Introduction
HER2 (human epidermal growth factor receptor 2),
which is also termed ErbB2/neu, is a membrane-bound tyrosine kinase
receptor of the epidermal growth factor receptor (EGFR) family
including EGFR, HER2, HER3 and HER4. Ligand binding and/or receptor
overexpression trigger homodimerization or heterodimerization of
HER receptors, which promotes autophosphorylation of the
intracellular tyrosine kinase domain, subsequently leading to
activation of downstream signaling cascades, including the PI3K/Akt
and Erk pathways (1). HER2 plays a
vital role in cell proliferation and survival in HER2-driven breast
cancer (2). Overexpression or
amplification of HER2 occurs in approximately 25% of malignant
breast tumors, and is associated with aggressive disease and
significantly decreased disease-free survival and overall survival
(3,4). Consequently, anti-HER2-targeted
therapies represent an attractive strategy in clinical practice.
Lapatinib, a small-molecule tyrosine kinase inhibitor of EGFR and
HER2, has been shown to inhibit cell proliferation and induce
apoptosis in HER2-overexpressing breast cancer cell lines (5–7). In
the clinic, lapatinib in combination with capecitabine or letrozole
is already successfully used in the treatment of HER2-amplified
locally advanced or metastatic breast cancer patients (8,9).
Nuclear factor κB (NF-κB) is a key mediator of a
variety of cellular processes involving cell cycle control,
differentiation, inflammation and survival (10). The NF-κB family consists of five
subunits: RelA (p65), RelB, c-Rel, p105/p50 and p100/p52. The NF-κB
subunits exist as either homodimers or heterodimers, and the
p65/p50 heterodimeric complex is the most abundant one in human
epithelial cells. In other cell types NF-κB dimers are sequestered
in the cytoplasm via association with the members of the inhibitors
of NF-κB (IκB) family, which includes structurally related proteins
identified as IκB-α, IκB-β, IκB-γ, IκB-ɛ, IκB-ζ and Bcl3. IκB-α is
the most extensively studied member of the IκB family. Upon
stimulation, the activated IκB kinase (IKK) phosphorylates IκB
proteins or IκB-like domains, directing the IκB to
ubiquitin-dependent degradation and releasing NF-κB homodimers or
heterodimers to enter the nucleus where they bind to specific DNA
sequences and promote the transcription of target genes (11,12).
In most canonical NF-κB signaling, IκB-α is phosphorylated by IKK
at Ser32 and Ser36, ubiquitylated at Lys21 and Lys22 and
subsequently degraded by the 26S proteasome (11).
NF-κB-associated pathways are tightly linked to
apoptosis, proliferation, invasion and angiogenesis in breast
cancer cells (13–15). Increasing evidence suggests that
constitutive activation of NF-κB appears to be a critical
determinant of chemoresistance (16,17),
endotherapy resistance (18) and
radioresistance (19) in breast
cancers.
It has been described that NF-κB can be activated by
HER2 in breast cancer cells (20,21).
However, the impact of lapatinib as a HER2 inhibitor on the
activity of NF-κB is still not completely clear. In the present
study, we aimed to investigate the effect of lapatinib on NF-κB
activity in breast cancer cells. In addition, we established a
lapatinib-resistant cell model by chronically exposing SKBR3 cells
to increasing concentrations of lapatinib and characterized the
activity of NF-κB in the resistant cells.
Materials and methods
Cell lines and cell culture
The human breast cancer cell lines, MDA-MB-435,
MDA-MB-231, MDA-MB-468, SKBR3 and MDA-MB-453, were obtained from
the American Type Culture Collection (Manassas, VA). Cells were
cultured in RPMI-1640 or DMEM supplemented with 10% fetal bovine
serum at 37°C with 5% CO2 in a humidified incubator.
Compounds and antibodies
Lapatinib was kindly provided by GlaxoSmithKline.
AZD6244 was purchased from Selleck Chemicals (Woburn, MA). LY294002
was purchased from Cell Signaling Technology (Beverly, MA). In
these cases, 10 mmol/l aliquots of drug in DMSO (dimethyl
sulfoxide) were stored at-20°C and diluted just before utilization.
All antibodies were purchased from commercial sources as indicated
below: anti-Neu (F-11), anti-p-Neu (Tyr1248)-R, anti-ErbB-3 (C-17),
anti-p-ErbB-3 (Tyr1328), anti-p-Akt1/2/3 (Ser 473)-R, anti-p-Erk
(E-4), anti-Akt (Ser 473)-R, anti-Erk (E-4), NF-κB p50 and β-actin
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). EGFR, p-EGFR, phospho-NF-κB p65 (Ser536),
NF-κB p65 (C22B4), IκB-α (L35A5), phospho-IκB-α (Ser32) and histone
H3 antibodies were obtained from Cell Signaling Technology.
HRP-conjugated goat-anti-rabbit IgG, goat-anti-mouse IgG and
donkey-anti-goat IgG antibodies were purchased from Santa Cruz
Biotechnology, Inc. or Cell Signaling Technology.
Development of lapatinib-resistant breast
cancer cells
To generate lapatinib-resistant SKBR3 cells
(rSKBR3), parental SKBR3 cells were initially grown in medium
containing 0.25 μmol/l lapatinib. The concentration of lapatinib in
medium was gradually increased to 5 μmol/l over 18 months. The
resistant cells were constantly pooled by collecting all viable
cells of several plates onto 1 plate. Single-cell clones were
isolated from the pooled resistant cells. Finally, the resistant
cells were maintained in medium supplemented with 5 μmol/l
lapatinib. Resistance to lapatinib was confirmed by a cell
viability assay as described below. The resistant cells were
cultured in medium without lapatinib for 2 or 3 days before each
experiment.
Cell viability assay
Cell viability was determined using the CCK-8 (Cell
Counting Kit-8; Dojindo, Japan) assay. Briefly, parental and
resistant cells were seeded at a density of 3–5×103
cells into a 96-well microplate and incubated overnight. The cells
were then treated with various concentrations of lapatinib. After
incubation for 48 h, 10 μl of CCK-8 solution was added to each
well, and the plates were further incubated for 3 h at 37°C. The
absorbance at 450 nm was measured with an absorbance microplate
reader. Cell survival for all experiments was expressed as the
percentage of viable cells compared with untreated cells.
Western blot analysis
Cells were washed twice in cold PBS and then lysed
in RIPA buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 0.25% (w/v)
deoxycholate, 1% NP-40, 5 mM sodium orthovanadate, 2 mM sodium
fluoride and a protease inhibitor cocktail]. The protein
concentration of the supernatants was determined using a
modification of the Bradford method. Equal amounts of proteins (40
μg) were resolved by SDS-PAGE. Proteins were transferred to PVDF
membranes and blocked with 5% non-fat milk in PBS-T. Membranes were
incubated overnight with specific antibodies. After four washes in
PBS-T, membranes were then incubated with horseradish
peroxidase-linked secondary antibody at a 1:2,000 dilution. Target
proteins were visualized with the SuperSignal West Femto Maximum
Sensitivity Substrate kit and subsequent exposure to X-OMAT X-ray
film according to the manufacturer’s instructions.
Nuclear extract preparation and
electrophoretic mobility shift assay
Nuclear extracts were prepared using a nuclear and
cytoplasmic proteins extract kit purchased from Beyotime Institute
of Biotechnology (Haimen, China). Electrophoretic mobility shift
assay (EMSA) was performed using a chemiluminescent assay kit
according to the protocol provided by the vendor (Beyotime
Institute of Biotechnology) with minor modifications. Briefly, for
each reaction, 20 μg of nuclear protein extracts was incubated with
biotin end-labeled probe. The protein-DNA complex was then resolved
by electrophoresis on 5% native polyacrylamide gel and transferred
to a nylon membrane. DNA on the membrane was immediately
cross-linked for 10 min using an UV cross-linker. The biotin
end-labeled DNA probe was detected using streptavidin conjugated to
horseradish peroxidase (HRP) and a chemiluminescent substrate.
Biotin-labeled double-strand NF-κB consensus oligonucleotide
(5′-AGT TGA GGG GAC TTT CCC AGG C-3′) and Oct-1 oligonucleotide
(5′-TGT CGA ATG CAA ATC ACT AGA A-3′) were obtained from Beyotime
Institute of Biotechnology.
Results
Lapatinib inhibits activation of NF-κB in
HER2-overexpressing breast cancer cells
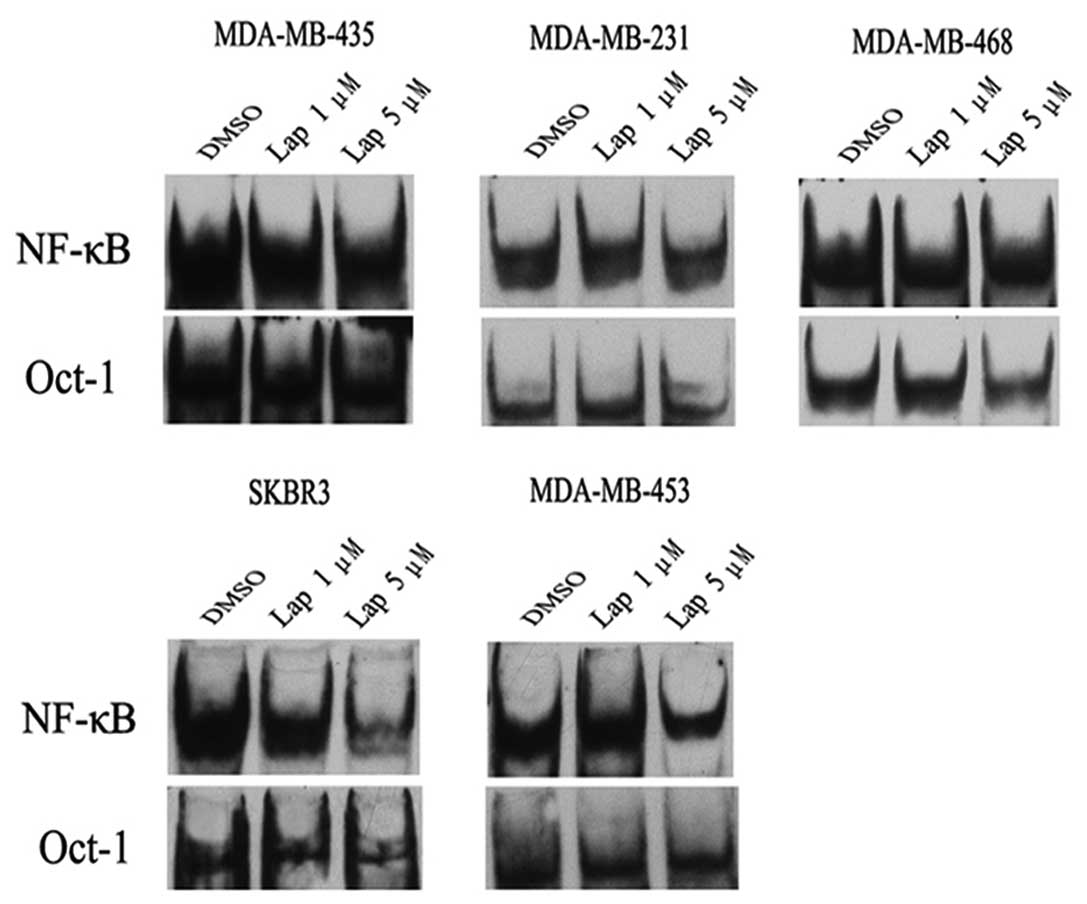
To determine the effect of lapatinb on the activity
of NF-κB, we performed EMSA. In SKBR3 cells, lapatinib inhibited
activation of NF-κB in a dose-dependent manner (Fig. 1). Similarly, a high dose of
lapatinib (5 μmol/l) markedly reduced the activity of NF-κB in
another HER2-overexpressing breast cancer cell line, MDA-MB-453. In
contrast, lapatinib had almost no impact on the activity of NF-κB
in non-HER2-overexpressing breast cancer cell lines, including
MDA-MB-231, MDA-MB-468 and MDA-MB-435 (Fig. 1). These results suggest that
lapatinib inhibits the activation of NF-κB in HER2-overexpressing
breast cancer cells.
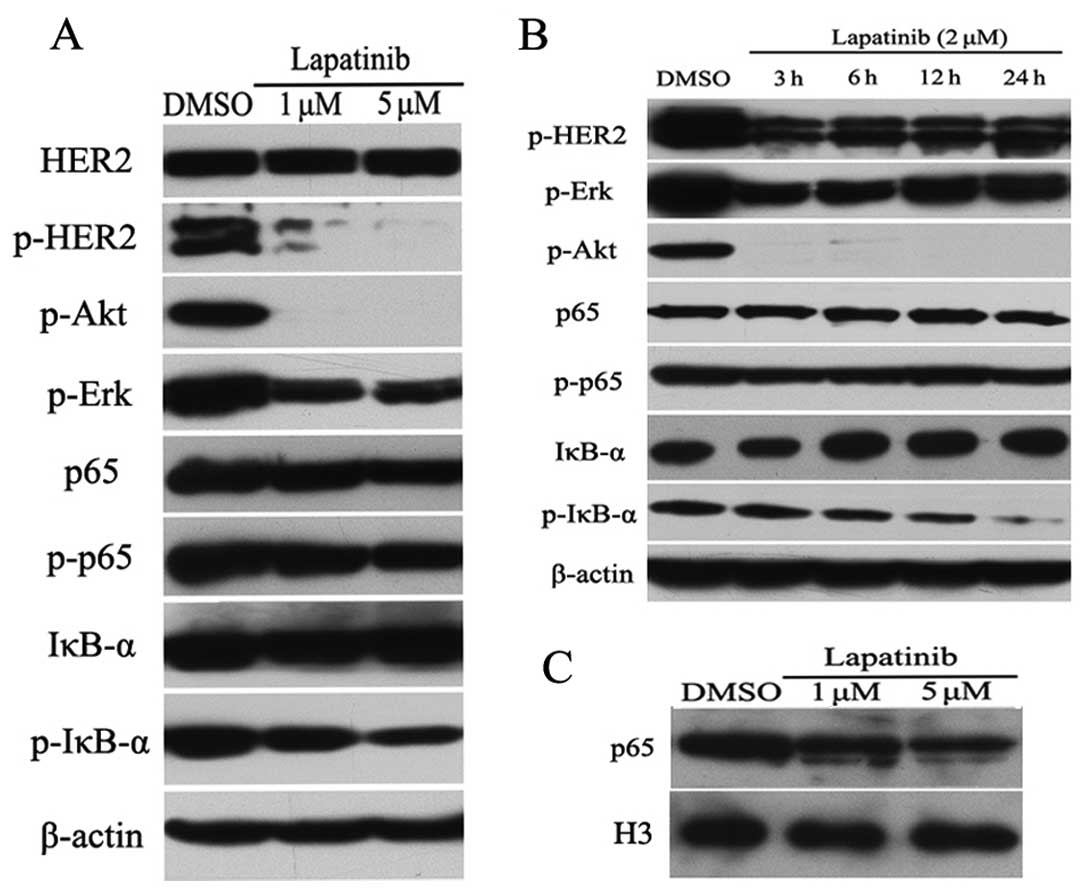
To better understand how lapatinib inactivates NF-κB
in HER2-positive breast cancer cells, we performed western blot
analysis. As expected, lapatinib reduced the expression levels of
p-HER2, p-Akt and p-Erk proteins in a dose-dependent manner in
SKBR3 cells (Fig. 2A). Importantly,
treatment with lapatinib resulted in a decreased phosphorylation of
IκB-α in SKBR3 cells, whereas lapatinib had almost no impact on the
expression levels of total HER2, Akt, Erk, p65 and p-p65 proteins
(Fig. 2A). We then treated SKBR3
cells with 2 μmol/l lapatinib for different times, and measured the
protein expression levels of HER2 and NF-κB pathways by western
blot analysis. As shown in Fig. 2B,
lapatinib reduced the phosphorylation of IκB-α in a time-dependent
manner, whereas lapatinib increased the expression level of total
IκB-α protein. To further confirm the inhibitory role of lapatinib
on NF-κB activity, we evaluated cellular localization of p65 by
western blot analysis. Interestingly, lapatinib inhibited p65
nuclear accumulation in a dose-dependent manner in SKBR3 cells
(Fig. 2C).
Establishment of lapatinib-resistant
SKBR3 cells
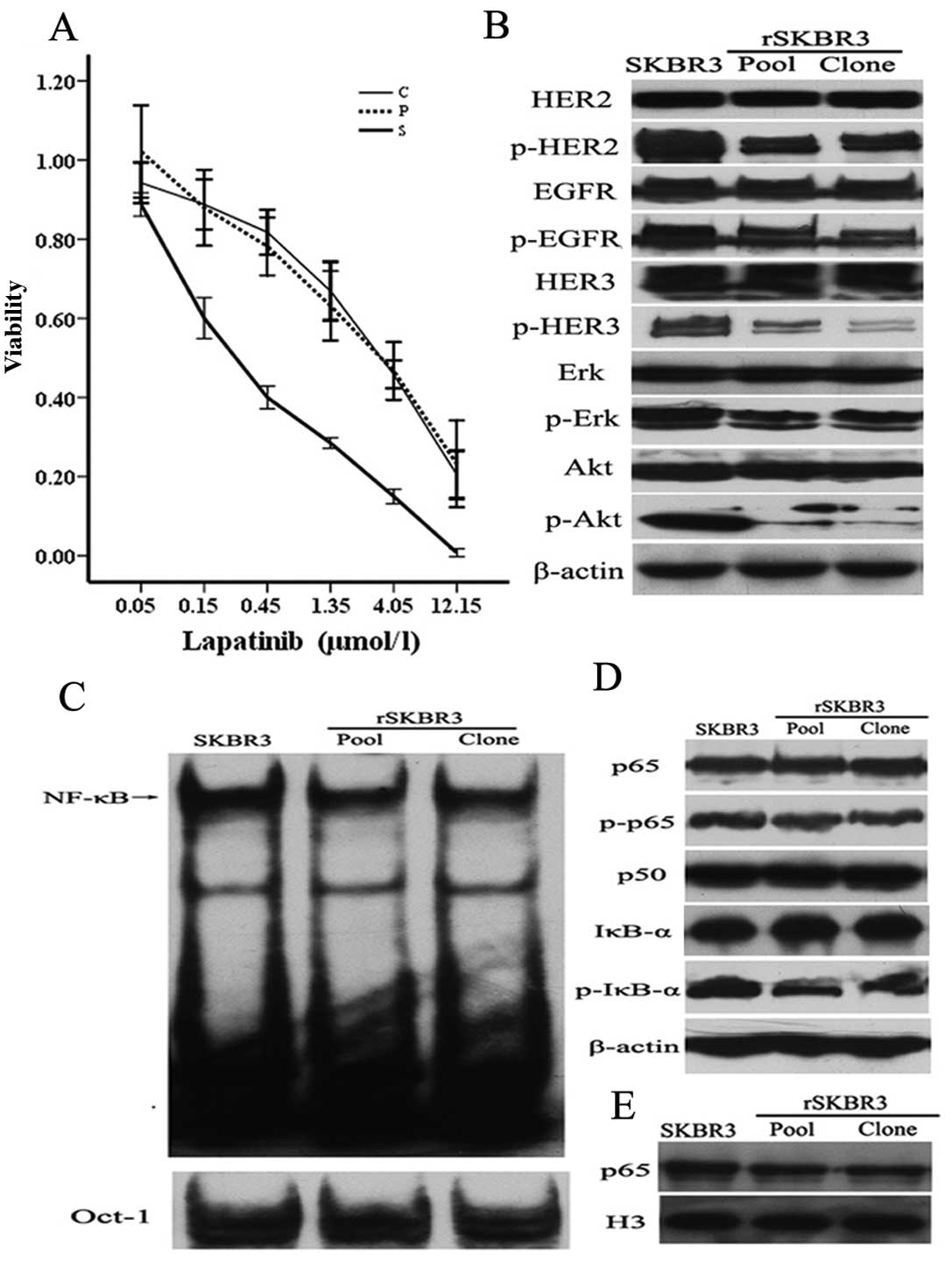
To measure the relative resistance of parental SKBR3
and rSKBR3 cells to lapatinib, we performed CCK-8 assays. As
indicated in Fig. 3A, the 50%
inhibitory concentrations (IC50) of lapatinib in
parental SKBR3 cells, pooled resistant cells and representative
clone were ~0.3, 3.7 and 3.8 μmol/l, respectively, suggesting that
SKBR3 cells gradually became insensitive to lapatinib. We next
assayed the expression levels of several HER signaling pathway
proteins in the parental and resistant rSKBR3 cells by immunoblot
analysis. The expression levels of p-HER2, p-HER3, p-EGFR, p-Erk
and p-Akt proteins decreased in rSKBR3 cells in comparison with
parental SKBR3 cells, while the expression levels of total HER2,
HER3, EGFR, Erk and Akt proteins had almost no change (Fig. 3B). This result demonstrated that
lapatinib still retained its inhibitory role of EGFR/HER2 signaling
in our resistant cell model.
Decreased NF-κB activity in
lapatinib-resistant SKBR3 cells
We next examined whether the activity of NF-κB
changed in our cell model. A moderately decreased NF-κB activity
was observed in rSKBR3 cells, compared with parental SKBR3 cells
(Fig. 3C).
We next assayed the expression levels of NF-κB
pathway proteins by western blot analysis. As showed in Fig. 3D, the expression level of the
p-IκB-α protein was markedly decreased in the pooled resistant
cells and representative clone, when compared with the parental
SKBR3 cells. The expression levels of p-p65, p65 and p50 had no
significant difference between the parental and resistant rSKBR3
cells (Fig. 3D). We further
evaluated the cellular localization of p65 by western blot
analysis. Nuclear accumulation of p65 was decreased in the
resistant cells, compared with the parental SKBR3 cells (Fig. 3E).
Lapatinib inhibits the activation of
NF-κB through the PI3K/Akt pathway
We previously showed that lapatinib inhibits
activation of the PI3K/Akt and Erk pathways in SKBR3 cells
(6). Thus it was tempting to
speculate that lapatinib may inhibit NF-κB activation through
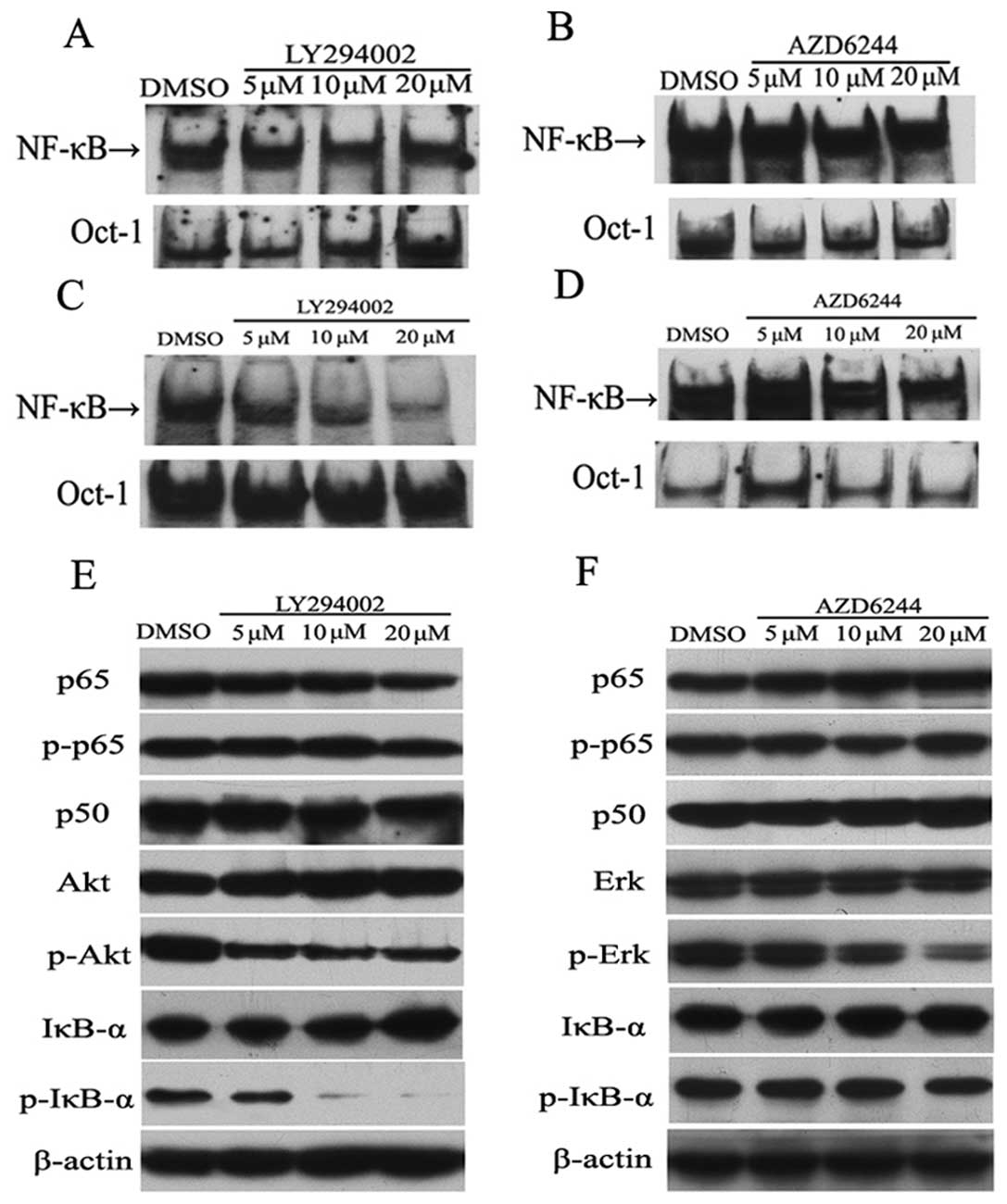
blocking these pathways. To address this possibility, we treated
SKBR3 and MDA-MB-453 cells with the PI3K inhibitor LY294002 and the
MEK inhibitor AZD6244, and measured the activity of NF-κB by EMSA.
As indicated in Fig. 4A, a deceased
NF-κB activity was observed in the SKBR3 cells treated with
LY294002. In MDA-MB-453 cells, LY294002 inhibited activation of
NF-κB in a dose-dependent manner (Fig.
4C). However, AZD6244 had relatively little impact on NF-κB DNA
binding in SKBR3 cells (Fig. 4B),
as well as in MDA-MB-453 cells (Fig.
4D).
We next examined the impact of LY294002 and AZD6244
on the expression level of NF-κB pathway proteins by western blot
analysis. In SKBR3 cells, LY294002 reduced phosphorylation of Akt
in a concentration-dependent manner (Fig. 4E). Importantly, phosphorylation of
IκB-α was potently inhibited by LY294002, while the expression
level of total IκB-α was increased, particularly in the presence of
the highest dose of LY294002 (Fig.
4E). As expected, phosphorylation of Erk was reduced by AZD6244
in a dose-dependent manner in SKBR3 cells, whereas there was no
detectable change in IκB-α phosphorylation in SKBR3 cells (Fig. 4F). AZD6244 had almost no effect on
the expression levels of p-p65, p65, p50 and total IκB-α.
Discussion
In the present study, we showed that lapatinib
potently inhibited activation of NF-κB in two HER2-overexpressing
breast cancer cell lines, SKBR3 and MDA-MB-453. We also found that
there was a decreased NF-κB activity in the lapatinib-resistant
SKBR3 cells. We further investigated the potential mechanism of
lapatinib-induced inactivation of NF-κB. We found that lapatinib
reduced the phosphorylation of IκB-α in SKBR3 cells, as well as in
the rSKBR3 cells. We also demonstrated that lapatinib inhibited
phosphorylation of IκB-α probably though blocking PI3K/Akt
activation.
It was previously shown that there was a positive
correlation between HER2 overexpression and constitutive activation
of NF-κB in breast cancer cells (13,20,21).
Biswas et al(13) revealed
that herceptin, a recombinant humanized monoclonal
antibody-directed HER2 receptor, blocked heregulin-induced NF-κB
activation in a concentration-dependent manner in SKBR3 cells. In a
separate study, it was reported that inhibition of HER-2 by AG825
or knockdown of HER-2 by RNA interference blocked TNFα-induced
NF-κB activation in breast cancer cells (20). Hence it is not entirely surprising
that lapatinib, a small-molecule inhibitor of HER2, has the ability
to inhibit the activation of NF-κB in HER2-overexpressing breast
cancer cells. Our finding is in keeping with a previous study
showing that lapatinib induced a 3-fold downregulation of NF-κB1
mRNA expression in LoVo cells (22).
Evidence suggests that the primary mechanism of
action of lapatinib is not compromised in cells with acquired
resistance (23–25). Liu et al(23) previously showed that lapatinib
inhibited tyrosine phosphorylation of HER1, HER2 and HER3 in both
lapatinib-sensitive BT474 cells and lapatinib-resistant BT474J4
cells. In this study, we showed that lapatinib still retained its
ability to inhibit phosphorylation of HER2, EGFR, Erk and Akt in
the rSKBR3 cells. Furthermore, we indicated that there was
decreased NF-κB activity and nuclear accumulation of p65 in our
rSKBR3 cells. Our findings in the resistant cell model further
confirmed the inhibitory role of lapatinib in NF-κB activation.
NF-κB activation is regulated primarily by two
pathways, the classical and the alternative pathways (26). In the canonical pathway, NF-κB is
retained in the cytoplasm as an inactive heterotrimer consisting of
three subunits: p50, p65 and IκB-α. On activation, IκB-α is
phosphorylated by the IKK holo-complex, leading to its degradation
by the ubiquitin/proteasome system, thereby releasing NF-κB dimers
to the nucleus (11,12). Degradation of IκB-α alters the
dynamic balance between cytosolic and nuclear localization signals
to favor nuclear localization of NF-κB (11). It has been reported that inhibition
of IκB-α phosphorylation leads to inactivation of NF-κB in glioma
cell lines (27). Additionally, Wu
et al(28) reported that
paeoniflorin inhibited NF-κB activation by reducing the degradation
of IκB-α through preventing its phosphorylation in gastric
carcinoma cells. In the present study, we convincingly demonstrated
that lapatinib reduced phosphorylation of IκB-α in a time- and
dose-dependent manner in SKBR3 cells. Furthermore, we revealed that
there was a decreased phosphorylation of IκB-α with subsequent
reduction in NF-κB activity in the lapatinib-resistant SKBR3 cells.
Thus, lapatinib appears to inhibit activation of NF-κB by reducing
phosphorylation of IκB-α.
Overwhelming evidence has indicated that PI3K/Akt
signaling triggers the activation of NF-κB and the underlying
molecular mechanisms are multifactorial (29,30).
Madrid et al(31)
demonstrated that the Akt-induced potentiation of p65
transactivation capacity requires the activation of IKKβ kinase and
MAPK p38. Recent data also described the ability of Akt to
stimulate IKK activity directed toward the phosphorylation of IκB-α
and RelA/p65 in prostate cancer cells (32). In the present study, we indicated
that the PI3K inhibitor LY294002 reduced phosphorylation of Akt,
and inhibited activation of NF-κB in HER2-overexpressing breast
cancer cells. Furthermore, we showed that LY294002 treatment
reduced phosphorylation of IκB-α in SKBR3 cells. Importantly, we
revealed that the MEK inhibitor AZD6244 had no ability to inhibit
phosphorylation of IκB-α in SKBR3 cells. The PI3K/Akt and Erk
pathways are putative downstream signals of HER2 (1,2). As
mentioned above, we convincingly demonstrated that lapatinib
reduced the phosphorylation of Akt and IκB-α in SKBR3 cells. Hence,
it is likely that lapatinib inhibits NF-κB activation through
reduction of IκB-α phosphorylation via blocking of the PI3K/Akt
cascade.
In conclusion, the current data presented herein
indicate that lapatinib potently inhibited the activation of NF-κB
in HER2-overexpressing breast cancer cells, and there was a
decreased NF-κB activity in lapatinib-resistant SKBR3 cells.
Lapatinib appears to inactivate NF-κB through reduction of the
phosphorylation of IκB-α via blocking of the PI3K/Akt cascade.
Acknowledgements
We thank GlaxoSmithKline for providing the lapatinib
(GW572016 and GSK572016). The study was supported by a grant (no.
ZR2010HM133) from the Natural Science Foundation of Shandong
Province (to M.C.) in 2011.
References
|
1
|
Citri A and Yarden Y: EGF-ERBB signaling:
towards the systems level. Nat Rev Mol Cell Biol. 7:505–516. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landgraf R: HER2 (ERBB2)-functional
diversity from structurally conserved building blocks. Breast
Cancer Res. 9:2022007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Slamon DJ, Clark GM, Wong SG, et al: Human
breast cancer: correlation of relapse and survival with
amplification of the HER-2/neu oncogene. Science. 235:177–182.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esteva F, Yu D, Hung M and Hortobagyi G:
Molecular predictors of response to trastuzumab and lapatinib in
breast cancer. Nat Rev Clin Oncol. 2:98–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rusnak DW, Lackey K, Affleck K, et al: The
effects of the novel, reversible epidermal growth factor
receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of
human normal and tumor-derived cell lines in vitro and in vivo. Mol
Cancer Ther. 1:85–94. 2001.
|
|
6
|
Ma C, Niu X, Luo J, Shao Z and Shen K:
Combined effects of lapatinib and bortezomib in human epidermal
receptor 2 (HER2)-overexpressing breast cancer cells and activity
of bortezomib against lapatinib-resistant breast cancer cells.
Cancer Sci. 10:2220–2226. 2010. View Article : Google Scholar
|
|
7
|
Konecny GE, Pegram MD, Venkatesan N, et
al: Activity of the dual kinase inhibitor lapatinib (GW572016)
against HER-2-overexpressing and trastuzumab-treated breast cancer
cells. Cancer Res. 66:1630–1639. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Geyer CE, Forster J, Lindquist D, et al:
Lapatinib plus capecitabine for HER2-positive advanced breast
cancer. N Engl J Med. 355:2733–2743. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Johnston S, Pippen J Jr, Pivot X, et al:
Lapatinib combined with letrozole versus letrozole and placebo as
first-line therapy for postmenopausal hormone receptor-positive
metastatic breast cancer. J Clin Oncol. 27:5538–5546. 2009.
View Article : Google Scholar
|
|
10
|
Ghosh S, May MJ and Kopp EB: NF-κB and Rel
proteins: evolutionarily conserved mediators of immune responses.
Annu Rev Immunol. 16:225–260. 1998.
|
|
11
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 3:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng C, Yin Q and Wu H: Structural
studies of NF-κB signaling. Cell Res. 1:183–195. 2011.
|
|
13
|
Biswas DK, Shi Q, Baily S, et al: NF-κB
activation in human breast cancer specimens and its role in cell
proliferation and apoptosis. Proc Natl Acad Sci USA.
101:10137–10142. 2004.
|
|
14
|
Merkhofer E, Cogswell P and Baldwin A:
Her2 activates NF-κB and induces invasion through the canonical
pathway involving IKKα. Oncogene. 8:1238–1248. 2010.
|
|
15
|
Liu M, Ju X, Willmarth N, et al: Nuclear
factor-κB enhances ErbB2-induced mammary tumorigenesis and
neoangiogenesis in vivo. Am J Pathol. 5:1910–1920. 2009.
|
|
16
|
Montagut C, Tusquets I, Ferrer B, et al:
Activation of nuclear factor-κB is linked to resistance to
neoadjuvant chemotherapy in breast cancer patients. Endocr Relat
Cancer. 2:607–616. 2006.
|
|
17
|
Manna S, Manna P and Sarkar A: Inhibition
of RelA phosphorylation sensitizes apoptosis in constitutive
NF-kappaB-expressing and chemoresistant cells. Cell Death Differ.
1:158–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
deGraffenried L, Chandrasekar B,
Friedrichs W, et al: NF-κB inhibition markedly enhances sensitivity
of resistant breast cancer tumor cells to tamoxifen. Ann Oncol.
15:885–890. 2004.
|
|
19
|
Papanikolaou V, Iliopoulos D, Dimou I, et
al: Survivin regulation by HER2 through NF-κB and c-myc in
irradiated breast cancer cells. J Cell Mol Med. 7:1542–1550.
2011.
|
|
20
|
Rivas M, Tkach M, Beguelin W, et al:
Transactivation of ErbB-2 induced by tumor necrosis factor α
promotes NF-κB activation and breast cancer cell proliferation.
Breast Cancer Res Treat. 122:111–124. 2010.
|
|
21
|
Guo G, Wang T, Gao Q, et al: Expression of
ErbB2 enhances radiation-induced NF-κB activation. Oncogene.
23:535–545. 2004.PubMed/NCBI
|
|
22
|
LaBonte M, Wilson P, Fazzone W, et al: The
dual EGFR/HER2 inhibitor lapatinib synergistically enhances the
antitumor activity of the histone deacetylase inhibitor
panobinostat in colorectal cancer models. Cancer Res. 10:3635–3648.
2011. View Article : Google Scholar
|
|
23
|
Liu L, Greger J, Shi H, et al: Novel
mechanism of lapatinib resistance in HER2-positive breast tumor
cells: activation of AXL. Cancer Res. 69:6871–6878. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aird K, Ghanayem R, Peplinski S, Lyerly H
and Devi G: X-Linked inhibitor of apoptosis protein inhibits
apoptosis in inflammatory breast cancer cells with acquired
resistance to an ErbB1/2 tyrosine kinase inhibitor. Mol Cancer
Ther. 5:1432–1442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang C, Park C, Hilsenbeck S, et al: β1
Integrin mediates an alternative survival pathway in breast cancer
cells resistant to lapatinib. Breast Cancer Res. 4:R842011.
|
|
26
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyakoshi J and Yagi K: Inhibition of
IκB-α phosphorylation at serine and tyrosine acts independently on
sensitization to DNA damaging agents in human glioma cells. Br J
Cancer. 1:28–33. 2000.
|
|
28
|
Wu H, Li W, Wang T, Shu Y and Liu P:
Paeoniflorin suppress NF-κB activation through modulation of IκBα
and enhances 5-fluorouracil-induced apoptosis in human gastric
carcinoma cells. Biomed Pharmacother. 9:659–666. 2008.
|
|
29
|
Salminen A and Kaarniranta K:
Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-κB
signaling. Cell Signal. 4:573–577. 2010.PubMed/NCBI
|
|
30
|
Romashkova JA and Makarov SS: NF-κB is a
target of AKT in anti-apoptotic PDGF signalling. Nature. 401:86–90.
1999.
|
|
31
|
Madrid L, Mayo M, Reuther J and Baldwin A:
Akt stimulates the transactivation potential of the RelA/p65subunit
of NF-κB through utilization of the IκB kinase and activation of
the mitogen-activated protein kinase p38. J Biol Chem.
22:18934–18940. 2001.
|
|
32
|
Dan H, Cooper M, Cogswell P, Duncan J,
Ting J and Baldwin A: Akt-dependent regulation of NF-κB is
controlled by mTOR and Raptor in association with IKK. Genes Dev.
11:1490–1500. 2008.
|