Introduction
Gallbladder cancer (GBC) is one of the most highly
malignant carcinomas with a low curative resection rate (10–30%)
and a low response rate to chemotherapy (20–40%) (1,2).
Although GBC is non-sensitive to routine chemotherapy, platinum
drugs have recently been proven effective for GBC treatment. The
most notable advance in the chemotherapy of biliary tract cancers
in the last 5 years is that compared with gemcitabine alone,
cisplatin (CDDP) plus gemcitabine have improved the overall
survival of patients by 3.6 months (3). Oxaliplatin plus gemcitabine have also
shown promising results in patients with advanced biliary tract
carcinoma (4,5).
The most common mechanism of multidrug resistance
(MDR) in cancer cells is frequently associated with the
overexpression of 4 ATP-binding cassette (ABC) transporters:
P-glycoprotein (MDR1), multidrug resistance-related protein 1
(MRP1), multidrug resistance-related protein 2 (MRP2), and the
breast cancer resistance protein (ABCG2), which may cause the
active efflux of a variety of anticancer drugs (6). MDR1, MRP1, MRP2 and ABCG2 in GBC
contribute to the MDR phenotype in vitro and in
vivo(7–9). The mechanisms of insensitivity to
platinum drugs have been mainly referred to glutathione (GSH)
conjugation and the transportation of GSH conjugates of platinum
drugs out of cancer cells (10).
Low reactive oxygen species (ROS) levels, increased GSH levels or
overexpressed GSH-related enzymes and pumps are observed in several
drug-resistant cancer cells (11).
We have previously found that the sensitivity of a number of cancer
cell lines, such as Du-145 (prostate carcinoma), to platinum drugs
is ABC transporter-dependent and/or ROS-dependent (8,12).
Thus, we hypothesized that the inhibition of ABC protein expression
or the suppression of GSH activity may enhance platinum
cytotoxicity in GBC.
In order to improve the effect of chemotherapy,
several chemosensitizers have been examined for GBC. Emodin
(1,3,8-trihydroxy-6-methylanthraquinone; a type of natural
anthraquinone enriched in the traditional Chinese herbal medicines)
and somatostatin (a regulator of numerous gastrointestinal
hormones) have been reported to enhance the chemosensitivity of GBC
cells (13,14). However, several of these
chemosensitizers remain in the experimental stage (15). Verapamil, a calcium channel blocker,
is a classic chemo-sensitizer which can enhance the antitumor
effect of CDDP in several cancer cells including neuroblastoma and
lung cancer in vitro or in vivo(16,17).
Its reported mechanisms include inhibiting the transport function
of MDR1 or stimulating GSH transport by MRP1 (18,19).
Recently, verapamil has been confirmed to enhance antitumor
efficacy in several clinical studies including metastatic
colorectal cancer (20,21); however, as regards GBC, its effects
have not been examined in preclinical or clinical studies.
In the present study, we aimed to clarify the
effects of verapamil on the sensitivity of GBC cells to platinum
drugs and its mechanisms. We discovered that verapamil enhanced the
anticancer efficacy of CDDP, carboplatin (CBP) and oxaliplatin to
GBC cells both in vitro and in vivo without obvious
systemic toxicity by decreasing GSH levels and inhibiting the
expression of MRP1.
Materials and methods
Cells and reagents
The SGC996 human GBC cell line was provided by the
Academy of Life Sciences, Tongji University (Shanghai, China) and
the GBC-SD cell line was provided by the Department of General
Surgery, Xinhua Hospital (Shanghai, China). SGC996 cells were
maintained in RPMI-1640 medium (Gibco-BRL, Gaitherburg, MD, USA)
and GBC-SD cells were maintained in DMEM medium (Gibco-BRL). These
media were supplemented with 10% fetal bovine serum. Cells were
cultured in a humidified atmosphere with 5% CO2 at 37°C.
CDDP and CBP were obtained from Qilu Pharmaceutical Co., Ltd.
(Jinan, China). Oxaliplatin was obtained from Jiangsu Hengrui
Medicine Co., Ltd. (Lian Yungang, China). Verapamil was purchased
from Sigma (St. Louis, MO, USA).
Cell viability and apoptosis
analysis
Cells were seeded in 96-microculture-well plates
(1.5×104/ml cells, 100 μl/well) and cultured overnight
before treatment. After exposure to the agents as indicated for 24
and 48 h, cell viability was assayed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
(Sigma) assay as previously described (22,23).
Cells were plated in 6-well plates
(5×105/ml cells, 2 ml/well) and cultured overnight prior
to treatment. After being treated with drugs for 24 and 48 h,
apoptotic rates were assessed by flow cytometry using the Annexin
V-fluorescein isothiocyanate (Annexin V-FITC)/propidium iodide (PI)
kit (BD Pharmingen, San Diego, CA, USA). All procedures were
performed according to the manufacturer’s instructions and analyzed
by flow cytometry using a FACS Calibur (Becton-Dickson, San Diego,
CA, USA) (22,23).
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
The expression levels of MDR1, ABCG2,
MRP1 and MRP2 were monitored by RT-PCR. After being
treated with drugs for 24 h, SGC996 and GBC-SD cells were lysed
with 1 ml of RNAse-clean TRIzol reagent (Invitrogen, Carlsbad, CA,
USA), and processed according to the manufacturer’s instructions to
obtain total cellular RNA. The isolated total RNA (1 μg) was
reverse-transcribed using random primers and AMV Reverse
Transcriptase (Promega, Madison, WI, USA) for 5 min at 70°C, 5 min
on ice and 60 min at 37°C. The single-stranded cDNA was amplified
by polymerase chain reaction using GoTaq DNA polymerase (Promega).
PCR of MDR1 gene was performed under the following
conditions: 20 sec at 94°C, 30 sec at 55°C and 60 sec at 72°C for
35 cycles. The sequences for MDR1 sense and antisense
primers were 5′-CCCATCATTGCAATAGCAGG-3′ and
5′-GTTCAAACTTCTGCTCCTGA-3′, respectively. PCR analysis of the
ABCG2 gene was performed under the following conditions: 30
sec at 94°C, 30 sec at 55°C and 30 sec at 72°C for 30 cycles. The
sequences for ABCG2 sense and antisense primers were
5′-TGGCTGTCATGGCTTCAGTA-3′ and 5′-GCCACGTGATTCTTCCACAA-3′,
respectively. PCR analysis of the MRP1 and MRP2 genes
was performed under the following conditions: 30 sec at 94°C, 30
sec at 58°C and 30 sec at 72°C for 34 cycles. The sequences for
MRP1 sense and antisense primers were
5′-TGGTGGGCCTCTCAGTGTCTTA-3′ and 5′-TCGGTAGCGCAGGCAGTAGTTC-3′,
respectively. The sequences for MRP2 sense and antisense
primers were 5′-ATGCTTCCTGGGGATAAT-3′ and 5′-TCAAAGGCACGGATAACT-3′,
respectively. Equal amounts of RT-PCR products were loaded on 1.5%
agarose gels. β-actin was used as the internal control. The
sequences for β-actin sense and antisense primers were 5′-GA
AGATGGTGATGGGGAT-3′ and 5′-GAAGGTGAAGGTCGGAGC-3′, respectively.
ABCG2 and MRP1 siRNA transfection
To determine the role of ABCG2 and MRP1 in cellular
sensitivity to CDDP, ABCG2 or MRP1 siRNA oligonucleotides were
transiently transfected, using the Lipofectamine 2000 reagent
(Invitrogen) according to the manufacturer’s instructions
(Invitrogen) with modifications as previously described (8,24). A
non-specific siRNA was also transfected as the mock control. After
48 h, SGC996 cells were lysed for RT-PCR or exposed to CDDP for an
additional 24 h prior to apoptosis assay. The siRNA sequences for
ABCG2 were 5′-UAAUGAUGUCCAAGAAGAAGUCUGC-3′ and
5′-GCAGACUUCUUCUUGGACAUCAUUA-3′; and for MRP1 were
5′-GGAGUGGAACCCCUCUCUG-3′ and 5′-CAGAGAGGGGUUCCACUCC-3′.
ROS and GSH measurement
2,7-Dichlorodihydrofluorescein diacetate (DCFH-DA)
(Sigma) was used for ROS capture in the cells. The average
fluorescence intensity of 2,7-dichlorofluorescein (DCF) stands for
intracellular ROS levels (8,22). The
cultured cells were then exposed to the various drugs and 10 μM of
DCFH-DA at 37°C for 15 min. After being washed once with ice-cold
PBS, cells were harvested and maintained on ice for an immediate
detection using the flow cytometer FACS Calibur.
The cells were treated with drugs for 12 h and then
assayed according to the instructions of the GSH Assay kit
(Jiancheng Bioengineering Institute, Nanjing, China) as previously
described (8). All results obtained
were normalized according to the cellular protein content, which
was measured using the BCA protein assay kit purchased from
Sigma.
In vivo study in tumor-bearing mice
SGC996 cells were harvested, washed, resuspended in
serum-free optimum medium and then injected subcutaneously into
6-week-old BALB/c-nu/nu mice, with 6×106 cells/mouse
(n=8 mice/group, purchased from the Shanghai Experimental Animal
Center, Shanghai, China). This experiment was undertaken after
obtaining approval from the institutional committee. Three days
after inoculation, the tumor-bearing mice were intraperitoneally
administered normal saline, verapamil (20 mg/kg/day), CDDP (2
mg/kg/day) and verapamil/CDDP, every 3 days. The mice were
sacrificed after 18 days, and body weight, as well as tumor volume
and weight were measured. The hearts, kidneys and livers of the
mice were histologically examined to determine the systemic
toxicity.
Immunohistochemistry for ABCG2 and MRP1
expression
All tumors from the mice and surgical samples from
51 patients with GBC treated at the Department of General Surgery,
Renji Hospital, Shanghai Jiao Tong University School of Medicine
were collected for immunohistochemistry. A total of 15 non-tumorous
gallbladder samples obtained from patients with cholecystolithiasis
that underwent cholecystectomy were also included for control in
this assay. The protocols for the use of animals were approved by
the Department of Laboratory Animal Sciences, Shanghai Jiao Tong
University School of Medicine.
Immunohistochemistry was performed following the
standard avidin/streptavidin-biotin peroxidase methods (25,26).
The tissue microarray slides (4 μM-slices) were deparaffinized,
rehydrated and boiled for antigen retrieval (30 m at 98°C in
citrate buffer pH 6.0). Primary antibodies against ABCG2 (1:150)
and MRP1 (1:20) proteins (Abcam, USA) were used on the sections of
the tumor tissue, with 1% BSA-PBS as the negative control. After
being incubated overnight at 4°C, the slides were incubated with
biotinylated anti-mouse immunoglobulin (LSAB) for 30 min and then
with horseradish peroxidase-conjugated streptavidin for 30 min.
Each step was followed by a washing with PBS. Staining was revealed
by 3,3′-diaminobenzidine (DAB) and counterstained with
hematoxylin.
Cells were scored on color micrographs of
characteristic lesions and the mean count of DAB-positive staining
intensity for ABCG2 or MRP1 signals was calculated using Adobe
Photoshop CS.
Statistical analysis
Data are presented as the mean values ± SE. SAS
software was used for statistical analysis. ANOVA (analysis of
variance) was applied for comparison of the means of 2 or multiple
groups, in which the Student-Newman-Keuls (SNK) was further used
for the comparison of 2 groups. A value of P<0.05 was considered
to indicate a statistically significant difference.
Results
Verapamil enhances platinum drug-induced
inhibition of cell viability in GBC cells by increasing
apoptosis
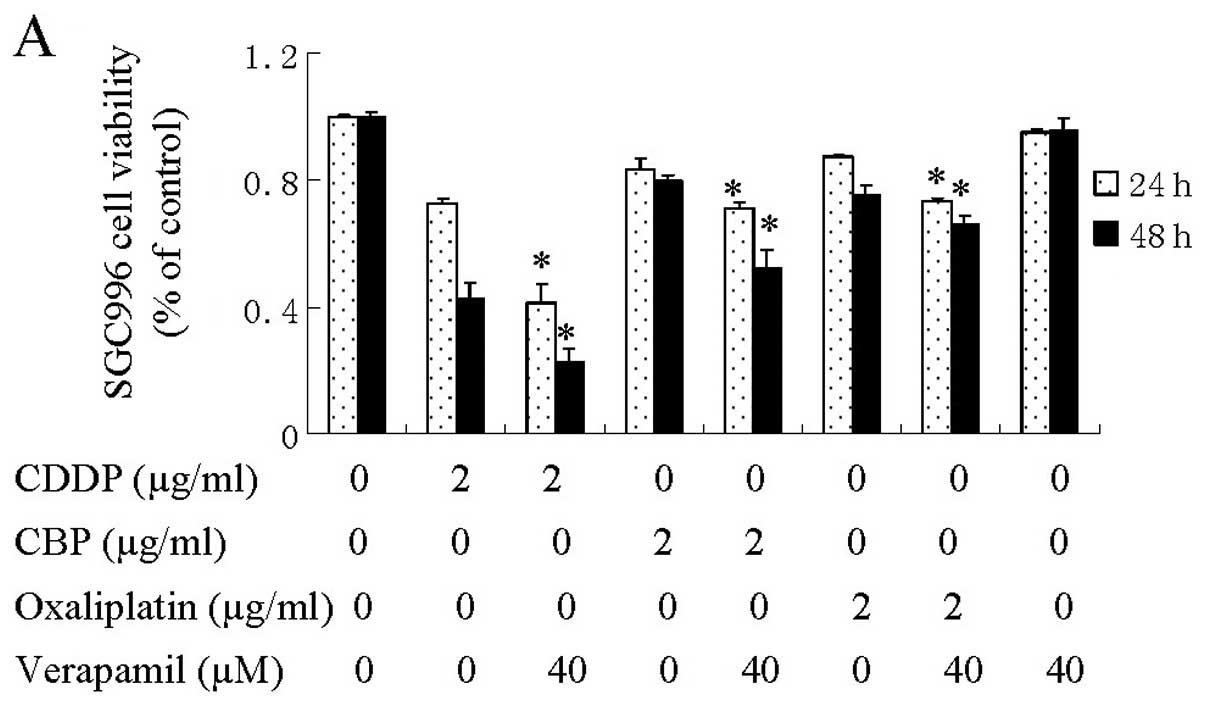
To examine the additive effect of verapamil on cell
viability, the SGC996 and GBC-SD human GBC cells were treated with
CDDP, CBP, oxaliplatin or verapamil alone, or co-treated. No
obvious reduction in viable cell number was observed in the group
treated with verapamil alone. Verapamil combination treatment with
CDDP, CBP and oxaliplatin led to a significant reduction in cell
viability for 24 and 48 h, compared with CDDP, CBP and oxaliplatin
treatment alone (Fig. 1).
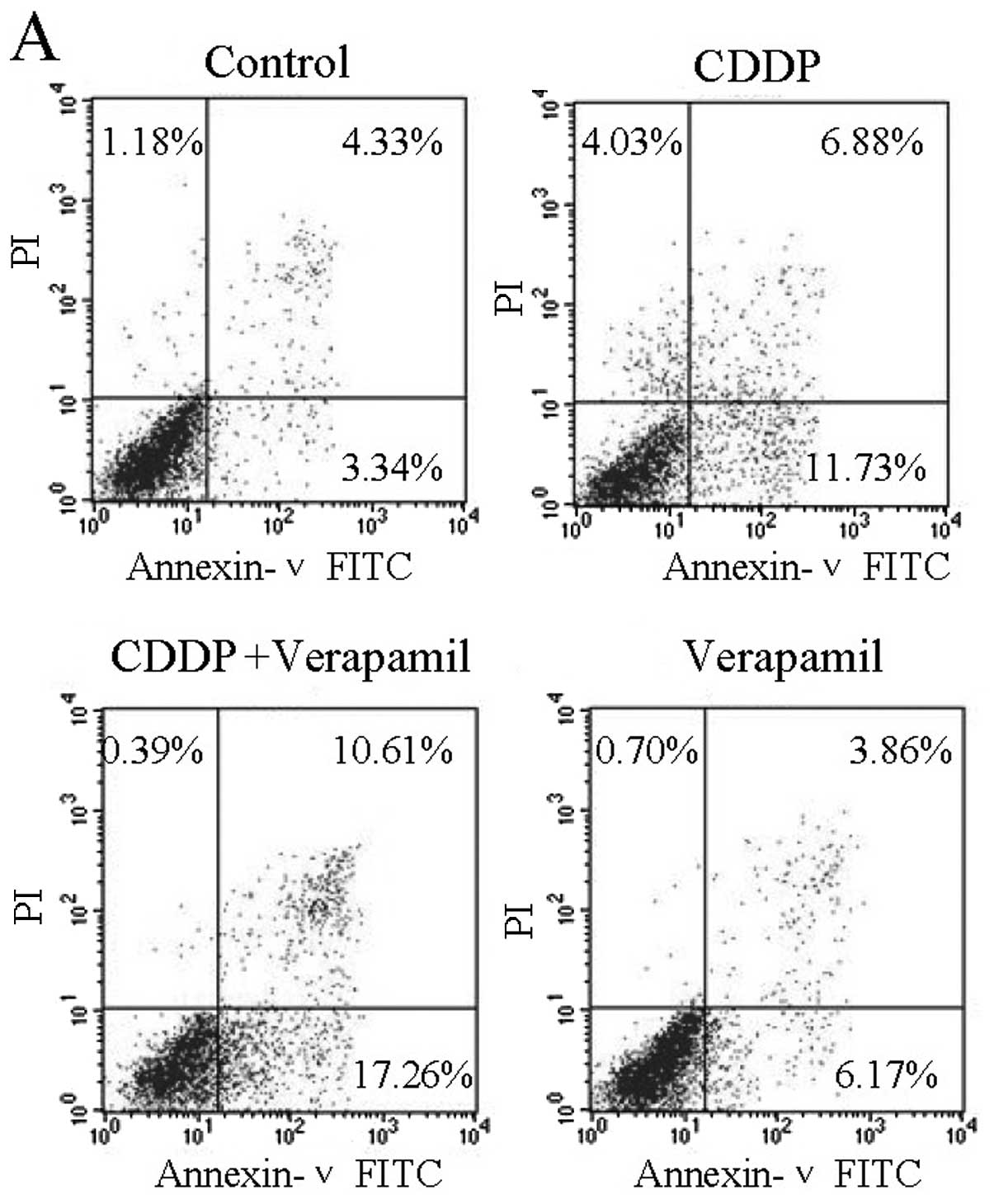
To determine whether the reduction in cell viability
was attributed to the increase in apoptosis, Annexin V-FITC/PI
double labeling flow cytometry was conducted. CDDP caused cell
apoptosis, and verapamil enhanced the CDDP-induced apoptosis of the
SGC996 (Fig. 2A–C) and GBC-SD cells
(Fig. 2D–F) at 24 and 48 h.
MRP1 is downregulated after
verapamil/platinum drug co-treatment and is responsible for
blockade of CDDP cytotoxicity
We investigated whether the enhanced apoptosis of
GBC cells to platinum drugs/verapamil correlated with the
regulation of the expression of the 4 MDR genes: MDR1, MRP1, MRP2
and ABCG2. The results from RT-PCR analysis demonstrated that the
SGC996 cells expressed ABCG2, MRP1 and MRP2 at high levels;
however, MDR1 expression was low, at an undetectable level. GBC-SD
cells expressed ABCG2, MDR1 and MRP1 at high levels; however, MRP2
expression was undetectable (Fig.
3A).
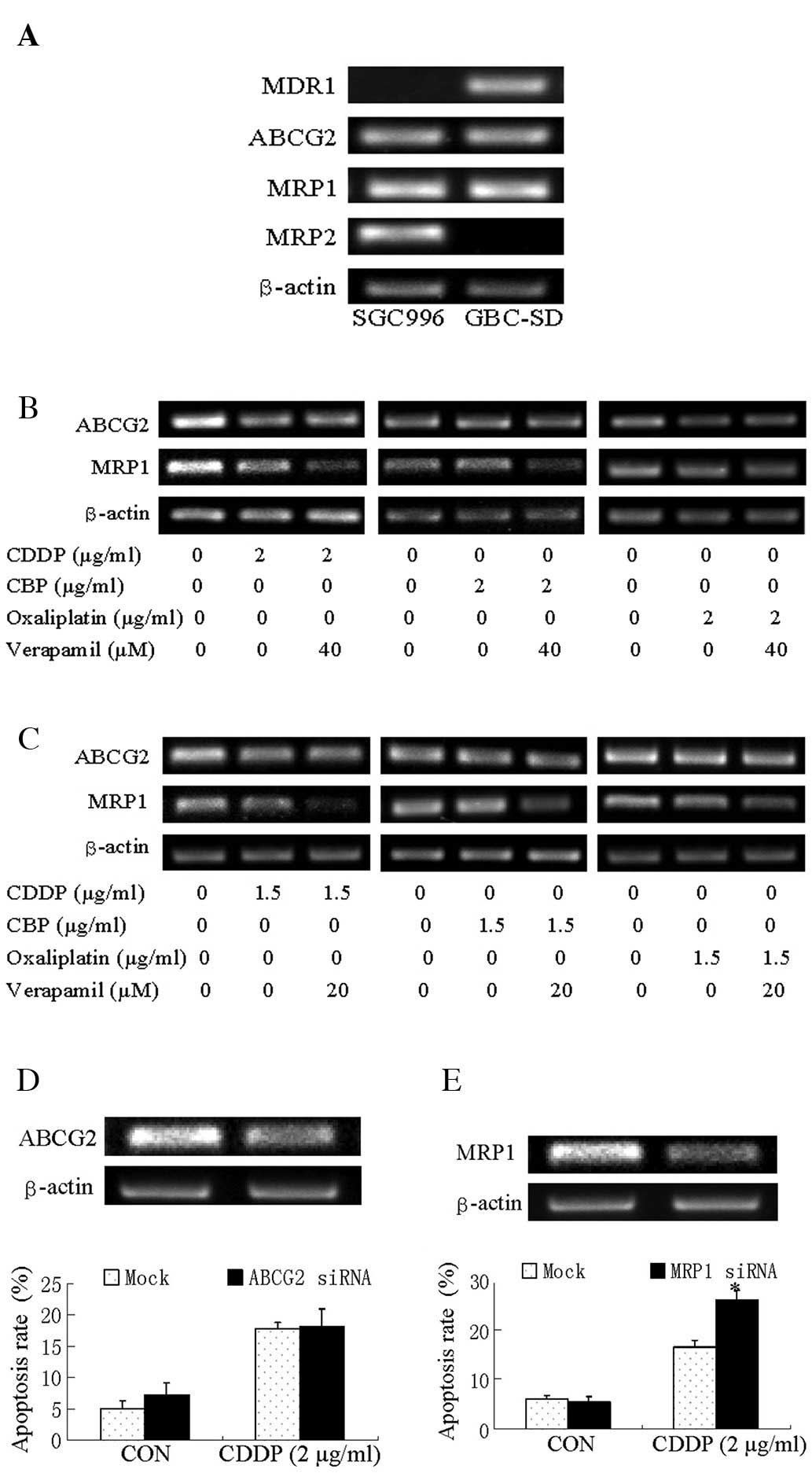 | Figure 3Expression of ABC transporters in
SGC996 and GBC-SD cells. (A) Expression of MDR1, ABCG2, MRP1 and
MRP2 in SGC996 and GBC-SD cells. (B) Expression of ABCG2 and MRP1
in SGC996 cells. SGC996 cells were exposed to CDDP, CBP or
oxaliplatin alone, verapamil alone and CDDP, CBP or
oxaliplatin/verapamil co-treatment for 24 h before being harvested
for RT-PCR. (C) Expression of ABCG2 and MRP1 in GBC-SD cells.
GBC-SD cells were exposed to CDDP, CBP or oxaliplatin alone,
verapamil alone and CDDP, CBP or oxaliplatin/verapamil co-treatment
for 24 h before being harvested for RT-PCR. (D) Apoptosis of SGC996
cells. Cells were transfected with non-specific siRNA (Mock) or
ABCG2 siRNA for 48 h. The apoptotic rate was analyzed by Annexin
V/PI flow cytometry in SGC996 cells transfected with ABCG2 siRNA
after being treated with CDDP for 24 h. (E) Apoptosis of SGC996
cells. Cells were transfected with non-specific siRNA (Mock) or
MRP1 siRNA for 48 h. The apoptotic rate was analyzed by Annexin
V/PI by flow cytometry in SGC996 cells transfected with MRP1 siRNA
after being treated with CDDP for 24 h. Columns, means of 3
experiments; bars, means ± SE. *P<0.05, MRP1
siRNA/CDDP group compared with the Mock/CDDP group. |
Since verapamil enhanced the platinum drug-induced
inhibition of cell viability in both GBC cell lines, we detected
the co-expressed MDR genes in the 2 GBC cell lines, ABCG2 and MRP1.
CDDP alone downregulated the expression of MRP1 and ABCG2 in the
SGC996 cells, while CDDP/verapamil co-treatment resulted in a
significant additive effect on the downregulation of the expression
of MRP1, but not that of ABCG2. CBP/verapamil and
oxaliplatin/verapamil slightly downregulated MRP1 expression in the
SGC996 cells (Fig. 3B).
Co-treatment also downregulated MRP1 expression in GBC-SD cells,
but ABCG2 did not (Fig. 3C).
To determine whether ABCG2 or MRP1 expression is
responsible for the cytotoxic sensitivity of the SGC996 cells to
CDDP, cells were transfected with siRNA oligonucleotide to silence
MRP1 expression, and were then treated with CDDP for 24 h. The
results demonstrated that the knockdown of ABCG2 had no significant
effect (Fig. 3D), while the
knockdown of MRP1 increased CDDP-induced cell apoptosis (Fig. 3E). These data suggest that MRP1 is
responsible for CDDP resistance in GBC cells, and that verapamil
may facilitate the cytotoxicity of CDDP by suppressing MRP1
expression.
Verapamil elevates intracellular ROS
levels and decreases GSH levels of GBC cells
We found that the exposure of SGC996 and GBC-SD
cells to verapamil or verapamil/platinum drugs resulted in a
significant elevation of intracellular ROS levels, while platinum
drug treatment alone had less of an effect (Fig. 4A and B). We measured the
intracellular GSH levels after exposing SGC996 and GBC-SD cells to
platinum drugs, verapamil, or platinum drugs in combination with
verapamil for 12 h. Verapamil alone or in combination with CDDP,
CBP and oxaliplatin markedly reduced the intracellular GSH levels.
These data indicated that the enhancement of verapamil on platinum
drug-induced cytotoxicity in both GBC cell lines was related to the
intracellular GSH reduction (Fig. 4C
and D).
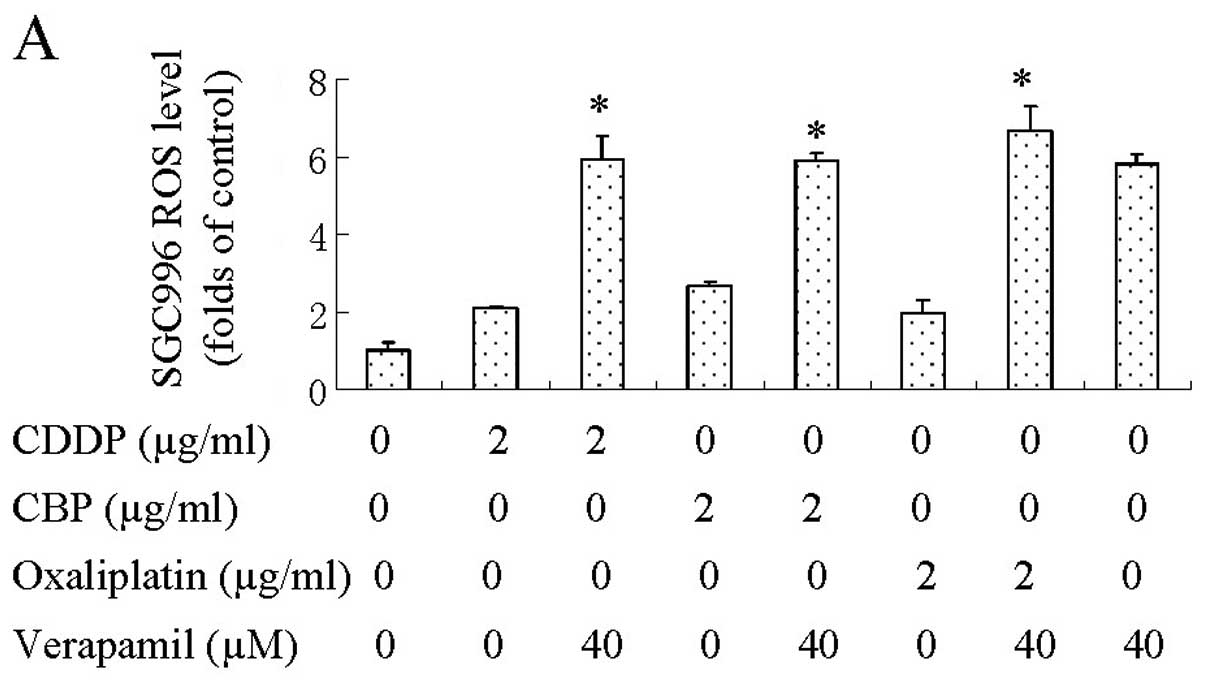 | Figure 4ROS and GSH in SGC996 and GBC-SD
cells. Cells were exposed to CDDP, CBP, oxaliplatin or verapamil
alone and CDDP, CBP, oxaliplatin/verapamil co-treatment. (A) ROS
level in SGC996 cells (DCF flow cytometry, drug treatments for 15
min). (B) ROS level in GBC-SD cells (DCF flow cytometry, drug
treatments for 15 min). (C) GSH levels in SGC996 cells (GSH
analysis kit, drug treatments for 12 h). (D) GSH levels in GBC-SD
cells (GSH analysis kit, drug treatments for 12 h). Columns, means
of 3 experiments; bars, means ± SE. *P<0.05,
combination treatment groups compared with the groups treated with
CDDP, CBP and oxaliplatin alone. |
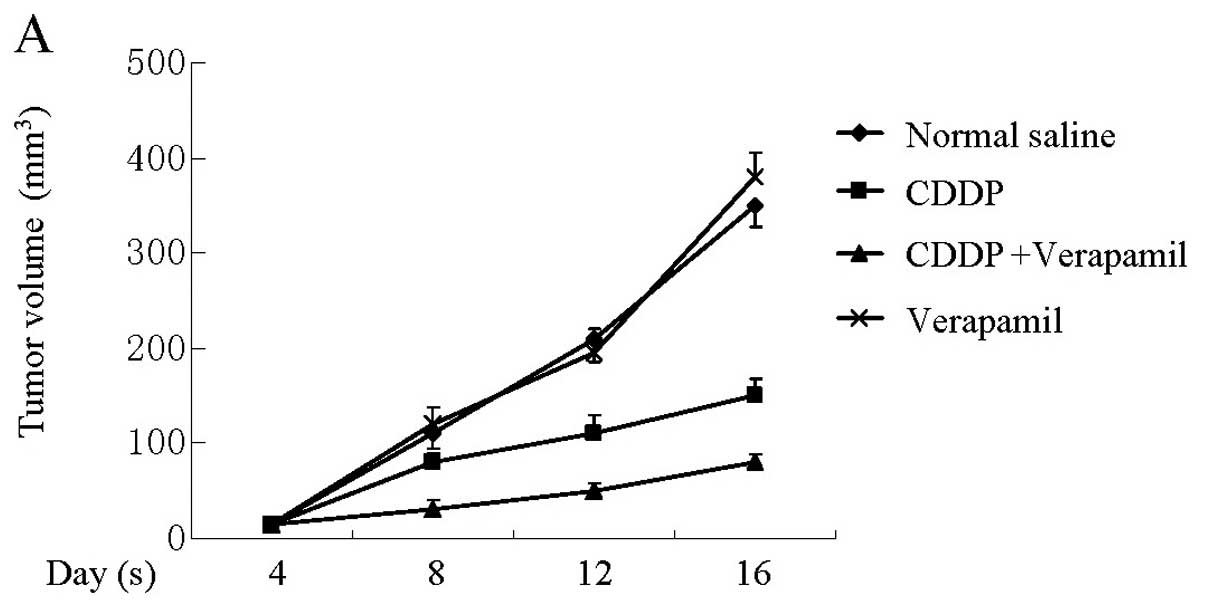
Verapamil enhances the sensitivity of
tumor xenografts to CDDP without systemic toxicity in vivo
In vitro experiments demonstrated that
verapamil enhanced the sensitivity of gallbladder cancer cells to
platinum drugs. To verify this effect in vivo and evaluate
its side-effects, SGC996 cells were transplanted into nude mice and
the mice were treated for 18 days. Our results demonstrated that
tumor xenografts exposed to the combination therapy were
significantly smaller compared to those from the other groups
(Fig. 5A). Systemic toxic effects
were evaluated by the body weight loss of mice and the pathological
changes in major organs including the heart, kidney and liver. No
notable differences were observed among these groups (Fig. 5B), demonstrating that verapamil/CDDP
co-treatment had no obvious toxic effects in vivo.
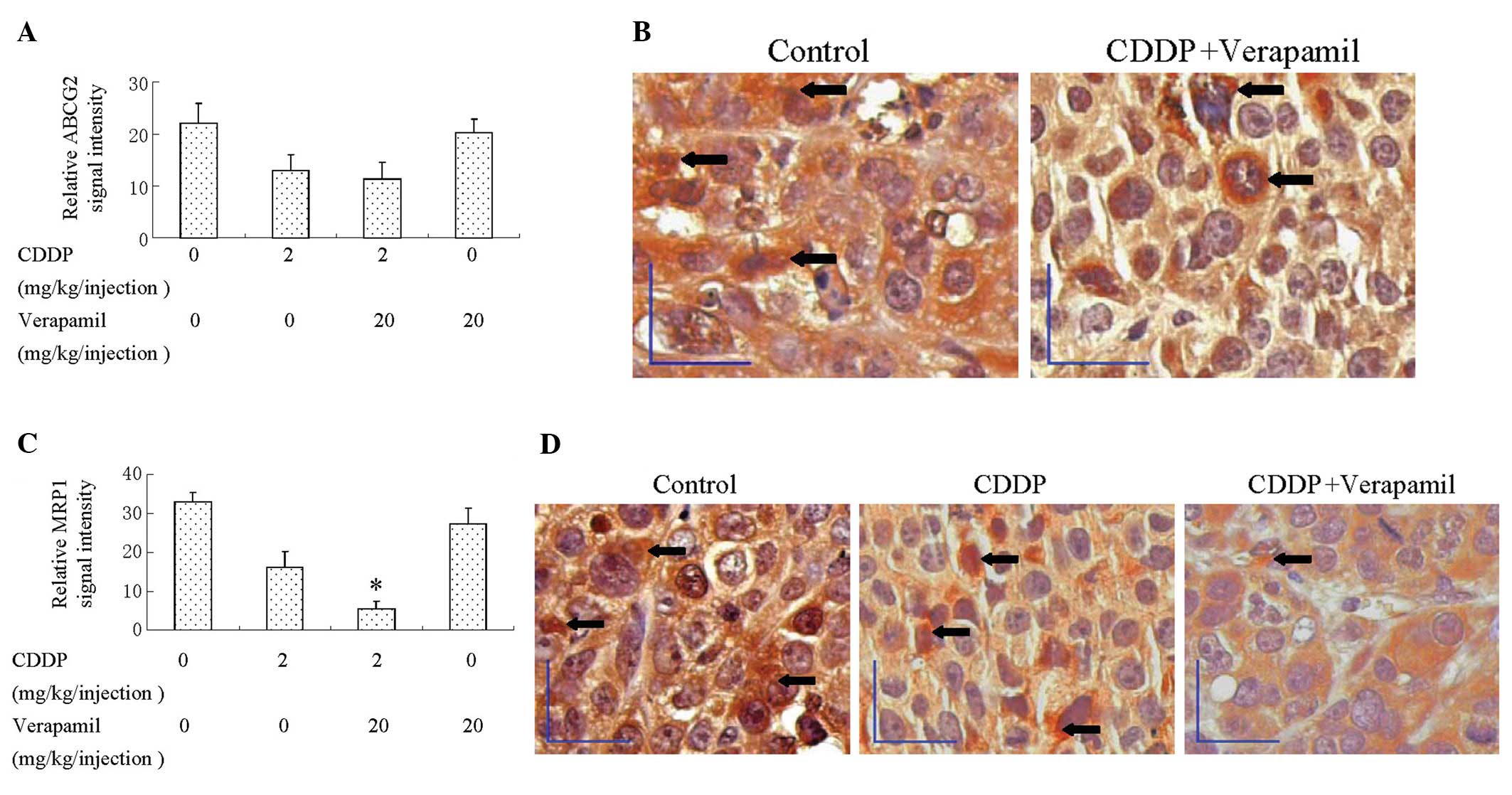
Verapamil/CDDP co-treatment represses the
expression of MRP1 in tumors xenografts
To ascertain the effect of verapamil/CDDP
co-treatment on ABCG2 and MRP1 expression in vivo,
immunohistochemistry for ABCG2 and MRP1 expression was performed on
paraffin-embedded tissue sections of tumors. The expression of
ABCG2 in tumors was downregulated by CDDP; however, co-treatment
with CDDP and verapamil did not lead to a more significant
downregulation (Fig. 6A and B). The
expression of MRP1 in tumors was downregulated by CDDP, and more
significantly by verapamil/CDDP combination treatment (Fig. 6C and D). These data demonstrated
that CDDP alone downregulated the expression of the MRP1 and ABCG2
proteins, while CDDP/verapamil co-treatment resulted in an additive
effect on the downregulation of the expression of MRP1, but not
that of ABCG2.
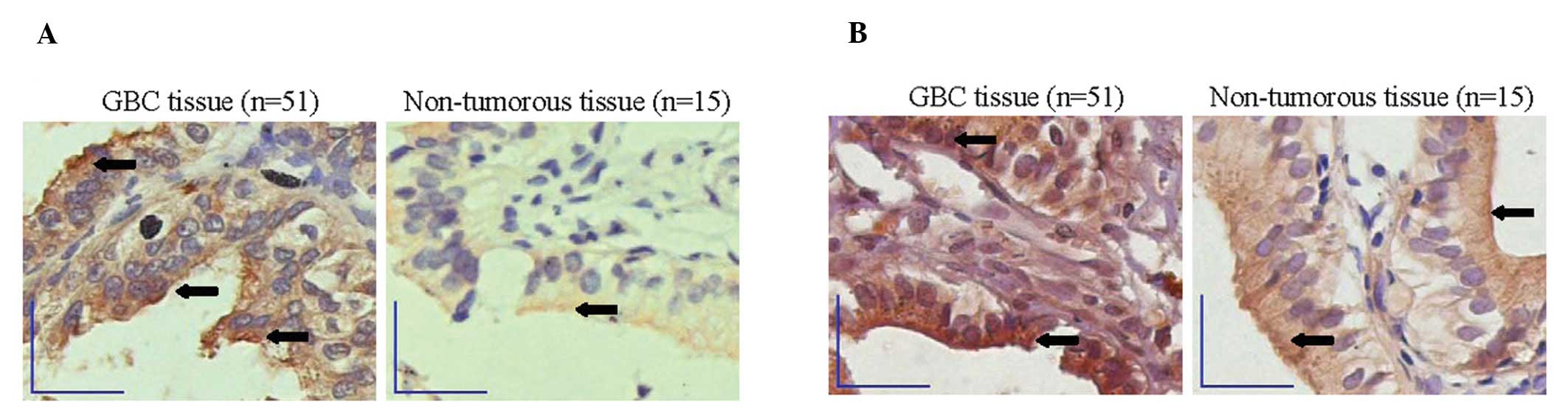
ABCG2 and MRP1 are highly expressed in
GBC tissue compared to non-tumorous gallbladder tissue
We used immunohistochemistry to determine the degree
of ABCG2 and MRP1 expression in cancer and non-tumorous gallbladder
tissue. The ABCG2 and MRP1 proteins were expressed predominantly in
the membrane and partly in the cytoplasm of epithelial cells in
both GBC and cholecystolithiasis specimens. There was stronger
staining of ABCG2 and MRP1 in GBC compared to non-tumorous
gallbladder tissue (signal intensity: 35.5±4.6 vs. 17.1±11.2,
P<0.05; 40.4±18.1 vs. 22.0±9.9, P<0.05) (Fig. 7).
Discussion
The 4 members of the ABC transporter family (MDR1,
ABCG2, MRP1 and MRP2) play an important role in MDR in cancer cells
(6,7,9). In
the present study, we investigated the expression of the 4 ABC
transporters in the 2 GBC cell lines, SGC996 (13) and GBC-SD (27), as well as in human tissue. We
discovered that ABCG2 and MRP1 were highly expressed in the SGC996
and GBC-SD cells; while the expression of MDR1 in the SGC996 cells
and that of MRP2 in the GBC-SD cells was undetectable. ABCG2 and
MRP1 are also highly expressed in GBC tissue compared to
non-tumorous tissue. Collectively, the results from previous
reports (8,9) and our present study allow us to
hypothesize that ABCG2 and MRP1 are 2 common denominators of the
intrinsic MDR phenotype in GBC cells. The accumulation of ABCG2 in
poorly differentiated GBC may coincide with side population cells
increasing and/or the malfunctioning of PI3K-signaling pathways
during tumor progression (9,28).
ABCG2 and MRP1 overexpression has also been documented in other MDR
cancer cells (6,29).
Verapamil is a specific first-generation MDR1
inhibitor and differs from other GBC chemosensitizers, such as
emodin or somatostatin. Its efficiency has been confirmed in
certain types of cancer, both in preclinical studies and in
clinical use. It has been reported that co-treatment with
verapamil-chemotherapeutic agents (including cisplatin)
downregulates MDR1 gene transcription (17,30).
We found that verapamil also enhanced the chemosensitivity of GBC
cells (SGC996) in which the expression of MDR1 was undetectable. To
the best of our knowledge, this is the first report demonstrating
the suppressive effect of verapamil on MRP1 expression in cancer
cells. A major fraction of the intracellular platinum drugs are
conjugated with GSH to form less toxic GS-platinum complexes and
are discharged from cancer cells via the glutathione conjugate
export pumps, such as MRP1 (10,31).
In addition, we demonstrated that silencing MRP1 expression may
increase CDDP-induced GBC cell apoptosis. These data support our
hypothesis that MRP1 downregulation causes the accumulation of
intracellular platinum drugs and enhances their cytotoxicity.
Transcription factors, such as p53 and Sp1 may regulate the
transcription of MRP1 (32,33). The upstream regulators that may
serve as the target of verapamil-platinum drug co-treatment to
mediate the downregulation of MRP1 require clarification in future
studies. MRP1 downregulation may be one of the mechanisms behind
the verapamil-enhanced cytotoxicity of platinum drugs on GBC cells.
Compared to CBP or oxaliplatin, CDDP inhibited GBC cell viability
more significantly either alone or in combination with verapamil
and it downregulated MRP1 more significantly. These findings may
have relevant clinical implications explaining why CDDP is a
superior choice to other platinum drugs in several clinical trials
(3). Although with a much less
significant effect on the downregulation of MRP1, CBP or
oxaliplatin also had a notable inhibitory effect on GBC cell
viability either alone or in combination with verapamil. Thus,
other more important mechanisms to enhance cytotoxicity may be
involved.
Depletion of intracellular GSH may restore the drug
sensitivity of various cancer cells (34). Thus, the reduction in GSH levels in
GBC cells may contribute to the enhanced cytotoxicity of platinum
drugs. However, verapamil, which stimulates GSH transport by MRP1,
has been reported to be only slightly, or not at all, effective in
restoring the drug sensitivity of several MRP1-overexpressing cells
(35,36). Only several types of
MRP1-overexpressing cells display hypersensitivity to verapamil,
which triggers apoptosis through MRP1-mediated GSH extrusion
(37,38). In the present study, it is notable
that verapamil enhanced the cytotoxicity of GBC cells to platinum
drugs and a remarkable reduction in intracellular GSH levels
followed the verapamil-platinum drug combination treatment. The
enhancement of apoptosis cannot be achieved through the simple
augmentation of ROS levels or a decrease in GSH levels, since a
single administration of verapamil has no effect in vitro
and in vivo. We suggest that intracellular GSH reduction in
GBC cells and thereby less platinum conjugates formed with less
cellular efflux of these conjugates may contribute to
verapamil-induced increased cytotoxicity. Since CBP or oxaliplatin
in combination with verapamil had a notable inhibitory effect on
GBC cell viability but a much less significant effect on the
downregulation of MRP1, verapamil is supposed to act primarily as a
functional modulator of GSH rather than as a modifier of MRP1
expression. Our findings may at least partly explain the apparent
controversial effect of verapamil on MRP1-associated drug
resistance and reflect the entire complexity of MDR, since GSH
reduction and MRP1 downregulation co-exist in reversing the MDR
phenotype in cancer cells.
In conclusion, our data present evidence that
verapamil may effectively enhance the anticancer effect of platinum
drugs on GBC cells with little systemic toxic effects. Verapamil or
more potent and safer verapamil analogs may be further developed in
the frame of a new therapeutic strategies aimed at eliminating MDR
in cancer cells with high MRP1 expression. In addition, we
demonstrate that GSH reduction and MRP1 downregulation are the
mechanisms involved in verapamil reversing the MDR phenotype in GBC
cells. Understanding the complex mechanisms of MDR may lead to
novel approaches in chemotherapy.
Acknowledgements
The authors thank Mrs. Yuying Chen, Mrs. Xiaojiao
Huo and Mrs. Guiying Shi, Shanghai Jiao Tong University School of
Medicine, for their technical assistance in this study. This study
was supported by grants from the Shanghai Science and Technology
Commission (09411960800, to J.W.).
Abbreviations:
|
MDR
|
multidrug resistance
|
|
ABC
|
ATP-binding cassette
|
|
MRP
|
multidrug resistance-related
protein
|
|
CDDP
|
cisplatin
|
|
CBP
|
carboplatin
|
|
GSH
|
glutathione
|
|
ROS
|
reactive oxygen species
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
Annexin V-FITC
|
Annexin V-fluorescein
isothiocyanate
|
|
PI
|
propidium iodide
|
|
DCFH-DA
|
2,7-dichlorodihydrofluorescein
diacetate
|
|
DCF
|
2,7-dichlorofluorescein
|
References
|
1
|
Gourgiotis S, Kocher HM, Solaini L,
Yarollahi A, Tsiambas E and Salemis NS: Gallbladder cancer. Am J
Surg. 196:252–264. 2008. View Article : Google Scholar
|
|
2
|
Zhu AX, Hong TS, Hezel AF and Kooby DA:
Current management of gallbladder carcinoma. Oncologist.
15:168–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Valle J, Wasan H, Palmer DH, et al:
Cisplatin plus gemcitabine versus gemcitabine for biliary tract
cancer. N Engl J Med. 362:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hollebecque A, Bouche O, Romano O, et al:
Experience of gemcitabine plus oxaliplatin chemotherapy in patients
with advanced biliary tract carcinoma. Chemotherapy. 56:234–238.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andre T, Reyes-Vidal JM, Fartoux L, et al:
Gemcitabine and oxaliplatin in advanced biliary tract carcinoma: a
phase II study. Br J Cancer. 99:862–867. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu CP, Calcagno AM and Ambudkar SV:
Reversal of ABC drug transporter-mediated multidrug resistance in
cancer cells: evaluation of current strategies. Curr Mol Pharmacol.
1:93–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao L, Duchrow M, Windhovel U, Kujath P,
Bruch HP and Broll R: Expression of MDR1 mRNA and encoding
P-glycoprotein in archival formalin-fixed paraffin-embedded gall
bladder cancer tissues. Eur J Cancer. 34:1612–1617. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang W, Sun YP, Huang XZ, et al: Emodin
enhances sensitivity of gallbladder cancer cells to platinum drugs
via glutathion depletion and MRP1 downregulation. Biochem
Pharmacol. 79:1134–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aust S, Obrist P, Jaeger W, et al:
Subcellular localization of the ABCG2 transporter in normal and
malignant human gallbladder epithelium. Lab Invest. 84:1024–1036.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suzuki T, Nishio K and Tanabe S: The MRP
family and anticancer drug metabolism. Curr Drug Metab. 2:367–377.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang K, Mack P and Wong KP:
Glutathione-related mechanisms in cellular resistance to anticancer
drugs. Int J Oncol. 12:871–882. 1998.PubMed/NCBI
|
|
12
|
Huang XZ, Wang J, Huang C, et al: Emodin
enhances cytotoxicity of chemotherapeutic drugs in prostate cancer
cells: the mechanisms involve ROS-mediated suppression of multidrug
resistance and hypoxia inducible factor-1. Cancer Biol Ther.
7:468–475. 2008. View Article : Google Scholar
|
|
13
|
Wang W, Sun Y, Li X, et al: Emodin
potentiates the anticancer effect of cisplatin on gallbladder
cancer cells through the generation of reactive oxygen species and
the inhibition of survivin expression. Oncol Rep. 26:1143–1148.
2011.
|
|
14
|
Quan ZW, Yang Y, Li JY, Gong W, Qin YY and
Li SG: The mechanisms of somatostatin induced enhanced
chemosensitivity of gallbladder cancer cell line to doxorubicin:
cell cycle modulation plus target enzyme up-regulation. Biomed
Pharmacother. 64:451–457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang JT, Fan YZ, Chen CQ, Zhao ZM and Sun
W: Norcantharidin: a potential antiangiogenic agent for gallbladder
cancers in vitro and in vivo. Int J Oncol.
40:1501–1514. 2012.PubMed/NCBI
|
|
16
|
Ikeda H, Nakano G, Nagashima K, et al:
Verapamil enhancement of antitumor effect of
cis-diamminedichloroplatinum(II) in nude mouse-grown human
neuroblastoma. Cancer Res. 47:231–234. 1987.PubMed/NCBI
|
|
17
|
Wang J, Wang H, Zhao L, et al:
Down-regulation of P-glycoprotein is associated with resistance to
cisplatin and VP-16 in human lung cancer cell lines. Anticancer
Res. 30:3593–3598. 2010.PubMed/NCBI
|
|
18
|
Perrotton T, Trompier D, Chang XB, Di
Pietro A and Baubichon-Cortay H: (R)- and (S)-verapamil
differentially modulate the multidrug-resistant protein MRP1. J
Biol Chem. 282:31542–31548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang XB, Hou YX and Riordan JR: ATPase
activity of purified multidrug resistance-associated protein. J
Biol Chem. 272:30962–30968. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koski A, Raki M, Nokisalmi P, et al:
Verapamil results in increased blood levels of oncolytic adenovirus
in treatment of patients with advanced cancer. Mol Ther.
20:221–229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Y, Lu Z, Fan P, et al: Clinical
efficacy of chemotherapy combined with verapamil in metastatic
colorectal patients. Cell Biochem Biophys. 61:393–398. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yi J, Yang J, He R, et al: Emodin enhances
arsenic trioxide-induced apoptosis via generation of reactive
oxygen species and inhibition of survival signaling. Cancer Res.
64:108–116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jing Y, Yang J, Wang Y, et al: Alteration
of subcellular redox equilibrium and the consequent oxidative
modification of nuclear factor kappaB are critical for anticancer
cytotoxicity by emodin, a reactive oxygen species-producing agent.
Free Radic Biol Med. 40:2183–2197. 2006. View Article : Google Scholar
|
|
24
|
Igarashi T, Izumi H, Uchiumi T, et al:
Clock and ATF4 transcription system regulates drug resistance in
human cancer cell lines. Oncogene. 26:4749–4760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mwakigonja AR, Kaaya EE, Heiden T, et al:
Tanzanian malignant lymphomas: WHO classification, presentation,
ploidy, proliferation and HIV/EBV association. BMC Cancer.
10:3442010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mwakigonja AR, Pak F, Pyakurel P, et al:
Oral Kaposi’s sarcoma in Tanzania: presentation, immunopathology
and human herpesvirus-8 association. Oncol Rep. 17:1291–1299.
2007.
|
|
27
|
Quan Z, Gu J, Dong P, et al: Reactive
oxygen species-mediated endoplasmic reticulum stress and
mitochondrial dysfunction contribute to cirsimaritin-induced
apoptosis in human gallbladder carcinoma GBC-SD cells. Cancer Lett.
295:252–259. 2010. View Article : Google Scholar
|
|
28
|
Ding XW, Wu JH and Jiang CP: ABCG2: a
potential marker of stem cells and novel target in stem cell and
cancer therapy. Life Sci. 86:631–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Abaan OD, Mutlu PK, Baran Y, Atalay C and
Gunduz U: Multidrug resistance mediated by MRP1 gene overexpression
in breast cancer patients. Cancer Invest. 27:201–205. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Donmez Y, Akhmetova L, Iseri OD, Kars MD
and Gunduz U: Effect of MDR modulators verapamil and promethazine
on gene expression levels of MDR1 and MRP1 in doxorubicin-resistant
MCF-7 cells. Cancer Chemother Pharmacol. 67:823–828. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hait WN and Yang JM: The individualization
of cancer therapy: the unexpected role of p53. Trans Am Clin
Climatol Assoc. 117:85–101. 2006.PubMed/NCBI
|
|
33
|
Tazzari PL, Cappellini A, Ricci F, et al:
Multidrug resistance-associated protein 1 expression is under the
control of the phosphoinositide 3 kinase/Akt signal transduction
network in human acute myelogenous leukemia blasts. Leukemia.
21:427–438. 2007. View Article : Google Scholar
|
|
34
|
Meurette O, Lefeuvre-Orfila L, Rebillard
A, Lagadic-Gossmann D and Dimanche-Boitrel MT: Role of
intracellular glutathione in cell sensitivity to the apoptosis
induced by tumor necrosis factor {alpha}-related apoptosis-inducing
ligand/anticancer drug combinations. Clin Cancer Res. 11:3075–3083.
2005.
|
|
35
|
Cole SP, Sparks KE, Fraser K, et al:
Pharmacological characterization of multidrug resistant
MRP-transfected human tumor cells. Cancer Res. 54:5902–5910.
1994.PubMed/NCBI
|
|
36
|
Cullen KV, Davey RA and Davey MW:
Verapamil-stimulated glutathione transport by the multidrug
resistance-associated protein (MRP1) in leukaemia cells. Biochem
Pharmacol. 62:417–424. 2001. View Article : Google Scholar
|
|
37
|
Barattin R, Perrotton T, Trompier D, et
al: Iodination of verapamil for a stronger induction of death,
through GSH efflux, of cancer cells overexpressing MRP1. Bioorg Med
Chem. 18:6265–6274. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Trompier D, Chang XB, Barattin R, du
Moulinet D’Hardemare A, Di Pietro A and Baubichon-Cortay H:
Verapamil and its derivative trigger apoptosis through glutathione
extrusion by multidrug resistance protein MRP1. Cancer Res.
64:4950–4956. 2004. View Article : Google Scholar : PubMed/NCBI
|