Introduction
Hepatocellular carcinoma (HCC) is one of the most
common cancers in the world and has an extremely poor prognosis.
Hepatectomy and liver transplantation are the best curative
procedures for patients with HCC. However, only ~10–15% of newly
detected cases of HCC can be treated by surgery (1). Adjuvant treatments, including
transarterial chemoembolization (TACE), percutaneous ethanol
injection (PEI), and radiofrequency ablation (RFA), have been shown
to be effective in reducing tumor bulk. Recently, sorafenib was
recommended for advanced HCC patients with compensated liver
function (2). For the majority of
patients presenting with advanced disease, systemic chemotherapy is
not the standard treatment option, as HCC has low sensitivity to
most chemotherapeutic agents. However, in some cases, it was
reported that combination chemotherapy might make some initially
unresectable tumors resectable and even induce a complete
pathologic response (3,4). 5-Fluorouracil (5-FU) is an important
chemotherapeutic agent for HCC; however, after several cycles of
5-FU-based chemotherapy, the tumors may develop drug resistance,
and the mechanism of which remains unclear (5).
The oncogene B cell-specific Moloney murine leukemia
virus integration site 1 (Bmi1) has been reported to play important
roles in stem cell pluripotency, cell proliferation, early
embryogenesis, and cancer initiation and progression (6). In addition, the Bmi1 gene is often
highly expressed in HCC, but its significance was not fully
clarified (7–9). In this study, we found that
downregulation of the Bmi1 gene by RNA interference (RNAi) could
inhibit the proliferation and invasiveness of human HCC cells, and
increase their sensitivity to 5-FU treatment in vitro.
Materials and methods
Cell culture
Two human HCC cell lines HepG2 and Bel-7402 were
obtained from the American Type Culture Collection (Manassas, VA,
USA) and cultured in RPMI-1640 (Invitrogen Co., Carlsbad, CA, USA)
supplemented with 10% heat-inactivated fetal bovine serum
(Invitrogen) as recommended by the supplier. All cultures were
maintained in humidified atmosphere containing 5% CO2 at
37°C.
siRNA transfection
For the RNAi analyses, human Bmi1 small interfering
RNA (siRNA) with the nucleotide sequence
5′-CCAAGAUAUUGUAUACAAATT-3′ (sense) and 5′-UUUGUAUACAAUAUCUUGGTT-3′
(antisense), corresponding to part of the Bmi1 mRNA, and the
negative control scrambled siRNA (NC-siRNA; sense,
5′-UUCUCCGAACGUGUCACGUTT-3′, antisense,
5′-ACGUGACAGGUUCGGAGAATT-3′) were designed and purchased from
Shanghai GenePharma Corp. (Shanghai, China). All of the siRNA
sequences were subjected to basic local alignment search tool
(BLAST) to confirm the absence of homology to any additional known
coding sequences in the human genome. Cells were transfected using
the Lipofectamine™ 2000 reagent (Invitrogen) according to the
manufacturer's protocol. Briefly, one day prior transfection, HepG2
and Bel-7402 cells (1.5×105/well) were cultured in
6-well tissue culture plates until they reached 50% confluence,
then the cells were transiently transfected with either Bmi1-siRNA
or NC-siRNA (100 nM).
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was isolated using RNAiso Plus reagent
according to the manufacturer's protocol (Takara, Tokyo, Japan).
cDNA was synthesized using the PrimeScript RT Reagent (Takara).
Portions of double-stranded cDNA were subjected to PCR with a
SYBR-Green Premix Ex Taq (Takara). Primer sets used for real-time
PCR amplification were shown in Table
I. As a control, the levels of glyceraldehyde phosphate
dehydrogenase (GAPDH) expression were also analyzed. The
amplification protocol comprised incubations at 95°C for 30 sec,
95°C for 5 sec and 65°C for 20 sec. Incorporation of the SYBR-Green
dye into PCR products was monitored in real-time with LightCycler
real-time PCR detection system (Roche Applied Science,
Indianapolis, IN, USA), thereby allowing determination of the
threshold cycle (Ct) at which exponential amplification of products
begins.
 | Table IPrimers for Bmi1, E-cadherin and
reference genes. |
Table I
Primers for Bmi1, E-cadherin and
reference genes.
| Gene | Primer | Sequence |
|---|
| Bmi1 | Forward |
5′-AGCAGCAATGACTGTGATGC-3′ |
| Reverse |
5′-CAGTCTCAGGTATCAACCAG-3′ |
| E-cadherin | Forward |
5′-CTGAGAACGAGGCTAACG-3′ |
| Reverse |
5′-GTCCACCATCATCATTCAATAT-3′ |
| GAPDH | Forward |
5′-GCACCGTCAAGGCTGAGAAC-3′ |
| Reverse |
5′-TGGTGAAGACGCCAGTGGA-3′ |
Western blot analysis
The cells (2×106/well) were washed twice
with ice-cold PBS (phosphate-buffered saline) and lysed on ice in
lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1%
SDS], 1% protease inhibitor phenylmethanesulfonyl fluoride (PMSF;
Sigma, St. Lois, MO, USA). Protein concentration was determined by
Lowry assay. Whole cell extracts (50 μg) were fractionated by 10%
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto polyvinylidene difluoride membrane
(Millipore Corp., Bedford, MA, USA). Proteins of interest were
revealed with specific antibodies as indicated: rabbit anti-Bmi1
polyclonal antibody in a final dilution of 1:300 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA), rabbit anti-E-cadherin
polyclonal antibody in a final dilution of 1:500 (Santa Cruz
Biotechnology), rabbit anti-N-cadherin polyclonal antibody in a
final dilution of 1:500 (Santa Cruz Biotechnology), rabbit
anti-vimentin polyclonal antibody in a final dilution of 1:500
(Santa Cruz Biotechnology), and mouse anti-α-tubulin monoclonal
antibody in a final dilution of 1:1,000 (Santa Cruz Biotechnology).
The membranes were further incubated with goat anti-rabbit
secondary antibody in a final dilution of 1:5,000, or goat
anti-mouse secondary antibody in a final dilution of 1:10,000
(Santa Cruz Biotechnology). Then the bound were visualized with the
enhanced chemiluminescence (ECL) system (Amersham, UK) and exposed
to X-ray film (Fuji, Dielsdorf, Switzerland).
Cell proliferation assay
The cells (5×103/well) were cultured in
96-well tissue culture plates until they reached 50% confluence,
then transfected with a final concentration of 100 nM. After
transfection (24, 48 and 72 h), viability of the cells were
determined using the Cell Counting Kit-8 (CCK-8) that was purchased
from the Dojindo Molecular Technologies (Gaithersburg, MD, USA).
Briefly, 10 μl of water soluble formazan dye was added to each well
and incubated for 2 h. The absorbance at 450 nm was measured by an
enzyme linked immunosorbent assay (ELISA) plate reader. The
absorbance of the negative control (OD) was considered to be
100%.
Cell invasion assay
The invasive activity of tumor cells was estimated
using transwells (6.5 mm in diameter, polycarbonate membrane, 8 μm
pore size) coated with extracellular matrix gel obtained from
Corning (Corning, NY, USA). Twenty-four hours after transfection,
an aliquot of 1×105 cells was placed in the upper
chamber with 0.1 ml serum-free medium, whereas the lower chamber
(24-well plate) was loaded with 0.5 ml of medium containing 10%
fetal bovine serum. After 24 h of incubation, the cells were fixed
with 4% paraformaldehyde and then counterstained with 0.1% crystal
violet. The cells that had migrated into the lower chamber were
observed and counted under a light microscope. Then the number of
migratory cells was calculated.
Measurement of cytotoxicity
Twenty-four hours after transfection, the cells were
treated with various concentrations (50, 100, 200 and 400 μg/ml,
respectively) of 5-FU (Sigma) for 48 h. Then the cell viability was
determined by CCK-8 assay. The rate of cell growth inhibition (IR)
was calculated according to the following equation: IR = [1-A570
(drug)/A570 (control)] × 100%, where A570 (drug) is the absorbance
of the cells exposed to 5-FU and A570 (control) is the absorbance
of the cells without 5-FU treatment.
DAPI staining
4,6-Diamidino-2-phenylindole (DAPI, Invitrogen)
staining was performed according to the manufacturer's protocol. In
brief, cells were fixed with 4% paraformaldehyde for 30 min at
25°C, washed three times with cold PBS, and exposed to 1 μg/ml DAPI
solution for 15 min in the dark at room temperature. Stained cells
were observed with a laser scanning microscope (Nikon, Japan).
Flow cytometry
To detect the apoptosis of HCC cells, the cells were
doubly stained with Annexin V-FITC (BD Bioscience) and propidium
iodide followed by flow cytometry (FCM) analysis. Apoptotic ratio
was determined on the basis of Annexin V+ PI+
and Annexin V+ PI− fractions.
Statistics
All experiments were performed at least in
triplicate. Statistical analysis was conducted with the SPSS
software package (version 13.0; SPSS Inc., Chicago, IL, USA). All
data were presented as the mean ± standard deviation (SD), and the
Student's t-test was used for evaluating the statistical
significance. For all tests, a P-value <0.05 was considered to
be statistically significant and indicated by asterisks in the
figures. All P-values given were the results of two-sided
tests.
Results
siRNAs targeting the Bmi1 gene
downregulated Bmi1 expression in HCC cells
To address the functional importance of the Bmi1
gene, we employed RNAi to deplete its expressions in HepG2 cells
and Bel-7402 cells, both of which were treated with negative
control (NC)-siRNA or siRNA targeting the Bmi1 gene. After 24 and
48 h, the cells were examined by real-time quantitative reverse
transcription-polymerase chain reaction (qRT-PCR) and western blot
analysis. The qRT-PCR analysis confirmed that the levels of
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were unaffected by
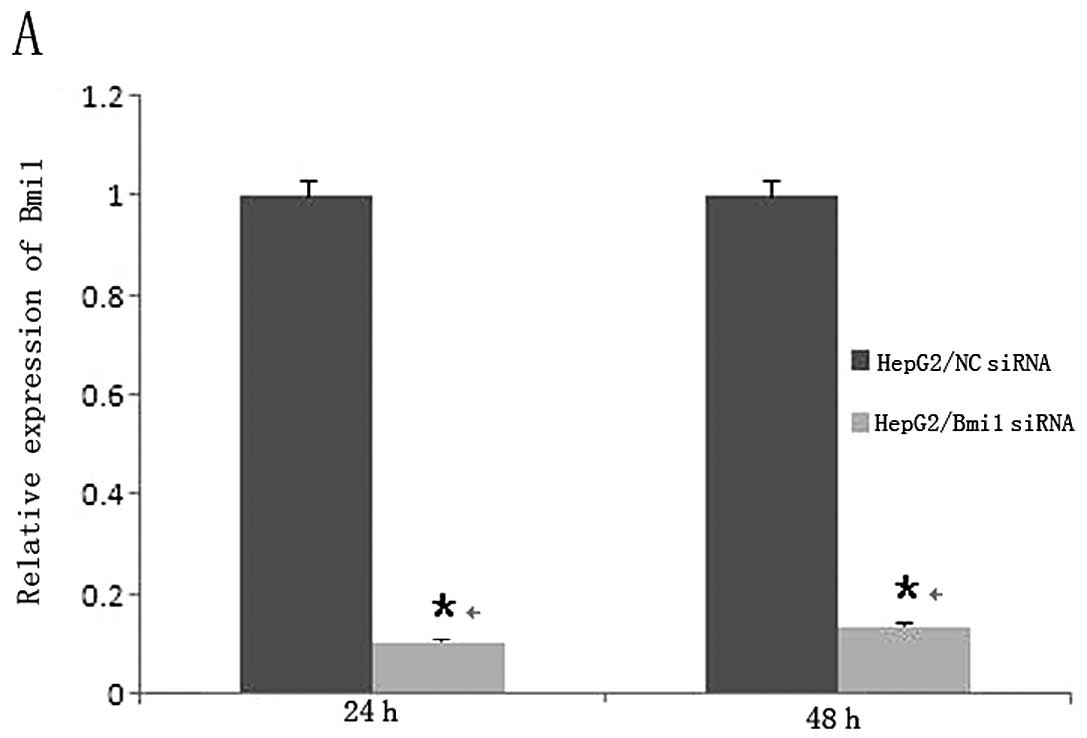
transfection of Bmi1-siRNA or NC-siRNA. As shown in Fig. 1A and B, qRT-PCR showed that
Bmi1-siRNA dowregulated the mRNA expression of Bmi1 (P<0.001).
Similar results were observed in the western blot analysis
(Fig. 1C and D; P<0.001). In
addition, the expression of Bmi1 in Bmi1-siRNA group was
significantly lower than that in the control group (P<0.001).
These data indicated that Bmi1-specific siRNA could effectively and
obviously suppress the expression of Bmi1 in HepG2 cells and
Bel-7402 cells.
Specific knockdown of Bmi1 expression by
RNAi inhibited the growth of HCC cells in vitro
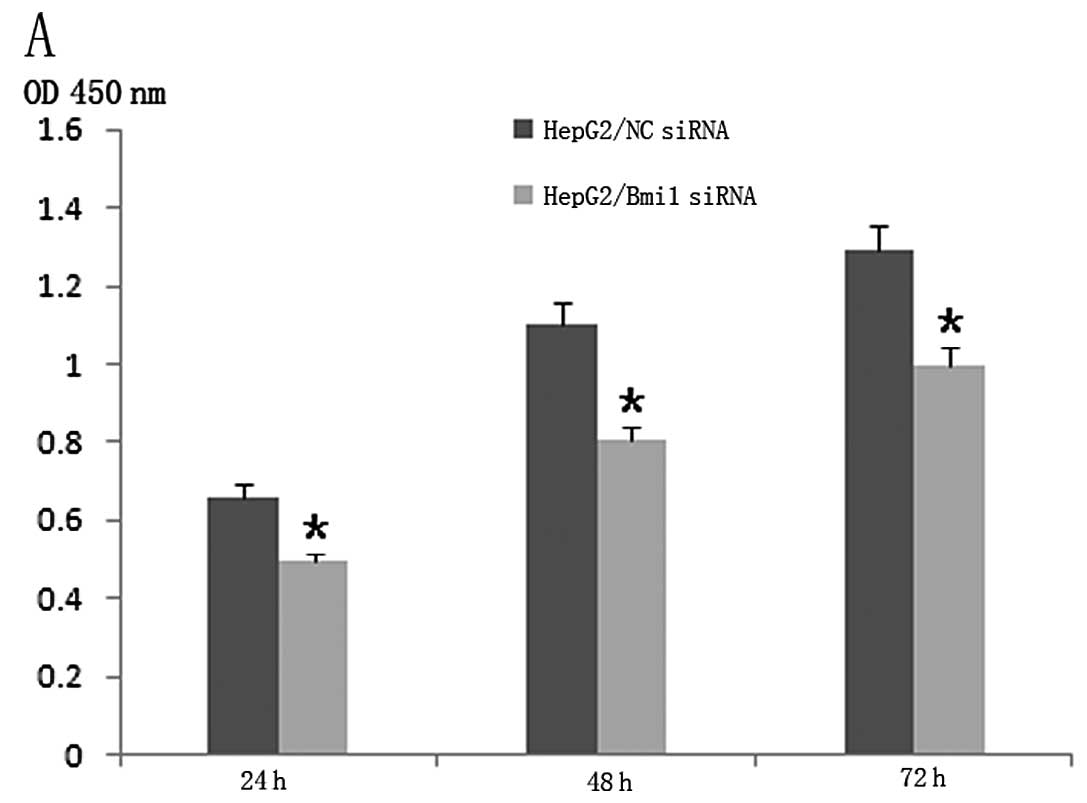
We then investigated the effect of Bmi1-siRNA on the
proliferation of HCC cells. To this end, the CCK-8 assay was
performed 24, 48 and 72 h after transfection. Compared to the
NC-siRNA, Bmi1-siRNA inhibited the growth of HepG2 cells and
Bel-7402 cells dramatically in vitro (Fig. 2A and B; P<0.001).
Bmi1-siRNA diminished the migration of
HepG2 cells in vitro
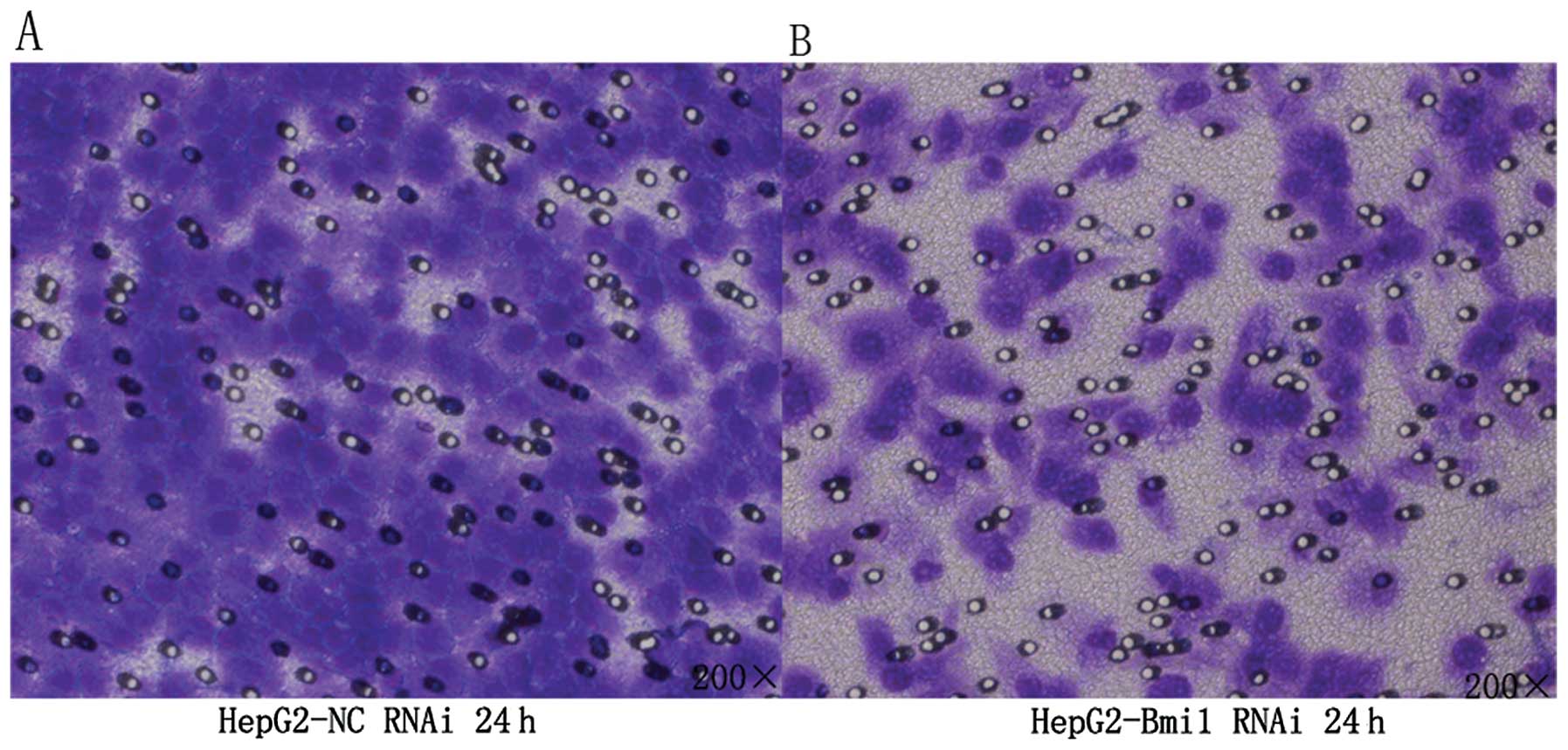
To examine whether targeted downregulation of Bmi1
in HepG2 cells affected the migration of tumor cells, in
vitro transwell migration assays were performed. The number of
tumor cells migrating through the filter in the Bmi1-siRNA group
was markedly lower than that in the NC-siRNA group (Fig. 3; P<0.001). Thus, Bmi1-siRNA
silencing can dramatically diminish the migration of HepG2 cells
in vitro.
Knockdown of Bmi1 can promote the
expression of E-cadherin and decrease the expression of vimentin
and N-cadherin
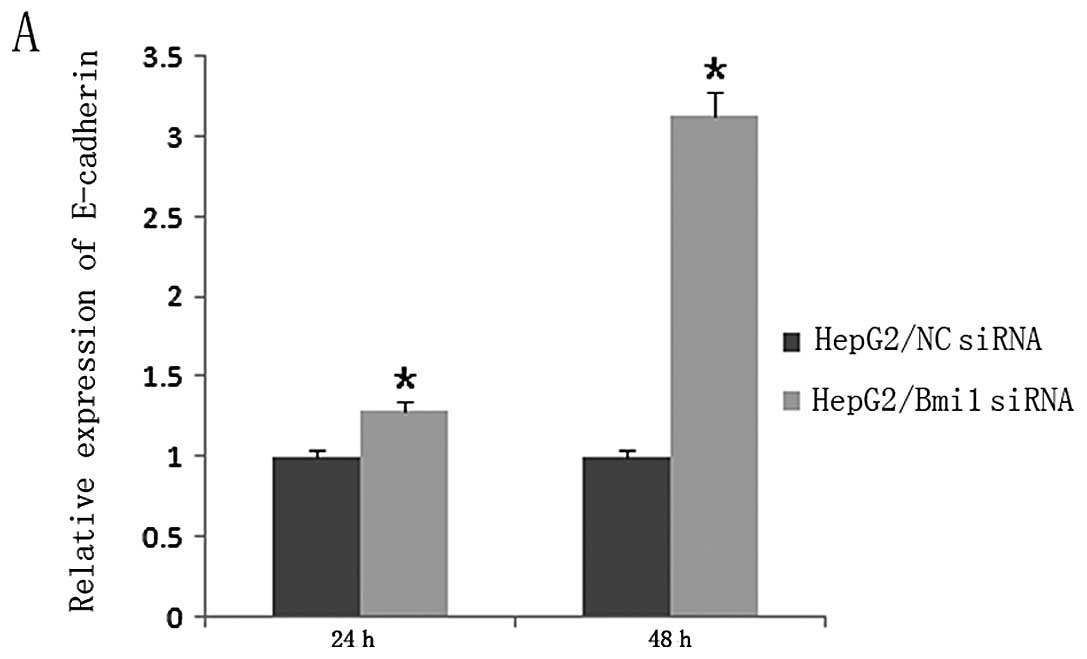
To further explore the mechanism underlying the
repression of HCC cells migration by the silencing of Bmi1, the
expression levels of the epithelial-mesenchymal transition (EMT)
marker E-cadherin in the HepG2-NC-siRNA and HepG2-Bmi1-siRNA cells
were examined. The results showed that knockdown of endogenous Bmi1
led to substantial enhancement in the levels of E-cadherin
(Fig. 4A; P<0.001), but
decreased the expressions of vimentin and N-cadherin in Bmi1
knockdown cells (Fig. 4B-D;
P<0.001).
Knockdown of Bmi1 sensitizes HCC cells to
5-FU-induced apoptosis
To further explore the role of Bmi1 in HCC, we
tested whether downregulation of Bmi1 by RNAi sensitizes HCC cells
to 5-FU chemotherapy. After transfection and treatment with various
concentrations of 5-FU, cell viability was determined by the CCK-8
assay. The results showed that both the Bmi1-siRNA transfected
cells (HepG2-Bmi1-siRNA and Bel-7402-Bmi1-siRNA cells) showed lower
cell viabilities than the control (HepG2-NC-siRNA and
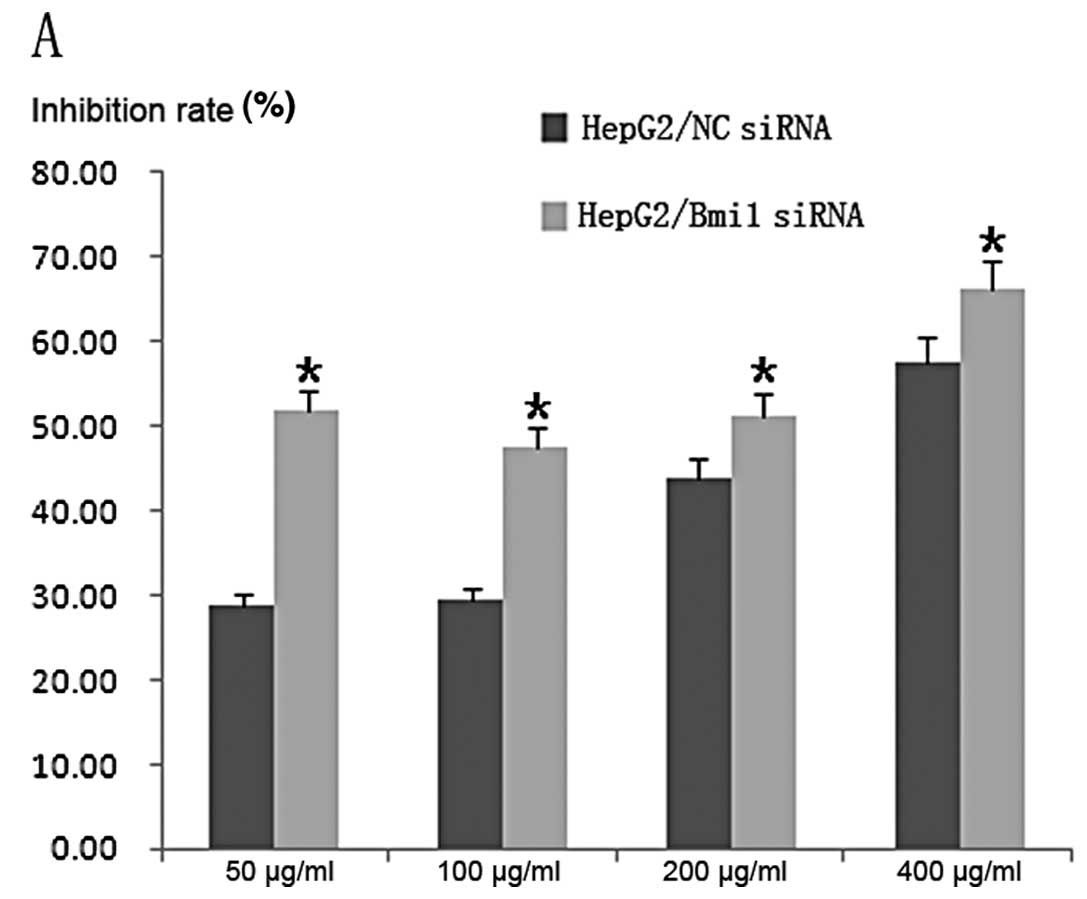
Bel-7402-NC-siRNA cells; P<0.001). The IC50 values of
5-FU in the HepG2-NC-siRNA and HepG2-Bmi1-siRNA cells were
215.23±0.89 and 47.67±0.70 μg/ml, respectively, and the
corresponding values for Bel-7402-NC-siRNA and Bel-7402-Bmi1-siRNA
cells were 106.63±0.63 and 26.21±0.38 μg/ml, respectively (Fig. 5).
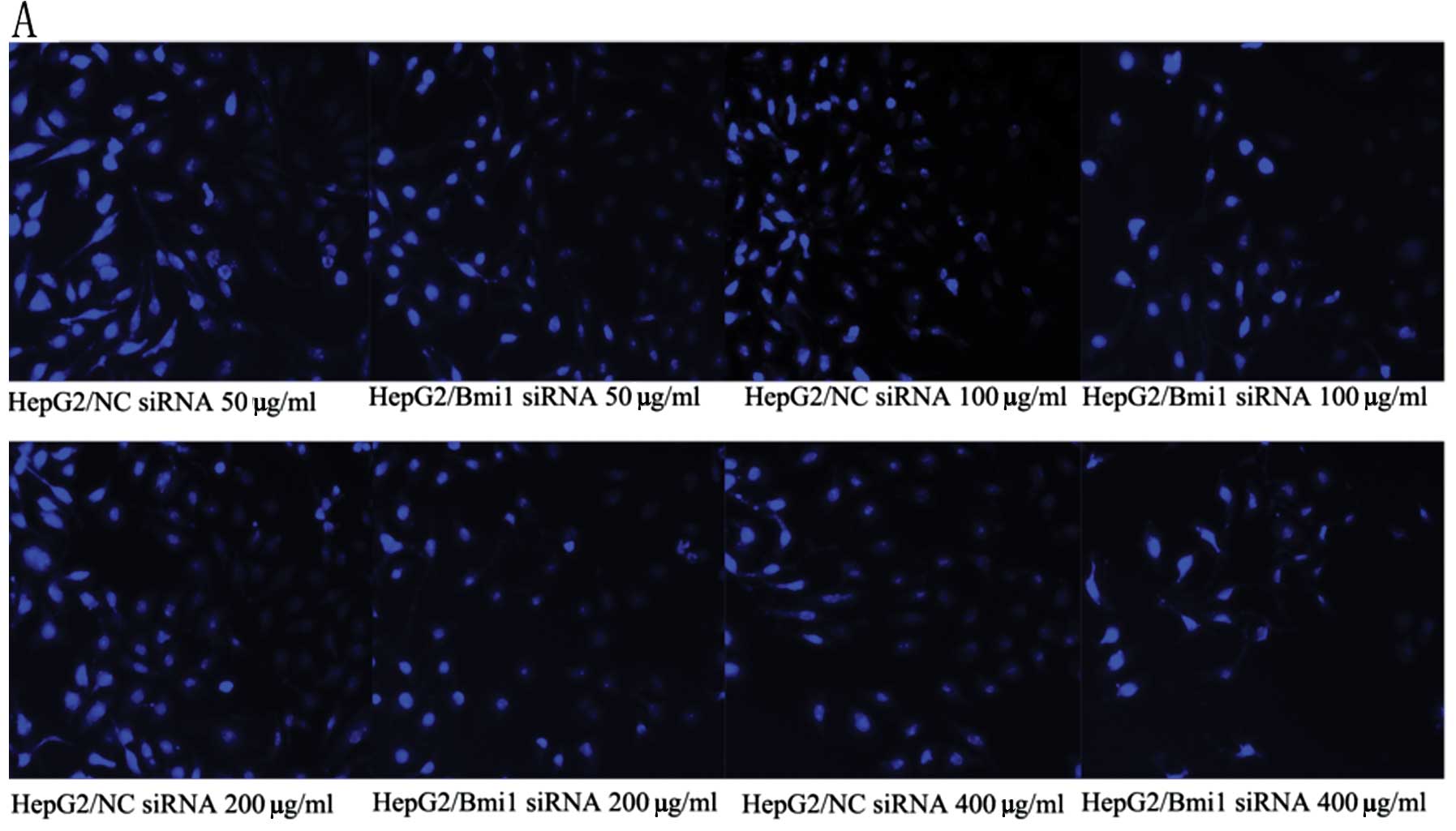
In order to confirm these results, the cells were
subjected to 4′,6-diamidino-2-phenylindole (DAPI) staining. As
shown in Fig. 6A and B, both
HepG2-Bmi1-siRNA and Bel-7402-Bmi1-siRNA cells showed lower cell
numbers than the control cells. We also examined the apoptotic rate
of tumor cells by flow cytometry. The results showed that the
apoptotic rates of the HepG2-Bmi1-siRNA and Bel-7402-Bmi1-siRNA
cells were much higher than those of the HepG2-NC-siRNA and
Bel-7402-NC-siRNA cells, respectively (Fig. 6C; P<0.001). These results
indicated that knockdown of Bmi1 could sensitize HCC cells to 5-FU
treatment.
Discussion
A majority of HCC patients who undergo chemotherapy
show multidrug resistance (MDR), which is often responsible for
therapy failure and poor outcome. However, the precise molecular
mechanisms of MDR remain unknown. One explanation for MDR is
overexpression of membrane transport proteins such as the
P-glycoprotein (P-gp) and the multidrug resistance protein isoform
1 (MRP1), which acts as an efflux pump for anticancer agents
(10). In addition, resistance to
apoptosis also contributes to chemoresistance (11).
Chemosensitization strategies involving gene
therapy, which aim to weaken the pathological activity of
MDR-related genes in cancer cells, are currently on the rise.
Gene-targeted therapies that enhance cancer cell sensitivity to
chemotherapeutic agents have the potential to increase drug
efficacy while reducing the toxic effects on untargeted cells
(12). This approach needs to be
improved and the methods to characterize the chemoresistance
profile of cancer need to be standardized. Therefore, the
development and improvement of methods to determine the
chemoresistance profile have become a crucial objective in
developing therapeutic strategies against cancer (13).
RNAi is a relatively new approach and represents a
prospective strategy to overcome MDR by selectively silencing the
target genes involved in the development of this deleterious
phenotype. RNAi technology can be directed against cancer by
inhibiting overexpressed oncogenes, blocking cell division by
interfering with cyclin E and related genes, or promoting apoptosis
by suppressing antiapoptotic genes (14). Duxbury et al(15) and Yu et al(16) reported that targeting the
ribonucleotide reductase M2 subunit and silencing the polo-like
kinase 1 gene by RNAi attenuated the cellular invasiveness and
gemtacibine chemoresistance of pancreatic adenocarcinoma. Singh
et al(17) reported that
RNAi-mediated silencing of the expression of nuclear factor
erythroid-2-related factor 2 in non-small cell lung cancer
inhibited tumor growth and increased the efficacy of chemotherapy.
Further, Dong et al(18)
reported that in human breast carcinoma cells, tumor-specific RNAi
targeting the eIF4E gene suppresses tumor growth, induces
apoptosis, and enhances cisplatin cytotoxicity. In contrast, in HCC
cells, downregulation of CD147 expression by RNAi-sensitized HCC
cells increased the sensitivity of HCC cells to curcumin (19). Chen et al(20) reported that siRNA-mediated
downregulation of the expression of x-linked inhibitor of apoptosis
reduced cellular viability and increased methotrexate
chemosensitivity in the human hepatoma cell line HepG2. However,
little is known about the effect of Bmi1 gene expression on tumor
progression and drug resistance in cases of HCC.
Bmi1, a member of the polycomb gene (PcG) family,
plays important roles in cell cycle regulation, cell
immortalization, cell senescence, maintenance of stem cell
pluripotency and early embryogenesis (6). In addition, numerous studies have
demonstrated that Bmi1 expression is frequently upregulated in
various types of human cancers, including lymphoma (21), lung cancer (22), ovarian cancer (23), nasopharyngeal carcinoma (24,25),
breast cancer (26), and HCC
(7–9), which indicate that the Bmi1 gene may
be implicated in tumor development and progression. The possible
molecular mechanism by which Bmi1 participates in tumor development
and progression is by suppressing p16/retinoblastoma protein (Rb)
and/or p19ARF/MDM2/p53 tumor suppressor pathways. Further, Bmi1
expression is also associated with the protection of tumor cells
from apoptosis, which is associated with inhibition of the
phosphoinositide-3 kinase (PI3K)/Akt pathway (27).
To examine the role of Bmi1 expression on the
proliferation, invasiveness, and chemotherapeutic response of HCC,
the Bmi1 gene was silenced by Bmi1-siRNA in 2 HCC cell lines, HepG2
and Bel-7402. After transfection, the cells were treated with
various concentrations of 5-FU. Then, the proliferation,
invasiveness, and 5-FU sensitivity of Bmi1-silenced tumor cells
were detected by CCK-8, transwell assays, DAPI staining, and flow
cytometry. The results showed that the proliferation and viability
of tumor cells in the Bmi1-silenced group were significantly lower
than those in the control group. Transwell migration assays showed
that downregulation of Bmi1 expression can significantly diminish
the migratory capacity of HCC cells in vitro. We also
observed that Bmi1 gene silencing significantly increased the
apoptotic rate of tumor cells treated by 5-FU and significantly
decreased the IC50 values of 5-FU. These results showed
that the knockdown of endogenous Bmi1 expression contributed to
sensitization of HCC cells to 5-FU, inhibited their proliferation
and invasiveness, and increased their apoptotic rates, which
suggested that the combination of conventional chemotherapy and
Bmi1-gene target therapy will be a potential clinical strategy for
HCC therapy.
To further investigate the mechanism underlying the
inhibition of invasiveness of human HCC cells by RNAi, we examined
the expression levels of the EMT markers, E-cadherin, N-cadherin,
and vimentin in the HepG2 cells after transfection by qRT-PCR
and/or western blot analysis. The results showed that knockdown of
endogenous Bmi1 expression led to significant promotion of
E-cadherin expression at both mRNA and protein levels, and the
expressions of vimentin and N-cadherin were significantly
downregulated. This means that Bmi1-RNAi inhibits the invasiveness
of human HCC cells, which may be partly mediated through EMT.
In conclusion, we showed the anticancer potential of
the combination of 5-FU treatment and Bmi1 depletion. We found that
knockdown of Bmi1 expression made HCC cells more sensitive to 5-FU
treatment and that depletion of Bmi1 enhanced 5-FU-induced
apoptosis. Our study suggests that a combination of 5-FU treatment
and Bmi1 depletion might be a potential novel therapeutic strategy
against drug resistance in HCC cells. However, further research is
required to better delineate the molecular mechanisms.
Acknowledgements
This study was supported by the Special Research
Foundation of the National Nature Science Foundation of China
(81172068).
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2009. CA Cancer J Clin. 59:225–249. 2009. View Article : Google Scholar
|
|
2
|
Morimoto M, Numata K, Kondo M, et al:
Higher discontinuation and lower survival rates are likely in
elderly Japanese patients with advanced hepatocellular carcinoma
receiving sorafenib. Hepatol Res. 41:296–302. 2011. View Article : Google Scholar
|
|
3
|
Meric F, Patt YZ, Curley SA, et al:
Surgery after downstaging of unresectable hepatic tumors with
intra-arterial chemotherapy. Ann Surg Oncol. 7:490–495. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Forner A, Reig ME, de Lope CR, et al:
Current strategy for staging and treatment: the BCLC update and
future prospects. Semin Liver Dis. 30:61–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park IK, Morrison SJ and Clarke MF: Bmi1,
stem cells, and senescence regulation. J Clin Invest. 113:175–179.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yonemitsu Y, Imazeki F, Chiba T, et al:
Distinct expression of polycomb group proteins EZH2 and BMI1 in
hepatocellular carcinoma. Hum Pathol. 40:1304–1311. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sasaki M, Ikeda H, Itatsu K, et al: The
overexpression of polycomb group proteins Bmi1 and EZH2 is
associated with the progression and aggressive biological behavior
of hepatocellular carcinoma. Lab Invest. 88:873–882. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiba T, Miyagi S, Saraya A, et al: The
polycomb gene product BMI1 contributes to the maintenance of
tumor-initiating side population cells in hepatocellular carcinoma.
Cancer Res. 68:7742–7749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Munoz M, Henderson M, Haber M, et al: Role
of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB
Life. 59:752–757. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baguley BC: Multidrug resistance in
cancer. Methods Mol Biol. 596:1–14. 2010. View Article : Google Scholar
|
|
12
|
Seth P: Vector-mediated cancer gene
therapy: an overview. Cancer Biol Ther. 4:512–517. 2005. View Article : Google Scholar
|
|
13
|
Baggetto LG, Gambrelle J, Dayan G, et al:
Major cytogenetic aberrations and typical multidrug resistance
phenotype of uveal melanoma: current views and new therapeutic
prospects. Cancer Treat Rev. 31:361–379. 2005. View Article : Google Scholar
|
|
14
|
Izquierdo M: Short interfering RNAs as a
tool for cancer gene therapy. Cancer Gene Ther. 12:217–227. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duxbury MS, Matros E, Ito H, Zinner MJ, et
al: Systemic siRNA-mediated gene silencing: a new approach to
targeted therapy of cancer. Ann Surg. 240:667–674. 2004.PubMed/NCBI
|
|
16
|
Yu C, Zhang X, Sun G, et al: RNA
interference-mediated silencing of the polo-like kinase 1 gene
enhances chemosensitivity to gemcitabine in pancreatic
adenocarcinoma cells. J Cell Mol Med. 12:2334–2349. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh A, Boldin-Adamsky S, Thimmulappa RK,
et al: RNAi-mediated silencing of nuclear factor
erythroid-2-related factor 2 gene expression in non-small cell lung
cancer inhibits tumor growth and increases efficacy of
chemotherapy. Cancer Res. 68:7975–7984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong K, Wang R, Wang X, et al:
Tumor-specific RNAi targeting eIF4E suppresses tumor growth,
induces apoptosis and enhances cisplatin cytotoxicity in human
breast carcinoma cells. Breast Cancer Res Treat. 113:443–456. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jia L, Wang H, Qu S, et al: CD147
regulates vascular endothelial growth factor-A expression,
tumorigenicity, and chemosensitivity to curcumin in hepatocellular
carcinoma. IUBMB Life. 60:57–63. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Xiao XQ, Deng CM, et al:
Downregulation of xIAP expression by small interfering RNA inhibits
cellular viability and increases chemosensitivity to methotrexate
in human hepatoma cell line HepG2. J Chemother. 18:525–531. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dutton A, Woodman CB, Chukwuma MB, et al:
Bmi-1 is induced by the Epstein-Barr virus oncogene LMP1 and
regulates the expression of viral target genes in Hodgkin lymphoma
cells. Blood. 109:2597–2603. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Breuer RH, Snijders PJ, Sutedja GT, et al:
Expression of the p16(INK4a) gene product, methylation of the
p16(INK4a) promoter region and expression of the polycomb-group
gene BMI-1 in squamous cell lung carcinoma and premalignant
endobronchial lesions. Lung Cancer. 48:299–306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhattacharya R, Kwon J, Ali B, et al: Role
of hedgehog signaling in ovarian cancer. Clin Cancer Res.
14:7659–7666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song LB, Li J, Liao WT, et al: The
polycomb group protein Bmi-1 represses the tumor suppressor PTEN
and induces epithelial-mesenchymal transition in human
nasopharyngeal epithelial cells. J Clin Invest. 119:3626–3636.
2009. View
Article : Google Scholar
|
|
25
|
Song LB, Zeng MS, Liao WT, et al: Bmi-1 is
a novel molecular marker of nasopharyngeal carcinoma progression
and immortalizes primary human nasopharyngeal epithelial cells.
Cancer Res. 66:6225–6232. 2006. View Article : Google Scholar
|
|
26
|
Kim JH, Yoon SY, Jeong SH, et al:
Overexpression of Bmi-1 oncoprotein correlates with axillary lymph
node metastases in invasive ductal breast cancer. Breast.
13:383–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qin L, Zhang X, Zhang L, et al:
Downregulation of BMI-1 enhances 5-fluorouracil-induced apoptosis
in nasopharyngeal carcinoma cells. Biochem Biophys Res Commun.
371:531–535. 2008. View Article : Google Scholar : PubMed/NCBI
|