Introduction
Ovarian cancer is the second more prevalent
gynecologic cancer and the fourth most common cause of death due to
cancer among women (1,2). The standard treatment for ovarian
cancer is surgical intervention followed by combination
chemotherapy. Emerging data suggest that ovarian cancer cells are
initially sensitive to chemotherapeutic drugs due to their genomic
instability, and exhibit a good initial response. However, acquired
resistance has become the most significant clinical problem and is
a main obstacle to successful therapy for ovarian cancer (2–4).
To date, considering DNA repair pathways as the
starting point with which to study tumorigenesis and resistance
with complicated causes has shown promise (5,6). The
Fanconi anemia (FA)/breast cancer susceptibility gene (BRCA)
pathway, a DNA damage repair pathway, mediates proliferation, the
cell cycle, apoptosis and invasiveness of tumor cells (7). FANCF, as an adaptor protein among 14
FA complementation (FANC) groups (FA-A, -B, -C, -D1, -D2, -E, -F,
-G, -I, -J, -L, -M, -N and -P) and one FA-like complementation
group (FA-O), is critically involved in regulating the function of
the FA/BRCA pathway by maintaining the stability of the FA core
complex and ubiquitin activation (monoubiquitination) of the FANCD2
protein (7–9). Epigenetic silencing of FANCF, such as
methylation-induced inactivation of FANCF, plays an
important role in the occurrence of several types of cancer
including ovarian cancer via disruption of the FA/BRCA pathway
(10–12). The disruption of the FA/BRCA pathway
may prevent acquired resistance to DNA cross-linking agents and
improve outcomes for cancer treatment. It has been reported that
silencing of FANCF in resistant myeloma cells with small
interfering RNA (siRNA) reversed resistance to melphalan (13). Taniguchi et al(10) found that FANCF demethylation
resulted in cisplatin (CDDP) resistance in ovarian cancer cells.
The FA pathway has also been reported to be critical in mediating
cellular resistance to temozolomide (TMZ) and
1,3-bis[2-chloroethyl]-1-nitroso-urea (BCNU) (14). Thus, the FA/BRCA pathway, via FANCF,
may represent a new target for preventing drug resistance and
improving cancer treatment.
Adriamycin (ADM) remains the second-line agent for
the treatment of patients with recurrent ovarian cancer after
first-line platinum-based chemotherapy (15,16).
However, the therapeutic effect of ADM has been significantly
influenced by the development of resistance in cancer cells during
treatment (17). Thus, it is
necessary to find new strategies to improve the efficacy of
chemotherapeutic agents and sensitize resistant cancer cells.
Here, we downregulated expression of FANCF by siRNA
in an OVCAR ovarian cancer cell line and evaluated the effects of
decreased FANCF expression on the function of the FA/BRCA pathway
in OVCAR cells and their chemosensitivity to ADM. The results
showed that downregulation of FANCF expression inhibited the
function of the FA pathway in OVCAR cells and enhanced their
susceptibility to ADM. It was also demonstrated that the enhanced
sensitivity to ADM was associated with induction of apoptosis
dependent on Jun N-terminal kinase (JNK) activation. Thus,
interference of FANCF expression may be a new approach to improve
chemosensitivity in the treatment for ovarian cancer.
Materials and methods
Cell culture
The human OVCAR ovarian cancer cell line was
obtained from the American Type Culture Collection. Cells were
maintained in RPMI-1640 (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 mg/ml streptomycin in a humidified atmosphere with 5%
CO2 at 37°C.
Antibodies and reagents
Antibodies against JNK, phospho-JNK, extracellular
signal-regulated kinase (ERK), phospho-ERK, p38, phospho-p38 and
β-actin were from Cell Signaling Technology (Beverly, MA, USA).
Antibodies against FANCF, FANCD2, cytochrome c
(cyt-c), cleaved-caspase-3 and cleaved-poly(ADP-ribose)
polymerase (PARP) were from Abcam, Inc. (Cambridge, MA, USA). ADM,
Annexin V, propidium iodide (PI), low melting point (LMP) and
normal melting point (NMP) agarose, and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The
JNK inhibitor SP600125 was from Calbiochem-Merck (Darmstadt,
Germany).
Design of the siRNA targeting the FANCF
gene and construction of the FANCF shRNA expression vector
According to the siRNA design guidelines, the RNA
interference (RNAi) target sequence to the FANCF gene was
designed (GeneBank accession no. NM022725.3) as follows: forward,
5′-GATCCGCTTCCTGAAGGTGATAGCGTTCAAGAGAC
GCTATCACCTTCAGGAAGTTTTTTGGAAA-3′ and reverse,
5′-AGCTTTTCCAAAAAACTTCCTGAAGGTGAT
AGCGTCTCTTGAACGCTATCACCTTCAGGAAGCG-3′.
FANCF small hairpin siRNA sequences were
synthesized, annealed, and cloned into the pSilencer™ 4.1-CMV
vector to generate the expression vector expressing FANCF shRNA.
After amplication using standard methods, the recombinant plasmid
was extracted and comfirmed by sequencing, then used throughout
this study. A scrambled shRNA with no significant homology to human
gene sequences was used as a negative control to detect nonspecific
effects.
FANCF shRNA transient transfection
Cells were seeded into 6-well plates
(3×105 cells/well) or 100 mm dishes (2×106
cells) and allowed to adhere for 24 h, then transfected with the
pSilencer™ 4.1-CMV control shRNA vector (control shRNA) or
pSilencer™ 4.1-CMV FANCF shRNA vector (FANCF shRNA) using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
instructions. After 4 h, the culture medium was replaced with fresh
media supplemented with 10% FBS, and the cells were harvested at 24
and 48 h after transfection and used for the functional assay. For
the determination of cell proliferation, cell numbers of viable
cells were measured using a cell counter after staining dead cells
with trypan blue.
Western blot analysis
Total protein extracts from cells were obtained
using RIPA lysis buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl,
1% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulphate
(SDS). Prior to cell lysis, 0.1% phenylmethyl sulfonylfluoride
(PMSF) and 1% phosphatase inhibitor were added to the lysis buffer.
After shaking for 20 min on ice, the complex was centrifuged at
12,000 × g for 10 min at 4°C. The supernatants were collected.
Protein quantification was carried out using a BCA kit (Walterson
Biotechnology, Inc., Beijing, China). Samples were boiled in the
presence of sample buffer (20% glycerol, 4% sodium dodecyl sulfate,
10% β-mercaptoethanol, 0.05% bromophenol blue and 1.25 M Tris-HCl,
pH 6.8; all were from Sigma). Thirty micrograms of proteins was
separated by electrophoresis on a 10% SDS-polyacrylamide gel and
transferred to PVDF membranes. Blocking was carried out with 5%
milk in Tris-buffered saline with 0.1% Tween-20 for 2 h at room
temperature. Then the blots were incubated overnight at 4°C with
the appropriate dilution of primary antibodies. After washing with
PBST, the blots were incubated for 1 h with horseradish
peroxidase-conjugated anti-IgG antibody (Santa Cruz Biotechnology,
Inc.). Immunocomplexes were visualized using enhanced
chemiluminescence (ECL) detection reagents (Santa Cruz
Biotechnology, Inc.). The results of the protein expression were
quantitatively analyzed with FluorChem v2.0 software (Alpha
Innotech Corp., USA). The density [integrated density value (IDV)]
of each protein expression band was normalized using the
corresponding β-actin density as an internal control.
Immunodetection of FANCD2 foci
OVCAR ovarian cancer cells were plated on glass
coverslips at 50% confluence, and 16 h later were exposed to
FANCF shRNA or control shRNA. At 24 and 48 h following
exposure, cells were washed with phosphate-buffered saline (PBS),
permeabilized with ice-cold 0.5% Triton X-100 in PBS, and then
fixed with 2% paraformaldehyde and blocked with 5% bovine serum
albumin at room temperature. FANCD2 was detected by incubation with
the anti-FANCD2 antibody (1:500) for 90 min at room temperature and
then with goat anti-rabbit antibody Alexa Fluor 488 (1:1,000;
Invitrogen). All slides were counterstained with DAPI and
visualized by fluorescence microscopy. The experiment was carried
out in triplicate.
Cell viability assay
Cell viability was assessed by MTT assay. Cells were
seeded in 96-well plates at a density of 1×104
cells/well and allowed to grow in the growth medium for 24 h. Cells
were transfected with control or FANCF shRNA for 24 h and
then treated with different concentrations of ADM for 24 h. After
drug treatment, cells were incubated with 5 mg/ml (10 μl) MTT for 4
h at 37°C, then the medium was replaced with 100 μl
dimethylsulfoxide (DMSO) and vortexed for 10 min. The absorbance
(A) was recorded at 492 nm using a microplate reader.
IC50 values were calculated from three independent
experiments.
Flow cytometry
Flow cytometric analysis was performed on a
FACSCalibur (Becton-Dickinson). Twenty four hours after plating
3×105 cells/well in 6-well plates, cells were
transfected with control shRNA or FANCF shRNA. Cells were collected
for the following studies at 24 h after transfection. Determination
of the percentage of apoptotic cells was carried out using
fluorescence isothiocyanate (FITC)-conjugated Annexin V (Annexin
V-FITC) and PI. Cells were collected by centrifugation and washed
twice with cold PBS, and the cell pellet was resuspended in 250 μl
Annexin V-binding buffer at a concentration of 1×106
cells/ml. The suspension (100 μl) was incubated in the dark at room
temperature for 15 min with a solution of Annexin V-FITC (2.5
μg/ml) and PI (5 μg/ml). After addition of 400 μl of binding buffer
to each tube, cells were analyzed for apoptosis by flow
cytometry.
For the measure of the fluorescence intensity of
intracelullar ADM, ADM was added to cells at a final concentration
of 0.1 μg/ml. The cells were incubated for 1, 12 and 24 h at 37°C
in 5% CO2 in the darkness. After the influx step, the
cells were washed with ice-cold PBS. The intracellular fluorescence
intensity of ADM was analyzed according to the fluorescence of ADM
by flow cytometry.
Determination of mitochondrial membrane potential
was carried out using a
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide
(JC-1) kit. Cells treated with or without ADM (0.1 μg/ml) for 24 h
after transfection were incubated for 15 min at 37°C with
mitochondrial membrane potential-sensitive fluorescent dye JC-1 (15
μg/ml) and used to assess changes in the mitochondrial membrane
potential by confocal laser scanning microscopy.
DNA fragmentation assays
The alkaline comet assay was performed according to
the procedure of Singh et al(18,19)
with modifications. A freshly prepared suspension of cells in 0.6%
LMP agarose dissolved in PBS was spread onto microscope slides
precoated with 1% NMP agarose, covered with coverslips and allowed
to set on ice for 10 min. After removing the coverslips, the slides
with cells were then lysed for 1.5 h at 4°C in a cold lysis buffer
consisting of 2.5 M NaCl, 100 mM EDTA, 10 mM Tris (pH 10), and 1%
Triton X-100 was added immediately before use. After lysis, the
slides were placed into an electrophoresis tank, and the DNA was
allowed to unwind for 40 min in the electrophoretic solution
consisting of 300 mM NaOH and 1 mM EDTA (pH >13.0).
Electrophoresis was conducted at 4°C (the temperature of the
running buffer did not exceeded 12°C) for 25 min at 300 mA. The
slides were then transferred to neutralization solution with 0.4 M
Tris-HCl (pH 7.5) for 3×5 min washes, then stained with 2.5 mM PI
and covered with coverslips. To prevent additional DNA damage, all
the steps described above were conducted under dimmed light or in
the dark. Five hundred randomly chosen cells/slide were scanned and
analyzed automatically using Casp1.01 software. Mean tail lengths
were calculated for ~400 cells.
Statistical analysis
Data obtained are representative of the averages of
at least three independent experiments. All data are presented as
means ± SD and analyzed using the one-way ANOVA with post-hoc
analysis. P<0.05 was considered to indicate a statistically
significant result.
Results
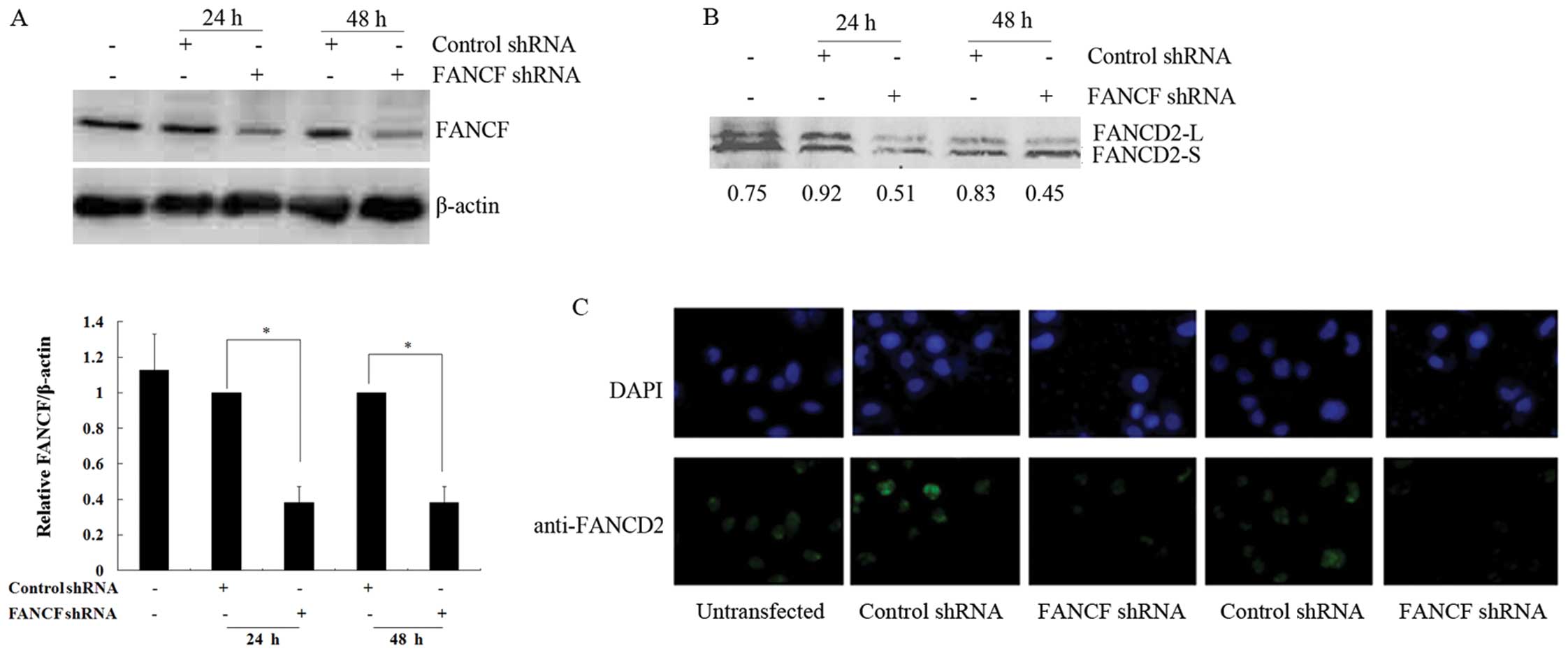
Silencing of FANCF reduces the function
of the FA/BRCA pathway in OVCAR3 ovarian cancer cells
In the present study, shRNA was used to knockdown
FANCF expression in OVCAR3 ovarian cancer cells. The
expression of FANCF protein was evaluated by western blot analysis
in OVCAR3 cells at 24 and 48 h after transfection with FANCF
shRNA. FANCF protein levels decreased to 38.1±9.2 and 38.1±9.1% of
the control at 24 and 48 h after transfection, respectively
(P<0.05) (Fig. 1A). The results
confirmed that FANCF expression was inhibited by transfection with
FANCF shRNA, and there was no obvious difference in the
results of FANCF silencing between the 24- and 48-h
transfection.
The monoubiquitination of FANCD2 is a key step in
activating the FA/BRCA pathway (20). Thus, in order to verify whether
FANCF silencing alters the function of the FA/BRCA pathway,
we detected the changes in FANCD2 monoubiquitination after
FANCF shRNA transfection in OVCAR3 cells by western blot
analysis. The result showed that the ratios of monoubiquitinated
FANCD2 (FANCD2-L)/non-ubiquitinated FANCD2 (FANCD2-S) were
obviously decreased at 24 h (0.51 vs. 0.92) and 48 h (0.45 vs.
0.83) after transfection, when compared with the negative control
(Fig. 1B). The data indicated that
FANCF silencing reduced the monoubiquitination of FANCD2.
Furthermore, we also observed reduced FANCD2 foci in FANCF
shRNA-transfected cells when compared with the negative control at
24 and 48 h after transfection by immunofluorescence (Fig. 1C). These results suggest that
FANCF silencing induces the inactivation of the FA/BRCA
pathway in OVCAR3 ovarian cancer cells.
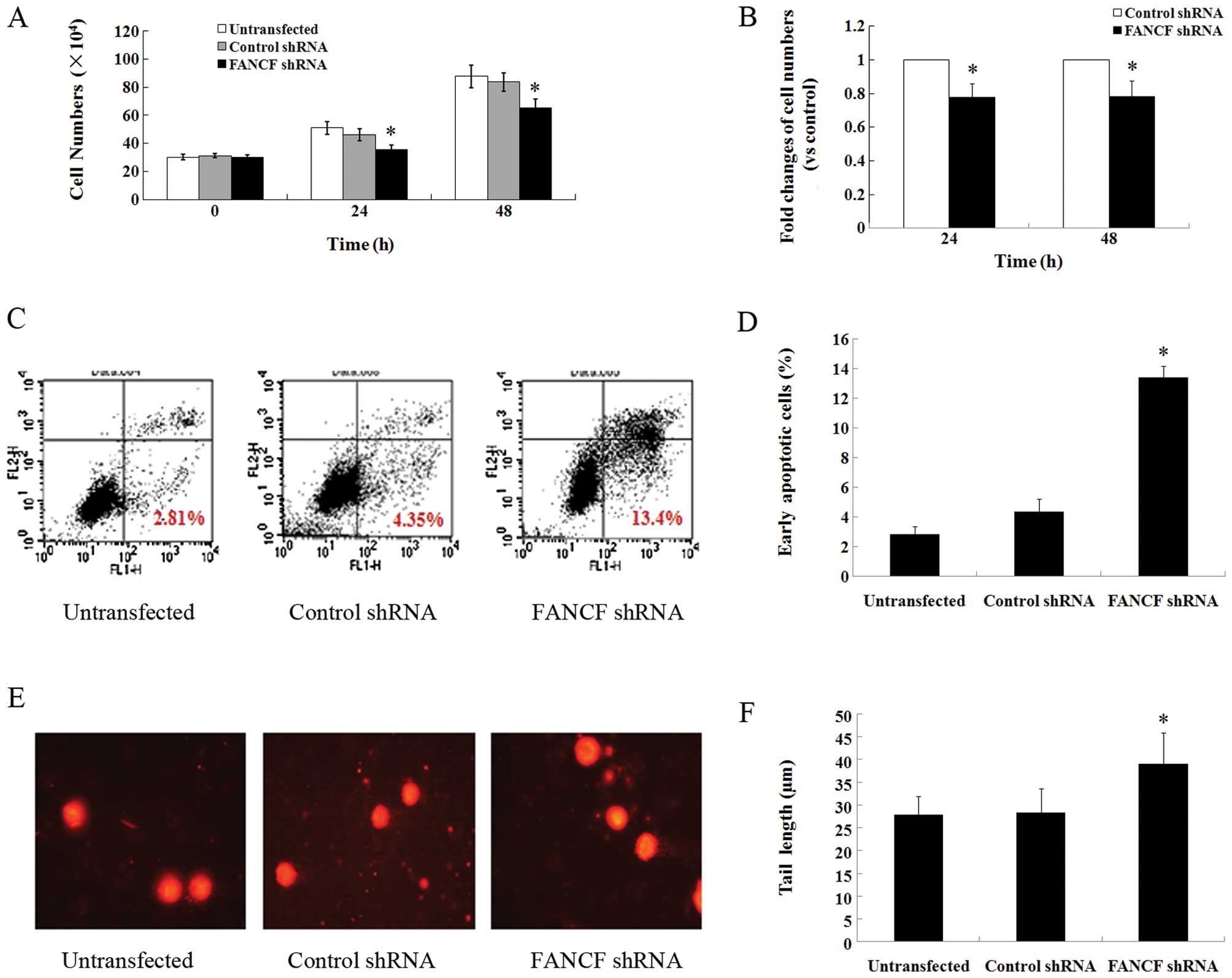
The main function of the FA/BRCA pathway is to
mediate cell proliferation, apoptosis, and DNA damage repair
(7). To further verify the blocked
function of the FA/BRCA pathway by FANCF silencing in OVCAR3
cells, we first measured the effects of FANCF silencing on
cell proliferation. FANCF shRNA decreased the total cell
number to 77.6±8.5% of the control at 24 h and 78.3±9.2% of the
control at 48 h after transfection in OVCAR3 cells (P<0.05)
(Fig. 2A and B). Since treatment
for different hours produced no obvious differences as revealed in
the two above experiments, we chose 24 h as the transfection time
in the subsequent experiments.
Flow cytometric analysis with Annexin V-FITC/PI
staining was carried out to explore the effects of FANCF
silencing on the apoptosis of OVCAR3 cells. The percentage of early
apoptotic cells in the FANCF shRNA-transfected OVCAR3 cells
was significantly higher (13.40±0.778%, P<0.05) than that in the
control cells (4.352±0.843%) at 24 h after transfection (Fig. 2C and D; in the lower right quadrant
of C). In addition, we explored the effects of FANCF
silencing on cellular DNA damage by comet assay, a biomarker of
apoptosis. The FANCF shRNA-transfected OVCAR3 cells were
found to exhibit DNA damage in the form of fragmentation and longer
tail length of the comet (39.1±6.79 μm, P<0.05) compared to the
control cells (28.4±5.17 μm) (Fig. 2E
and F). Taken together, these findings suggest that
FANCF silencing reduces the function of the FA/BRCA pathway
and inhibits proliferation, induces cell apoptosis and DNA damage
in OVCAR3 cells.
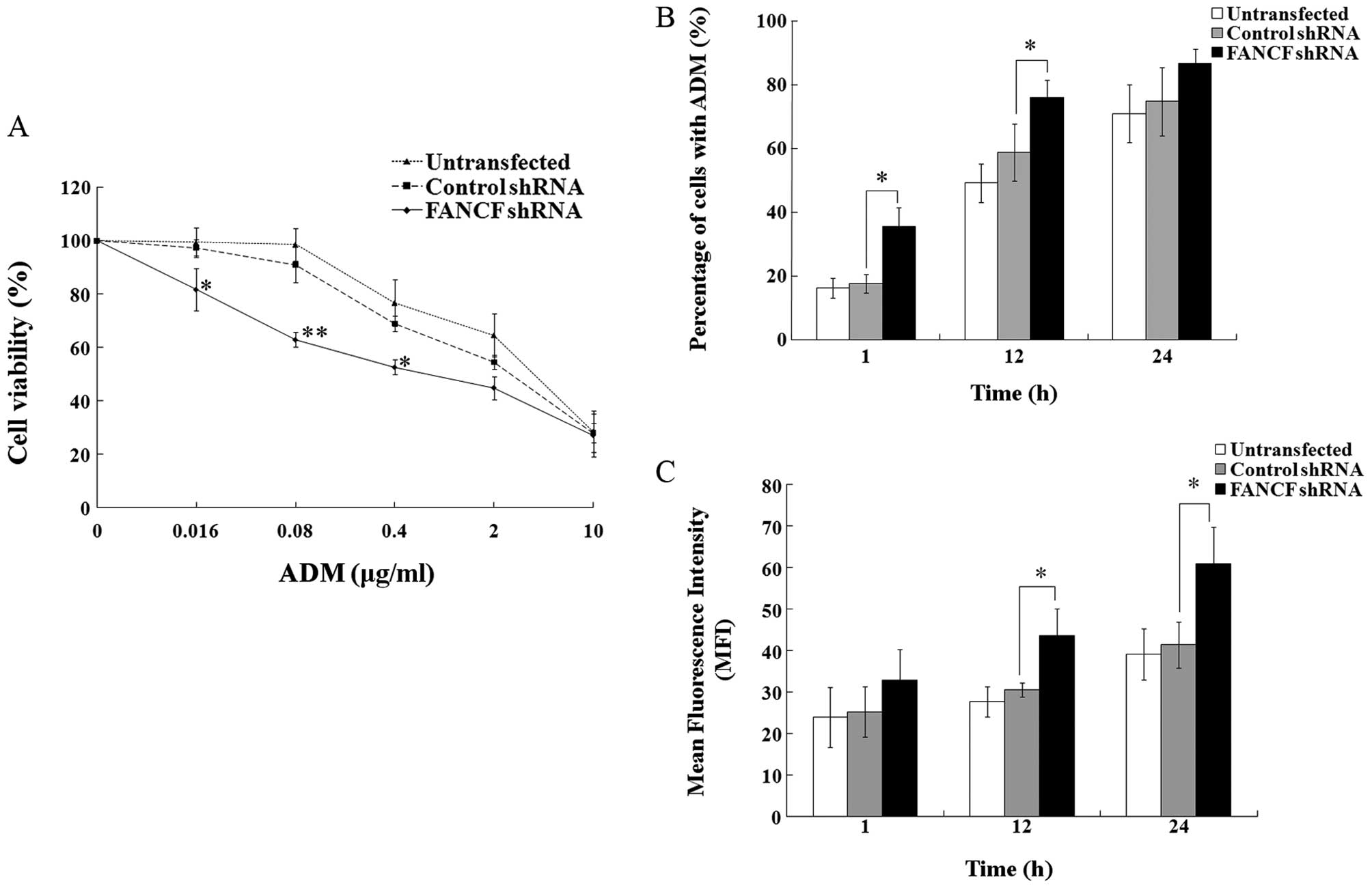
FANCF silencing sensitizes OVCAR3 ovarian
cancer cells to ADM
Since it was found that FANCF silencing
inhibits the function of the FA/BRCA pathway in OVCAR3 cells, we
aimed to ascertain whether FANCF silencing affects the
antitumor effects of ADM on OVCAR3 cells. First, we evaluated the
sensitivity of OVCAR3 cells to ADM by MTT assay. The dose-response
curves showing the relationship between concentrations of ADM and
cell viability revealed that cell viability was obviously decreased
in the FANCF-silenced cells at ADM concentrations of 0.016,
0.08 and 0.4 μg/ml (P<0.05 or P<0.01) (Fig. 3A). The IC50 values
(0.640±0.386 μg/ml) for ADM were markedly decreased after
FANCF silencing, compared with the negative control
(1.760±0.514 μg/ml, P<0.01). The results indicate that silencing
of FANCF significantly enhances the antiproliferative effect
of ADM in the OVCAR3 cells.
We next tested the effects of FANCF silencing
on the intracellular accumulation of ADM in OVCAR3 cells by
determining the percentage of cells containing ADM and the
fluorescence intensity of intracellular ADM by flow cytometry. The
percentages of cells containing ADM were obviously increased in the
FANCF-silenced cells compared with the percentage in the
control cells following treatment of ADM (0.1 μg/ml) for 1 h
(35.52±5.93 vs. 17.58±3.01%, P<0.05) and 12 h (75.89±5.62 vs.
58.79±8.92%, P<0.05) (Fig. 3B).
When compared with the control cells, the mean fluorescence
intensity (MFI) was markedly increased in the FANCF-silenced
cells following treatment of ADM (0.1 μg/ml) for 12 h (43.60±6.48
vs. 30.54±1.73, P<0.05) and 24 h (60.89±8.96 vs. 41.37±5.55,
P<0.05) (Fig. 3C). These data
further suggest that FANCF silencing sensitizes OVCAR3
ovarian cancer cells to ADM through increased ADM intracellular
accumulation. Considering the greater accumulation of ADM following
the treatment of ADM for 24 h, we treated the OVCAR3 cells with ADM
for 24 h in the subsequent experiments.
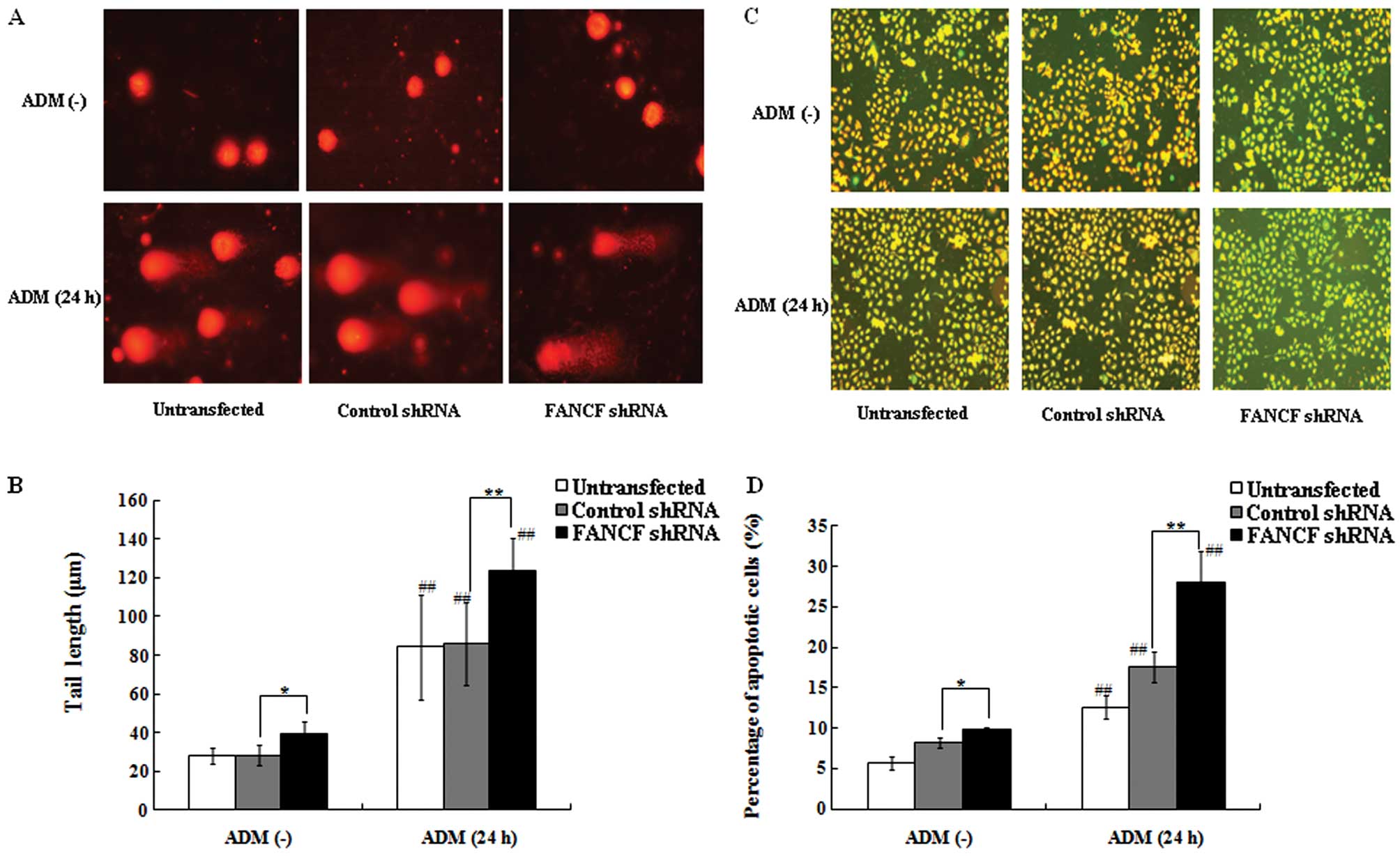
FANCF silencing increases ADM-induced
apoptosis via JNK activation
Since FANCF silencing enhanced the
antiproliferative effect of ADM in OVCAR3 cells, we hypothesized
that FANCF silencing alters ADM-induced DNA damage, the main
cytotoxic effect of ADM. Using comet assay again, we found that
FANCF-silenced OVCAR3 cells and the control cells following
treatment with ADM (0.1 μg/ml, 24 h) exhibited extensive DNA damage
reflected by the tail length of the comet when compared with DNA
damage in the cells without ADM treatment (P<0.01) (Fig. 4A and B). In addition, the
FANCF-silenced cells were found to have increased DNA damage
as evident from fragmentation and the longer tail length of the
comet (123.46±17.35 μm) compared with the control cells
(85.91±21.59 μm, P<0.01) following treatment of ADM (0.1 μg/ml,
24 h). These findings suggest that FANCF silencing increases
the ADM-induced cellular DNA damage.
Decreased mitochondrial membrane potential (MMP) is
a marker of early apoptosis and one of the reasons for DNA damage.
We evaluated whether FANCF silencing increases the
ADM-induced cellular DNA damage via decreased MMP in OVCAR3 cells
by flow cytometry with JC-1 staining, a lipophilic and cationic
dye. In normal cells, JC-1 concentrates in the mitochondrial
matrix, where it forms red fluorescent aggregates (J-aggregates).
In apoptotic cells with decreased MMP, JC-1 stays in the cytoplasm
as monomers and fluoresces green (21). Without ADM treatment, there was an
obvious increase in the percentage of apoptotic cells that emitted
only green fluorescence, representing cells with depolarized
mitochondrial membranes, in the FANCF-silenced cells
(9.91±0.29%) when compared with the control cells (8.17±0.64%,
P<0.05) (Fig. 4C and D). This
result was consistent with the above findings by Annexin V-FITC/PI
assay. Moreover, it was found that the treatment of ADM (0.1 μg/ml,
24 h) significantly increased the percentage of apoptotic cells,
particularly in the FANCF-silenced cells (28.06±3.85%) when
compared with the control cells (17.58±1.85%, P<0.01). These
results indicate that FANCF silencing sensitizes OVCAR3
cells to ADM-induced apoptosis.
Activation of the mitogen-activated protein kinase
(MAPK) pathway mediates ADM-induced cell apoptosis in multiple
cancer cell lines (22). Using
western blot analysis, we examined protein expression of genes
related to the MAPK pathway, including JNK, ERK, and p38, in OVCAR3
cells following the treatment of ADM (0.1 μg/ml, 24 h) after
FANCF silencing. These results showed that the expression of
JNK and its phosphorylation level, but not the expression of ER and
p38 proteins or their phosphorylation levels, was increased in
OVCAR3 cells following the treatment of ADM (0.1 μg/ml, 24 h)
compared with cells without ADM treatment. It was also found that
only FANCF silencing without ADM treatment did not alter the
expression of JNK, ERK, p38 proteins or their phosphorylation
levels in OVCAR3 cells, indicating that FANCF silencing in
OVCAR3 cells did not activate the MAPK pathway. However,
FANCF silencing with ADM treatment (0.1 μg/ml, 24 h) notably
increased the expression of JNK and its phosphorylation level
compared with control cells (Fig. 5A
and B). These results demonstrated that FANCF silencing
increases ADM-induced JNK activation.
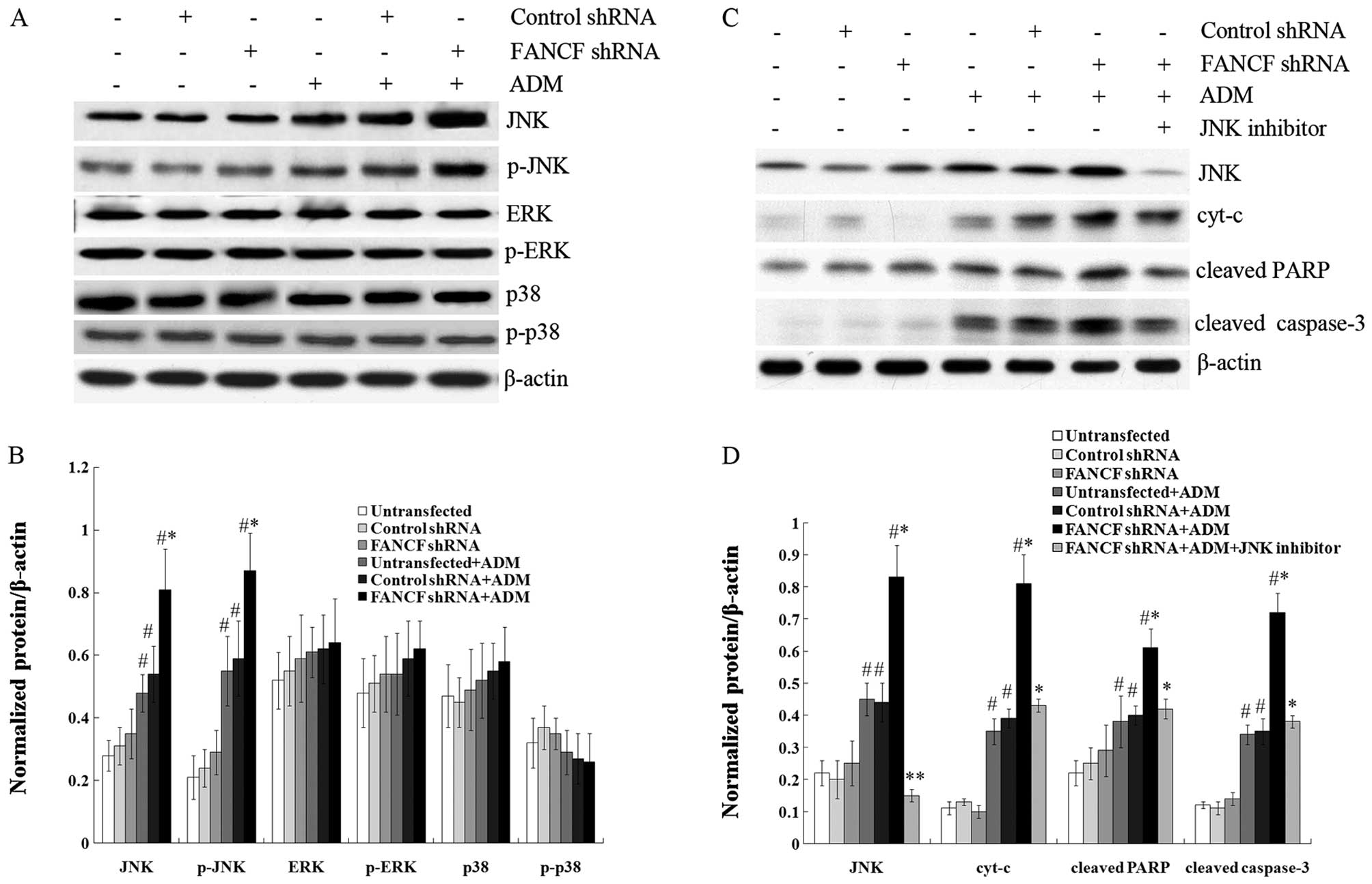 | Figure 5FANCF silencing increases
ADM-induced apoptosis via JNK activation in OVCAR3 ovarian cancer
cells. (A) Representative image of a western blot showing the
ADM-induced changes in protein expression of JNK, p-JNK, ERK,
p-ERK, p38 and p-p38 in OVCAR3 cells after FANCF silencing.
(B) Protein quantification was carried out by densitometric
analysis. Normalized proteins of JNK, p-JNK, ERK, p-ERK, p38 and
p-p38 were relative to the internal control β-actin. (C)
Representative image of a western blot indicating the ADM-induced
changes in protein expression of JNK, cyt-c, cleaved caspase-3 and
cleaved PARP in OVCAR3 cells after FANCF silencing and the
treatment of JNK inhibitor SP600125. (D) Protein quantification was
carried out by densitometric analysis. Normalized proteins of JNK,
cyt-c, cleaved caspase-3 and cleaved PARP were relative to the
internal control β-actin. *P<0.05,
**P<0.01, FANCF shRNA-transfected cells
compared with control shRNA-transfected cells, or FANCF
shRNA-transfected cells with the treatment of JNK inhibitor
compared with FANCF shRNA-transfected cells without
treatment of JNK inhibitor. #P<0.05, cells with
treatment of ADM compared with cells without ADM treatment. ADM,
adriamycin. |
We also found that treatment with ADM (0.1 μg/ml, 24
h) significantly increased the expression of cyt-c from the
release of mitochondria, cleaved caspase-3, and PARP in OVCAR3
cells by western blot analysis. Furthermore, FANCF-silenced
OVCAR3 cells notably exhibited increased expression of these
proteins induced by ADM. It was also shown that the increase in
expression of these proteins was blocked by the JNK inhibitor
SP600125 (Fig. 5C and D). These
results indicate that FANCF silencing increases ADM-induced
apoptosis via JNK activation.
Discussion
FANCF protein is an important adaptor protein
involved in the stabilizing component of a larger FA complex and
maintains the biological functions of the FA/BRCA pathway (7–9).
FANCD2 is expressed in normal human cells as two isoforms: FANCD2-S
and FANCD2-L. DNA cross-linking agents, such as CDDP and mitomyclin
C (MMC), and ionizing radiation (IR) can activate the conversion of
FANCD2-S to FANCD2-L. The activated FANCD2 protein accumulates in
nuclear foci in response to DNA-damaging agents and colocalizes
with BRCA1. Central to the FA/BRCA pathway is the
monoubiquitination of FANCD2, which connects upstream signaling
with downstream enzymatic repair steps and activates the function
of this pathway (23–25). Thus, the monoubiquitination and
focus formation of FANCD2 are surrogate markers for FA/BRCA pathway
activation (24,25). In the present study, we silenced the
FANCF gene by RNA interference in OVCAR3 ovarian cancer
cells and found decreased expression of FANCF protein, ratio of
FANCD2-L/FANCD2-S and FANCD2 foci, which suggests inactivation of
the FA/BRCA pathway by FANCF silencing in OVCAR3 ovarian
cancer cells.
The main functions of the FA/BRCA pathway involve
the cell cycle, DNA damage and repair, apoptosis, gene
transcription and gene stability. Moreover, this pathway is
necessary for cells to respond to DNA damage caused by IR,
mitoxantrone (MX), CDDP and ADM (26,27).
In the present study, we found that FANCF silencing
inhibited proliferation, induced cell apoptosis and DNA damage in
OVCAR3 cells, indicating that the function of the FA/BRCA pathway
was blocked. Previous studies have reported that changes in the
function of the FA/BRCA pathway affects the sensitivity of cancer
cells to DNA-damaging agents (13,14).
We also found that FANCF silencing increased the sensitivity
of OVCAR3 ovarian cancer cells to ADM. To the best of our
knowledge, this is the first evidence that blockage of the function
of the FA/BRCA pathway by FANCF silencing in ovarian cancer
cells increases the sensitivity of cancer cells to ADM. Although it
has been reported that the TOV-21G cells absent of FANCF function
with FANCF cDNA are resistant to MMC and CDDP (10), our study is the first to investigate
the sensitivity of OVCAR3 ovarian cancer cells to another
therapeutic drug ADM through loss of function of FANCF.
Following assessment of the cell viability at
different concentrations of ADM, we found that FANCF
silencing significantly decreased the IC50 values of ADM
in OVCAR3 cells, suggesting the increased sensitivity of OVCAR3
cells to ADM. This enhanced antiproliferative effect of ADM in
OVCAR3 cells by FANCF silencing was more obvious at
concentrations from 0.08 to 0.4 μg/ml. Therefore, we selected the
concentration of 0.1 μg/ml between the two concentrations to treat
cells in subsequent experiments. Furthermore, we demonstrated that
FANCF silencing increased the intracellular accumulation of
ADM by measuring the percentage of cells containing ADM and the
fluorescence intensity of intracellular ADM by flow cytometry and
determined that it was one of the reasons for the increased
sensitivity of OVCAR3 cells to ADM. Since the effect of the
increased intracellular accumulation of ADM was more pronounced
after treatment of ADM for 24 h, we chose this condition in
subsequent experiments.
ADM is an active agent used for the treatment of
patients with ovarian cancer (15,16).
The main molecular target of ADM cytotoxicity is topoisomerase II
that catalyzes a change in DNA topology via a concerted mechanism
of transient DNA strand cleavage and religation. ADM can stabilize
a transient DNA-topoisomerase II complex in which DNA strands are
cut and covalently linked to the enzyme subunits. The stabilized
complex results in DNA damage that is associated with the cytotoxic
effect of ADM (28). In the present
study, we found that ADM (0.1 μg/ml, 24 h) induced DNA damage of
OVCAR3 cells through comet assay, and FANCF silencing
increased the ADM-induced cellular DNA damage. One possible reason
is that the function of the FA/BRCA pathway, in terms of DNA damage
repair, was disrupted by FANCF interference in OVCAR3 cells,
resulting in the decreased repair function of ADM-induced DNA
damage.
Decreased MMP is one of the reasons for DNA damage.
Many studies have shown that ADM accumulates in both the cellular
nucleus and mitochondria and interferes with mitochondrial function
and initiates the pathway of apoptosis by reducing MMP and
releasing cyt-c(29–31). We observed that ADM induced a
decrease in MMP and cell apoptosis, and FANCF silencing
increased ADM-induced cell apoptosis in OVCAR3 cells. Activation of
the MAPK pathway has been known to mediate ADM-induced cell
apoptosis in multiple cancer cell lines (22). In the present study, treatment with
ADM (0.1 μg/ml, 24 h) increased the expression of JNK and its
phosphorylation level, induced the release of cyt-c, and
increased the expression of cleaved caspase-3 and PARP dependent on
JNK activation in OVCAR3 cells. These results were consistent with
previous reports (32,33). Caspase-3 is an important executioner
of apoptosis among the caspase family members. One of the
substrates of caspase-3, PARP, a DNA repair enzyme, when cleaved,
is inactivated and unable to repair DNA breaks or fragmentation,
and plays a critical role in early apoptosis. The release of
mitochondrial cyt-c is an important event in the apoptotic
process and caspase-3 activation (34–36).
Our data demonstrated that FANCF silencing increased the
expression of ADM-induced cleaved caspase-3, cleaved PARP and
cyt-c dependent on JNK activation, leading to cell apoptosis
in OVCAR3 cells.
In conclusion, our findings demonstrated that
FANCF silencing-induced dysfunction of the FA/BRCA pathway
increased the sensitivity of the human ovarian cancer cell line,
OVCAR, to ADM, by increased cell apoptosis dependent on JNK
activation. We propose that FANCF may represent a novel target for
enhancing the response of ovarian cancer cells to ADM, thus
improving ovarian cancer treatment.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 30873097 and
81102472). This study was also supported by the Specialized
Research Fund for the Doctoral Program of Higher Education (no.
20092104110020) and the Science and Technology Project of Shenyang
(no. F11-264-1-19).
References
|
1
|
Permuth-Wey J and Sellers TA: Epidemiology
of ovarian cancer. Methods Mol Biol. 472:413–437. 2009. View Article : Google Scholar
|
|
2
|
Marchetti C, Pisano C, Facchini G, et al:
First-line treatment of advanced ovarian cancer: current research
and perspectives. Expert Rev Anticancer Ther. 10:47–60. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orth K, Hung J, Gazdar A, et al: Genetic
instability in human ovarian cancer cell lines. Proc Natl Acad Sci
USA. 91:9495–9499. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsuo K, Eno ML, Im DD, et al: Clinical
relevance of extent of extreme drug resistance in epithelial
ovarian carcinoma. Gynecol Oncol. 116:61–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jacquemont C, Simon JA, D’Andrea AD and
Taniguchi T: Non-specific chemical inhibition of the Fanconi anemia
pathway sensitizes cancer cells to cisplatin. Mol Cancer.
11:262012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hucl T and Gallmeier E: DNA repair:
exploiting the Fanconi anemia pathway as a potential therapeutic
target. Physiol Res. 60:453–465. 2011.PubMed/NCBI
|
|
7
|
Su X and Huang J: The Fanconi anemia
pathway and DNA interstrand cross-link repair. Protein Cell.
2:704–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leveille F, Blom E, Medhurst AL, et al:
The Fanconi anemia gene product FANCF is a flexible adaptor
protein. J Biol Chem. 279:39421–39430. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kowal P, Gurtan AM, Stuckert P, et al:
Structural determinants of human FANCF protein that function in the
assembly of a DNA damage signaling complex. J Biol Chem.
282:2047–2055. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taniguchi T, Tischkowitz M, Ameziane N, et
al: Disruption of the Fanconi anemia-BRCA pathway in
cisplatin-sensitive ovarian tumors. Nat Med. 9:568–574. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lim SL, Smith P, Syed N, et al: Promoter
hypermethylation of FANCF and outcome in advanced ovarian cancer.
Br J Cancer. 98:1452–1456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Li M, Lu S, et al: Promoter
hypermethylation of FANCF plays an important role in the occurrence
of ovarian cancer through disrupting Fanconi anemia-BRCA pathway.
Cancer Biol Ther. 5:256–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Q, Van der Sluis PC, Boulware D, et
al: The FA/BRCA pathway is involved in melphalan-induced DNA
interstrand cross-link repair and accounts for melphalan resistance
in multiple myeloma cells. Blood. 106:698–705. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen CC, Taniguchi T and D’Andrea A: The
Fanconi anemia (FA) pathway confers glioma resistance to DNA
alkylating agents. J Mol Med. 85:497–509. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valerio MR, Tagliaferrri P, Raspagliesi F,
et al: A phase II study of pegylated liposomal doxorubicin
oxaliplatin and cyclophosphamide as second-line treatment in
relapsed ovarian carcinoma. Int J Gynecol Cancer. 16:79–85. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sehouli J, Camara O, Schmidt M, et al:
Pegylated liposomal doxorubicin (CAELYX) in patients with advanced
ovarian cancer: results of a German multicenter observational
study. Cancer Chemother Pharmacol. 64:585–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lei T, Srinivasan S, Tang Y, et al:
Comparing cellular uptake and cytotoxicity of targeted drug
carriers in cancer cell lines with different drug resistance
mechanisms. Nanomedicine. 7:324–332. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar
|
|
19
|
Klaude M, Eriksson S, Nygren J and
Ahnström G: The comet assay: mechanisms and technical
considerations. Mutat Res. 363:89–96. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gordon SM, Alon N and Buchwald M: FANCC,
FANCE, and FANCD2 form a ternary complex essential to the integrity
of the Fanconi anemia DNA damage response pathway. J Biol Chem.
280:36118–36125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Liu Y, Fu Y, et al: The triggering
of apoptosis in macrophages by pristine graphene through the MAPK
and TGF-beta signaling pathways. Biomaterials. 33:402–411. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ji C, Yang B, Yang YL, et al: Exogenous
cell-permeable C6 ceramide sensitizes multiple cancer cell lines to
doxorubicin-induced apoptosis by promoting AMPK activation and
mTORC1 inhibition. Oncogene. 29:6557–6568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
D’Andrea AD: The Fanconi anemia/BRCA
signaling pathway: disruption in cisplatin-sensitive ovarian
cancers. Cell Cycle. 2:290–292. 2003.PubMed/NCBI
|
|
24
|
Kim H and D’Andrea AD: Regulation of DNA
cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev.
26:1393–1408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Garcia-Higuera I, Taniguchi T, Ganesan S,
et al: Interaction of the Fanconi anemia proteins and BRCA1 in a
common pathway. Mol Cell. 7:249–262. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bogliolo M, Lyakhovich A, Callén E,
Castellà M, et al: Histone H2AX and Fanconi anemia FANCD2 function
in the same pathway to maintain chromosome stability. EMBO J.
26:1340–1351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Joenje H and Patel KJ: The emerging
genetic and molecular basis of Fanconi anaemia. Nat Rev Genet.
2:446–457. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Burden DA and Osheroff N: Mechanism of
action of topoisomerase II and drugs targeted to the enzyme.
Biochim Biophys Acta. 1400:139–154. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clementi ME, Giardina B, Di Stasio E, et
al: Doxorubicin-derived metabolites induce release of cytochrome C
and inhibition of respiration on cardiac isolated mitochondria.
Anticancer Res. 23:2445–2450. 2003.PubMed/NCBI
|
|
30
|
Green PS and Leeuwenburgh C: Mitochondrial
dysfunction is an early indicator of doxorubicin-induced apoptosis.
Biochim Biophys Acta. 1588:94–101. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bar-Joseph H, Ben-Aharon I, Rizel S, et
al: Doxorubicin-induced apoptosis in germinal vesicle (GV) oocytes.
Reprod Toxicol. 30:566–572. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Naci D, El Azreq MA, Chetoui N, et al:
α2β1 integrin promotes chemoresistance against doxorubicin in
cancer cells through extracellular signal-regulated kinase (ERK). J
Biol Chem. 287:17065–17076. 2012.
|
|
33
|
Aroui S, Mili D, Brahim S, et al:
Doxorubicin coupled to penetratin promotes apoptosis in CHO cells
by a mechanism involving c-Jun NH2-terminal kinase. Biochem Biophys
Res Commun. 396:908–914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Budihardjo I, Oliver H, Lutter M, et al:
Biochemical pathways of caspase activation during apoptosis. Annu
Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Velagapudi C, Bhandari BS, Abboud-Werner
S, et al: The tuberin/mTOR pathway promotes apoptosis of tubular
epithelial cells in diabetes. J Am Soc Nephrol. 22:262–273. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li M, Wang AJ and Xu JX: Redox state of
cytochrome c regulates cellular ROS and caspase cascade in
permeablized cell model. Protein Pept Lett. 15:200–205. 2008.
View Article : Google Scholar : PubMed/NCBI
|