fold upregulated genes in
response to nickel exposure in Nrf2 knockdown cells. |
Table I
List of 1.5-fold upregulated genes in
response to nickel exposure in Nrf2 knockdown cells.
| Gene symbol | Accession no. | Gene name | Con-Nia | Ni-Nrf2,Nib |
|---|
|
|
|---|
| Fold-change | P-value | Fold-change | P-value |
|---|
| CAV1 | NM_001753 | Caveolin 1,
caveolae protein, 22 kDa | 1.54 | 0.000167 | 1.52 | 0.00015 |
| FOSL2 | NM_005253 | FOS-like antigen
2 | 1.68 | 0.00235 | 1.68 | 0.00289 |
| MICA | NM_000247 | MHC
classIpolypeptide-related sequence A | 2.13 | 0.0000063 | 2.13 | 0.00000368 |
| PIM2 | NM_006875 | Pim-2 oncogene | 1.57 | 0.00000675 | 1.62 | 0.00165 |
| RUNX1 | NM_001001890 | Runt-related
transcription factor 1 (acute myeloid leukemia 1; aml1
oncogene) | 1.73 | 0.000165 | 1.51 | 0.00239 |
| SLC7A6 | NM_001076785 | Solute carrier
family 7 (cationic amino acid transporter, y+ system), member
6 | 1.77 | 0.0000165 | 1.58 | 0.00011 |
| Table IIList of 1.5-fold downregulated genes
in response to nickel exposure in Nrf2 knockdown cells. |
Table II
List of 1.5-fold downregulated genes
in response to nickel exposure in Nrf2 knockdown cells.
| Gene symbol | Accession no. | Gene name | Con-Nia | Ni-Nrf2,Nib |
|---|
|
|
|---|
| Fold-change | P-value | Fold-change | P-value |
|---|
| APLP1 | NM_005166 | Amlyoid β (A4)
precursor-like protein 1 | 0.47 | 0.00101 | 0.66 | 0.000268 |
| CLSPN | NM_022111 | Claspin homolog
(Xenopus laevis) | 0.57 | 0.00278 | 0.62 | 0.04623 |
| FANCB | NM_001018113 | Fanconi anemia,
complementation group B | 0.64 | 0.00136 | 0.58 | 0.03481 |
| H2AFX | NM_002105 | H2A histone family,
member X | 0.35 | 0.0000521 | 0.6 | 0.000443 |
| HRK | NM_003806 | Hara-kiri, BCL2
interacting protein (contains only BH3 domain) | 0.35 | 0.000758 | 0.59 | 0.00671 |
| HSP90AB1 | NM_007355 | Heat shock protein
90kDa α (cytosolic), class B member 1 | 0.51 | 0.00213 | 0.53 | 0.00998 |
| KLK3 | AF335478 | Kallikrein-related
peptidase 3 | 0.2 | 0.0000151 | 0.55 | 0.00176 |
| PCAF | NM_003884 | P300/CBP-associated
factor | 0.6 | 0.00606 | 0.52 | 0.0128 |
| PRAME | NM_206956 | Preferentially
expressed antigen in melanoma | 0.62 | 0.00456 | 0.4 | 0.000403 |
| RBMX | NM_002139 | RNA binding motif
protein, X-linked | 0.65 | 0.00641 | 0.4 | 0.00971 |
| TSC22D3 | NM_004089 | TSC22 domain
family, member 3 | 0.47 | 0.00277 | 0.53 | 0.00855 |
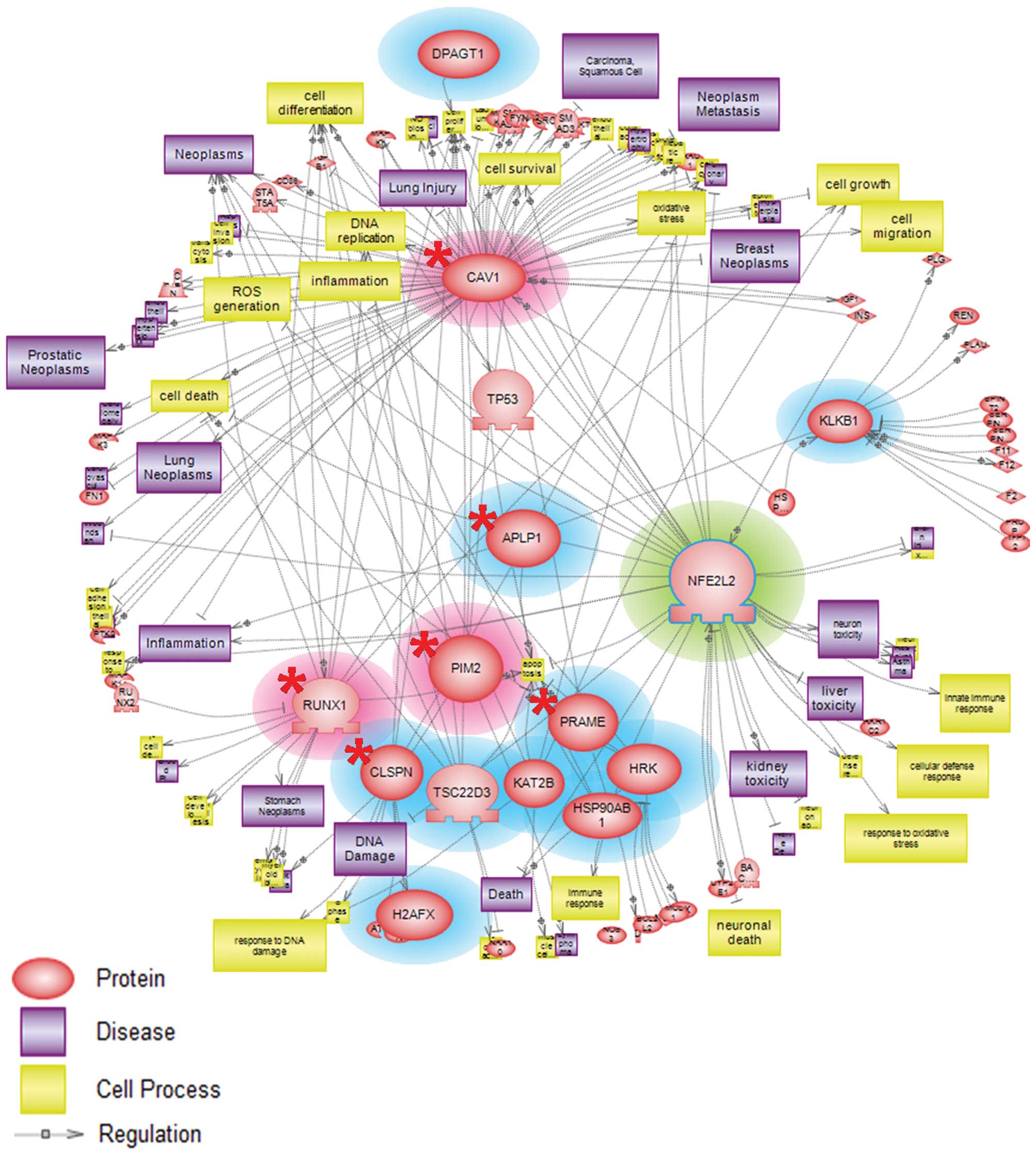
Nrf2-responsive genes under nickel
exposure were involved in toxicity-prone and cytoprotective
pathway
Networks are used ubiquitously throughout biology to
represent the relationships between genes and gene products.
Pathway Studio software is capable of establishing a more dynamic
model of cellular processes via the incorporation of gene
expression data. We employed Pathway Studio (version 7.1) to
evaluate possible interactions among Nrf2- and nickel-responsive
genes obtained from our microarray data analysis. Our analyzed
results showed that potential nickel- and Nrf2-target genes from
microarray data were mainly associated with oxidative stress,
inflammation, apoptosis, cell cycle and cell survival processes
(Fig. 4). These results provide
insights into the manner in which these upregulated and
downregulated genes were critical in mediating toxicity-prone
responses and protection of the cells.
Discussion
The toxicity of nickel has become a matter of some
interest because of the widespread environmental release of nickel
and the incidence of accidental poisoning in nickel workers
(24). One possible mechanism
underlying the toxicity and carcinogenicity of nickel compounds in
humans and animals evolves the induction of DNA damage related to
oxidative stress. The Nrf2-regulated signaling pathway plays an
important role in protection against oxidative stress-mediated
cytotoxicity after exposure to oxidants. Increased oxidative stress
occurring as the result of the destruction of redox homeostasis has
been implicated in the etiology of a number of acute and chronic
diseases linked to exposures to environmental toxicants such as
heavy metals. In this study, we investigated the protective roles
of Nrf2 against nickel-induced toxicity at sub-lethal dose in terms
of the suppression of oxidative stress and DNA damage.
Additionally, we suggested the cytotoxic and protective cellular
mechanisms among the selected nickel and Nrf2-related genes using
microarray analysis.
In general, nickel exposure causes accumulation in
human at low concentrations over the long-term. Therefore, we
selected 20 μM of nickel as a sub-lethal dose, and detected high
cell survival rates and unchanged cell cycles (data not shown). Our
data showed that under nickel treatment, Nrf2 siRNA-treated cells
produced higher levels of ROS and DNA strand breakage, in
comparison to Nrf2 wild-type cells (Figs. 1 and 2). Nickel enhanced oxidative stress in
plasma by stimulating the generation of ROS, including
•OH, O2•−,
H2O2(25).
Gong and Cederbaum (26) have
previously shown that the knockdown of Nrf2 in a human liver cell
line caused an increase in ROS levels and a reduced expression of
antioxidative enzymes. Several reports have mentioned the
protective effects of Nrf2 in response to various environmental
toxic chemicals. Aoki et al(27) showed an increase in the levels of
8-oxodeoxyguanosine (8-oxo-dG) in bronchial epithelial cells from
Nrf2 knockout mice compared to wild-type sub-chronically exposed to
diesel exhaust particles, thereby indicating that the absence of
Nrf2 could contribute to an increase in oxidative DNA damage and
adduct formation. Additionally, our previous study has demonstrated
an increased level of DNA strand breaks, following the induction of
oxidative stress in Nrf2 siRNA-treated cells under carcinogenic
metal cadmium exposure conditions (12).
We can explain these toxic molecular mechanisms by
analyzing differentially expressed genes using microarray and
qRT-PCR. In our study, six genes (CAV1, FOSL2, MICA, PIM2, RUNX1
and SLC7A6) were notably upregulated in nickel-treated Nrf2
knockdown cells as compared to nickel-treated wild-type cells
(Fig. 3A and Table I). Caveolin 1 (CAV1) and FOS-like
antigen 2 (FOSL2) expressions have been shown to be elevated in
many human cancer cell lines and numerous human tumor specimens
(28,29), thereby indicating that CAV1 and
FOSL2 play important roles in the promotion of mammary
tumorigenesis. Particularly, these genes have been associated with
the development and progression of breast cancer (30,31).
MHC class I polypeptide-related sequence A (MICA) is overexpressed
as the result of a DNA damage response involving the ATM (ataxia
telangiectasia mutated) and the ATR (ATM- and Rad3-related) protein
kinases (32). Del Toro-Arreola
et al(33) have also
reported that MICA was strongly upregulated after genotoxic stress
in human cervical cancer cell lines. The Pim-2 (PIM2) is a novel
oncogene and its protein has a profound inhibitory effect on the
apoptosis of tumor cells without cell specificity (34). PIM2 and runt-related transcription
factor 1 (RUNX1) oncogenes have critical role in the development of
several kinds of tumors, including prostatic carcinoma (35,36).
Additionally, the expression of LAT family members, including the
solute carrier family 7, member 6 (SLC7A6; LAT3) gene has been
observed in a variety of malignant cells (37,38).
Pathway analysis results using Pathway Studio software (version
7.1) demonstrated that upregulated genes observed by microarray
analysis were involved in the regulation of gene expression
associated with oxidative stress, inflammation, and necrosis
processes (Fig. 4). Hereby, we
might postulate that CAV1, FOSL2, MICA, PIM2, RUNX1 and SLC7A6
genes could be regarded as potential targets of nickel and
Nrf2-regulated genes.
We also discovered downregulated genes in response
to nickel and redox status. The expressions of APLP1, CLSPN, PCAF
and PRAME were downregulated in nickel and Nrf2 siRNA-treated cells
as compared to cells treated only with nickel (Fig. 3B and Table II). Amyloid-β precursor-like
protein (APLP1) is a member of the amyloid precursor protein (APP)
family, harboring the copper (Cu) binding domain. APP modulates the
toxic reactive oxygen intermediates generated by unregulated redox
reactivity of Cu (39). Tang et
al(40) have reported that
APLP1 might be a novel transcriptional target of p53, according to
the results of in vivo and in vitro characterization
of a p53 responsive element found in the first intron of the APLP1
gene locus. Similarly, p300/CBP-associated factor (PCAF) has been
shown to acetylate p53 in response to DNA damage, resulting in the
increased transcription of p53-regulated genes (41). Additionally, the claspin homolog
(CLSPN) is an essential upstream regulator of checkpoint kinase 1
and triggers a checkpoint arrest of the cell cycle in response to
replicative stress or DNA damage (42). The gene is also required for
efficient DNA repair system (43)
and DNA replication during a normal S phase (44). The preferentially expressed antigen
of melanoma (PRAME) gene is involved in the regulation of cell
death or cell cycle. Furthermore, this gene induces
caspase-independent cell death in vitro and reduces
tumorigenicity in vivo(45).
From pathway results, downregulated genes were shown to mediate
cell cycle arrest, and cell survival (Fig. 4). These results implied that the
abovementioned downregulated genes might be possible markers useful
for assessing the relationship between nickel and the Nrf2
gene.
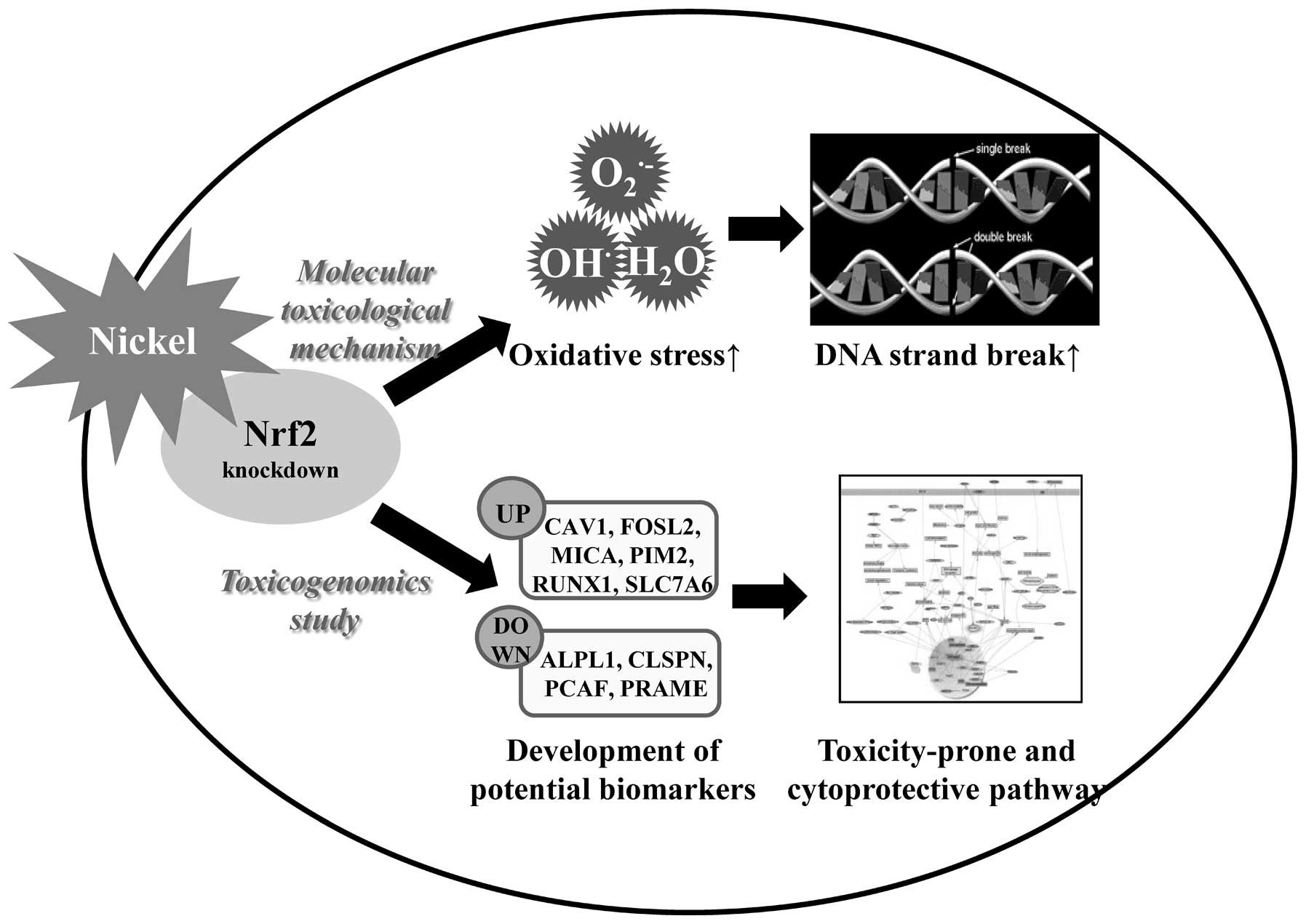
In summary, we demonstrated the synergistic toxic
effects of Nrf2 gene knockdown following exposure to the
environmental toxicant nickel, in terms of gene-environment
interaction (Fig. 5). Nrf2 gene
silencing accelerated an increase in nickel-induced oxidative
stress and DNA damage. Using microarray and qRT-PCR, we found
several genes (CAV1, FOSL2, MICA, PIM2, RUNX1, SLC7A6, APLP1,
CLSPN, PCAF and PRAME) markedly enhanced or reduced expression
levels in nickel-exposed Nrf2 knockdown cells as compared to
nickel-treated wild-type cells. These up- and downregulated genes
perform a crucial function in toxicity-prone and cytoprotection,
respectively. Thereby, the nickel and Nrf2-responsive genes
detected in our study might be useful in the development of
strategies targeting the Nrf2 pathway for the purposes of
attenuating environmental diseases, including cancer.
Acknowledgements
This study was supported in part by a grant
(091-091-083) and ‘The Ecoinnovation Project’ (412-112-011) from
the Korea Ministry of Environment.
References
|
1
|
Kasprzak KS, Sunderman FW Jr and Salnikow
K: Nickel carcinogenesis. Mutat Res. 533:67–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kasprzak KS: Possible role of oxidative
damage in metal-induced carcinogenesis. Cancer Invest. 13:411–430.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynn S, Yew FH, Chen KS and Jan KY:
Reactive oxygen species are involved in nickel inhibition of DNA
repair. Environ Mol Mutagen. 29:208–216. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aust AE and Eveleigh JF: Mechanisms of DNA
oxidation. Proc Soc Exp Biol Med. 222:246–252. 1999. View Article : Google Scholar
|
|
5
|
Chakrabarti SK, Bai C and Subramanian KS:
DNA-protein crosslinks induced by nickel compounds in isolated rat
lymphocytes: role of reactive oxygen species and specific amino
acids. Toxicol Appl Pharmacol. 170:153–165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen XL and Kunsch C: Induction of
cytoprotective genes through Nrf2/antioxidant response element
pathway: a new therapeutic approach for the treatment of
inflammatory diseases. Curr Pharm Des. 10:879–891. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen T, Nioi P and Pickett CB: The
Nrf2-antioxidant response element signaling pathway and its
activation by oxidative stress. J Biol Chem. 284:13291–13295. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JY and Seo YR: Enhancement of
mitomycin C-induced apoptosis in Nrf2-deficient human colon cancer
cells. Mol Cell Toxicol. 6:51–56. 2010. View Article : Google Scholar
|
|
9
|
Giudice A and Montella M: Activation of
the Nrf2-ARE signaling pathway: a promising strategy in cancer
prevention. Bioessays. 28:169–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K,
Yamamoto M, Talalay P and Kensler TW: Sensitivity to carcinogenesis
is increased and chemoprotective efficacy of enzyme inducers is
lost in nrf2 transcription factor-deficient mice. Proc Natl Acad
Sci USA. 98:3410–3415. 2001. View Article : Google Scholar
|
|
11
|
Osburn WO and Kensler TW: Nrf2 signaling:
an adaptive response pathway for protection against environmental
toxic insults. Mutat Res. 659:31–39. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park JY and Seo YR: The protective role of
Nrf2 in cadmium-induced DNA damage. Mol Cell Toxicol. 7:61–66.
2011. View Article : Google Scholar
|
|
13
|
d’Errico A, Malats N, Vineis P and
Boffetta P: Review of studies of selected metabolic polymorphisms
and cancer. IARC Sci Publ. 148:323–393. 1999.PubMed/NCBI
|
|
14
|
Masuko H, Sakamoto T, Kaneko Y, et al: An
interaction between Nrf2 polymorphisms and smoking status affects
annual decline in FEV1: a longitudinal retrospective cohort study.
BMC Med Genet. 20:12–97. 2011.PubMed/NCBI
|
|
15
|
Koizumi S and Yamada H: DNA microarray
analysis of altered gene expression in cadmium-exposed human cells.
J Occup Health. 45:331–334. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carinci F, Pezzetti F, Volinia S,
Francioso F, Arcelli D, Farina E and Piattelli A: Zirconium oxide:
analysis of MG63 osteoblast-like cell response by means of a
microarray technology. Biomaterials. 25:215–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boverhof DR and Zacharewski TR:
Toxicogenomics in risk assessment: applications and needs. Toxicol
Sci. 89:352–360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Werner T: Bioinformatics applications for
pathway analysis of microarray data. Curr Opin Biotechnol.
19:50–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Snow ET: Metal carcinogenesis: mechanistic
implications. Pharmacol Ther. 53:31–65. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ercal N, Gurer-Orhan H and Aykin-Burns N:
Toxic metals and oxidative stress part I: mechanisms involved in
metal-induced oxidative damage. Curr Top Med Chem. 1:529–539. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar
|
|
22
|
Peng X, Wood CL, Blalock EM, Chen KC,
Landfield PW and Stromberg AJ: Statistical implications of pooling
RNA samples for microarray experiments. BMC Bioinformatics. 4:1–9.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nikitin A, Egorov S, Daraselia N and Mazo
I: Pathway studio - the analysis and navigation of molecular
networks. Bioinformatics. 19:2155–2157. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sunderman FW Jr, Dingle B, Hopfer SM and
Swift T: Acute nickel toxicity in electroplating workers who
accidently ingested a solution of nickel sulfate and nickel
chloride. Am J Ind Med. 14:257–266. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen CY, Su YJ, Wu PF and Shyu MM:
Nickel-induced plasma lipid peroxidation and effect of antioxidants
in human blood: involvement hydroxyl radical formation and
depletion of alpha-tocopherol. J Toxicol Environ Health A.
65:843–852. 2002. View Article : Google Scholar
|
|
26
|
Gong P and Cederbaum AI: Nrf2 is increased
by CYP2E1 in rodent liver and HepG2 cells and protects against
oxidative stress caused by CYP2E1. Hepatology. 43:144–153. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aoki Y, Sato H, Nishimura N, Takahashi S,
Itoh K and Yamamoto M: Accelerated DNA adduct formation in the lung
of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl
Pharmacol. 173:154–160. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shatz M and Liscovitch M: Caveolin-1: a
tumor-promoting role in human cancer. Int J Radiat Biol.
84:177–189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Milde-Langosch K: The Fos family of
transcription factors and their role in tumourigenesis. Eur J
Cancer. 41:2449–2461. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Milde-Langosch K, Janke S, Wagner I, et
al: Role of Fra-2 in breast cancer: influence on tumor cell
invasion and motility. Breast Cancer Res Treat. 107:337–347. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Syeed N, Husain SA, Abdullah S, Sameer AS,
Chowdri NA, Nanda MS and Siddiqi MA: Caveolin-1 promotes mammary
tumorigenesis: mutational profile of the Kashmiri population. Asian
Pac J Cancer Prev. 11:689–696. 2010.PubMed/NCBI
|
|
32
|
Gasser S, Orsulic S, Brown EJ and Raulet
DH: The DNA damage pathway regulates innate immune system ligands
of the NKG2D receptor. Nature. 436:1186–1190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Del Toro-Arreola S, Arreygue-Garcia N,
Aguilar-Lemarroy A, et al: MHC class I-related chain A and B
ligands are differentially expressed in human cervical cancer cell
lines. Cancer Cell Int. 11:152011.PubMed/NCBI
|
|
34
|
Fox CJ, Hammerman PS, Cinalli RM, Master
SR, Chodosh LA and Thompson CB: The serine/threonine kinase Pim-2
is a transcriptionally regulated apoptotic inhibitor. Genes Dev.
17:1841–1854. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen WW, Chan DC, Donald C, Lilly MB and
Kraft AS: Pim family kinases enhance tumor growth of prostate
cancer cells. Mol Cancer Res. 3:443–451. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pratap J, Lian JB, Javed A, Barnes GL, van
Wijnen AJ, Stein JL and Stein GS: Regulatory roles of Runx2 in
metastatic tumor and cancer cell interactions with bone. Cancer
Metastasis Rev. 25:589–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yanagida O, Kanai Y, Chairoungdua A, et
al: Human L-type amino acid transporter 1 (LAT1): characterization
of function and expression in tumor cell lines. Biochim Biophys
Acta. 1514:291–302. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakanishi K, Matsuo H, Kanai Y, et al:
LAT1 expression in normal lung and in atypical adenomatous
hyperplasia and adenocarcinoma of the lung. Virchows Arch.
448:142–150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
White AR, Multhaup G, Galatis D, et al:
Contrasting, species-dependent modulation of copper-mediated
neurotoxicity by the Alzheimer’s disease amyloid precursor protein.
J Neurosci. 22:365–376. 2002.PubMed/NCBI
|
|
40
|
Tang X, Milyavsky M, Goldfinger N and
Rotter V: Amyloid-beta precursor-like protein APLP1 is a novel p53
transcriptional target gene that augments neuroblastoma cell death
upon genotoxic stress. Oncogene. 26:7302–7312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu L, Scolnick DM, Trievel RC, Zhang HB,
Marmorstein R, Halazonetis TD and Berger SL: p53 sites acetylated
in vitro by PCAF and p300 are acetylated in vivo in response to DNA
damage. Mol Cell Biol. 19:1202–1209. 1999.PubMed/NCBI
|
|
42
|
Lindsey-Boltz LA, Serçin O, Choi JH and
Sancar A: Reconstitution of human claspin-mediated phosphorylation
of Chk1 by the ATR (ataxia telangiectasia-mutated and rad3-related)
checkpoint kinase. J Biol Chem. 284:33107–33114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Praetorius-Ibba M, Wang QE, Wani G,
El-Mahdy MA, Zhu Q, Qin S and Wani AA: Role of Claspin in
regulation of nucleotide excision repair factor DDB2. DNA Repair.
6:578–587. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chini CC and Chen J: Claspin, a regulator
of Chk1 in DNA replication stress pathway. DNA Repair. 3:1033–1037.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tajeddine N, Gala JL, Louis M, Van Schoor
M, Tombal B and Gailly P: Tumor-associated antigen preferentially
expressed antigen of melanoma (PRAME) induces caspase-independent
cell death in vitro and reduces tumorigenicity in vivo. Cancer Res.
65:7348–7355. 2005. View Article : Google Scholar
|