Introduction
Normal human cells have a limited life span due to
the loss of telomeric sequences after each DNA replication
(1). A critically short telomere
length causes the cells to move into the stage of senescence and
ultimately to pursue the path of apoptosis (2). This mechanism helps to protect the
entire organism (3). Early studies
of hTERT expression revealed that telomerase is suppressed in human
somatic tissues, but it is expressed robustly in germ and tumor
cells (4). However, germ and
embryonic cells are able to maintain their telomere stability to
clone adult cell nuclei into their shortened telomeres (5,6).
Telomerase activity alone is not enough to maintain stable telomere
lengths, since telomeres continue to shorten (7,8). Even
hTERT expression and telomerase activation in early passages of
human fibroblasts are mainly detected in the S phase (9). This indicates that the regulation of
hTERT and telomerase in normal cells is important for the
proliferation of normal human cells.
Evidence for the role of telomeres in tumorigenesis
has been provided in studies of primary human fibroblasts. These
primary human cells reach a limited potential of replication of
approximately 60–80% population doubling per day before moving into
the senescence stage (10). In
contrast, tumor cells establish indefinite devision in culture.
Observations of primary and tumor cells indicate that this
difference was associated with the shortening of telomeres. There
is a loss of telomeric sequences in normal primary cells after each
cell division, but not in tumor cells (1). However, tumor cells achieve a
stabilization of telomeres through the activation of telomerase or
alternative lengthening of telomere (ALT) mechanism, and thus
overcome the senescence stage. Immortalization is an important
prerequisite for unimpeded tumor growth and thus is an essential
step in the malignant transformation of cells (11). Thus, immortalization allows an
unhindered proliferation of tumor cells and malignant
transformation by the accumulation of genetic alterations.
The ALT mechanism plays an important role in
malignant transformation. In a carcinogenesis model, cell cycle
immortalization based on ALT induced an oral-esophageal squamous
epithelial origin after inactivation of the tumor-suppressor p53
and overexpression of cyclin D1 protein (12). In addition, overexpression of
epidermal growth factor receptor (EGFR) resulted in the in
vitro transformation of these cells through concomitant
activation of telomerase (13,14).
Since a crucial step in the pathway to malignant transformation of
cells and tumor formation is immortalization, which essentially
depends on telomere maintenance, the present study aimed to
investigate the role of telomerase in the progression of esophageal
adenocarcinoma arising from Barrett’s esophagus.
Materials and methods
Cell line and culture
The Barrett’s esophageal cell line CP-A was used in
this study. CP-A cells were cultured in serum-free keratinocyte
medium (Invitrogen Life Technologies) and passaged when reaching
30–40% confluence. Subsequently, the medium was removed, and the
cells were washed with 5 ml PBS and resolved with a Trypsin/PBS
mixture (1:1). After a 10- to 20-min incubation in an incubator at
37°C in 5% CO2, the dissolved cells were taken up in 6
ml medium containing Dulbecco’s modified Eagle’s medium F-12 (DMEM;
Invitrogen) and centrifuged for 5 min at 138 × g. Subsequently, the
supernatant was discarded, and the cells were diluted to 1:10–1:80
in fresh medium according to the thickness of the cell pellets.
These cells were then transferred into new 10-cm culture dishes.
The cells were maintained in an incubator at 37°C in 5%
CO2 in a moisture-saturated atmosphere. In order to wash
the cells, only the medium was removed. The cells were washed with
5 ml of PBS, and 8–10 ml of fresh cells suitable for the respective
medium was added.
DNA extraction
DNA extraction was performed using the
QIAamp® DNA Mini kit (Qiagen) according to the
manufacturer’s protocol. After trypsinization and subsequent
centrifugation, the cells were resuspended in 1 ml of PBS and
centrifuged for 1 min at 5.9 × g. The entire supernatant was
carefully removed, and the pellet was resuspended in 200 μl PBS.
This was followed by addition of 20 ml proteinase K (Qiagen) and
lysis buffer AL (Qiagen), and mixed for 15 sec and then incubated
at 56°C for 10 min. After incubation, the tubes were centrifuged
briefly, and 200 μl 100% ethanol was added. The mixtures were mixed
again for 15 sec and finally applied to a well QIAamp Mini spin
column and centrifuged at 5.8 × g for 1 min. The column was placed
in a new collection tube, 500 μl of AW1 buffer (Qiagen) was added
to the column and centrifugation was carried out for 1 min at 5.9 ×
g. Then 500 μl AW2 buffer (Qiagen) was added to the column and
centrifugation was carried out for 3 min at 15.7 × g. DNA was
placed in a 1.5-ml reaction vessel, pipetted with 150–200 μl AE
buffer (Qiagen) and centrifuged for 1 min with incubation at RT at
5.8 × g.
Transduction
The cells were seeded in a 6-well plate at
2.5×105/well and incubated overnight in an incubator at
37°C in 5% CO2. Two days later the cells were washed and
spread onto a 6-well plate with the infection medium containing 2
ml of optimal media and 8 mg/ml Polybrene®. The cells
were then centrifuged for 2 h at 800 × g at 20°C, and then
incubated overnight in an incubator at 37°C in 5% CO2.
On the next day, the infection medium was removed and replaced with
2 ml of fresh medium. A few days after transduction, when cells
were grown to a sufficient density, the cells were analyzed and
sorted using a MoFlo high-speed cell sorter (Dako). The cells were
then trypsinized, centrifuged and resuspended in 1 ml of DMEM and
PBS and maintained on ice during the sorting process. After
sorting, the cells were added to a fresh 10 cm plate, which was
filled with fresh medium. The plasmids used for transduction are
listed in Table I.
 | Table IList of the plasmids used for
trasduction. |
Table I
List of the plasmids used for
trasduction.
| ID | Name | Description |
|---|
| Plasmid 1 | WT-hTER | Wild-type RNA
template |
| Plasmid 2 | MT-hTER/AU5 | Mutated RNA
template |
| Plasmid 3 | MT-hTER/47/A | Mutated RNA
template |
| Plasmid 6 |
MT-hTER/AU5+siRNA | Mutated RNA template
in combination with an siRNA against the RNA template |
| Plasmid 7 |
MT-hTER/47A+siRNA | Mutated RNA template
in combination with an siRNA against the RNA template |
| Plasmid 8 | Empty vector | Control |
Terminal restriction fragment (TRF)
analysis
The DNA digestion was performed using the
RsaI/HinfI (Roche, Indianapolis, IN, USA) restriction
enzymes. The restriction mixture (50 ml) contained 2–5 g DNA, 1X
NEB buffer 2 or 4, 10 U of RsaI and 10 U of
HinfI.
The DNA extraction was digested overnight at 37°C.
Each 4 μl of digested or undigested DNA was mixed with 16 μl TE
buffer (Qiagen) and 4 l 6X loading buffer (Fermentas), loaded
alternately onto a 1% agarose TAE gel, boiled in a microwave oven
and treated with 0.1 μg/ml ethidium bromide. The mixture was poured
into cooled horizontal gel chambers for 1 h. The separation time
was 1 h at 120 V. Then it was poured in a Protean XL horizontal
chamber (Bio-Rad Laboratories). Samples (20 ml) were mixed with 4
μl 6X loading buffer. The running time was 14 h at 40 V. The gel
was then viewed under UV light. In preparation for the Southern
blot analysis, the gel was placed for 15 min in 0.25 M hydrochloric
acid and finally washed with neutralizing buffer. Southern blot
analysis was performed.
Fluorescence in situ hybridization
The quantitative fluorescence in situ
hybridization (Q-FISH) was performed by metaphase chromosome spread
preparation with a telomeric PNA probe, which was labeled with the
fluorescent dye Cy3 hybridized. After counterstaining the
chromosomes with DAPI, they were observed under a fluorescence
microscope and the fluorescence intensities of the telomeres was
determined. The advantage of this method is that telomere
measurements may be performed on individual cells. Q-FISH method
was performed as previously described (15). In chromosome orientation FISH
(CO-FISH), the exchange of sister chromatids at telomeres and
chromosomes may be demonstrated in single cells.
Results
Telomerase inhibition was observed in the
genetically transduced Barrett’s and control cells with telomerase
inhibitors. These cells were transduced with the telomerase
inhibitors at a very low initial passage (passage 32).
There was a reduction in telomerase activity after
CP-A cells were transduced with plasmid 7, which led to an almost
complete disappearance of telomerase activity. This inhibition was
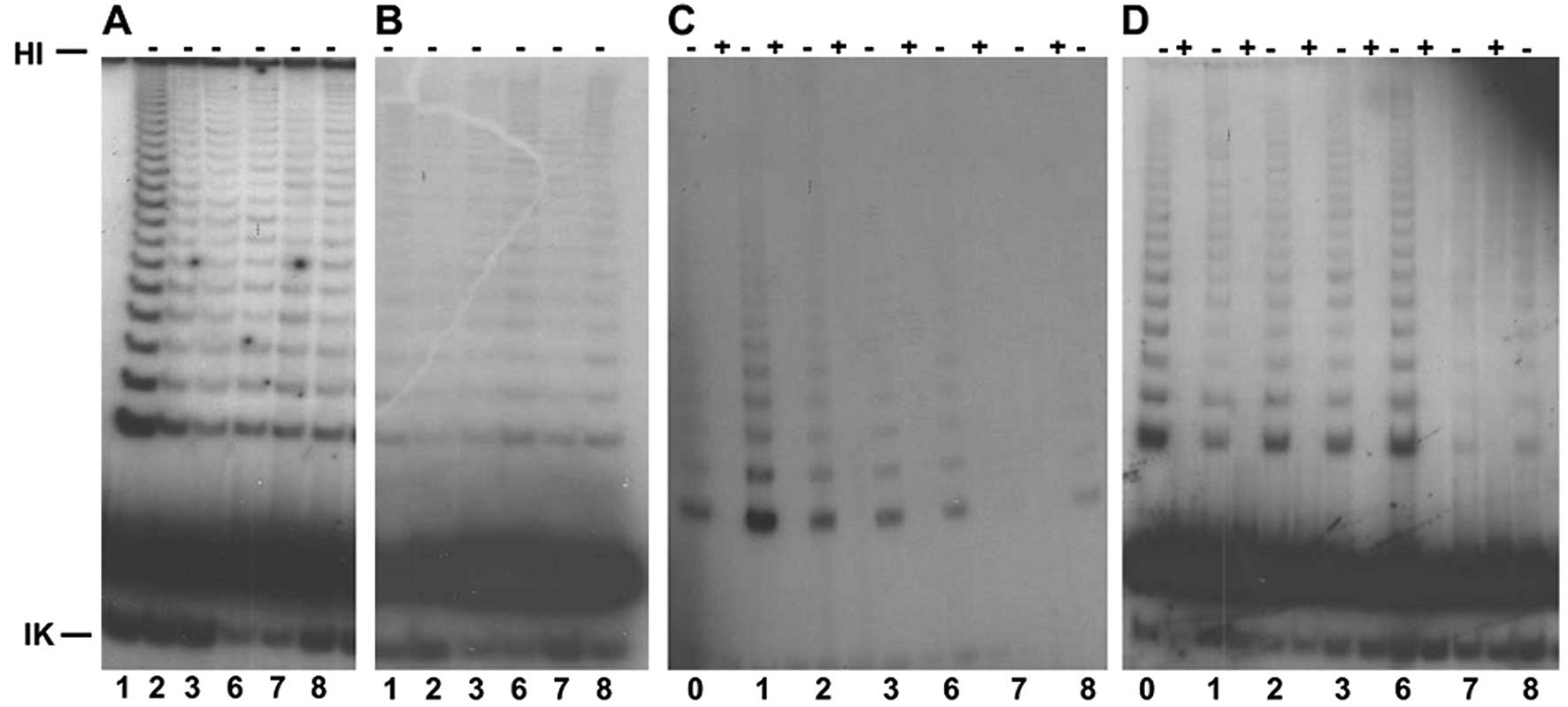
demonstrated in different passages (Fig. 1).
The telomere length of the control cells was reduced
compared with the untransduced wild-type CP-A cells as determined
by TRF analysis. The telomeres were stable and were 12 kbp in 3
passages after transduction with plasmids 1–8 (Fig. 2A). After transduction with plasmid 1
(WT-hTER), analysis of the telomere lengths at passage 50
demonstrated an extension of telomeres, which was ultimately >21
kbp.
The telomere length of the telomerase-inhibited
cells initially remained the same length as the control cells,
although a significant reduction in telomerase activity was noted
after transduction with plasmid 7 (MT-hTER/47A) over further
passages 36 h later. The telomere length of the
telomerase-inhibited cells remained the same length as the control
cells in passage 86 (Fig. 2C). In
contrast, the telomere lengths of the CP-A cells transduced with
plasmid 7 were slightly shorter (Fig.
2C). This telomere heterogeneity remained constant over a
further passage 23 (Fig. 2D),
indicating an ALT mechanism.
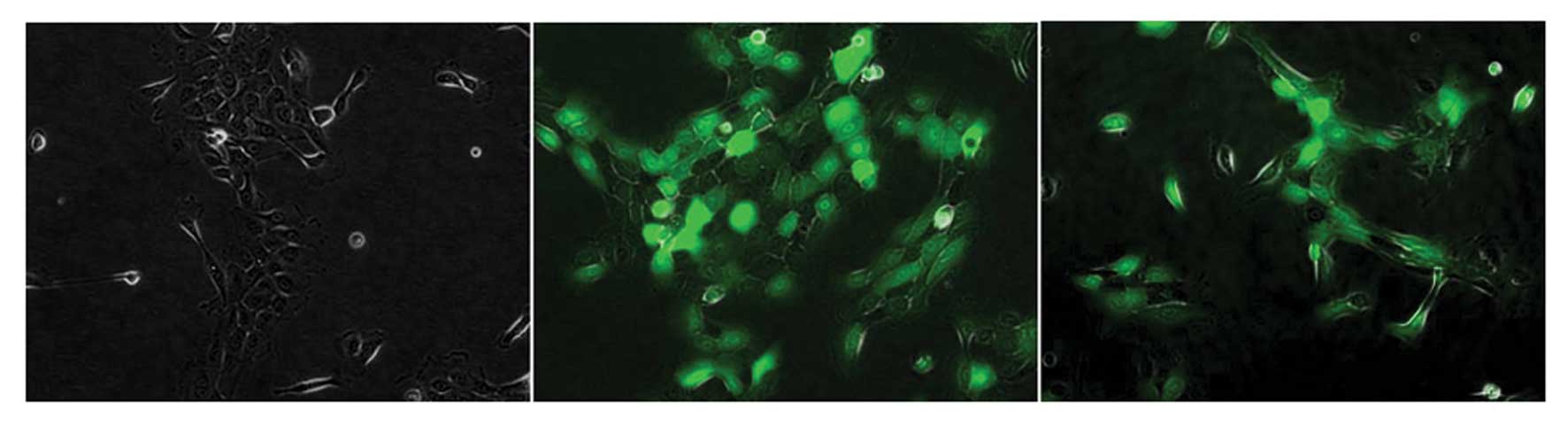
Following the successful reduction in telomerase by
the telomerase inhibitory plasmids, the cells were examined for
morphological changes. The different telomerase-inhibited cells
were morphologically indistinguishable from the untransduced and
WT-hTER-transduced cells (Fig. 3).
The cells underwent division normally and showed no cellular
morphology for further changes of apoptosis. The GFP-positive cells
differed only in their brightness.
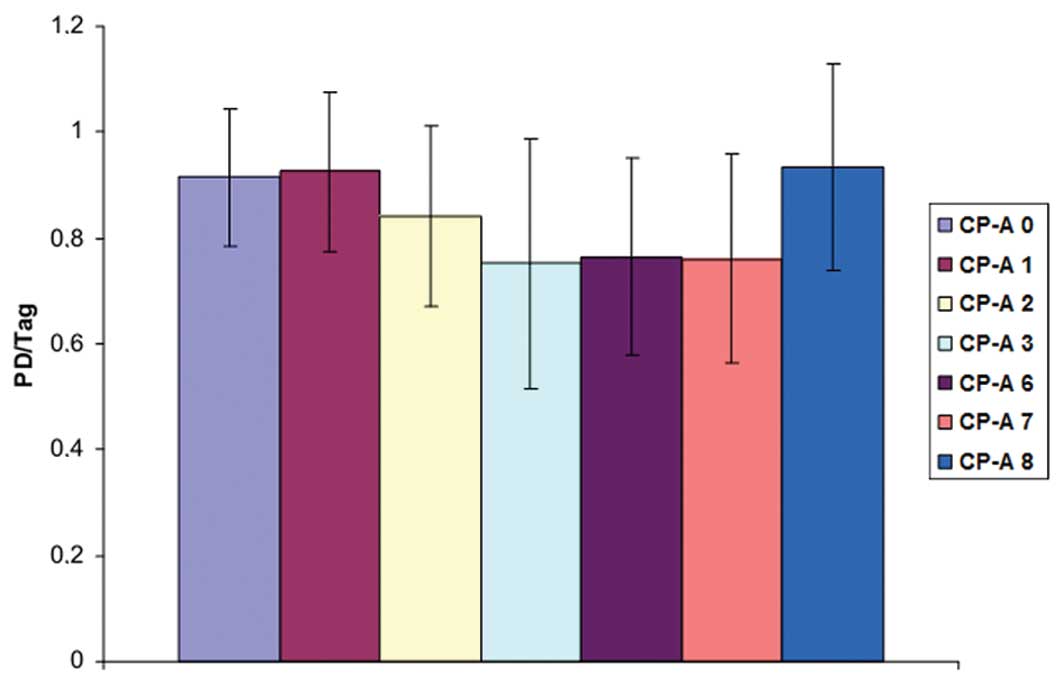
To create the growth curve of the transduced CP-A
cells, CP-A cells transduced with plasmid 1–8 were plated at
1×105 cells/well and trypsinized for 2–3 days. Then the
cells were counted. This procedure was repeated 4 to 6 times. In
the untransduced cells and the cells transduced with plasmid 1 or
8, the growth rate was between 0.9 to 1.1 with the population
doubling per day (Fig. 4). A
similar situation was also noted in the CP-A cells transduced with
telomerase-inhibitory plasmids 2–7. Again, the transduction with
the telomerase inhibitors did not cause a significant reduction in
cell growth. However, these transduced cells grew generally slower
compared with the untransduced cells and the cells transduced with
plasmid 1 or 8.
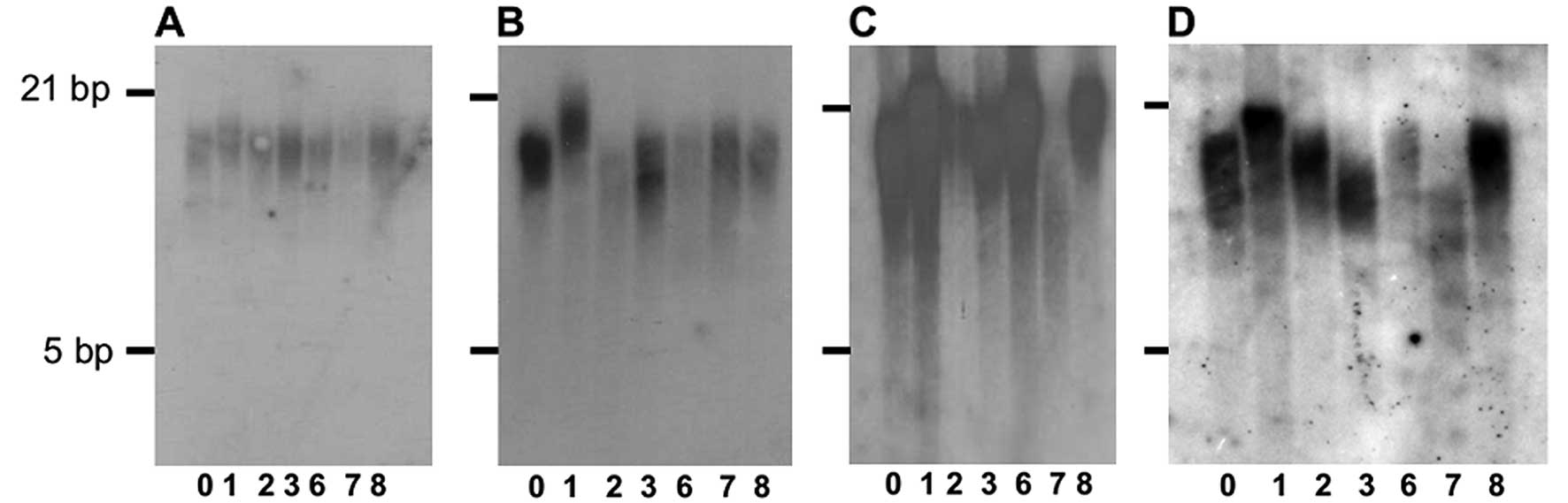
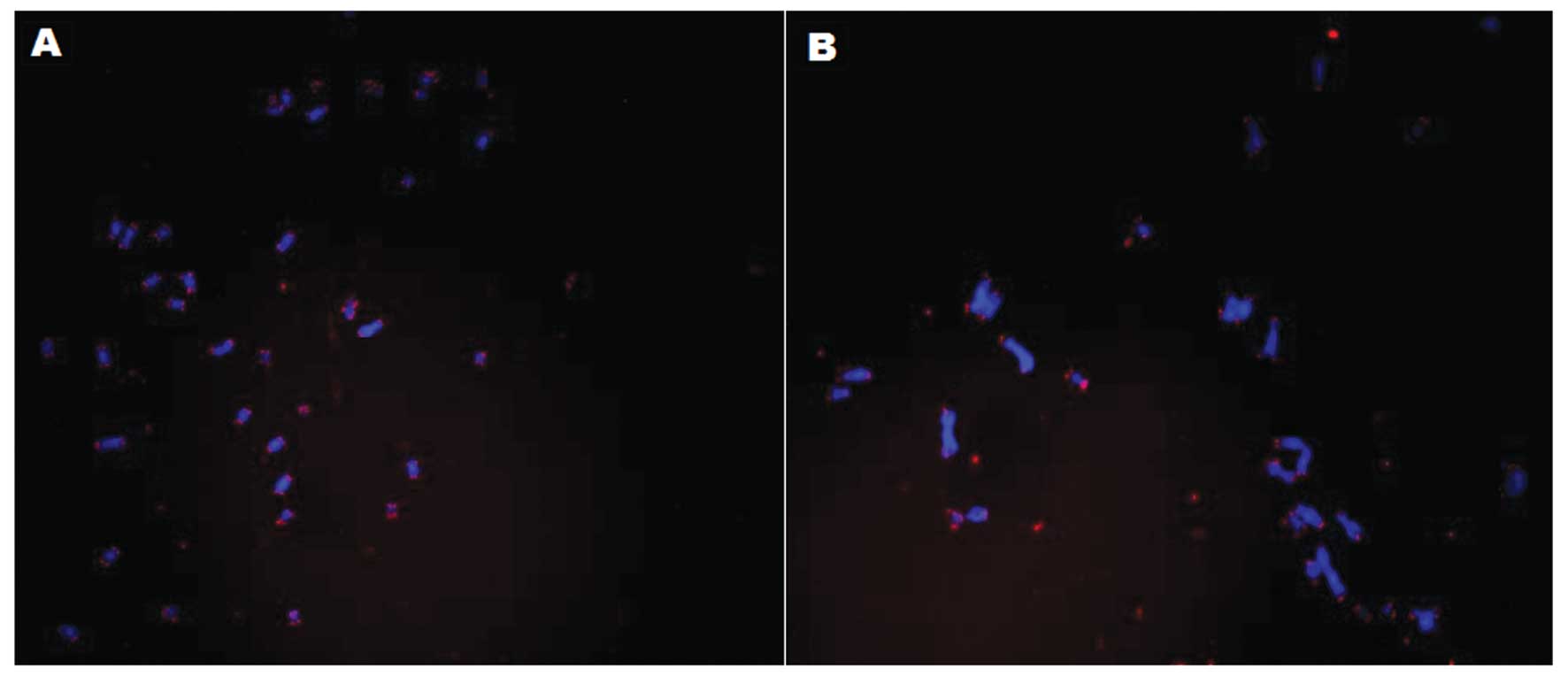
A heterogeneous telomere length was also detected in
the telomerase-inhibited CP-A cells (Fig. 5B). However, the telomere length
remained homogeneous in the control cells (Fig. 5A). The untransduced cells and the
cells transduced with plasmid 1–8 were seeded for 6 h, and on the
next day cells were treated with colcemid to arrest cells in the
metaphase. After the KCl treatment and subsequent fixation, the
swollen cells were dropped onto a slide and thus burst. The
metaphase spreads were stained with DAPI (blue) and the telomeres
with a Cy3-labeled probe (red), and the telomeric sequence was
hybridized.
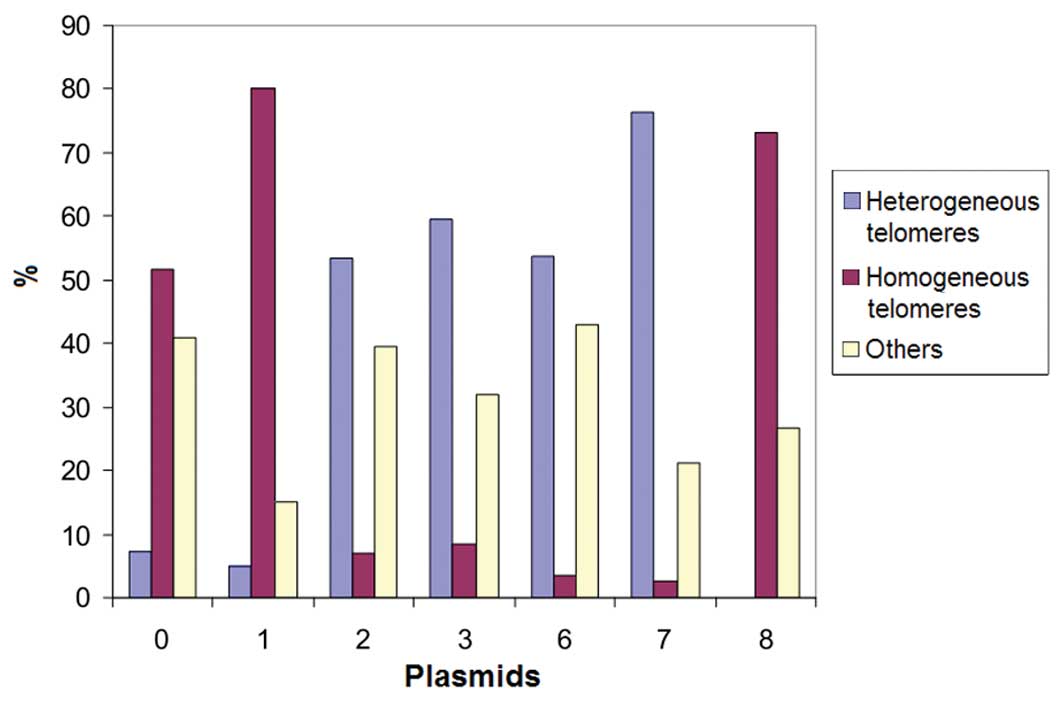
The metaphases of CP-A cells transduced with plasmid
7 showed 70% heterogeneous telomeres (Fig. 6). This correlated with the decrease
in telomerase activity in CP-A cells transduced with plasmid 7 as
determined by TRF analysis and the increasing heterogeneous
telomere lengths.
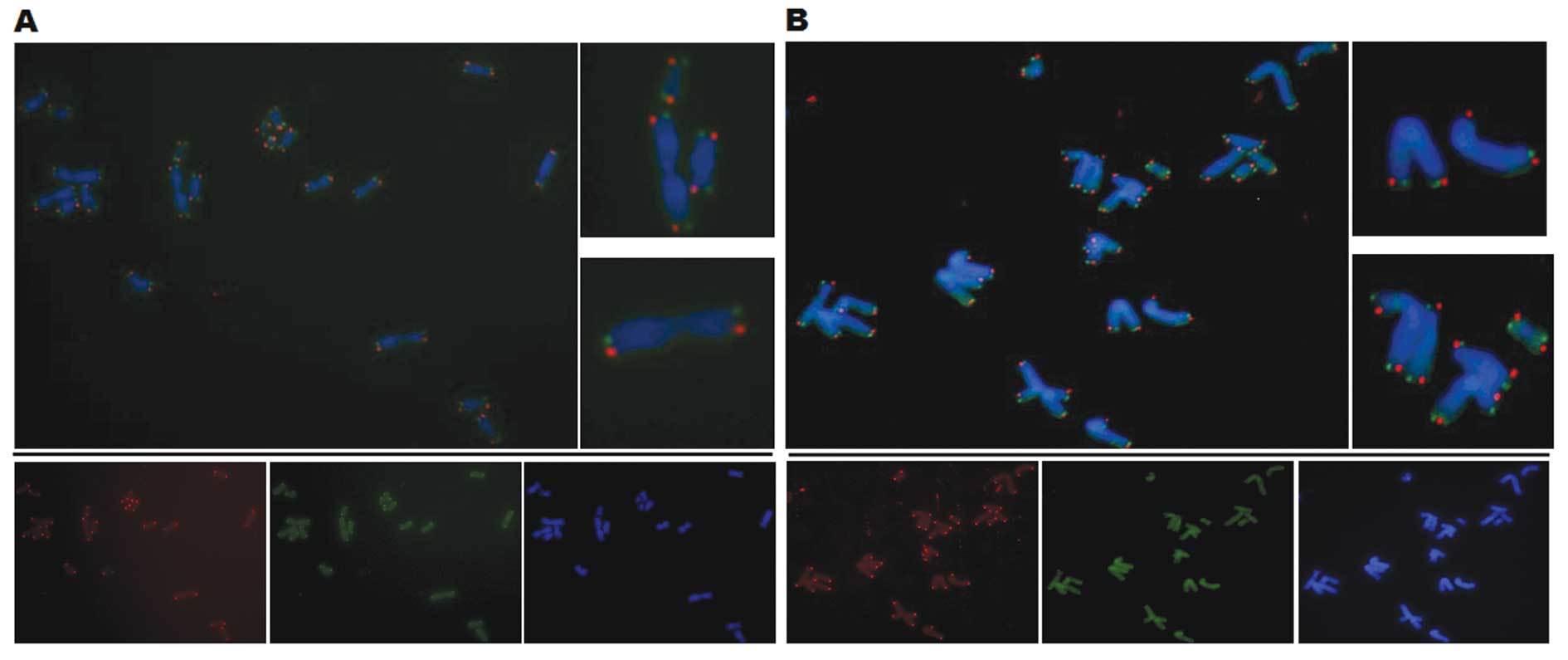
To investigate the exchange of sister chromatids,
the chromosome orientation FISH (CO-FISH) was performed. The newly
replicated DNA strand was degraded by metaphase, leading to a
single-stranded target DNA with either a C-rich or G-rich telomere
strand. The next step was the hybridization with specific probes,
performed either against the C-rich (green) and G-rich (red)
strand. In the event there was a recombination between sister
chromatids, a colocalization of two probes was found at the
telomeres, which led to a yellow color.
However, no increased recombination was observed
between sister chromatids in the transduced CP-A cells (Fig. 7B) compared with the control cells
(Fig. 7A). Thus, there existed an
ALT mechanism that was not based directly on the exchange of sister
chromatids.
Discussion
All telomerase-inhibited cells were compared with
the untransduced and plasmid 1- and 8-transduced cells. To confirm
whether the telomerase inhibition led to a reduction in telomerase,
the enzyme activity was determined. A reduction in telomerase was
observed in all CP-A cells transduced with the
telomerase-inhibiting plasmids, similar to that noted in other
tumor cells after telomerase inhibition (16). This inhibition remained stable.
There was an increased telomerase inhibition in Barrett’s cells
after transduction with plasmid 7, which is a combination of
mutated RNA template and an siRNA against the RNA template. This is
considered logical, since the mutated version is present in the
cells and the RNA template is present and may be inserted in the
telomerase, thus leading to altered telomere sequences. In
contrast, endogenous RNA can be inhibited by the complementary
siRNA. The two plasmids (6 and 7) were also found to cause almost
complete inhibition of growth in the HCT116 colon carcinoma cell
line with telomerase inhibition after 10 days (16).
After transduction with the control plasmid 1,
increased telomerase activity was observed. This amplification of
telomerase activity by exogenous overexpression of hTER was also
observed in other tumor cells, such as HeLa cells and HT1080 cells
(17). Previously it was shown in
various species that the cells were inhibited in their growth after
telomerase inhibition (16,18–20),
while the telomere lengths did not decrease (16). The telomere lengths were observed
over many passages in the Barrett’s cells. The first stable
telomere length was shorter in Barrett’s cells after transduction
with the telomerase inhibitors. Heterogeneous telomere lengths are
an essential characteristic that results following the activation
of the ALT mechanism (21). It has
also been shown that immortalized esophageal keratinocytes maintain
their telomere lengths in spite of the reduced telomerase activity
after telomerase inhibition (22).
In the Barrett’s cells, the telomere lengths were
increased after transduction with plasmid 1. Researchers have
observed that the exogenous expression of hTERT and telomerase
activity led to increased and lengthened telomeres in
hTER-overexpressing HT1080 cells and HeLa cells (23). However, the telomere lengths were
noted with the increasing population doubling per day of a
heterogeneous character, as described in ALT cells (17). The telomeres were lengthened, but
only up to a certain length, which was not further exceeded. This
may be because telomerase-positive cells receive a balance between
telomere erosion and erect extension. This is a negative feedback
by telomere length on telomerase activity, where each individual
telomere is regulated by telomere-binding protein. Thus, a plateau
is reached, which does not lead to infinite telomeres (24–26).
This feedback mechanism controls the number of telomere-binding
protein, and leads to changes in telomere accessibility (17).
The growth behavior of the cells was observed. It
was noted that there was only a slight reduction in cell growth
after telomerase inhibition with plasmids 2, 6 and 7. However,
telomerase inhibition had no significant effect on cell growth.
These results differed from a study of mutants with hTER-transduced
melanoma cells and bladder carcinoma cells. Telomerase inhibition
led to a rapid growth arrest or even apoptosis (16). This may have been due to the lack of
downregulation of telomerase in the telomerase-inhibitory
plasmid-transduced cells. Another explaination for why the
telomerase-inhibitory plasmids resulted in no significant
inhibition of growth could be that the cells were able to quickly
switch to an ALT mechanism. Compared with the controls, the cell
morphology remained unchanged in Barrett’s cells transduced with
the telomerase inhibitors. The cells after transduction with the
telomerase inhibitors were neither senescent nor apoptotic. In
constrast, transduction with telomerase inhibitors in epithelial
keratinocytes resulted in a strong inhibition of growth with
several senescent and apoptotic-looking cells observed (22). This difference could arise from
genetic alterations. In addition to exogenous overexpression of
hTERT, there were no further genetic alterations noted in the
esophageal keratinocytes. Inactivation of the tumor-suppressor p16
was noted in Barrett’s cells, which is an important component for
the activation of replicative senescence (27).
A conceptual model of recombination is that it
occurs in ALT-positive cells by an exchange of telomeric sequences
between sister chromatids or chromosomes, since it was observed
that sister chromatid exchanges in ALT cells occur at a higher
frequency when compared to telomerase-positive cells (28,29).
Since 85% of immortalized and tumor cells are
telomerase-positive, this is the most common telomere maintenance
mechanism (4). This strong presence
in human tumors makes this enzyme an attractive target for the
development of new cancer therapeutics. Several telomerase
inhibitors have been developed to eliminate telomerase in the hope
of telomerase-positive tumor cells undergoing a shortening of
telomeres, or the rupture of the T-loop structure to promote
senescence or apoptosis. Telomerase induces a high proliferation
rate not only in tumor cells, but also in normal tissues, such as
hematopoietic stem and germ line cells. Thus, telomerase has the
attractiveness of the target in question (30). Telomerase and ALT in the same cell
can be active. ALT can be induced even after telomerase inhibition,
reducing the attractiveness of telomerase as a therapeutic target.
This was demonstrated in immortalized esophageal keratinocytes
(31). A switch from telomerase to
an ALT mechanism was also observed in esophageal keratinocytes
after genetic telomerase inhibition (22).
In summary, our findings suggest that an alternative
lengthening of telomere (ALT) mechanism was induced by telomerase
inhibitors in Barrett’s cells. In addition, the telomerase
inhibitors may have high potency in the treatment of Barrett’s
esophagus. However, further studies and analyses are warranted.
References
|
1
|
Harley CB, Futcher AB and Greider CW:
Telomeres shorten during ageing of human fibroblasts. Nature.
345:458–460. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hahn WC and Meyerson M: Telomerase
activation, cellular immortalization and cancer. Ann Med.
33:123–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wright WE and Shay JW: Cellular senescence
as a tumor-protection mechanism: the essential role of counting.
Curr Opin Genet Dev. 11:98–103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim NW, Piatyszek MA, Prowse KR, et al:
Specific association of human telomerase activity with immortal
cells and cancer. Science. 266:2011–2015. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schaetzlein S, Lucas-Hahn A, Lemme E, et
al: Telomere length is reset during early mammalian embryogenesis.
Proc Nat Acad Sci USA. 101:8034–8038. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dahse R, Fiedler W and Ernst G: Telomeres
and telomerase: biological and clinical importance. Clin Chem.
43:708–714. 1997.PubMed/NCBI
|
|
7
|
Allsopp RC, Cheshier S and Weissman IL:
Telomere shortening accompanies increased cell cycle activity
during serial transplantation of hematopoietic stem cells. J Exp
Med. 193:917–924. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Son NH, Murray S, Yanovski J, et al:
Lineage-specific telomere shortening and unaltered capacity for
telomerase expression in human T and B lymphocytes with age. J
Immunol. 165:1191–1196. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Masutomi K, Yu EY, Khurts S, et al:
Telomerase maintains telomere structure in normal human cells.
Cell. 114:241–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayflick L and Moorhead PS: The serial
cultivation of human diploid cell strains. Exp Cell Res.
25:585–621. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hahn WC and Weinberg RA: Rules for making
human tumor cells. N Engl J Med. 347:1593–1603. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Opitz OG, Suliman Y, Hahn WC, et al:
Cyclin D1 overexpression and p53 inactivation immortalize primary
oral keratinocytes by a telomerase-independent mechanism. J Clin
Invest. 108:725–732. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goessel G, Quante M, Hahn WC, et al:
Creating oral squamous cancer cells: a cellular model of
oral-esophageal carcinogenesis. Proc Natl Acad Sci USA.
102:15599–15604. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heeg S, Hirt N, Queisser A, et al: EGFR
overexpression induces activation of telomerase via
PI3K/AKT-mediated phosphorylation and transcriptional regulation
through Hif1-alpha in a cellular model of oral-esophageal
carcinogenesis. Cancer Sci. 102:351–360. 2011. View Article : Google Scholar
|
|
15
|
Lansdorp PM, Verwoerd NP, van de Rijke, et
al: Heterogeneity in telomere length of human chromosomes. Hum Mol
Genet. 5:685–691. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li S, Rosenberg JE, Donjacour AA, et al:
Rapid inhibition of cancer cell growth induced by lentiviral
delivery and expression of mutant-template telomerase RNA and
anti-telomerase short-interfering RNA. Cancer Res. 64:4833–4840.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pickett HA, Cesare AJ, Johnston RL, et al:
Control of telomere length by a trimming mechanism that involves
generation of t-circles. EMBO J. 28:799–809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McEachern MJ and Blackburn EH: Runaway
telomere elongation caused by telomerase RNA gene mutations.
Nature. 376:403–409. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Singer MS and Gottschling DE: TLC1:
template RNA component of Saccharomyces cerevisiae
telomerase. Science. 266:404–409. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu GL, Bradley JD, Attardi LD, et al: In
vivo alteration of telomere sequences and senescence caused by
mutated Tetrahymena telomerase RNAs. Nature. 344:126–132.
1990. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bryan TM, Englezou A, Gupta J, et al:
Telomere elongation in immortal human cells without detectable
telomerase activity. EMBO J. 14:4240–4248. 1995.PubMed/NCBI
|
|
22
|
Döbele M, von Werder A, Fulda C, et al:
Inhibition of telomerase by mutant template telomerase RNA and
anti-telomerase short interfering RNA induces ALT in immortalized
human epithelial cells. Z Gastroenterol. 44:P2812006.PubMed/NCBI
|
|
23
|
Cristofari G and Lingner J: Telomere
length homeostasis requires that telomerase levels are limiting.
EMBO J. 25:565–574. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bianchi A and Shore D: How telomerase
reaches its end: mechanism of telomerase regulation by the
telomeric complex. Mol Cell. 31:153–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smogorzewska A and de Lange T: Regulation
of telomerase by telomeric proteins. Annu Rev Biochem. 73:177–208.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smogorzewska A, van Steensel B, Bianchi A,
et al: Control of human telomere length by TRF1 and TRF2. Mol Cell
Biol. 20:1659–1668. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Collado M, Blasco MA and Serrano M:
Cellular senescence in cancer and aging. Cell. 130:223–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bechter OE, Zou Y, Walker W, et al:
Telomeric recombination in mismatch repair deficient human colon
cancer cells after telomerase inhibition. Cancer Res. 64:3444–3451.
2004. View Article : Google Scholar
|
|
29
|
Londono-Vallejo JA, Der-Sarkissian H,
Cazes L, et al: Alternative lengthening of telomeres is
characterized by high rates of telomeric exchange. Cancer Res.
64:2324–2327. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Broccoli D, Young JW and de Lange T:
Telomerase activity in normal and malignant hematopoietic cells.
Proc Natl Acad Sci USA. 92:9082–9086. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Von Werder A: Immortalized human
esophageal squamous epithelial cells maintain their telomerases by
either telomerase or ALT in a cell cycle-dependent fashion.
http://www.biomedsearch.com/sci/immortalized-human-esophageal-squamous-epithelial/0040149415.html.
2007
|