Introduction
Integrins, cell surface receptors that mediate
cell-extracellular matrix (ECM) and cell-cell interactions,
function to modulate cell behaviors including cell adhesion,
migration, proliferation, survival, invasion and angiogenesis
(1–3). Integrin-mediated signaling cascades
include the activation of a variety of protein kinases such as
focal adhesion kinase, Src-family kinases, extracellular
signal-regulated kinase (ERK), and phosphatidylinositol 3-kinase
(PI3K)/Akt (1,3). These signaling events are complex and
very similar to those triggered by growth factor receptors,
demonstrating cross-talk between ECM- and growth factor-induced
signal transduction pathways, which are involved in physiological
and pathological processes (3–5).
Integrin α3β1, a major receptor for laminin, is expressed on many
types of cancer cells and plays the pivotal roles in regulation of
cancer progression (3,6). Reduced integrin α3 expression in lung
cancer is probably associated with increased aggressiveness and
poor prognosis in lung cancer patients (7,8).
Although significant advances have been made in understanding the
function of integrin α3β1, the roles and molecular mechanisms in
mediating the regulation of malignant cell behavior still remain
unexplored.
The epidermal growth factor receptor (EGFR) is a
receptor tyrosine kinase which is highly expressed or activated in
a variety of human cancers including lung, ovary, stomach, brain,
breast and colon cancer, and it has therefore been known as the
main therapeutic target for cancer treatment (9–11).
EGFR-dependent signaling pathways include the activation of Akt and
ERK, which are implicated in the cell proliferation, survival,
migration and invasion (9). In
addition, recent studies have established that nuclear factor-κB
(NF-κB) is closely associated with regulation of cell proliferation
and anti-apoptosis as well as inflammation and immune responses
(12,13). NF-κB activation can either promote
or suppress the proliferation and survival of cancer cells,
depending on the cell and tissue types as well as the expression
status of tumor suppressor proteins such as p53 or phosphatase and
tensin homolog (12).
In the present study, we evaluated the biological
effects and molecular mechanisms of integrin α3β1 on cell
proliferation and migration in p53-deficient human lung cancer
H1299 cells. Enhanced proliferation and migration in integrin
α3-silenced cells are mediated by activation of Akt and
ERK-dependent pathways and induction of cyclin-dependent kinases,
EGFR, and NF-κB-inducible anti-apoptotic protein Bcl-2. These
findings indicate that reduced integrin α3 expression in
p53-deficient NSCLC cells results in multiple phenotypic changes
that enhance the aggressiveness.
Materials and methods
Cell culture conditions
Human lung carcinoma cells (H1299) from American
Type Culture Collection (Manassas, VA, USA) were grown in 10% fetal
bovine serum-Dulbecco’s modified Eagle’s medium (FBS-DMEM) (HyClone
Laboratories, Logan, UT, USA).
Reagents
PD98059 (MEK 1/2 inhibitor) and LY294002 (PI3K
inhibitor) were obtained from Upstate Biotechnology (Lake Placid,
NY, USA). The following antibodies were purchased from commercial
sources: anti-phospho-ERK (T202/Y204), anti-phospho-Akt (Ser473),
anti-lamin A/C (Cell Signaling, Beverly, MA, USA); anti-integrin β1
(BD Biosciences, Bedford, MA, USA); anti-integrin α3, anti-EGFR,
anti-ERK, anti-Akt, anti-Cdk4, anti-Cdk2, anti-p27Kip1,
anti-NF-κB, anti-Bcl-2, anti-actin antibodies, and mouse, rabbit
and goat IgG-horseradish peroxidase conjugates (Santa Cruz
Biotechnology, Santa Cruz, CA, USA).
RNA purification and RT-PCR
Total RNA was purified with easy Blue™ Total RNA
extraction kit (iNtRON Biotechnology, Sungnam, Gyeonggi, Korea).
Integrity of RNA was checked by agarose gel electrophoresis and
ethidium bromide staining. RNA (1 μg) was used as template for each
reverse-transcriptase (RT)-mediated polymerase chain reaction (PCR)
by using the ImProm-II™ Reverse Transcription System (Promega,
Madison, WI). Primers for PCR were synthesized by Bioneer
Corporation (Daejeon, Korea). Primer sequences were: integrin α3,
forward 5′-AAGCCAAGTCTGAGACT-3′ and reverse
5′-GTAGTATTGGTCCCGAGTCT-3′; glyceraldehyde-3-phosphate
dehydrogenase (GAPDH), forward 5′-GAAGGTGAAGGTCGGAGTC-3′ and
reverse 5′-GAAGATGGTGATGGGATTTC-3′ (14).
siRNA preparation and transfection
For design of siRNA inserts, a cDNA sequence of
integrin α3 AGCAACACAGACTACCTGGAG was selected according to the
InvivoGen siRNAWizard program based on a BLAST search. As a control
we used the scrambled oligonucleotide sequence
AGCATATGTGCGTACCTAGCT, available prepackaged in the psiRNA-hH1zeo
vector (InvivoGen, San Diego, CA, USA). Both the siRNA-targeting
integrin α3 gene and scrambled sequences cloned into psiRNA-hH1zeo
vector were designated α3 shRNA and control shRNA, respectively.
These vector constructs were transfected into H1299 cells using
LyoVec (InvivoGen) according to the manufacturer’s instructions.
After 48 h, the cells were selected with zeocin in 10% FBS-DMEM for
1–2 weeks until positive colonies formed (15).
Adhesion assay
Subconfluent cells were detached with trypsin and
allowed to recover in 10% FBS-DMEM for 1 h at 37°C with gentle
rocking. After recovery, the cells were collected by low-speed
centrifugation and resuspended in fresh 10% FBS-DMEM. The cells
were plated on non-coated, laminin-coated, or collagen-coated
96-well plates (1.5×104 cells/well), and further
incubated for 2 h at 37°C. Following incubation unattached cells
were removed by washing the wells three times with ice-cold
phosphate buffered saline (PBS, pH 7.4). Attached cells were fixed
with methanol, and then stained with 0.04% Giemsa staining solution
(Sigma-Aldrich Co., St. Louis, MO, USA). The cells were
photographed and counted. The results (mean ± standard deviation)
are presented as the fold-increase of the untreated adherent cells
(16).
Cell growth assay
Subconfluent control shRNA or integrin α3
shRNA-transfected H1299 cells, plated on 6-well plates
(2×104 cells/well), were serum-starved for 48 h to
synchronize cells in G1/G0 phase of cell
cycle, and further incubated with 10% FBS-DMEM for the indicated
time points in the presence or absence of MEK or PI3K inhibitor.
The cells were then washed in ice-cold PBS, detached with trypsin,
and counted using trypan blue exclusion method. The results from
triplicate determinations (mean ± standard deviation) are presented
as the numbers of cells per culture or the fold-increase of the
untreated controls.
Migration assay
Cell migration was quantified in the in vitro
wound-healing assay as previously described (17). After cells were plated on 48-well
plates, grown to confluence, and a single wound was created in the
center of the cell monolayer by the gentle removal of the attached
cells with a sterile plastic pipette tip. Cells were pretreated
with or without MEK or PI3K inhibitor for 30 min, followed by serum
stimulation for 18 h. Cells were fixed with methanol, and then
stained with 0.04% Giemsa staining solution. The migration of the
cells into the wound was observed with still images taken at the
indicated time point.
Western blot analysis
Subconfluent cells in 100-mm dishes (BD Biosciences)
were serum-starved for 48 h in DMEM and replaced with fresh media,
followed by treatments for different time points, as indicated.
Cells were rinsed twice with ice-cold PBS and lysed by incubation
in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, 1% Triton
X-100, 1 mM EDTA, 100 μg/ml 4-(2-aminoethyl)benzenesulfonyl
fluoride, 10 μg/ml aprotinin, 1 μg/ml pepstatin A, 0.5 μg/ml
leupeptin, 80 mM β-glycerophosphate, 25 mM NaF and 1 mM sodium
orthovanadate for 30 min at 4°C. Cell lysates were clarified at
13,000 × g for 20 min at 4°C, and the supernatants were subjected
to western blot analysis as described previously (18).
Subcellular fractionation
Following treatments as indicated, cells were rinsed
twice with ice-cold PBS, and cytoplasmic and nuclear extracts were
prepared using Nuclear/Cytosol Fractionation kit (BioVision Inc.,
Mountain View, CA, USA), according to the manufacturer’s
instructions.
Zymogram analysis
Activities of matrix metalloproteinases were
measured by zymography (19).
Aliquots of conditioned medium were diluted in sample buffer,
applied to 10% polyacrylamide gels containing 1 mg/ml gelatin
(Sigma-Aldrich Co.) as a substrate. After electrophoresis, the gels
were incubated in 2.5% Triton X-100 for 1 h to remove SDS and allow
re-naturalization of MMPs, and further incubated in developing
buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM CaCl2,
and 150 mM NaCl for 20 h at 37°C. The gels were stained with 0.5%
Coomassie brilliant blue R-250 in 30% methanol-10% acetic acid for
2 h and followed by destaining with 30% methanol-10% acetic acid.
Gelatinolytic activities were detected as unstained bands against
the background of the Coomassie blue-stained gelatin.
Statistical analysis
Statistical analysis was performed using Student’s
t-test, and was based on at least three different experiments. The
results were considered to be statistically significant at
p<0.05.
Results
Integrin α3 silencing-induced cell
proliferation is mediated by enhanced expression and nuclear
localization of cyclin-dependent kinases
It has been reported that integrin α3β1 plays the
pivotal roles in cell growth and migration (3,6). To
investigate the biological roles of integrin α3β1 in p53-deficient
NSCLC, H1299 cells were transfected with psiRNA-hH1zeo vector
including siRNA-targeting integrin α3 or scrambled sequences
(15,18), and the cells were designated α3
shRNA and control shRNA, respectively. RT-PCR and western blot
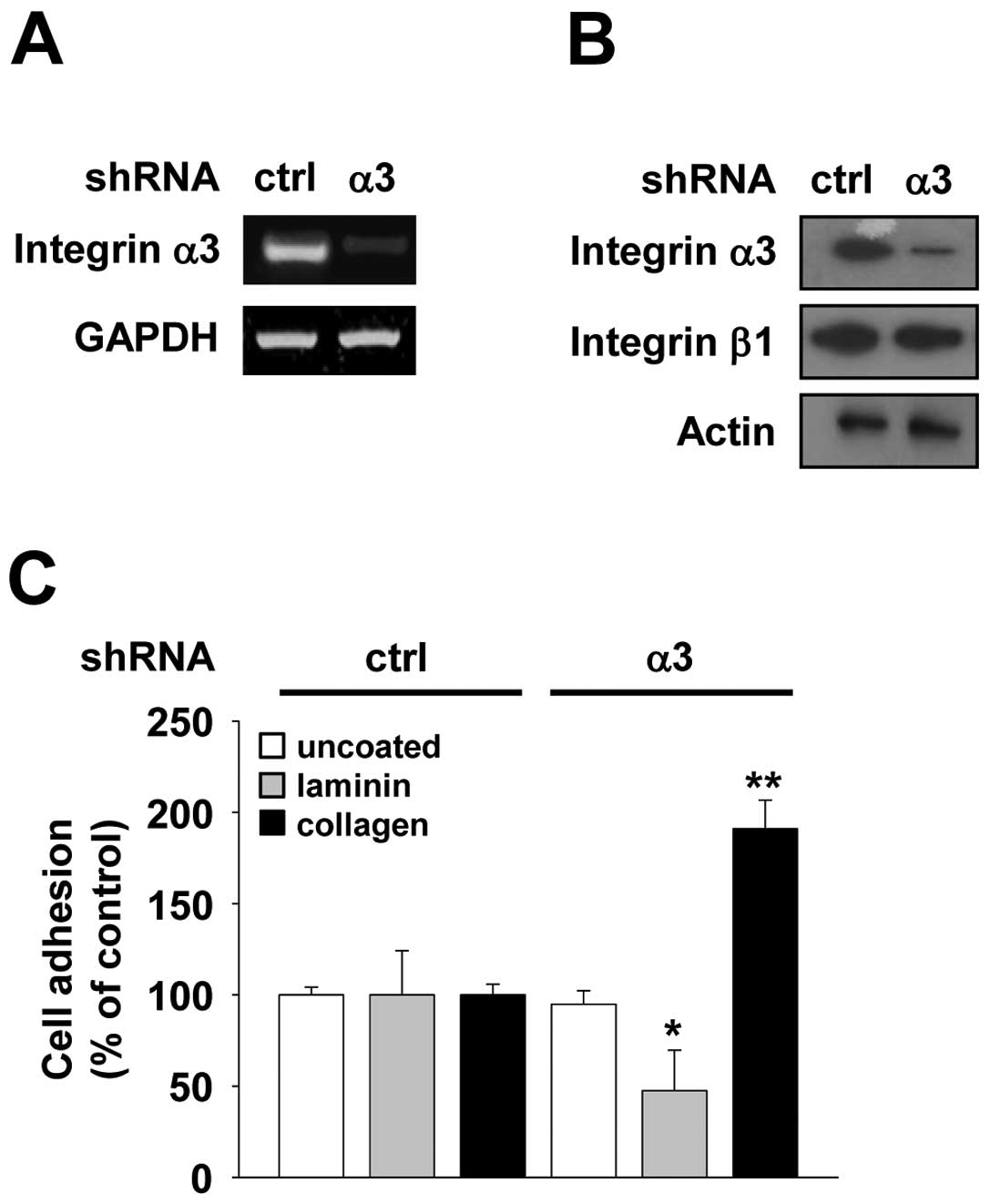
analysis demonstrated that α3 shRNA dramatically suppressed
expression of integrin α3 subunit in H1299 cells when compared with
control cells transfected with control shRNA, and did not affect β1
subunit expression (Fig. 1A and B).
Integrin α3β1 is abundantly expressed on the epithelial cell
membranes and is known to be a major receptor for laminin-5
(3). To test whether integrin α3
functions to regulate the degree of cell adhesion, we first
examined the adhesion of integrin α3-silenced H1299 cells to
extracellular matrix proteins. As expected, reduction in integrin
α3 subunit expression markedly blocked the adhesion of cells to
laminin, while did not alter the adhesion to tissue culture plastic
(Fig. 1C). In contrast, integrin α3
silencing resulted in a two-fold increase in cell adhesion to
collagen when compared with control shRNA (Fig. 1C), consistent with previous reports
demonstrating that collagen receptors are more active in integrin
α3-deficient mice keratinocytes (20). In addition, microscopic analysis
revealed that integrin α3 silencing did not affect morphological
changes in H1299 cells (data not shown).
We next examined the effect and molecular mechanisms
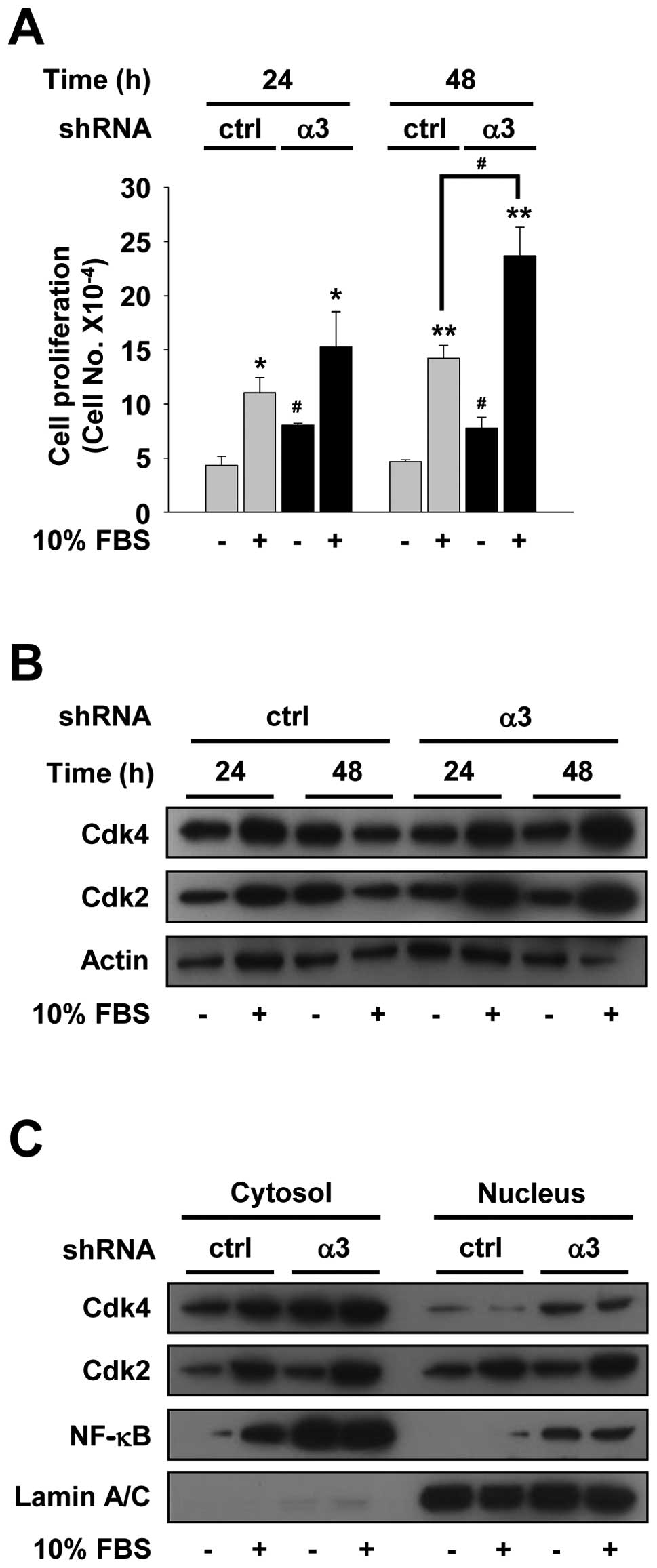
of integrin α3 silencing on cell proliferation. Control or integrin
α3-silenced H1299 cells were serum-starved for 48 h to allow
synchronization and growth arrest (G1/G0), and then stimulated with
10% FBS-DMEM for 24 or 48 h. After mitogenic stimulation for 48 h,
integrin α3 silencing resulted in a significant increase (>70%)
in cell proliferation, as compared with controls (Fig. 2A). Of note, integrin α3 silencing
also induced cell proliferation in the absence of mitogenic
stimuli. After serum starvation for 48 h this increase was
maintained for additional 48 h culture. These findings suggest that
disruption of integrin α3 expression results in induction of cell
proliferation and survival, enhancing the aggressiveness of lung
cancer and leading to shorter survival of lung cancer patients
(8,21). Based on these findings, we next
analyzed the changes of cell cycle-related proteins in H1299 cells.
It has been reported that cell cycle progression specifically
requires activation of cyclin-dependent kinases (Cdks) through
formation with cyclins, and is associated with subcellular
localization of Cdks from the cytoplasm to nucleus (22). In control shRNA-transfected cells,
mitogenic stimulation increased the protein levels of both Cdk4 and
Cdk2 for 24 h, while showed little or no change of protein
expression under 48 h cell culture conditions, compared with those
in unstimulated controls (Fig. 2B).
However, integrin α3 silencing dramatically induced the levels of
Cdks until the end time point of this experiment. The levels of
total Cdks in integrin α3-silenced cells were higher than those in
control shRNA-transfected cells. These results are consistent with
initial findings that integrin α3 silencing functions to increase
cell proliferation (Fig. 2A). To
investigate whether integrin α3 silencing mediates induction of
cell proliferation through the changes of Cdk localization, Cdks in
the nuclear compartments were directly examined by western blot
analysis of nuclear and cytosolic extracts of H1299 cells with or
without integrin α3 shRNA. As shown in Fig. 2C, reduction in integrin α3
expression markedly enhanced nuclear localization of Cdk4,
regardless of mitogenic stimulation. Furthermore,
p21WAF1/Cip1, one of the best known Cdk inhibitors, was
not detected in the nuclear compartments of integrin α3-silenced
cells without mitogenic stimulation (data not shown). These changes
of Cdk4 and p21WAF1/Cip1 in the nuclear compartments of
integrin α3-silenced cells are well correlated with previous
observations on cell proliferation (Fig. 2A).
NF-κB is well known as a transcription factor that
regulates the expression of a variety of genes in response to
inflammation, adhesion, cell cycle progression, survival, and
anti-apoptosis (12,13,23).
Reduced expression of integrin α3 markedly induced the levels of
NF-κB in the cytosolic compartments in both unstimulated and
stimulated culture conditions (Fig.
2C). Moreover, nuclear localization patterns of NF-κB by
integrin α3 silencing were similar to those of Cdk4. Collectively,
these results demonstrate that knockdown of integrin α3 expression
induces cell cycle progression and survival through enhanced
expression and nuclear localization of Cdks and NF-κB.
Knockdown of integrin α3 enhances cell
migration
Cell migration is controlled by the coordination of
integrin-mediated interactions from the ECM to the intracellular
components of the migrating cells, and plays the important roles in
tumor invasion and metastasis (1,2,5). To
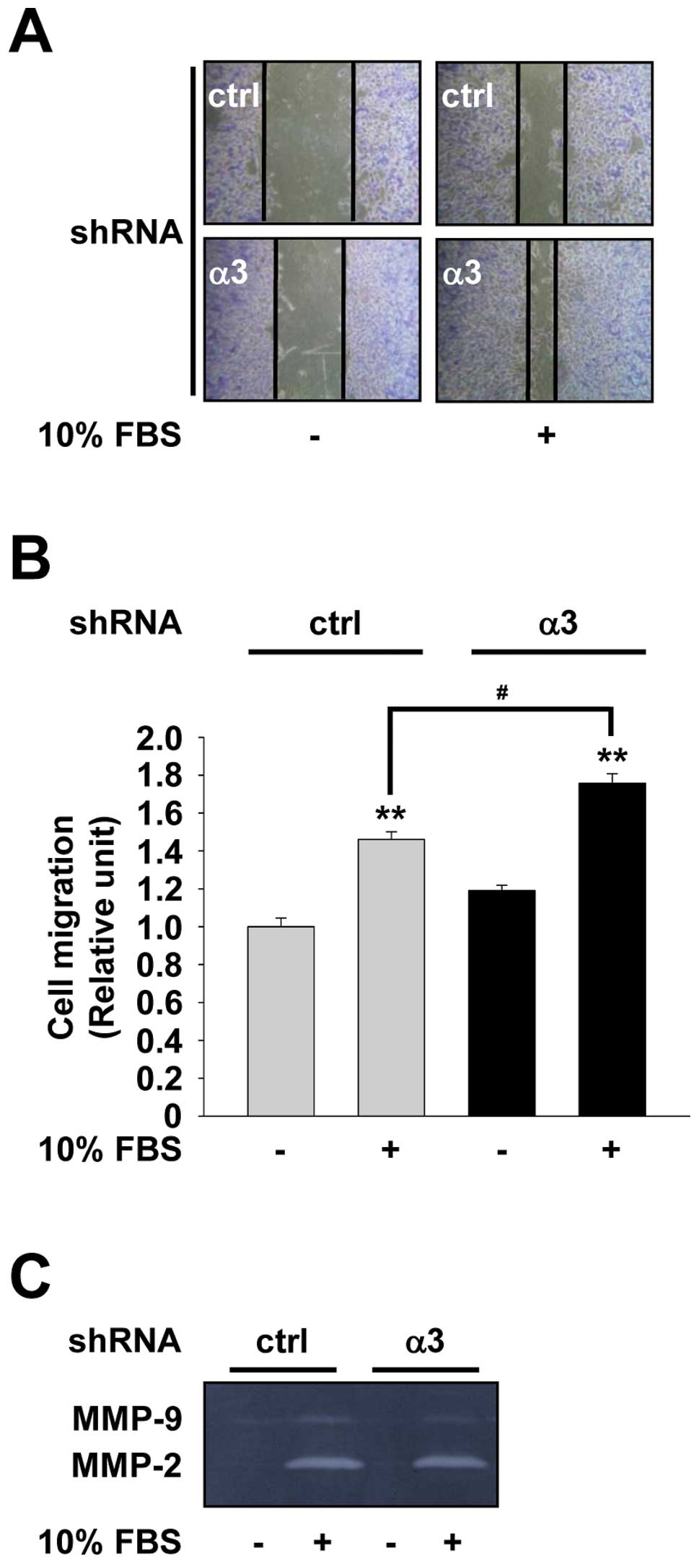
study the effect of integrin α3 expression on cell migration, we
next performed wound-healing assay using H1299 cells transfected
with control or integrin α3 shRNA. As shown in Fig. 3A and B, integrin α3-silenced cells
readily migrated into the wounded area within 18 h, consistent with
previous reports (20). Expression
and activation of matrix metalloproteinases (MMPs) are associated
with enhanced cell migration and invasion by selective proteolysis
of extracellular matrix components (24). Based on integrin α3
silencing-mediated induction of cell migration, we next analyzed
the activities of MMP-2 and MMP-9 in H1299 cells. As shown in
Fig. 3C, the conditioned media from
cell cultures had high levels of MMP-2 activity relative to those
of MMP-9. Knockdown of integrin α3 expression significantly reduced
MMP-9 activity, but not MMP-2, similar to previous reports that
integrin α3β1 expression is required for the production of MMP-9 in
immortalized mouse keratinocytes (25). These observations suggest that
integrin α3 silencing-mediated induction of H1299 cell migration
may not require the expression and activity of MMPs.
Roles of ERK and Akt activation in
integrin α3 silencing-induced cell proliferation and migration
To further investigate the molecular mechanism by
which knockdown of integrin α3 expression induces cell
proliferation and migration, we analyzed the changes in the
expression of EGFR, which is overexpressed or constitutively
activated in a variety of human cancers including lung cancer
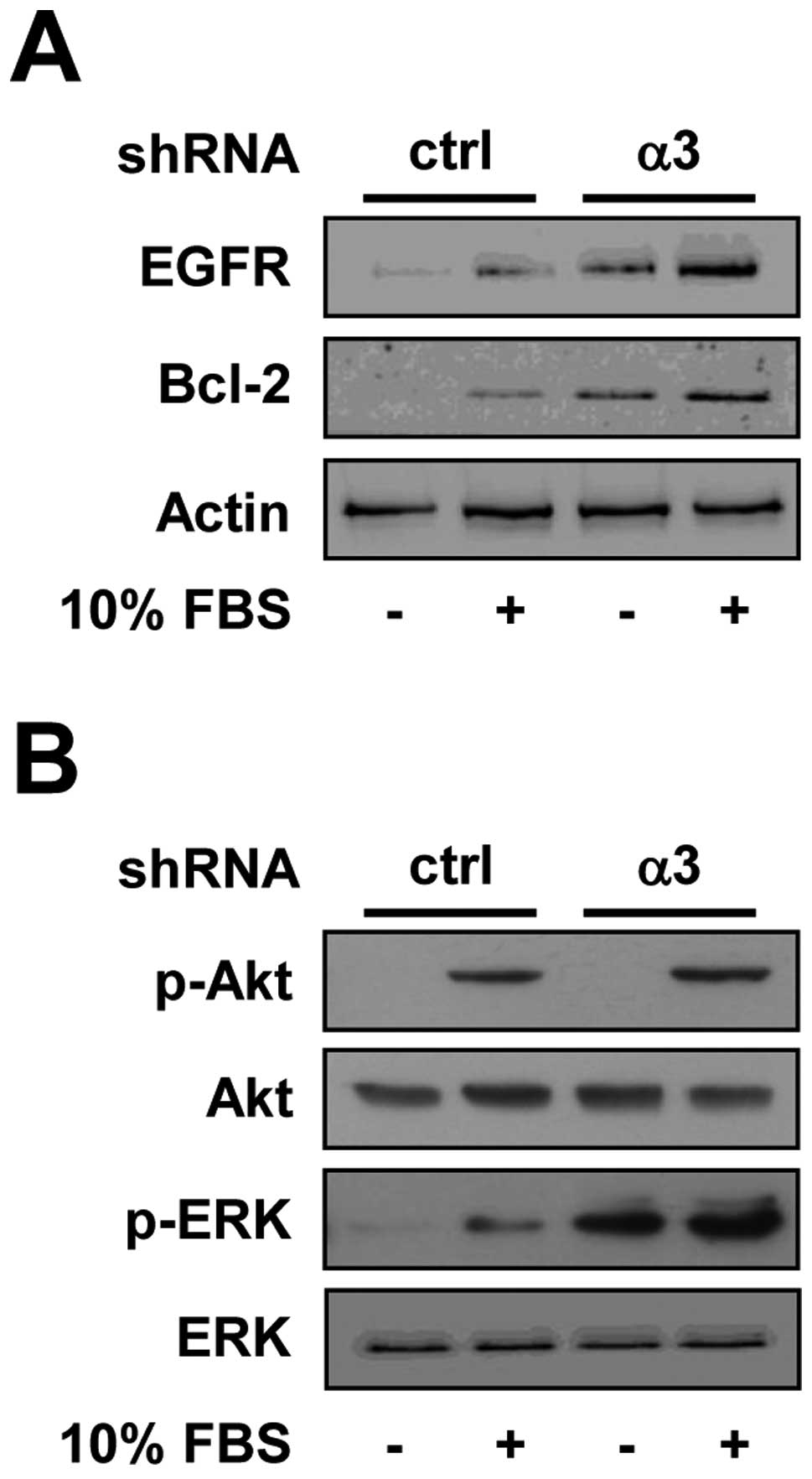
(9–11). As shown in Fig. 4A, the expression of EGFR was induced
in integrin α3-silenced H1299 cells, as compared with control
cells. Moreover, in integrin α3-silenced cells anti-apoptotic
protein Bcl-2 was highly expressed, consistent with previous
findings that NF-κB activation results in an increase in
anti-apoptotic regulators such as Bcl-2, Bcl-xL,
Bfl-1/A1, and inhibitor of apoptosis (13,26,27).
We next examined the changes in activation of PI3K/Akt and ERK,
which are key down-stream molecules of EGFR signaling pathways
(9,28). As shown in Fig. 4B, integrin α3-silenced cells showed
little or no change in phosphorylation status of Akt as compared
with control cells at the 24 h time point.
In contrast, activation of ERK in integrin
α3-silenced cells was sustained up to 24 h in the absence or
presence of mitogenic stimuli, indicating the cell mitogenesis and
survival might be mediated through ERK rather than Akt activity.
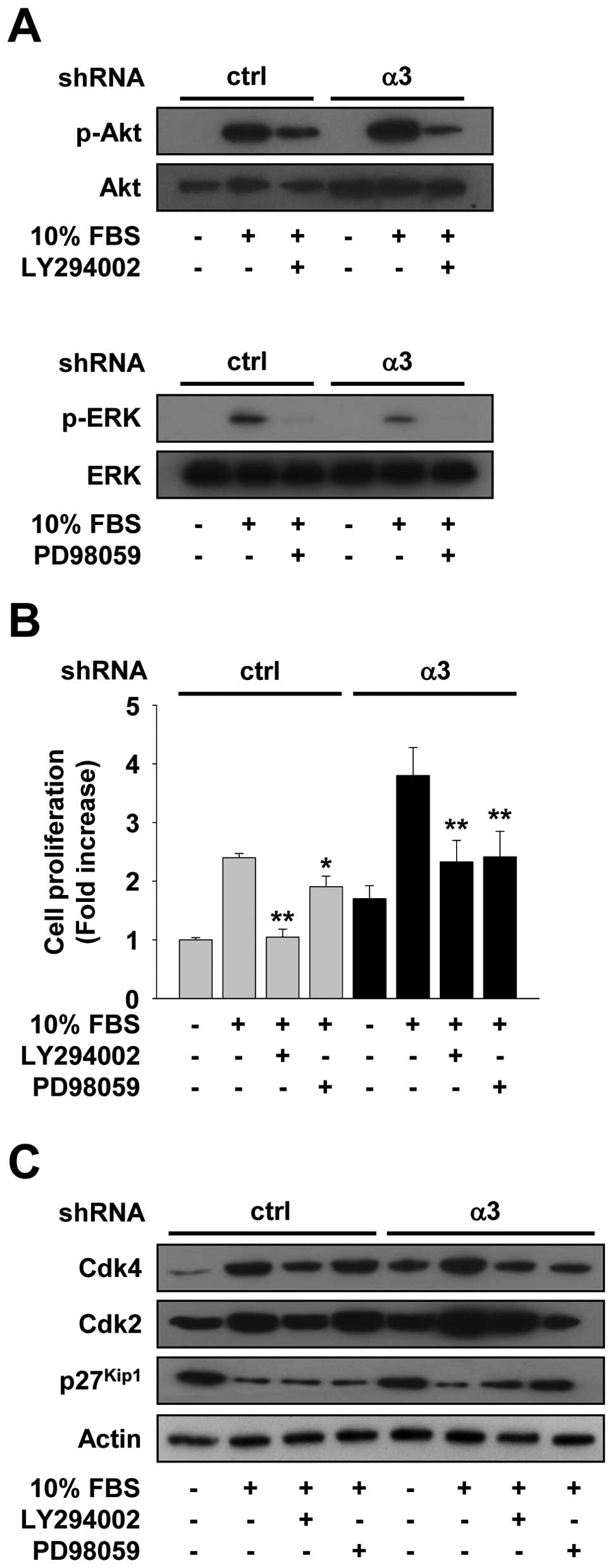
Pretreatment with PD98059, an inhibitor of ERK pathway or LY294002,
an inhibitor of PI3K-Akt pathway, significantly inhibited the
activation of ERK and Akt, and the proliferation of both control
and integrin α3-silenced cells (Fig. 5A
and B). Integrin α3-silenced cells are more responsive to
PD98059 inhibition of cell proliferation as compared with control
cells. Furthermore, PD98059 treatment decreased the protein levels
of both Cdk4 and Cdk2, and increased a Cdk inhibitor
p27Kip1 levels in integrin α3-silenced cells, but not in
control cells (Fig. 5C), in a good
agreement with our findings on cell proliferation (Fig. 5B). Finally, we examined the roles of
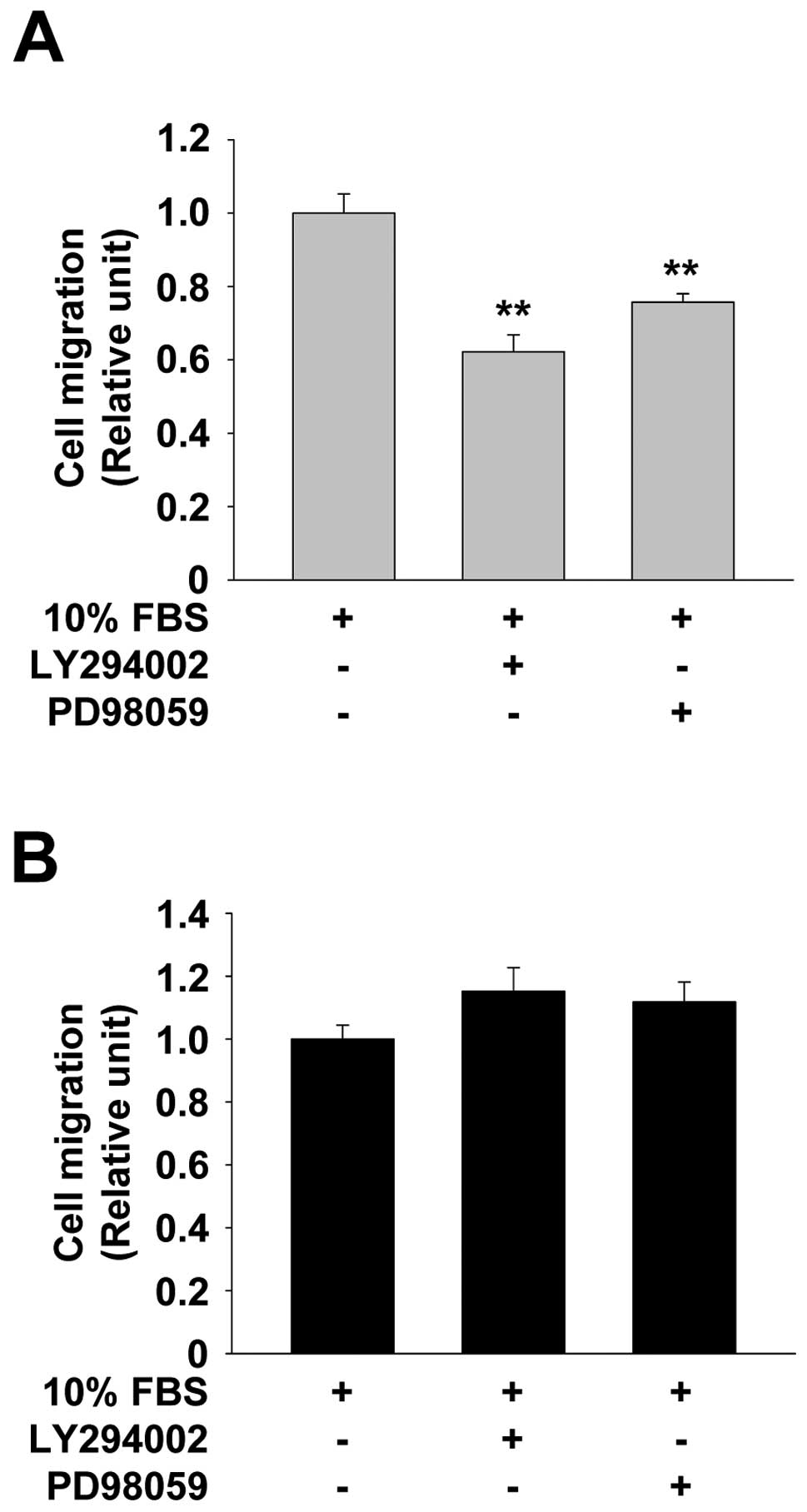
Akt and ERK in integrin α3 silencing-induced cell migration.
Pretreatment with LY294002 or PD98059 significantly reduced the
migration of control cells (Fig.
6A). In contrast, in integrin α3-silenced cells neither
LY294002 nor PD98059 treatment blocked cell migration (Fig. 6B). Taken together, these
observations suggest that knockdown of integrin α3 expression in
p53-deficient NSCLC cells induces cell proliferation through Akt-
and ERK-dependent pathways, whereas enhanced cell migration is
independent of the activation of Akt and ERK.
Discussion
In the present study we demonstrate that reduced
expression of integrin α3β1 induces the proliferation and migration
of p53-deficient NSCLC cells. In addition, we show that
upregulation of EGFR, Cdks, NF-κB, and Bcl-2 and sustained
activation of ERK in integrin α3-silenced cells are responsible for
the accelerated cell mitogenesis and survival. Our study is the
first demonstration that p53 functions to regulate integrin
α3β1-EGFR-mediated signaling pathways and invasive phenotypes of
NSCLC cells.
Integrin α3β1 is positively or negatively correlated
with cancer invasion and metastasis (6,29).
Expression of integrin α3β1 is highly induced in the process of
brain metastasis of NSCLC (30). In
contrast, reduced expression of integrin α3β1 has been reported to
be associated with the highly invasive and metastatic behavior of
small cell lung cancer (7) and poor
prognosis in patients with NSCLC (8). These conflicting observations may be
due to heterogeneity of cancer types and stages as well as
differences in cancer microenvironment.
Overexpressed, constitutively activated EGFR, and
deregulation of EGFR signaling are closely related to highly
aggressive behaviors and therapeutic resistance of lung cancers
(10,11), and these events are very complex.
Increasing evidence indicates that cross-talk between integrin α3β1
and RTKs plays pivotal roles in angiogenesis and cancer progression
(4,31,32).
Although potential roles of integrin α3β1 or EGFR in cancer
progression have been reported, however, no relationship between
integrin α3β1 and EGFR in regulating NSCLC cell proliferation and
migration has been clearly investigated to date. A recent study
demonstrates that integrin β1 silencing inactivates the EGFR
signaling pathways, resulting in suppression of tumorigenic
properties of lung cancer cells (33). NF-κB has been reported to be
activated by EGFR signaling events and positively associated with
breast cancer cell proliferation and anti-apoptosis, indicating
linkage between EGFR and NF-κB signaling (34). Mutations of the p53 tumor suppressor
gene occur in a high percentage of lung cancer, resulting in cancer
progression (35).
Our data show that integrin α3 silencing in
p53-deficient H1299 cells induces the expression of EGFR and
activation of ERK, and these events appear to be related to
induction of Cdks, NF-κB, and Bcl-2, leading to cell proliferation
and migration. These findings are correlated with a previous report
that loss of p53 can drive NF-κB toward cancer-promoting activity
by activation of signaling pathways including Akt and ERK, and
induction of anti-apoptotic gene expression, leading to cancer
growth, invasion and metastasis (12). In conclusion, our findings
demonstrate that alterations in integrin α3β1 expression and
subsequent signaling pathways may provide a molecular basis for the
coordination of cell proliferation and migration in p53-deficient
NSCLC, and suggest that expression of integrin α3β1 and regulation
of EGFR-NF-κB signaling pathways result in suppression of invasive
phenotypes of lung cancer.
Acknowledgements
This work was supported by Basic Science Research
Program (2009-0065896) (2010-0021913) through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education,
Science and Technology (MEST).
References
|
1
|
Cox D, Brennan M and Moran N: Integrins as
therapeutic targets: lessons and opportunities. Nat Rev Drug
Discov. 9:804–820. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hood JD and Cheresh DA: Role of integrins
in cell invasion and migration. Nat Rev Cancer. 2:91–100. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hynes RO: Integrins: bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eliceiri BP: Integrin and growth factor
receptor crosstalk. Circ Res. 89:1104–1110. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kreidberg JA: Functions of α3β1 integrin.
Curr Opin Cell Biol. 12:548–553. 2000.
|
|
7
|
Barr LF, Campbell SE, Bochner BS and Dang
CV: Association of the decreased expression of α3β1 integrin with
the altered cell: environmental interactions and enhanced soft agar
cloning ability of c-myc-overexpressing small cell lung cancer
cells. Cancer Res. 58:5537–5545. 1998.
|
|
8
|
Adachi M, Taki T, Huang C, et al: Reduced
integrin α3 expression as a factor of poor prognosis of patients
with adenocarcinoma of the lung. J Clin Oncol. 16:1060–1067.
1998.
|
|
9
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hynes NE and Lane HA: ERBB receptors and
cancer: the complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perkins ND: The diverse and complex roles
of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132. 2012.
|
|
13
|
Kim YK, Lee EK, Kang JK, et al: Activation
of NF-κB by HDAC inhibitor apicidin through Sp1-dependent de novo
protein synthesis: its implication for resistance to apoptosis.
Cell Death Differ. 13:2033–2041. 2006.
|
|
14
|
Cho Y-R, Choi S and Seo D-W: Sepiapterin
regulates cell proliferation and migration: its association with
integrin α3β1 and p53 in human lung cancer cells. Genes Genom.
33:577–582. 2011.
|
|
15
|
Seo D-W, Li H, Qu C-K, et al: Shp-1
mediates the antiproliferative activity of tissue inhibitor of
metalloproteinase-2 in human microvascular endothelial cells. J
Biol Chem. 281:3711–3721. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim S, Cho Y-R, Kim M-D, Kim H, Choi S and
Seo D-W: Inhibitory effects of sepiapterin on vascular endothelial
growth factor-A-induced proliferation and adhesion in human
umbilical vein endothelial cells. Arch Pharm Res. 34:1571–1577.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho Y-R, Kim SH, Ko HY, Kim M-D, Choi SW
and Seo D-W: Sepiapterin inhibits cell proliferation and migration
of ovarian cancer cells via down-regulation of
p70S6K-dependent VEGFR-2 expression. Oncol Rep.
26:861–867. 2011.PubMed/NCBI
|
|
18
|
Seo D-W, Kim SH, Eom S-H, et al: TIMP-2
disrupts FGF-2-induced downstream signaling pathways. Microvasc
Res. 76:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hong SY, Cho JY and Seo D-W: Ginsenoside
Rp1 inhibits proliferation and migration of human lung cancer
cells. Biomol Ther. 19:411–418. 2011. View Article : Google Scholar
|
|
20
|
Hodivala-Dilke KM, DiPersio CM, Kreidberg
JA and Hynes RO: Novel roles for α3β1 integrin as a regulator of
cytoskeletal assembly and as a trans-dominant inhibitor of integrin
receptor function in mouse keratinocytes. J Cell Biol.
142:1357–1369. 1998.
|
|
21
|
Gogali A, Charalabopoulos K and
Constantopoulos S: Integrin receptors in primary lung cancer. Exp
Oncol. 26:106–110. 2004.
|
|
22
|
Sherr CJ: The Pezcoller lecture: cancer
cell cycles revisited. Cancer Res. 60:3689–3695. 2000.PubMed/NCBI
|
|
23
|
Mayo MW, Denlinger CE, Broad RM, et al:
Ineffectiveness of histone deacetylase inhibitors to induce
apoptosis involves the transcriptional activation of NF-κB through
the Akt pathway. J Biol Chem. 278:18980–18989. 2003.PubMed/NCBI
|
|
24
|
Stetler-Stevenson WG and Seo D-W: TIMP-2:
an endogenous inhibitor of angiogenesis. Trends Mol Med. 11:97–103.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
DiPersio CM, Shao M, Di Costanzo L,
Kreidberg JA and Hynes RO: Mouse keratinocytes immortalized with
large T antigen acquire α3β1 integrin-dependent secretion of
MMP-9/gelatinase B. J Cell Sci. 113:2909–2921. 2000.PubMed/NCBI
|
|
26
|
Heckman C, Mehew J and Boxer L: NF-κB
activates Bcl-2 expression in t(14;18) lymphoma cells. Oncogene.
21:3898–3908. 2002.
|
|
27
|
Lee HH, Dadgostar H, Cheng Q, Shu J and
Cheng G: NF-κB-mediated up-regulation of Bcl-x and Bfl-1/A1 is
required for CD40 survival signaling in B lymphocytes. Proc Natl
Acad Sci USA. 96:9136–9141. 1999.
|
|
28
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stipp CS: Laminin-binding integrins and
their tetraspanin partners as potential antimetastatic targets.
Expert Rev Mol Med. 12:e32010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshimasu T, Sakurai T, Oura S, et al:
Increased expression of integrin α3β1 in highly brain metastatic
subclone of a human non-small cell lung cancer cell line. Cancer
Sci. 95:142–148. 2004.
|
|
31
|
Seo D-W, Li H, Guedez L, et al: TIMP-2
mediated inhibition of angiogenesis: an MMP-independent mechanism.
Cell. 114:171–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seo D-W, Saxinger WC, Guedez L, Cantelmo
AR, Albini A and Stetler-Stevenson WG: An integrin-binding
N-terminal peptide region of TIMP-2 retains potent angio-inhibitory
and anti-tumorigenic activity in vivo. Peptides. 32:1840–1848.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Morello V, Cabodi S, Sigismund S, et al:
β1 integrin controls EGFR signaling and tumorigenic properties of
lung cancer cells. Oncogene. 30:4087–4096. 2011.
|
|
34
|
Biswas DK and Iglehart JD: Linkage between
EGFR family receptors and nuclear factor kappaB (NF-κB) signaling
in breast cancer. J Cell Physiol. 209:645–652. 2006.PubMed/NCBI
|
|
35
|
Fuster JJ, Sanz-González SM, Moll UM and
Andrés V: Classic and novel roles of p53: prospects for anticancer
therapy. Trends Mol Med. 13:192–199. 2007. View Article : Google Scholar : PubMed/NCBI
|