Introduction
Cyclin-dependent kinases (Cdks) and the cyclins are
the major regulators of cell cycle progression. Their activities
are controlled by a complex system directing stepwise
phosphorylation and dephosphorylation events. For example, the
activation of Cdk2 requires binding to cyclin A and the
phosphorylation of Cdk2 at a conserved threonine, T160 (1,2). On
the other hand, the phosphorylation of the other 2 conserved
residues in the catalytic cleft (tyrosine 15 and threonine 14)
inhibits the activity of activated Cdk2 (3,4). Such
regulatory phosphorylation pathways are conserved among various
species, from yeast to human (5).
It has been shown that several kinases, such as Cdk7-cyclin H, a
component of TFIIH in humans, and Cdk-activating kinase 1, a
serine/threonine kinase in budding yeast, have the ability to
phosphorylate T160 (6,7). On the other hand, the
dephosphorylation of T160 can be achieved by various enzymes, such
as protein phosphatase 2A and a dual-specific phosphatase, termed
Cdk-associated protein phosphatase (KAP) (8,9). Since
these enzymes can phosphorylate or dephosphorylate T160, they have
been considered to play a role in regulating Cdk activity.
KAP, also known as cyclin-dependent kinase
interactor 1 (Cdi1), is the product of the Cdk inhibitor 3
(CDKN3) gene (10–12). The expression of KAP increases at
the G1-S phase transition. It forms stable complexes
with Cdk2 and counteracts the stimulatory effect of Cdk-activating
kinase on Cdk2 activity (9). It has
been shown that the overexpression of KAP delays cell cycle
progression in yeast and HeLa cells. On the other hand, the
KAP-mediated dephosphorylation can be abolished by the binding of
cyclin A to Cdk2, despite the fact that KAP can still bind to the
cyclin A-Cdk2 complex. KAP can also bind to 2 other cell cycle
regulators, Cdc2 and Cdk3. However, direct evidence is still
lacking regarding the regulatory effect of KAP on these proteins
(10,11).
The fact that KAP is one of the important regulators
of cell cycle progression raises the possibility that it may
participate in carcinogenesis. However, few reports have addressed
the role of KAP in cancer and the results were somewhat
contradictory. Our previous studies have demonstrated that various
aberrant KAP mRNA transcripts, which encode truncated KAP mutants
lacking phosphatase activity, can be found in hepatocellular
carcinoma (HCC) and in cultured hepatoma cells (13). These truncated KAP mutants are
capable of inhibiting protein interaction in vitro between
wild-type KAP and Cdk2 (14). The
aberrantly-spliced KAP transcripts have also been found in
glioblastoma (15). In this type of
cancer, aberrant splicing leads to the generation of a
dominant-negative KAP variant that increases cell proliferation and
tumor migration. By contrast, KAP has been reported to be
overexpressed in breast and prostate cancers, suggesting a
growth-promoting effect (16). Our
recent study also demonstrated that the expression of KAP was
associated with poorly differentiated human renal cell carcinoma
and that the overexpression of KAP in vitro enhanced cell
proliferation, resistance to apoptosis and xenograft tumor
formation (17). However, these
findings were difficult to explain, since they were in conflict
with the established role of KAP in cell cycle inhibition.
HCC is the sixth most common cancer and the third
most frequent cause of cancer-related mortality worldwide (18). More than 70% of HCCs develop within
an established background of chronic liver disease. In eastern
Asia, the dominant risk factor is chronic hepatitis B virus (HBV)
infection, while in North America, Europe and Japan, hepatitis C
virus (HCV) infection is the major risk factor, in conjunction with
alcohol abuse (19). The
mechanistic events in HCC carcinogenesis are complex and, to date,
few molecular markers that correlate with the etiology and
prognosis of cancer have been reported (20,21).
Despite the aberrant KAP mRNA transcripts found in HCC, the role of
KAP expression in HCC remains unclear. In this study, we
investigated whether KAP expression correlates with
clinicopathological factors in HCC, including etiology,
pathological staging and clinical prognosis. Furthermore, the
growth-regulatory effects of KAP in HCC were evaluated in
vitro by antisense-mediated knockdown in Huh-7 cells. We aimed
to elucidate the possible role of KAP in hepatocarcinogenesis.
Materials and methods
Patients and HCC tissues
Under the approval of the Institutional Review
Board, Chang Gung Memorial Hospital, a total of 117 HCC patients
undergoing surgical resection from January 1996 to January 2002 at
Linkou Chang Gung Memorial Hospital, Taoyuan Hsien, Taiwan were
included in this study. The samples were retrieved from the Chang
Gung Tissue Bank. Clinicopathological information (including
gender, age, etiology, pathological subtype, histological grading,
biochemistries, tumor number and size) was retrospectively
reviewed. All cancerous and non-cancerous liver samples derived
from the periphery of the primary cancers were collected for
analysis. Immunohistochemistry and western blot analysis were
performed using mouse anti-KAP antibody (BD Biosciences, San Jose,
CA, USA). Actin was detected using mouse anti-β-actin antibody
(clone mAbcam 8226; Abcam Inc., Cambridge, MA, USA). The
quantification of the tumor-to-non-tumor KAP expression ratio (T/N
ratio) on the western blots was measured by ImageJ software
(developed by NIH, USA).
Plasmid construction, cell culture,
transfection and establishment of stable transformants
To knock down KAP expression, a plasmid capable of
producing an antisense transcript of KAP was constructed by
inserting the EcoRI and BamH1 (reverse) fragment of
pB42AD-KAP (14) into the pcDNA
3.1/V5-His A (Clontech) BamH1-EcoRI site. The
construct, pcDNA-KAPr, was sequence-verified and transfected into
human Huh-7 HCC cells. The cells were maintained in Dulbecco's
modified Eagle's medium containing 10% fetal bovine serum (FBS) and
stable clones were selected by neomycin (G-418, Geneticin;
Invitrogen).
Cell proliferation assay
Cell proliferation was assessed by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Cells were grown in a 96-well plate at an initial density of
5×103 cells per well. On the day of the assay, cells
were incubated at 37°C for 4 h in a culture medium containing 0.5
mg/ml MTT and then lysed by dimethyl sulfoxide (DMSO). The
absorbance was measured by a spectrophotometer at 570 nm. Three
independent experiments were performed for each measurement.
FACS analysis of cell cycle
Cells were plated at a density of 2×105
per 6-mm dish in complete medium for 24 h. 5-Bromo-2′-deoxy-uridine
(BrdU) was diluted to a concentration of 1 mM with 1X Dulbecco's
phosphate-buffered saline (DPBS) according to the BD Pharmingen
BrdU Flow kit instruction manual. BrdU (10 μl/ml medium) was added
to the plates (final concentration, 10 μM of BrdU) except for the
unpulsed control plate. Cells were incubated at 37°C with 5%
CO2 for 1 h. BrdU detection in the cells was performed
with a fluorescein anti-BrdU antibody (BD Biosciences) according to
the manufacturer's instructions.
BrdU incorporation assay to determine DNA
synthesis in the cell cycle
Cells were plated at a density of 1×104
cells per well in a 96-well plate in serum-free DMEM for 48 h to
synchronize the cells at the G0 phase. The medium was
replaced with DMEM containing 10% FBS to initiate the cell cycle.
Cells were then pulsed with 10 μM BrdU for 1 h at a 3-h interval
from 0 to 24 h. The amount of incorporated BrdU at each time-point
was measured by a chemiluminescence immunoassay, the BrdU Cell
Proliferation ELISA (Roche Applied Science, Indianapolis, IN, USA)
according to the manufacturer's instructions. All assays were
carried out in triplicate.
Terminal deoxynucleotidyl transferase
dUTP nick end-labeling (TUNEL) assay for detection of
apoptosis
The measurement of fragmented DNA from apoptotic
cells by incorporating flourescein 12-dUTP at the 3′-OH DNA end,
using the enzyme terminal deoxynucleotidyl transferase (TdT), was
used to detect apoptosis. To perform this assay, cells in
5×104 seeding density were plated on coverslips in a
12-well plate. Recombinant human tumor necrosis factor (TNF)-α (25
ng/ml; 2.8×107 U/mg, R&D Systems, Minneapolis, MN,
USA) and actinomycin-D (1 μg/ml, Boehringer Ingelheim, Mannheim,
Germany) were then added to the growth medium. Cells were cultured
for a further 8 h in a 37°C incubator and then fixed in 4%
paraformaldehyde. The DeadEnd™ Fluorometric TUNEL System (Promega,
Madison, WI, USA) was applied according to the manufacturer's
instructions. The TUNEL-stained coverslips were mounted onto slides
with Vectashield Mounting Medium with DAPI (Vector Laboratories,
Inc., Burlingame, CA, USA) and immediately examined under a
fluorescence microscope. The ratios of TUNEL-positive apoptotic
cells to DAPI-positive cells in 10 random fields at ×200
magnification were calculated. The assay was carried out in
triplicate.
Colony-forming assay
Soft agar assay was performed in 6-well plates by
growing 1×103 cells per well in DMEM with 10% FBS and
0.35% low melting agarose on top of a 0.8% agarose base layer. The
colonies were counted after staining with crystal violet 21 days
following incubation.
Tumorigenicity experiments in nude
mice
Male athymic BALB/c nude mice were obtained from the
National Animal Experimental Center (Taipei, Taiwan). The
procedures for animal experiments were approved by our local
(Linkou Chang Gung Memorial Hospital, Taoyuan, Taiwan) Animal
Ethics Committee. The mice were maintained under specific
pathogen-free conditions and used for the experiments when they
reached 4 weeks of age. Cells were harvested by trypsinization and
1×106 cells with >95% viability were injected
subcutaneously into the right side of the backs of nude mice. Tumor
formation was monitored daily until week 10. Tumor size was
calculated as 1/2 × ab2, where a is the longest diameter
and b is the shortest diameter of the tumor. Ten weeks later, the
mice were sacrificed and the tumors were isolated for
examination.
Immunoprecipitation by anti-Cdk2
The Huh-7-pcDNA-KAPr and Huh-7-mock cells were lysed
in 1 ml RIPA buffer [150 mM NaCl, 1.0% NP-40, 0.5% sodium
deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris (pH 7.5), 1
mM phenylmethylsulfonyl fluoride (PMSF) and 10 g/ml leupeptin] for
immunoprecipitation by anti-Cdk2 (Cdk2-Ab4, NeoMarkers, Fremont,
CA, USA). The precipitate was then analyzed by polyacrylamide gel
electrophoresis and subsequently electrotransferred onto a
nitrocellulose membrane for western blot analysis. KAP was detected
by mouse monoclonal anti-KAP antibody (BD Biosciences).
Western blot analysis of
proliferation-and apoptosis-related proteins
To investigate whether a growth regulatory signaling
pathway was involved, western blot analysis was performed. The
following antibodies were used: rabbit anti-phosphatase and tensin
homolog (PTEN) antibody and anti-phospho-PTEN (Ser380) antibody
(Cell Signaling Technology, Inc., Beverly, MA, USA); rabbit
anti-Akt antibody (Abcam Inc.); rabbit anti-phospho-AKT (Ser473)
antibody (Cell Signaling Technology, Inc.); rabbit anti-glycogen
synthase kinase (GSK)3 antibody (Imgenex Corp., San Diego, CA);
rabbit anti-phospho-GSK3 (Ser9) antibody (Cell Signaling
Technology, Inc.); rabbit anti-p44/42 mitogen-activated protein
kinase (MAPK) antibody (Cell Signaling Technology, Inc.); and
rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibody (Cell
Signaling Technology, Inc.). Actin was detected using mouse
anti-β-actin antibody (clone mAbcam 8226, Abcam Inc.).
Results
KAP expression in HCC
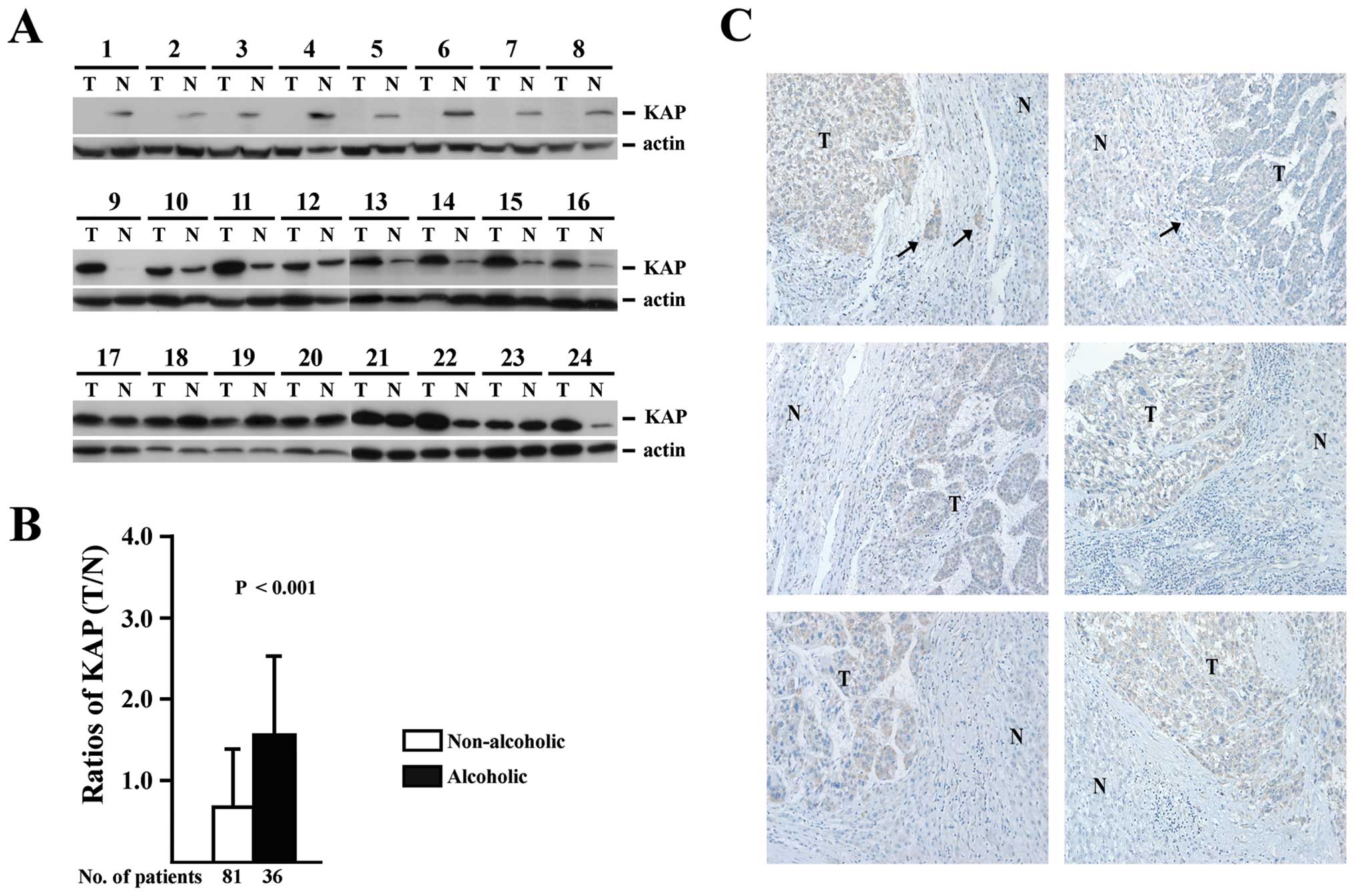
To identify the expression pattern of KAP in HCC,
117 HCC tissue sampes obtained by surgical resection were subjected
to western blot analysis for KAP expression. All patients had
clinically localized disease and were treated with partial
hepatectomy. Adjuvant chemotherapy and immunotherapy were not
administered to the patients prior to surgery. The basic
clinicopathological data are listed in Table I. Tissues obtained from tumor and
non-tumor sections of the same patient were analyzed by western
blot analysis, as shown in Fig. 1A.
To investigate the association between KAP expression and
clinicopathological parameters, the ratios of KAP expression in
tumor and non-tumor sections (T/N ratios) were measured and
correlated with clinicopathological parameters as well as
histopathological features by univariate and multivariate
statistical analysis, as shown in Table II. The histopathological features
(including micro-and macrovascular invasion, Edmondson's
histological grading, encapsulation, microsatellite lesions, tumor
number and size) were not associated with the T/N ratios of KAP
expression.
 | Table IBasic clinicopathological
characteristics of the 117 HCC patients. |
Table I
Basic clinicopathological
characteristics of the 117 HCC patients.
| Parameter | Value |
|---|
| Age (years) | 54.2±15.0a |
| Gender
(male/female) | 90/27 |
| Cirrhosis | 48 |
| HBsAg-positive | 80 |
|
Anti-HCV-positive | 31 |
| Tumor number |
| 1 | 76 |
| 2 | 15 |
| 3 | 20 |
| 4 | 6 |
| Size (diameter,
cm) | 7.0±4.7a |
| Microvascular
invasion | 39 |
| Edmondson's
grading |
| 1–2 | 29 |
| 3 | 70 |
| 4 | 18 |
| Encapsulation | 86 |
| Macrovascular
invasion | 0 |
| Ascites | 9 |
| α-fetoprotein
(ng/ml) | 14
(3–327500)b |
| Albumin (g/dl) | 3.8±0.7a |
| Bilirubin
(mg/dl) | 1.9±1.9a |
| Prothrombin time
(sec) | 12.3±1.4a |
| Creatinine
(mg/dl) | 1.2±1.5a |
| AST (U/l) | 100.8±128.3a |
| ALT (U/l) | 82.3±104.6a |
| Alcoholism | 36 |
| Time to last
follow-up or death (months) | 25 (2–127)b |
| Table IIUnivariate and multivariate analysis
of clinicopathological parameters associated with T/N ratio of KAP
in HCC. |
Table II
Univariate and multivariate analysis
of clinicopathological parameters associated with T/N ratio of KAP
in HCC.
| Factors | Groups | Pt. no. (Mean ±
SD) | KAP T/N ratio (95%
CI) | Unadjusted β (95%
CI) | Adjusted β |
|---|
| Gender | Male | 90 | 1.05±0.89 | 0.28 (−0.09,
0.65) | |
| Female | 27 | 0.77±0.71 | | |
| Age (years) | >54 | 62 | 0.90±0.81 | −0.19 (−0.50,
0.12) | |
| ≤54 | 55 | 1.09±0.91 | | |
| Cirrhosis | Yes | 48 | 1.03±0.99 | 0.74 (−0.25,
0.39) | |
| No | 69 | 0.96±0.76 | | |
| HBsAg | Positive | 80 | 1.02±0.86 | 0.11 (−0.22,
0.45) | |
| Negative | 37 | 0.91±0.85 | | |
| Anti-HCV | Positive | 31 | 1.10±1.05 | 0.15 (−0.21,
0.50) | |
| Negative | 86 | 0.95±0.78 | | |
| Microvascular
invasion | Yes | 39 | 1.12±1.00 | 0.21 (−0.13,
0.54) | |
| No | 78 | 0.92±0.77 | | |
| Macrovascular
invasion | Yes | 9 | 1.26±0.75 | 0.30 (−0.29,
0.89) | |
| No | 108 | 0.97±0.86 | | |
| Edmondson's
histological grading | >II | 87 | 0.94±0.83 | −0.19 (−0.55,
0.17) | |
| ≤II | 30 | 1.13±0.93 | | |
| Capsule | Yes | 86 | 0.95±0.75 | −0.16 (−0.52,
0.19) | |
| No | 31 | 1.11±1.10 | | |
| Microsatellite
lesions | Yes | 20 | 1.25±0.88 | 0.32 (−0.10,
0.73) | |
| No | 97 | 0.93±0.85 | | |
| Tumor number | >1 | 41 | 0.95±0.88 | −0.95 (−0.43,
0.24) | |
| 1 | 74 | 1.04±0.85 | | |
| Size (cm) | >7.0 | 37 | 1.02±0.67 | 0.01 (−0.33,
0.36) | |
| ≤7.0 | 77 | 1.00±0.94 | | |
| Ascites | Yes | 9 | 1.13±0.76 | 0.16 (−0.43,
0.75) | |
| No | 108 | 0.98±0.87 | | |
| AFP (ng/ml) | >400 | 31 | 0.79±0.75 | −0.27 (−0.62,
0.09) | |
| ≤400 | 86 | 1.06±0.89 | | |
| AST (U/l) | >100 | 32 | 1.30±1.00 | 0.43 (0.08,
0.77) | 0.00 (−0.00,
0.00)a |
| ≤100 | 85 | 0.87±0.77 | | |
| ALT (U/l) | >80 | 31 | 1.18±0.77 | 0.19 (−0.17,
0.55) | |
| ≤80 | 86 | 0.94±0.88 | | |
| Alcoholism | Yes | 36 | 1.58±0.93 | 0.86 (0.56,
1.16) | 0.87 (057,
1.18)b |
| No | 81 | 0.72±0.68 | | |
| Child-Pugh
classification | A | 100 | 0.96±0.82 | −0.20 (−0.64,
0.25) | |
| B | 17 | 1.16±1.06 | | |
On the other hand, while most of the clinical
parameters, including age, gender, cirrhosis, HBV surface antigen
(HBsAg) serological positivity, anti-HCV serological positivity,
ascites, α-fetoprotein (AFP), alanine aminotransferase (ALT) and
Child-Pugh classification, were not associated with KAP expression,
alcoholism and AST showed a positive correlation with increased KAP
expression in the tumor sections (higher T/N ratios). However,
since AST is usually higher in alcoholic patients, it was not
associated with KAP expression following adjustment for the
alcoholism factor (P<0.001). The T/N ratios of KAP between
alcoholic and non-alcoholic patients are shown in Fig. 1B and KAP expression in the tumor
sections was found to be significantly higher in HCC samples from
alcoholic patients.
The association between KAP T/N ratios and tumor
number in the 36 alcoholic patients is shown in Table III. An even higher expression of
KAP was observed in the tumor sections from alcoholic HCC patients
with <3 tumors. To confirm this finding, KAP
immunohistochemistry staining was performed on HCC tissues from
alcoholic HCC patients with <3 tumors, as shown in Fig. 1C. Again, a stronger staining for KAP
was observed in the tumor sections compared to the adjacent
non-tumor sections. The finding that KAP expression was
significantly increased in the tumor sections of alcoholic HCC
patients with a smaller number of tumors (<3) suggested that KAP
may play a role in alcohol-related carcinogenesis.
| Table IIIAssociation betwen KAP T/N ratios and
tumor number in the 36 alcoholic HCC patients. |
Table III
Association betwen KAP T/N ratios and
tumor number in the 36 alcoholic HCC patients.
| Tumor number | No. of
patients | KAP T/N ratios |
|---|
| <3 | 26 | 1.21±0.38a |
| ≥3 | 10 | 0.90±0.29a |
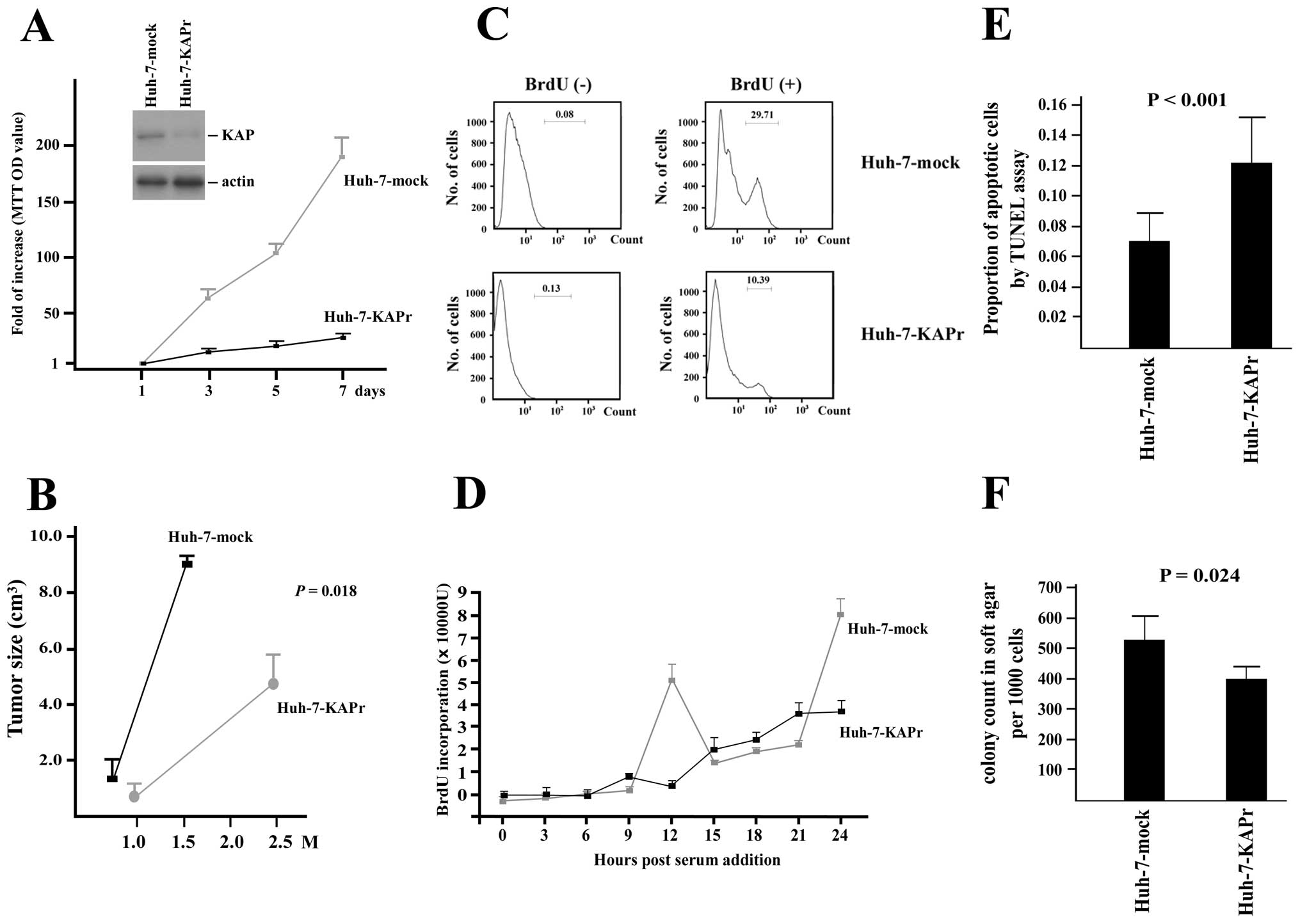
Antisense-mediated suppression of KAP in
Huh-7 cells results in decreased proliferation and increases
apoptosis
To evaluate the role of KAP expression in HCC cells,
KAP knockdown was achieved by the transfection of pDR2-KAPr, which
expressed an antisense KAP RNA fragment. Western blot analysis of
KAP expression showed a significant decrease in KAP expression in
the Huh-7-KAPr cells compared to the Huh-7-mock cells, as shown in
Fig. 2A. MTT cell proliferation
assays showed a decreased cell growth rate in the Huh-7-KAPr cells
(Fig. 2A). To confirm the
association between KAP and the cell proliferation of Huh-7 cells,
a FACS analysis of BrdU expression in the Huh-7-KAPr and Huh-7-mock
cells was performed and the results are shown in Fig. 2C. The number of BrdU-positive cells
was significantly decreased among the Huh-7-KAPr cells compared to
the Huh-7-mock cells, suggesting that the inhibition of KAP
expression resulted in decreased cell proliferation.
In order to determine whether KAP affected cell
apoptosis, TUNEL assay was performed on the Huh-7-KAPr and
Huh-7-mock cells. The proportion of apoptotic cells was
significantly higher among the Huh-7-KAPr cells, suggesting that
the inhibition of KAP enhanced Huh-7 cell apoptosis (Fig. 2E).
Inhibition of KAP in Huh-7 cells affects
normal cell cycle progression
The Huh-7-KAPr and Huh-7-mock cells were
synchronized by serum starvation for 48 h. The amount of BrdU
incorporation was assessed every 3 h after the addition of FBS to
the culture medium. The first peak of BrdU incorporation was
detected 12 h after cell cycle initiation in the Huh-7-mock cells,
while no obvious peak was observed in the Huh-7-KAPr cells within
24 h, suggesting that the KAP knockdown affected the normal cell
cycle progression in the Huh-7 cells (Fig. 2D).
Soft agar colony formation assay and
tumorigenicity experiments
To investigate whether the anti-proliferation,
apoptosis enhancement and cell cycle disturbance caused by KAP
knockdown affected the colony-forming ability of the cells, the
soft agar colony formation assay was performed. As shown in
Fig. 2F, the Huh-7-KAPr cells
exhibited decreased colony-forming ability compared to the
Huh-7-mock cells. To further demonstrate the tumorigenic ability
in vivo, the mock-transfected cells and the Huh-7-KAPr cells
were injected subcutaneously into nude mice. The xenograft tumors
grew quickly in the 6 weeks following the injection of Huh-7-mock
cells, while the tumor size was significantly smaller even at 10
weeks following the injection of Huh-7-KAPr cells (8.9±0.2
cm3 vs. 4.3±1.3 cm3; P=0.018) (Fig. 2B).
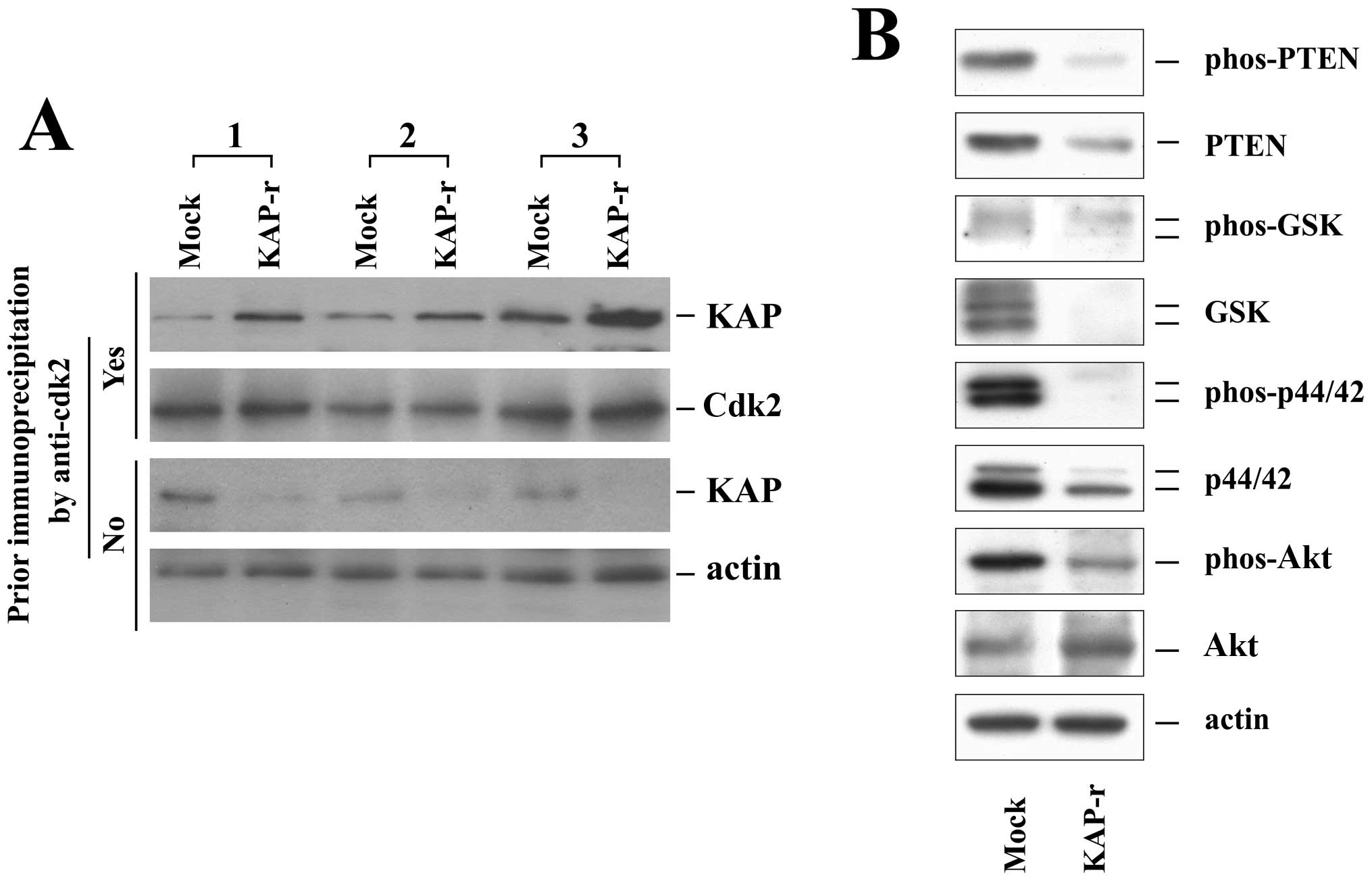
Increased Cdk2-KAP binding ability in
Huh-7-KAPr cells
Cdk2 is the essential protein for the cell cycle
G1/S phase transition. It is not only regulated by the
regulatory cyclin subunits but also by phosphorylation. Binding to
KAP can inactivate Cdk2 and disturb the cell cycle. Since cell
proliferation was decreased in Huh-7-KAPr cells, we examined the
Cdk2-KAP binding ability of the Huh-7-mock and Huh-7-KAPr cells by
immunoprecipitation (Fig. 3A).
Without prior immunoprecipitation by an anti-Cdk2 antibody, the KAP
protein was expressed in smaller amounts in the Huh-7-KAPr cells
than in the Huh-7-mock cells, as shown by the western blot analysis
results. With prior co-immunoprecipitation by the anti-Cdk2
antibody, the amount of KAP protein was significantly higher in the
Huh-7-KAPr than in Huh-7-mock cells in all 3 independent
experiments. These results suggested that, despite the smaller
amount of KAP protein produced by Huh-7-KAPr cells, the Huh-7-KAPr
cells exhibited a stronger Cdk2-KAP binding ability.
KAP affects PTEN, GSK, p44/42 and Akt
activities
To determine whether KAP affects proteins associated
with cell proliferation and apoptosis, PTEN, GSK, p44/42, Akt and
their phosphorylated forms were analyzed in the Huh-7-mock and
Huh-7-KAPr cells (Fig. 3B). While
the expression of Akt was increased, the expression of PTEN, GSK
and p44/42 was decreased in the Huh-7-KAPr cells. All the active
phosphorylated forms of PTEN, GSK, p44/42 and Akt were decreased in
the Huh-7-KAPr cells, suggesting that the activity of these
proteins was suppressed in the Huh-7-KAPr cells.
Discussion
In this study, KAP was overexpressed mainly during
the early stages of alcohol-related HCC. The major risk factors of
HCC include chronic HBV and HCV infection, exposure to aflatoxins
and alcohol abuse. It is generally believed that HCC develops in
the cirrhotic liver resulting from chronic inflammation and
fibrosis caused by the above-mentioned factors (22). However, some unique mechanisms have
been demonstrated which contribute to the development of HCC,
specifically in patients with alcoholic liver disease (23,24).
Acetaldehyde, the oxidized form of alcohol, has been identified as
a toxic compound with mutagenic properties (25). It can form DNA adducts, such as
N2-ethyl-2′deoxyguanosine (N2-ethyl-dG) and
1,N2-propano-2′-deoxyguanosine (1,N2-PdG),
resulting in alterations in DNA integrity. Furthermore, chronic
alcohol consumption causes the induction of cytochrome P450 2E1
(CYP2E1), which leads to the increased generation of acetaldehyde
and reactive oxygen species (ROS) (26,27).
Furthermore, CYP2E1 also metabolizes several toxic substrates,
including pro-carcinogenic compounds in alcoholic beverages. The
induction of CYP2E1 promotes carcinogenesis not only through its
own CYP2E1-dependent metabolism, but also through affecting the
rate of metabolism of other substrates (28–30).
Long-term alcohol consumption has been demonstrated
to decrease hepatocyte growth in vitro(31,32)
and hepatic regeneration following surgical ablation in
vivo(33,34). It has also been demonstrated that
alcohol consumption upregulates the expression and activity of
guanine nucleotide regulatory proteins (Gi-proteins) (35). The Gi-protein activation further
increases MAPK activity and cellular mitogenesis in HCC. Despite
these findings, the actual role of alcohol in HCC carcinogenesis is
not yet completely understood. In this study, we demonstrate that
KAP is overexpressed in alcohol-related HCC, particularly in
patients with <3 tumors, suggesting a potential role of KAP in
early alcohol-related carcinogenesis.
The studies on the role of KAP in carcinogenesis
have been somewhat contradictory. Initially, it was suggested that
KAP is an inhibitor of cell cycle progression and thus inhibits
cell growth (9). However, ensuing
studies demonstrated that KAP was overexpressed in breast, prostate
and renal cell cancers, suggesting a growth-promoting role
(16,17). Despite the fact that
dominant-negative mutant transcripts of KAP have been identified in
HCC and glioblastoma, these findings are not sufficient to explain
why KAP overexpression promotes tumor cell growth. In this study,
we found that the suppression of KAP in HCC in vitro
resulted in decreased proliferation and reduced colony-forming
ability of the cells, changes in the cell cycle, increased
apoptosis and decreased tumorigenicity. These findings support
those from previous studies on breast, prostate and renal cell
carcinoma, which suggested that KAP plays a growth-promoting role
in cancer. Our results also demonstrated that the phosphorylation
of proliferation- and apoptosis-related proteins, including PTEN,
GSK, p44/42 and Akt, was decreased in the Huh-7-KAPr cells,
suggesting that the decrease in KAP expression suppressed the
activity of these proteins and affected cell proliferation and
apoptosis. Furthermore, our data showed that the Cdk2-KAP binding
ability was increased in the KAP-suppressed HCC cells. The
increased Cdk2-KAP binding ability may theoretically result in the
decrease in the levels of phosphorylated Cdk2, leading to the
inhibition of cell proliferation. However, the reason why the
binding ability was increased remains unknown and further studies
are warranted to resolve this issue.
In conclusion, our study demonstrates that KAP is
overexpressed mainly during the early stages of alcohol-related
HCCs. The suppression of KAP expression resulted in decreased cell
proliferation, changes in the cell cycle, reduced colony-forming
ability of the cells and the suppression of tumorigenicity, at
least partly through the increased physical interaction with Cdk2
and thus the reduction of the active forms of growth-related
proteins.
Acknowledgements
This study was supported by a grant from the Chang
Gung Medical Research Program (CMRPG370694).
References
|
1
|
Brown NR, Noble ME, Lawrie AM, et al:
Effects of phosphorylation of threonine 160 on cyclin-dependent
kinase 2 structure and activity. J Biol Chem. 274:8746–8756. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Solomon MJ: Activation of the various
cyclin/cdc2 protein kinases. Curr Opin Cell Biol. 5:180–186. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Atherton-Fessler S, Hannig G and
Piwnica-Worms H: Reversible tyrosine phosphorylation and cell cycle
control. Semin Cell Biol. 4:433–442. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Den Haese GJ, Walworth N, Carr AM and
Gould KL: The Wee1 protein kinase regulates T14 phosphorylation of
fission yeast Cdc2. Mol Biol Cell. 6:371–385. 1995.PubMed/NCBI
|
|
5
|
Coleman TR and Dunphy WG: Cdc2 regulatory
factors. Curr Opin Cell Biol. 6:877–882. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaldis P, Sutton A and Solomon MJ: The
Cdk-activating kinase (CAK) from budding yeast. Cell. 86:553–564.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nigg EA: Cyclin-dependent kinase 7: at the
cross-roads of transcription, DNA repair and cell cycle control?
Curr Opin Cell Biol. 8:312–317. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lew DJ and Kornbluth S: Regulatory roles
of cyclin dependent kinase phosphorylation in cell cycle control.
Curr Opin Cell Biol. 8:795–804. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poon RY and Hunter T: Dephosphorylation of
Cdk2 Thr160 by the cyclin-dependent kinase-interacting phosphatase
KAP in the absence of cyclin. Science. 270:90–93. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gyuris J, Golemis E, Chertkov H and Brent
R: Cdi1, a human G1 and S phase protein phosphatase that associates
with Cdk2. Cell. 75:791–803. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hannon GJ, Casso D and Beach D: KAP: a
dual specificity phosphatase that interacts with cyclin-dependent
kinases. Proc Natl Acad Sci USA. 91:1731–1735. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song H, Hanlon N, Brown NR, Noble ME,
Johnson LN and Barford D: Phosphoprotein-protein interactions
revealed by the crystal structure of kinase-associated phosphatase
in complex with phosphoCDK2. Mol Cell. 7:615–626. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yeh CT, Lu SC, Chen TC, Peng CY and Liaw
YF: Aberrant transcripts of the cyclin-dependent kinase-associated
protein phosphatase in hepatocellular carcinoma. Cancer Res.
60:4697–4700. 2000.PubMed/NCBI
|
|
14
|
Yeh CT, Lu SC, Chao CH and Chao ML:
Abolishment of the interaction between cyclin-dependent kinase 2
and Cdk-associated protein phosphatase by a truncated KAP mutant.
Biochem Biophys Res Commun. 305:311–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu Y, Jiang X, Schoch BS, Carroll RS,
Black PM and Johnson MD: Aberrant splicing of cyclin-dependent
kinase-associated protein phosphatase KAP increases proliferation
and migration in glioblastoma. Cancer Res. 67:130–138. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SW, Reimer CL, Fang L, Iruela-Arispe
ML and Aaronson SA: Overexpression of kinase-associated phosphatase
(KAP) in breast and prostate cancer and inhibition of the
transformed phenotype by antisense KAP expression. Mol Cell Biol.
20:1723–1732. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lai MW, Chen TC, Pang ST and Yeh CT:
Overexpression of cyclin-dependent kinase-associated protein
phosphatase enhances cell proliferation in renal cancer cells. Urol
Oncol. 30:871–878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar
|
|
19
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Villanueva A, Newell P, Chiang DY,
Friedman SL and Llovet JM: Genomics and signaling pathways in
hepatocellular carcinoma. Semin Liver Dis. 27:55–76. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morgan TR, Mandayam S and Jamal MM:
Alcohol and hepatocellular carcinoma. Gastroenterology. 127(5 Suppl
1): S87–S96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McKillop IH and Schrum LW: Role of alcohol
in liver carcinogenesis. Semin Liver Dis. 29:222–232. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brooks PJ and Theruvathu JA: DNA adducts
from acetaldehyde: implications for alcohol-related carcinogenesis.
Alcohol. 35:187–193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Koop DR: Oxidative and reductive
metabolism by cytochrome P450 2E1. FASEB J. 6:724–730.
1992.PubMed/NCBI
|
|
27
|
Lieber CS: Cytochrome P-4502E1: its
physiological and pathological role. Physiol Rev. 77:517–544.
1997.PubMed/NCBI
|
|
28
|
Badger TM, Ronis MJ, Seitz HK, Albano E,
Ingelman-Sundberg M and Lieber CS: Alcohol metabolism: role in
toxicity and carcinogenesis. Alcohol Clin Exp Res. 27:336–347.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tanaka E, Terada M and Misawa S:
Cytochrome P450 2E1: its clinical and toxicological role. J Clin
Pharm Ther. 25:165–175. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang CS, Yoo JS, Ishizaki H and Hong JY:
Cytochrome P450IIE1: roles in nitrosamine metabolism and mechanisms
of regulation. Drug Metab Rev. 22:147–159. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carter EA and Wands JR: Ethanol inhibits
hormone stimulated hepatocyte DNA synthesis. Biochem Biophys Res
Commun. 128:767–774. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lad PJ, Shier WT, Skelly H, de Hemptinne B
and Leffert HL: Adult rat hepatocytes in primary culture. VII.
Proliferative and functional properties of cells from
ethanol-intoxicated animals: evidence for a reversible albumin
‘production defect’. Alcohol Clin Exp Res. 6:72–79. 1982.PubMed/NCBI
|
|
33
|
Duguay L, Coutu D, Hetu C and Joly JG:
Inhibition of liver regeneration by chronic alcohol administration.
Gut. 23:8–13. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wands JR, Carter EA, Bucher NL and
Isselbacher KJ: Inhibition of hepatic regeneration in rats by acute
and chronic ethanol intoxication. Gastroenterology. 77:528–531.
1979.PubMed/NCBI
|
|
35
|
McKillop IH, Vyas N, Schmidt CM, Cahill PA
and Sitzmann JV: Enhanced Gi-protein-mediated mitogenesis following
chronic ethanol exposure in a rat model of experimental
hepatocellular carcinoma. Hepatology. 29:412–420. 1999. View Article : Google Scholar
|